

Abstract
The extracellular matrix (ECM) in skeletal muscle plays an integral role in tissue development, structural support, and force transmission. For successful adaptation to mechanical loading, remodeling processes must occur. In a large cohort of older adults, transcriptomics revealed that genes involved in ECM remodeling, including matrix metalloproteinase 14 (MMP14), were the most upregulated following 14 weeks of progressive resistance exercise training (PRT). Using single‐cell RNA‐seq, we identified macrophages as a source of Mmp14 in muscle following a hypertrophic exercise stimulus in mice. In vitro contractile activity in myotubes revealed that the gene encoding cytokine leukemia inhibitory factor (LIF) is robustly upregulated and can stimulate Mmp14 expression in macrophages. Functional experiments confirmed that modulation of this muscle cell‐macrophage axis facilitated Type I collagen turnover. Finally, changes in LIF expression were significantly correlated with MMP14 expression in humans following 14 weeks of PRT. Our experiments reveal a mechanism whereby muscle fibers influence macrophage behavior to promote ECM remodeling in response to mechanical loading.
Keywords: extracellular matrix remodeling, macrophage, muscle hypertrophy, scRNA‐seq
Abbreviations
- BMDM
bone marrow‐derived macrophages
- CM
conditioned media
- CSA
cross‐sectional area
- DAPI
4′,6‐diamidino‐2‐phenylindole
- DEG
differentially expressed gene
- DM
differentiation media
- ECM
extracellular matrix
- EPS
electrical pulse stimulation
- FACS
fluorescent activated cell sorting
- FAP
fibrogenic‐adipogenic progenitor
- FBS
fetal bovine serum
- IHC
immunohistochemistry
- LIF
leukemia inhibitory factor
- MMP
matrix metalloproteinase
- MOV
mechanical overload
- NHS
normal horse serum
- PBS
phosphate‐buffered saline
- PFA
paraformaldehyde
- PRT
progressive resistance exercise training
- scRNA‐seq
single‐cell RNA‐seq
1. INTRODUCTION
Macrophage recruitment is necessary for skeletal muscle hypertrophy in mice following mechanical overload (MOV). 1 Our laboratory as well as others have recently reported a significant increase in skeletal muscle macrophage number following exercise training in middle‐aged and older adults using pan (CD11b/CD68) and anti‐inflammatory (MRC1/CD163) macrophage markers. 2 , 3 , 4 Macrophages produce factors that may directly promote growth, such as insulin‐like growth factor‐1, 5 but also factors such as transforming growth factor beta‐1 that regulate extracellular matrix (ECM) production. 6 Although extensive investigations have characterized the role of macrophages in response to muscle injury, 7 to date, the potential relationship between macrophages and ECM remodeling has not been explored in the setting of load‐induced hypertrophy in adult muscle.
The ECM in skeletal muscle is dominated by type I and III collagens, which are fibrous proteins that converge with a specialized basement membrane composed mainly of type IV collagen, laminin, and fibronectin surrounding each muscle fiber. 8 The degradation of ECM is mediated primarily by the production of matrix metalloproteinases (MMPs) and cathepsins. In a cohort of older adults who participated in 14 weeks of progressive resistance exercise training (PRT), ECM remodeling pathways were the most enriched following training. 9 Genes of interest included matrix metalloproteinase 14 (MMP14), a potent membrane‐tethered collagenase that can cleave intact collagen fibrils, which has been previously shown to correlate with changes in macrophage number following exercise training. 2 , 10
Although very little is known about macrophage‐specific MMP expression in skeletal muscle, upstream regulators of MMP14 expression include leukemia inhibitory factor (LIF) and interleukin 6 (IL6), both of which are expressed during skeletal muscle adaptations to exercise, as well as regeneration. 11 , 12 , 13 , 14 In other cell types, both cytokines induce a similar signaling network that involves the targeted degradation of tumor protein p53 (p53), a known repressor of MMP14 expression. 15 The degradation of p53 enables specificity protein 1 (Sp1) to access the promoter region of MMP14, increasing MMP14 expression. Transgene overexpression of LIF in macrophages has been shown to reduce type I collagen content in mdx mice compared to wild‐type mdx control animals; however, gene or protein expression levels of Mmp14/Mmp14 were not reported. 14 Using single‐cell RNA‐sequencing (scRNA‐seq), we hypothesized that MOV would induce a transcriptome signature of ECM remodeling in macrophages. We further hypothesized that MOV would induce the expression of LIF by skeletal muscle fibers, driving MMP14 expression in macrophages, thereby promoting the degradation of collagen. In the present study, we used several in vivo and in vitro analyses to test these hypotheses in both human and mouse muscles. Our findings highlight an underappreciated and understudied crosstalk between skeletal muscle fibers and macrophages that may be important for ECM remodeling to facilitate myofiber hypertrophy.
2. METHODS
2.1. Human subjects and intervention
Data were acquired from the MASTERS trial (NCT02308228), which was conducted to determine whether metformin could augment the response to 14‐weeks of PRT in healthy older men and women, age >65 years old. Full details of the study protocol, including participant inclusion/exclusion criteria and the PRT paradigm designed to optimize muscle mass and strength gains, can be found elsewhere. 16 The trial was conducted at the University of Kentucky and the University of Alabama at Birmingham, and all participants signed an informed consent approved by university IRBs prior to study enrollment. Thirty‐one participants from the placebo‐only group were included in this analysis. Muscle biopsies from the vastus lateralis were collected two weeks before the 14‐week PRT program began and then a final biopsy 3 days after their last bout of exercise.
2.2. Muscle biopsies
For all MASTERS participants, muscle biopsies were obtained from the vastus lateralis muscle, under local anesthetics (1% lidocaine), using the percutaneous needle biopsy technique of Bergström, with 5–6 mm needles and manual suction. For RNA extraction, 35 mg of muscle tissue was snap‐frozen in liquid nitrogen and stored at −80°C. For immunohistochemical analysis, approximately 50–100 mg of tissue was embedded in tragacanth gum on cork, frozen in liquid nitrogen‐cooled isopentane, and stored at −80°C.
2.3. Animals
Mice from a Pax7CreER/+‐tdTomatofl/+ strain were used for all scRNA‐seq in vivo studies as part of a study exploring the role of satellite cell communication during MOV. 17 Experiments and animal care were performed in accordance with the University of Kentucky Institutional Animal Care and Use Committee. All mice were housed in a temperature‐ and humidity‐controlled room and maintained on a 14:10 light:dark cycle, with standard chow and water ad libitum. All mice were females >4 months old at the time of experimentation.
2.4. Synergist ablation surgery‐induced MOV of muscle
Mice underwent bilateral synergist ablation surgery to induce hypertrophy of the plantaris muscle, as previously described by our laboratory. 17 Briefly, mice were anesthetized using 95% oxygen and 5% isoflurane gas, then approximately 1/3 of the lower gastrocnemius/soleus complex was removed, careful not to disturb neural or vascular supply. Sham mice were also anesthetized, and a similar incision was made followed by an immediate suture. Following 4 days of MOV (4DMOV), mice were euthanized via lethal dosage of sodium pentobarbital and cervical dislocation.
2.5. Cell culture
Human myogenic progenitor cells were isolated from the gracilis of two patients (1 female and 1 male) undergoing anterior cruciate ligament reconstruction (age 30–34 years) using a human anti‐CD56 (1:20, Cat# 355503, Biolegend, San Diego, CA, USA) antibody for fluorescent activated cell sorting (FACS). Cells were passaged 2–3 times in growth media consisting of Ham's F‐10 (ThermoFisher, Waltham, MA, USA), 20% fetal bovine serum (FBS, Atlanta Biological, Minneapolis, MN, USA), 1% penicillin/streptomycin, and 10 ng/ml basic fibroblast growth factor (bFGF, Peprotech, Rocky Hill, NJ, USA). Cells were differentiated into myotubes for 5 days using MyoCult differentiation media (DM, Stemcell Technologies, Vancouver, Canada). On the fifth day, fresh media was added just prior to electrical pulse stimulation (E). Cells were then either stimulated at 12 V, 1 Hz, 2 ms for 24 h (IonOptix C‐Pace EP, Westwood, MA, USA) or had electrodes placed in wells with no stimulation (E(+) or E(−)), followed by immediate RNA extraction using Qiazol (Qiagen, Hilden, Germany). Conditioned media (CM) was pooled from each 6‐well plate and centrifuged at 600 g for 10 min to remove debris and frozen at −80°C.
Bone marrow‐derived macrophages (BMDM) were obtained from bone marrow (BM) precursor cells as previously described. 18 Briefly, total BM was obtained from mice by flushing femur BM with Dulbecco's modified eagle's media (DMEM, ThermoFisher). Cells were cultured in DMEM medium with 20% FBS and 10 ng/ml colony‐stimulating factor‐ 1 (CSF‐1, Stemcell Technologies) for 6 days; CSF‐1 free media was added on day 7 for 12 h. CM from myotubes was incubated with goat anti‐human LIF neutralizing antibody (0.1 mg/ml, ThermoFisher) or control IgG overnight at 4°C with gentle agitation and then diluted 1:1 with Myocult DM. The CM was added on day 7 for 24 h. Cells were then immediately collected in Qiazol (Qiagen) for RNA extraction.
2.6. DQ type I collagen assay
The fluorogenic DQ collagen was used to directly monitor collagenase activity in vitro. Briefly, myotube CM from either E(−) or E(+) was added to BMDM seeded in 12‐well plates. 100 μg/ml of DQ type I collagen (ThermoFisher) was added to 5 different conditions; DM only (control), 1:1 CM/DM from E(−) and E(+) myotubes with no BMDM, and 1:1 CM/DM from E(−) and E(+) myotubes with BMDM. Following 120 min, 200 ul was removed from each well and added to a 96‐well flat‐bottom plate. To measure the fluorescence intensity, a fluorescence microplate reader equipped with standard fluorescein filters was used. Digestion products from the DQ collagen were measured at absorption maxima of ~495 nm and fluorescence emission maxima of ~515 nm and corrected for background by subtracting the control from the value derived.
2.7. Immunohistochemistry
Human skeletal muscle macrophage identification and quantification were performed according to a detailed and validated method. 19 Briefly, macrophages were identified by stepwise incubation with antibodies against the pan macrophage marker CD11b (Cat# MON1019‐1, Cell Sciences, Newburyport, MA, USA) followed by CD206 (Cat# AF2535, R&D Systems, Minneapolis, MN, USA). Slides were fixed in −20°C acetone and blocking steps were taken to prevent nonspecific background and cross‐reactivity between amplification reagents. Slides were incubated with mouse IgG1 anti‐CD11b (1:100, Cell Sciences) in 2.5% normal horse serum (NHS, Vector Laboratories, Burlingame, CA, USA) overnight at 4°C. On the second day, the sections were rinsed in 1× phosphate‐buffered saline (PBS), followed by incubation with goat anti‐mouse biotin secondary (1:1000, Jackson ImmunoResearch, West Grove, PA, USA) for 90 min at room temperature, followed by streptavidin‐HRP (ThermoFisher) for 1 h and then tyramide signal amplification with Alexa Fluor 488 (ThermoFisher) for 15 min. Sections were placed back in streptavidin/biotin blocking kit (Vector Laboratories) and 2.5% NHS for 1 h and incubated overnight with goat anti‐ CD206 (1:200, R&D Systems) in 2.5% NHS at 4°C. On the fourth day, sections were incubated in rabbit anti‐goat biotinylated secondary antibody (1:1000, Jackson ImmunoResearch) for 90 min at room temperature. Streptavidin Alexa Fluor 594 was then added to the section for 1 h, followed by nuclei visualization with 4′,6‐diamidino‐2‐phenylindole (DAPI, ThermoFisher). Slides were coverslipped with Vectashield (Vector Laboratories) and stitched images of whole muscle cross‐sections were acquired with a 20× objective on a Zeiss upright microscope (Carl Zeiss, Thornwood, NY, USA). The abundance of all CD11b+/CD206+ cells was manually quantified using the event count tool in Zen software (Carl Zeiss) and normalized to the number of fibers per cross‐section.
Mouse skeletal muscle macrophage and Mmp14 visualization were performed on frozen muscle sections as follows. In brief, slides were fixed in 4% paraformaldehyde and PBS for 5 min, and blocking steps were taken to prevent nonspecific background and cross‐reactivity between amplification reagents. Slides were incubated with rabbit anti‐Mmp14 (1:20, Abcam) and rat anti‐Cd206 (1:200, Bio‐rad, Hercules, California, USA) in 2.5% normal goat serum (NGS, Vector Laboratories) overnight at 4°C. On the second day, sections were rinsed in PBS, followed by incubation with goat anti‐rabbit HRP secondary (1:1000, Jackson ImmunoResearch) and goat anti‐rat secondary Alexa Flour 488 (1:500, ThermoFisher) in PBS for 90 min at room temperature, followed by tyramide signal amplification with Alexa Fluor 594 (ThermoFisher) for 15 min. Slides were coverslipped in Prolong Diamond anti‐fade with DAPI mounting medium (ThermoFisher) and images were acquired with a 63× oil immersion objective on a Zeiss LSM 710 upright confocal microscope (Carl Zeiss).
To visualize capillaries, frozen muscle sections were dried and rehydrated for 5 min with PBS, followed by two 5 min washes with PBS. Sections were blocked for 1 h in 2.5% NHS then incubated for 90 min with a mixture of 2 lectins: biotinylated Ulex europaeus (1:200, Cat# B‐1065‐2, Vector Laboratories) and biotinylated Griffonia Simplicifolia (1:200, Cat# B‐1215‐2, Vector Laboratories), and anti‐laminin (1:200, Cat# L9393, Millipore Sigma, St. Louis, MO, USA). Sections were then washed and incubated for 1 h with streptavidin Alexa Fluor 594 (ThermoFisher) to visualize capillaries and anti‐rabbit Alexa Fluor 488 (ThermoFisher) to visualize laminin to demarcate fiber borders. After 3 final washes, the slides were coverslipped with Vectashield and imaged. Capillaries were counted manually and expressed per muscle fiber.
Analysis of satellite cell abundance was performed as follows. After drying, sections were fixed in −20°C acetone, washed with PBS, incubated with 3% hydrogen peroxide, and blocked for 1 h in 2.5% NHS.
Satellite cells were labeled by overnight incubation with anti‐paired box 7 (Pax7) (Developmental Studies Hybridoma Bank, Iowa City, Iowa, USA). Following PBS washes, sections were incubated with biotinylated anti‐mouse IgG1 (1:1000 Jackson ImmunoResearch) for 90 min. Washes were repeated followed by a 1 h incubation with streptavidin‐HRP (ThermoFisher). Fluorescent labeling was achieved by incubating with SuperBoost tyramide signal amplification Alexa Fluor 488 (ThermoFisher) in PBS for 20 min. Primary antibody against laminin (Millipore Sigma) was added followed by anti‐rabbit Alexa Fluor 594 secondary antibody (ThermoFisher) to visualize and quantify muscle fibers. Sections were washed, incubated for 10 min with DAPI to label nuclei, and coverslipped. Whole cross‐section images were acquired with a 20× objective, satellite cells were identified as Pax7+/DAPI+ and expressed per fiber. Laminin immuno‐stained images were previously used to determine fiber size. 3 The myonuclear number was determined using rabbit anti‐dystrophin (Abcam) followed by anti‐rabbit Alexa Fluor 488 secondary antibody (ThermoFisher) and DAPI to visualize and quantify nuclei within the dystrophin border. Fiber cross‐sectional area (CSA) and myonuclear number were analyzed using MyoVision. 20
To analyze ECM, muscle sections were fixed for 15 min in Bouin's fixative in a humidified chamber at 37°C, washed, then incubated in Picosirius Red (PSR) solution (0.1% in saturated picric acid) for 2 h at room temperature. After washing and dehydration with ethanol, sections were mounted using a xylene‐based media. PSR area normalized to muscle area was quantified using a semi‐automated macro for hue, saturation, and lightness in the Zen software.
2.8. Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assay was performed using the Zymo‐Spin ChIP Kit (Zymo Research, Irvine, CA, USA) with the anti‐Sp1 antibody (5 μg/Reaction, Cat# 17‐601, Millipore Sigma). Briefly, BMDMs were treated for 24 h with either E(+) or E(−) myotube CM (1:1 with DM) and then cross‐linked with chromosomal DNA by 1% formaldehyde followed by sonication. Lysates were immunoprecipitated with the anti‐Sp1 antibody or rabbit IgG isotype control (Millipore Sigma) at 4°C overnight. Protein‐A beads (Millipore Sigma) were then added for 1 h at 4°C with gentle rotation. Immunoprecipitated DNA was amplified by real‐time PCR (qPCR) using primers; forward, 5′‐TCGAGCTGAAACACCACGTC; reverse, 3′‐GGAGGTGAGGGAATTGCTCC, which flanked the −66 nt through −59 nt sequence (distance from TSS) in the Mmp14 promoter region previously identified in HCT116 cells. Immunoprecipitated DNA was calculated according to the bound (immunoprecipitated chromatin)/input DNA ratio minus rabbit IgG isotype control.
2.9. Gene expression experiments in myotubes and BMDM
For RNA extraction, cells and tissue were lysed in Qiazol reagent (Qiagen), and RNA was extracted using the Zymo MiniPrep kit (Zymo Research) with slight modification; the aqueous phase from Qiazol (Qiagen) combined with 3‐bromo‐chloropropane following phase separation was added directly to the spin column. RNA concentration and quality were assessed via NanoDrop (ThermoFisher) and Bioanalyzer using Nano chips (Agilent, Santa Clara, CA, USA). For qPCR, 10 ng of complementary DNA (cDNA) was used per reaction, and reactions were performed using TaqMan chemistry (ThermoFisher) and Taqman probes; LIF‐ Hs01055668_m1; MMP14‐Hs00237119_m1; Mmp14‐Mm00485054_m1; β2‐Microglobulin (B2M)‐ Hs00187842_m1; (B2m)‐ Mm00437762_m1, on a QuantStudio 3 (ThermoFisher) in duplicate with B2M as the housekeeping gene.
For RNA sequencing (RNA‐seq), myotube RNA was prepared as a 250–300 bp nonstrand‐specific library using the NEBNext Ultra RNA Library Prep Kit (NEB) followed by paired‐end sequencing using an Illumina NovaSeq 6000 s4 platform (Novogene, Sacramento, CA). Mapping to the human—hg19 transcriptome (release 100) was performed using STAR. Data were normalized to counts per million (CPM) add 1.0E‐4 per Partek recommendation. Quality was assessed and reads were filtered by removing reads that contained adapters, reads that contained N > 0.1% (N represents base that could not be determined), and low‐quality reads (quality value of over 50% of bases of the read ≤20), and differential analysis was performed using Partek GSA tool with default report options.
2.10. Mononuclear cell isolation from skeletal muscle
To isolate mononuclear cells from mouse muscle, we used a modified version of the FACS protocol from the Rando laboratory. 21 After a single‐cell suspension was made from fresh muscle using collagenase followed by collagenase and dispase, it was filtered through a 40 µm strainer. DAPI (ThermoFisher) was added to gate out dead cells using an iCyt FACS machine (Sony Biotechnology, Champaign, IL, USA). In mice, we removed satellite cells before scRNA‐seq, which were identified as Vcam+ (Cat# 105703, Biolegend), and all other DAPI cells were collected.
2.11. Single‐cell RNA‐sequencing
The single‐cell suspension from mouse muscles was cleared of debris, dead cells, and doublets via FACS, as described above. The cells were sorted directly into reaction buffer (10X Genomics, Pleasanton, CA, USA). Cell concentration was determined via hemocytometer prior to being loaded on the 10X Chromium chip. After cells were loaded into the 10X Chromium Controller, the Single Cell 3′ reagent kit was applied according to the manufacturer's protocol. Libraries were prepared using version 3.0 chemistry and were sequenced on the Illumina NextSeq 500 platform for the mouse. The Cell Ranger Single Cell Software Suite was used to perform sample demultiplexing, barcode processing, and single‐cell 3′ gene counting (http://software.10xgenomics.com/single‐cell/overview/ddddome). The cDNA insert was aligned to an appropriate reference genome using STAR. For mouse muscle, mm10 transcriptome (release 103) was used. Partek Flow was used for all downstream analysis from Filtered_Barcode_Matrix.h5 files. For all mouse samples, 96.89% of reads were within cells, and the estimated number of cells was 7432 with a median gene/feature per cell of 1829. Filter criteria included number of reads (Min: 1500, and Max 200 000), expressed genes/features (Min: 500, Max: 5000), mitochondrial reads percent (Min: 0%, Max: 12%), and ribosomal read percent (Min: 0%, Max: 20%) were used based on silhouette distribution (Figure S1A–D). Batch effects were observed between Sham and 4DMOV samples upon data integration and batch corrections were employed using Harmony 22 with the batch attribute set to Sample name and default configurations present in Partek. Additional filtering was performed which excluded features where values were <3 in all cells of the samples yielding a total of 9111 genes for downstream analysis. Samples were normalized using CPM and log transformation. Principal component analysis was used, and batch corrections could not be implemented reliably as no replicate conditions were used. Uniform Manifold Approximation and Projection for Dimension Reduction was performed for data visualization. K‐means sub clustering was performed on macrophages from Sham and 4DMOV aggregate samples using the Euclidean distance metric with the Davies‐Bouldin index (Figure S1E). All statistical comparisons were conducted in Partek using the Hurdle Model.
2.12. Data availability and statistical analysis
Integration of Human RNA‐seq data sets (GSE157585) and mouse 4DMOV scRNA‐seq (GSE168872) were done using Partek Flow. Raw data sets for human myotube RNA‐seq are deposited in ArrayExpress (E‐MTAB‐10833). Filtered barcoded Sham.h5 data are available upon request. Results are presented as mean ± SEM. Data were analyzed with GraphPad Prism software, via a one‐way repeated measures ANOVA with Sidak's correction for multiple comparisons or a paired or unpaired two‐tailed Student's t‐test. Linear regressions employed the Pearson product‐moment correlation coefficient when the two continuous variables were normally distributed (including all figures depicting regressions). Significance was set at a p < .05. False‐discovery rates were set to Adj‐p < .05 were implemented for all scRNA‐seq data analyzed.
2.13. Study approval
All animal studies were approved by the Institutional Animal Care and Use Committees at the University of Kentucky. Human samples and accompanying clinical and genetic data were used for this study. Sample and data collection were previously approved by Institutional Review Boards at the University of Kentucky and University of Alabama‐Birmingham. All human subjects provided written informed consent and authorized use of their biospecimens and data for future research.
3. RESULTS
3.1. Positive correlation of muscle adaptative responses to PRT with an increase in macrophage abundance
We previously published results of immunohistochemical analyses of muscle biopsies from the MASTERS clinical trial (NCT02308228) demonstrating an increase in macrophages, muscle fiber CSA, and satellite cells after 14 weeks of PRT. 3 In the present study, we determined if the change in macrophage abundance was correlated to changes in the other muscle properties. We found positive associations between the percent change in macrophage number and percent change in fiber CSA (r = .578, p < .005, Figure 1A), myonuclear number (r = .467, p < .05, Figure 1B), satellite cells (r = .421, p < .05, Figure 1C), and capillaries (r = .507, p < .01, Figure 1D). Representative immunohistochemical images were used for quantifications of macrophages (CD11b and CD206, Figure 2A), satellite cells (Pax7) and fiber CSA (Laminin, Figure 2B), myonuclear density (Dystrophin/DAPI, Figure 2C), and capillaries (Lectin, Figure 2D) are shown.
FIGURE 1.
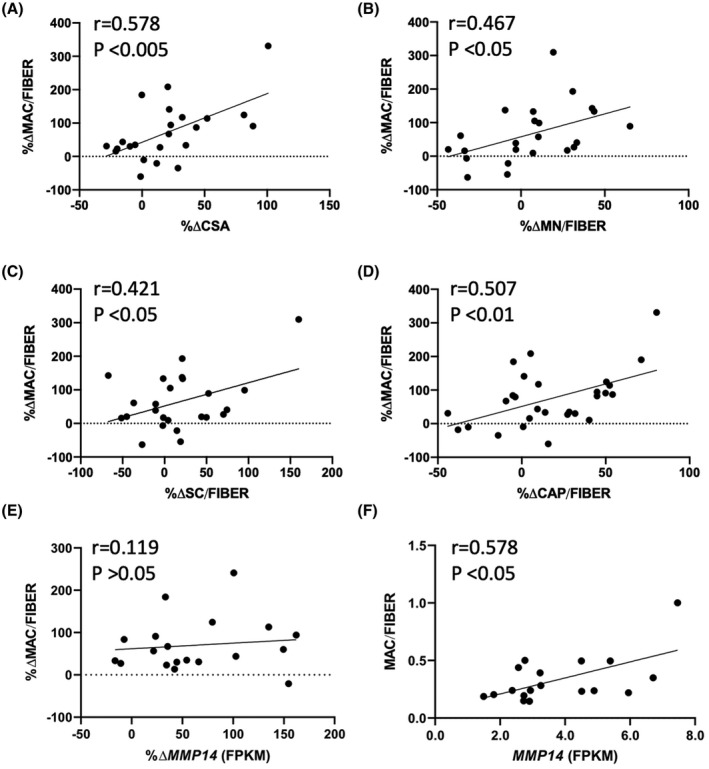
In human vastus lateralis, a significant association between changes in macrophage abundance and changes in (A) muscle fiber CSA (r = .578, p < .005, N = 23), (B) MN (r = .467, p < .05, N = 23), (C) SC (r = .421, p < .05, N = 24), (D) CAP (r = .507, p < .01, N = 24) following 14‐weeks of PRT. (E) The change in macrophage content with PRT does not associate with the change in MMP14 expression (r = .1190, p > .05). N = 19. (F) Absolute number of macrophages correlates to MMP14 expression at the end of PRT (r = .578, p < .05) N = 18. Pearson r correlations were used for all analysis. CAP, capillaries; CSA, cross‐sectional area; FPKM, fragments per kilobase of transcript per million mapped reads; MMP14, matrix metalloproteinase 14; MN, myonuclei; PRT, progressive resistance exercise training; SC, satellite cells
FIGURE 2.
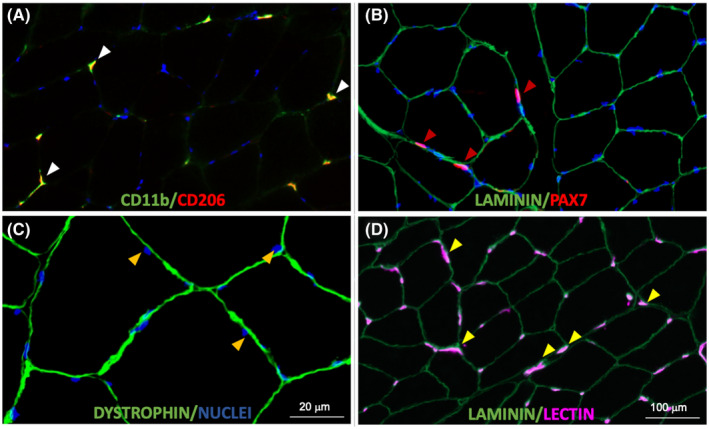
Representative images of skeletal muscle immunohistochemistry. (A) Skeletal muscle macrophages were identified using antibodies against CD11b (green) and CD206 (red); CD11b+/CD206+ cells were counted (white arrows). (B) Satellite cells were identified using a Pax7 antibody (red) within the laminin border (green); satellite cells were expressed per fiber. Muscle fiber cross‐sectional area was measured in these images. (C) Myonuclei (gold arrows) were identified as DAPI+ (blue) nuclei within the dystrophin border (green). (D) Capillaries were identified using fluorescently conjugated lectin (pink) with a laminin antibody to outline fibers (green) and measured per fiber (yellow arrows). Scale bar = 100 μm for (A), (B), and (D); scale bar = 20 μm in (C)
We previously demonstrated that ECM remodeling genes, including MMP14, are increased in muscle following 14 weeks of PRT in participants from this investigation. 9 Although the change in macrophage abundance was not correlated to the change in MMP14 expression with PRT (Figure 1E, r = .119, p > .05), macrophage number/fiber and MMP14 mRNA abundance were correlated in the post‐PRT biopsy (Figure 1F, r = .578, p < .05). Despite ECM remodeling being one of the top upregulated pathways following PRT, we did not observe any significant changes in fibrous collagen abundance by PSR staining between pre (Figure S2A) and post‐ (Figure S2B) muscle biopsies, which was highly variable across participants (Figure S2C). Correspondingly, we did not observe any association with change in macrophage number to change in fibrous collagen content by PSR staining (Figure S2D). We also did not observe any significant association with change in MMP14 expression and change in collagen content (Figure S2E).
3.2. Four days of MOV in mice resulted in macrophage accretion and robust transcriptional differences
We sought to explore macrophage dynamics in response to a hypertrophic stimulus in greater detail. Synergist ablation is a widely employed hypertrophic stimulus in mice that is associated with pronounced macrophage infiltration in the overloaded muscle in the early stages. 1 Sham and 4‐day MOV (4DMOV) mouse plantaris muscles were enzymatically digested and sorted on all live cells excluding satellite cells. Cell annotation was performed using a curated list of markers derived from the scRNA‐seq atlas of mouse skeletal muscle which identified fibro‐adipogenic progenitors (FAPs), endothelial cells, macrophages, myonuclei, and neutrophils for Sham and 4DMOV aggregated samples (Figure 3A,B and Table S1). 23 Increased macrophage content was visually apparent following 4DMOV when compared to the Sham (Figure 4A). The proportion of macrophages versus total mononuclear cells was also increased following 4DMOV (Sham = 8.3% vs. 4DMOV = 50.9%, Figure S3). At the 4‐day time point, changes in the macrophage transcriptome comprised 2105 differentially expressed genes (DEG, FDR < 0.05, Fold Change (FC) <−1.5 or >1.5) as visualized by volcano plot (Figure 4B and Table S2). The list of upregulated DEGs with 4DMOV include Arg1 (27‐fold), Mmp14 (22‐fold), Chil3 (10‐fold), and Vegfa (9 fold). Those genes downregulated in 4DMOV compared to Sham included Retnla (909‐fold), Mgl2 (336‐fold), Cd163 (134‐fold), and Lyve1 (83‐fold). Furthermore, tissue inhibitors of metalloproteinases 2 and 3 (Timp2/3, 11‐fold, and 1.5‐fold, respectively), were significantly lower in 4DMOV macrophages relative to Sham. In terms of cell‐type expression of Mmp14, it was clear that under resting conditions the majority of cells expressing Mmp14 were FAPs, whereas, at the 4DMOV time point, macrophages were the major source of Mmp14 expression (Figure 5A). To confirm that the transcriptional changes resulted in translational changes to Mmp14 expression in macrophages, we performed immunohistochemistry on Sham and 4DMOV mouse muscle cross‐sections. Using antibodies against macrophage marker CD206 and Mmp14, we were able to visually confirm both the absence of Mmp14 co‐staining in macrophages in sham plantaris muscles (Figure 5B, left panel) and the co‐localization of Mmp14 and CD206 staining in 4DMOV macrophages (Figure 5B, right panel). Changes in macrophage heterogeneity were also apparent by k‐means clustering, which generated 3 subclusters of macrophages between Sham and 4DMOV aggregates (Figure 5C and Table S3). K‐means clustering coupled with pathway analysis illustrated a divergence in macrophage metabolic signature between clusters 1 (oxidative, Table S4) and 2 (glycolytic, Table S5). Classical macrophage subtypes such as inflammatory or anti‐inflammatory were less apparent at the day 4 time point. Cluster 3, which was composed entirely of Sham macrophages, demonstrated the highest expression levels for resident macrophage markers Lyve1 and Timd4 (Table S3). We were not able to confidently identify a subcluster of macrophages that expressed the highest levels of Mmp14 in the 4DMOV group (Figure 5D) as both KM1 and KM2 clusters expressed similar levels of Mmp14 and, therefore, sought to explore the muscle‐macrophage crosstalk in vitro for a greater mechanistic understanding of how MOV was influencing macrophage‐specific expression of Mmp14.
FIGURE 3.
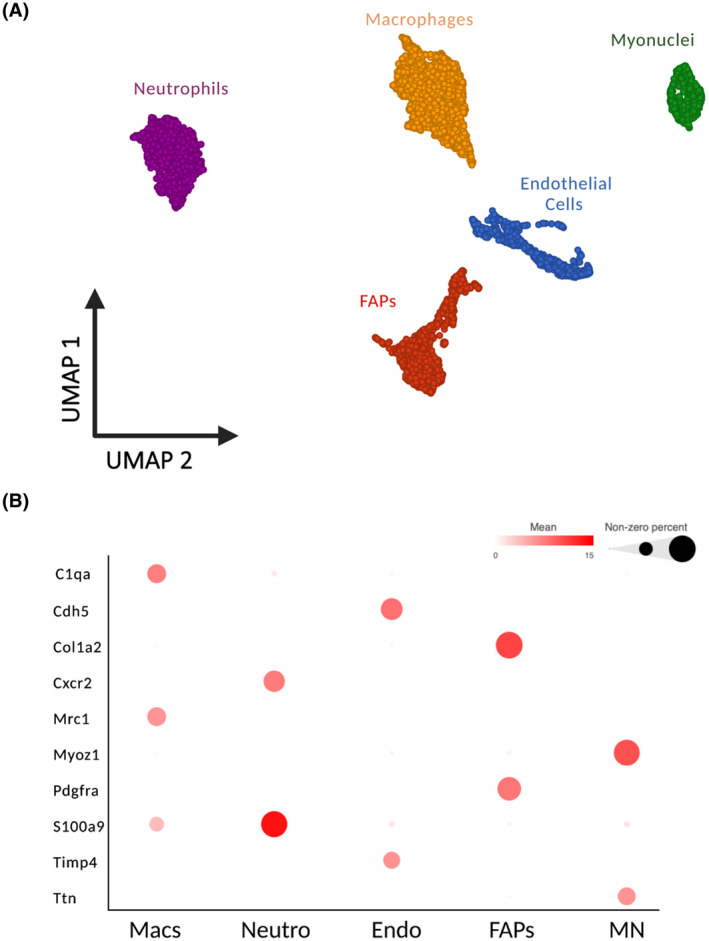
scRNA‐seq demonstrates cell populations of combined Sham and 4 days of mechanical overload (4DMOV) mouse plantaris muscles using UMAP. (A) Cell types were classified as either endothelial cells (Endo, blue), myonuclei (MN, green), fibro‐adipogenic progenitors (FAPs, red), macrophages (Macs, gold), or neutrophils (Neutro, purple). (B) Biomarkers illustrated as bubble plots for identifying cell types in Sham and 4DMOV samples. N = 6528 total cells (Sham = 2430 cells and 4DMOV = 4098 cells). scRNA‐seq, single cell RNA‐sequencing; UMAP, uniform manifold approximation and projection
FIGURE 4.
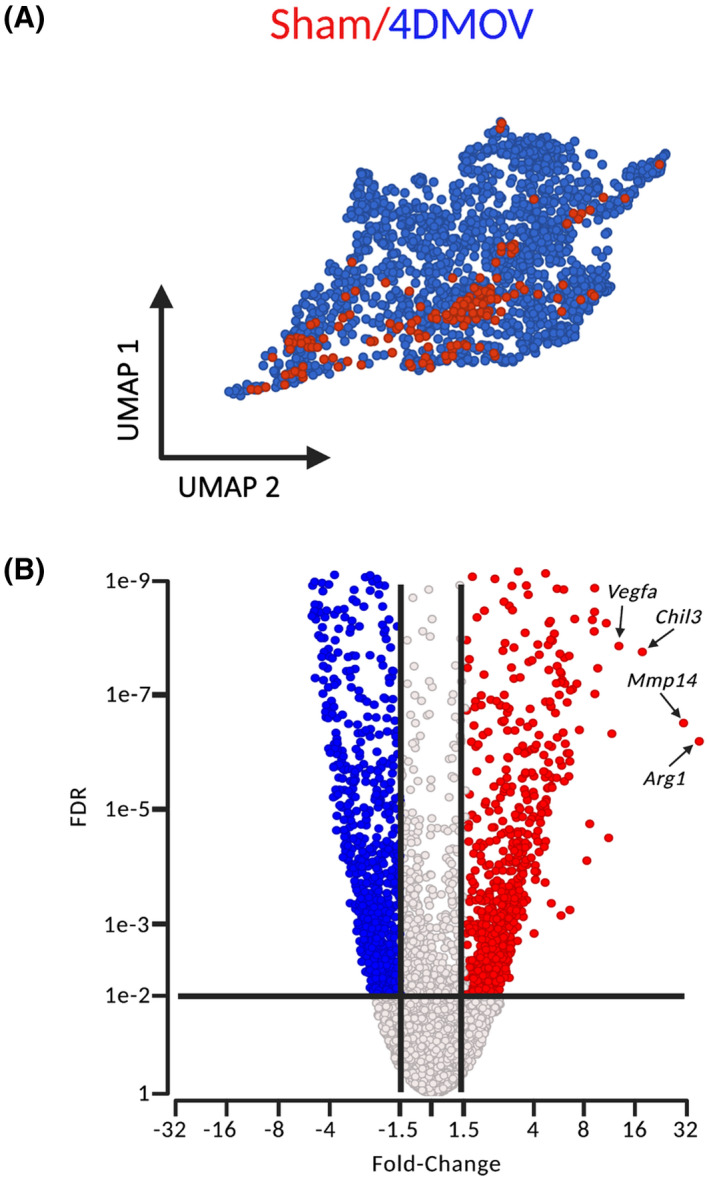
scRNA‐seq of mouse macrophage populations under Sham and 4‐day mechanical overload (4DMOV) conditions. (A) UMAP plot of macrophages under Sham conditions (red) compared to after 4DMOV (blue) in mouse plantaris muscles. (B) Volcano plot demonstrating the number of differentially expressed genes (DEG) significantly increased (red), decreased (blue), or unchanged (black) when comparing 4DMOV versus Sham (FDR < 0.05). Arg1, Arginase 1; Chil3, chitinase‐like 3; FDR, false discovery rate; Mmp14, matrix metalloproteinase 14; scRNA‐seq, single‐cell RNA‐sequencing; UMAP, uniform manifold approximation and projection; Vegfa, vascular endothelial growth factor alpha
FIGURE 5.
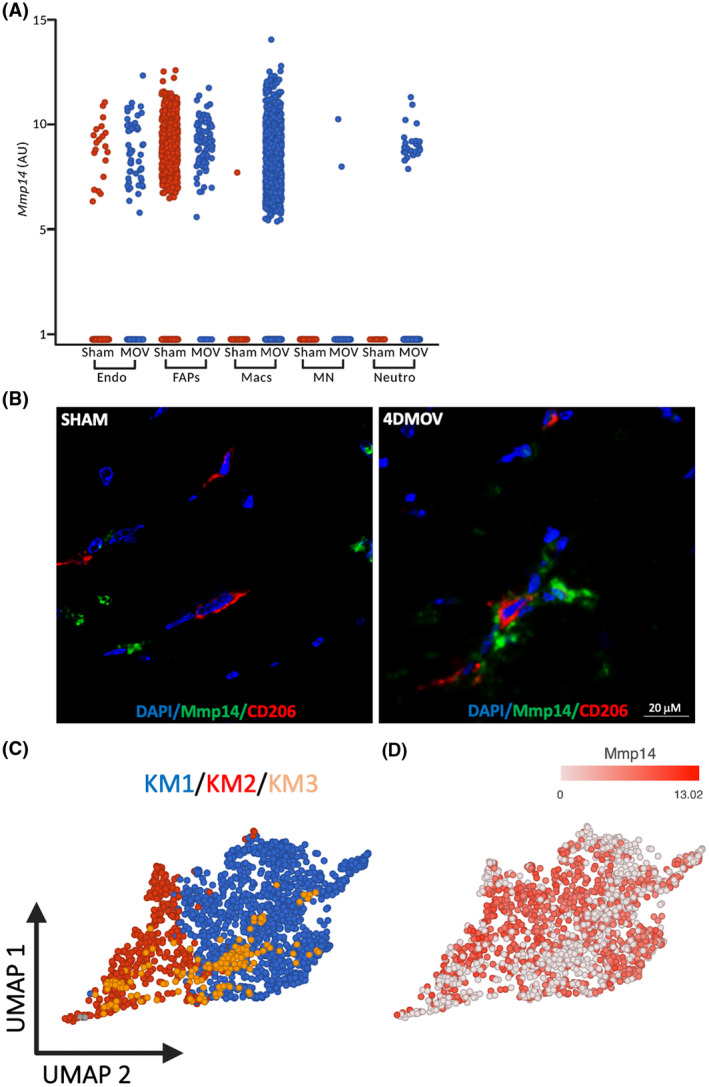
Assessing Mmp14 expression across cell types in Sham and 4‐day mechanical overloaded (4DMOV) muscles and macrophage subclusters. (A) Under Sham conditions (red), FAPs are the predominant source of Mmp14. Following 4DMOV (blue) macrophages present as the majority of Mmp14 expressing cells. (B) Representative images of mouse skeletal muscle staining of pan macrophage marker CD206 (red), Mmp14 (green), and DAPI (blue) under Sham or 4DMOV conditions. Scale bar = 20 μm. (C) K‐means clustering of macrophages from both sham and 4DMOV muscles reveals 3 sub clusters with Sham clustering separately (cluster 3, gold) and 4DMOV macrophage clusters consisting of cluster 1 (blue) and cluster 2 (red). (D) Feature plot of Mmp14 expression across the macrophage cluster. Endo, endothelial; FAPs, fibro‐adipogenic progenitors; Macs, macrophages; Mmp14, matrix metalloproteinase 14; MN, myonuclei; Neutro, Neutrophils
3.3. CM from myotubes following an in vitro exercise mimetic (E) in BMDM
Muscle fibers are the largest cells in muscle and release cytokines in response to contraction. We speculated that a factor or factors released by muscle cells in response to contraction could affect Mmp14 production by macrophages. We used an in vitro exercise mimetic, electrical pulse stimulation (E+/−), to induce chronic myotube contractions for a 24‐h period. Following 24 h of E(+) or E(−), we collected myotube CM and treated mouse BMDM for 24 h using both E(+) and E(−) conditions. We then performed qPCR for Mmp14 expression between treatment groups. Following 24 h of E(+) CM treatment, BMDM had a nearly twofold higher Mmp14 expression relative to E(−) CM treatment (Figure 6A, p < .01).
FIGURE 6.
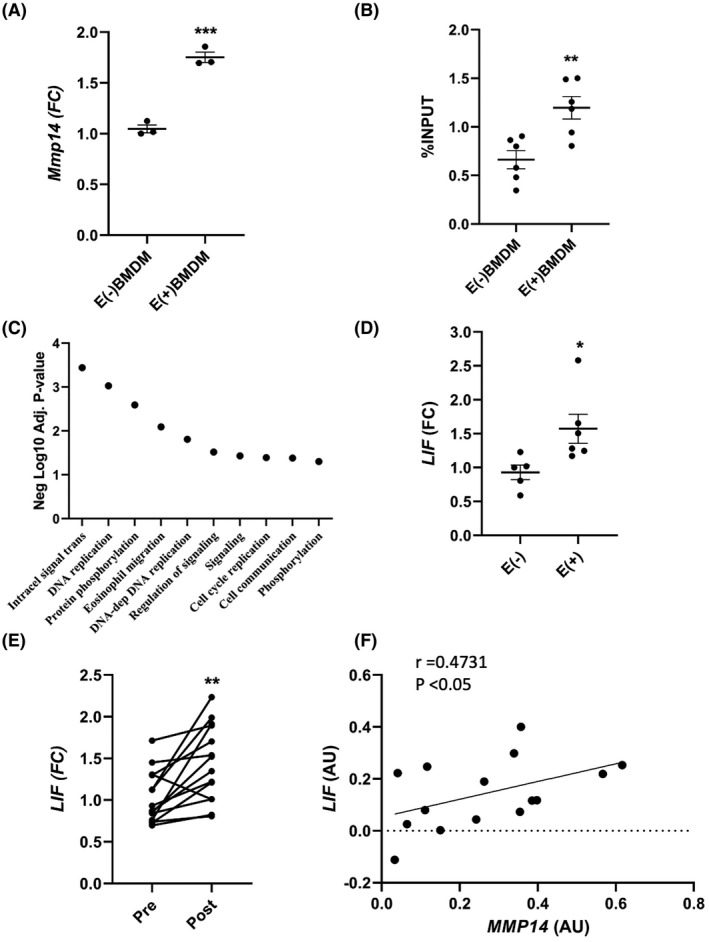
Effects of E(+) and PRT on BMDM and myotube gene expression. (A) BMDM expression levels of Mmp14 were quantified by qPCR following treatment with E(+/−) CM for 24 h (**p < .01) using an unpaired student's t‐test. N = 5. (B) ChIP of Sp1 binding activity to Mmp14 promoter in BMDM following 24 h of E(+/−) CM treatment (**p < .01). The percent Sp1 fraction over input was compared between treatments using an unpaired student's t‐test. N = 6. (C) Gene set analysis of biological processes upregulated in E(+) myotubes compared to E(−) myotubes using a negative log10 adjusted p‐value. N = 2. (D) RNA was isolated from human primary myotubes subjected to E(+/−) for 24 h. LIF mRNA was quantified by qPCR (*p < .05), and compared between treatments using an unpaired student's t‐test. N = 5–6. (E) LIF mRNA was quantified in pre‐ and post‐14‐week PRT muscle from older adults using qPCR (*p < .05), and compared across treatments using a paired student's t‐test. N = 16. Correlative studies also demonstrate that (F) changes in LIF expression positively associate with changes in MMP14 expression following 14 weeks of PRT by qPCR (r = .4731, p < .05). N = 16. Data presented as mean ± SEM. CM, conditioned media; E(+/−), electrical pulse stimulation; FC, fold change; FPKM, fragments per kilobase of transcript per million mapped reads; LIF, leukemia inhibitory factor; Mmp14/MMP14, matrix metalloproteinase 14; Sp1, specificity protein 1; qPCR, real‐time PCR
3.4. CM from E(+) myotubes promotes Sp1 binding to the Mmp14 promoter in BMDM
Previous research in vitro has demonstrated that p53 binds to the MMP14 promoter region between the transcriptional start site −66 nt and −59 nt to inhibit Sp1 mediated transcription in HCT‐116 cells. 15 In analyzing the mouse Mmp14 promoter, we identified a conserved sequence (5′‐GGGGCGGGG‐3′) 4 nt upstream of the previously mentioned human promoter binding site. To determine whether changes in Sp1 binding to the Mmp14 promoter occurred with E(+) CM treatment in mouse BMDM, we performed ChIP on BMDM using an antibody against the mouse Sp1 protein following 24 h of E(+) or E(−) CM treatment. We generated primers that flanked the response element for p53/Sp1 and performed qPCR. We found a twofold enrichment in Sp1 binding to the Mmp14 promoter in E(+) CM‐treated BMDM compared to E(−) CM‐treated BMDM (Figure 6B, p < .01).
3.5. Elevated LIF expression in human primary myotubes with electrical stimulation and skeletal muscle following MOV
First, we sought to identify muscle‐derived cytokines that were upregulated in vitro by electric pulse stimulation. We performed RNA‐seq on two different primary human cell lines comparing E(+) to E(−) myotubes. We used fold change cut‐offs >1.5 and unadjusted p < .05 to identify potential myokines that were upregulated by 24 h of E(+/−) (Table S6). Pathway analysis of biological processes demonstrated an upregulation of intracellular signal transduction, DNA replication, and protein phosphorylation (Figure 6C and Table S7). Investigation of candidate myokines showed that LIF mRNA was upregulated with E(+), and we validated these findings in a separate experiment by qPCR (Figure 6D, p < .05). IL6 transcript was expressed at very low levels and was not significantly different in the RNA‐seq analysis (data not shown). To translate our in vitro findings, we analyzed LIF expression from a subset of MASTERS participants. LIF expression by qPCR was significantly higher following PRT in skeletal muscle (Figure 6E). Correlative studies demonstrated that there was a significant association between the changes in LIF and MMP14 expression following PRT (Figure 6F, r = .4731, p < .05).
3.6. Inhibition of LIF blunts the increase in macrophage Mmp14 expression following E(+) CM treatment
We next assessed if LIF was directly responsible for the increase in macrophage Mmp14 expression. Using a human anti‐LIF neutralizing antibody (LIFAb), we treated BMDM for 24 h with either E(+) or E(−) CM, with or without LIFAb (LIFAb+/−). Following the incubation period, RNA isolated from BMDM was subjected to qPCR using probes for Mmp14. Treatment of BMDM with E(+)LIFAb(−) CM resulted in a significant increase in Mmp14 mRNA compared to E(−)LIFAb(−) CM (Figure 7A, p < .05). Conversely, E(+)LIFAb(+) CM failed to induce a significant increase in Mmp14 expression in BMDM (Figure 7A), implicating muscle‐derived LIF as the primary mediator of the Mmp14 gene expression in macrophages.
FIGURE 7.
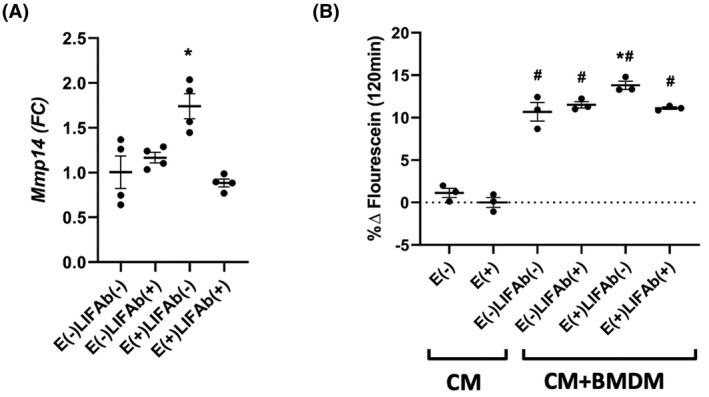
LIF modulates Mmp14 expression and type I collagen degradation rates in vitro. (A) CM from E(+) and E(−) myotubes was pre‐treated with 0.1 μg/ml of human anti‐LIF neutralizing antibody or IgG control antibody (LIFAb(+/−) followed by incubation with BMDM for 24 h. Cells were collected, RNA isolated, and Mmp14 expression quantified by qPCR (*p < .05). N = 6. Results are expressed as fold change from the control group; E(−)LIFAb(−). (B) DQ type I collagen substrate was used to measure the degradation rates by BMDM in the presence of myotube CM. BMDMs had a significant effect on type I collagen degradation compared to CM alone (# p < .05). E(+) CM treatment of BMDM (E(+)LIFAb(−)) had a significant effect above that observed in control group (E(−)LIFAb(−), *p < .05, # p < .05) compared to CM (*p < .05). N = 3. All analyses were performed using one‐way repeated measures ANOVA with Sidak's correction for multiple comparisons. Data presented as mean ± SEM. BMDM, bone marrow‐derived macrophages; CM, conditioned media; E(+/−), electrical pulse stimulation; LIF, leukemia inhibitory factor; LIFAb, LIF neutralizing antibody; Mmp14, matrix metalloproteinase 14; qPCR, real‐time PCR
3.7. BMDM treated with E(+) CM display enhanced type I collagen degradation mediated by LIF
To determine if elevated Mmp14 expression in BMDM by E(+)CM results in collagen degradation, we used an in vitro model to assess type I collagen degradation using a DQ type I collagen assay, which emits fluorescein as collagen is degraded. We first assessed if CM from myotubes following E(+) or E(−) induced any significant changes above control (unconditioned media). We detected very little fluorescein emission when the collagen was treated with E(+/−) CM in the absence of BMDM (CM, Figure 7B). In the presence of BMDM cells, DQ collagen degradation was higher than CM alone (Figure 7B, p < .0001). E(+)LIFAb(−) CM‐treated BMDM showed a significant increase in degradation above E(−)LIFAb(−) CM‐treated BMDM (Figure 7B, p < .05). CM treatment with the anti‐LIF neutralizing antibody (E(+)LIFAb(+)) prevented the increased degradation in E(+)LIFAb(−) CM‐treated BMDM (Figure 7B).
4. DISCUSSION
ScRNA‐seq is an attractive approach to identify heterogeneity within cell types, especially macrophages, which have classically been defined in a binary fashion as pro‐ or anti‐inflammatory. In skeletal muscle regeneration studies, macrophages have been proposed to transition along a continuum from a pro‐inflammatory phenotype at the onset of injury toward an anti‐inflammatory and regenerative phenotype over 7–14 days. 24 To date, several RNA‐seq and scRNA‐seq manuscripts have gone into great detail describing the transcriptomic alterations of macrophages during regeneration. 23 , 25 While macrophage‐derived growth factors have obvious implications for muscle regeneration, the role of macrophages is less clear in the context of hypertrophy. Here, we utilized scRNA‐seq to describe the macrophage response to MOV to add to the understanding of macrophage biology within adult non‐regenerating skeletal muscle. We also provide insight on the potential role of macrophages in ECM remodeling with loading and identification of a novel muscle‐cell macrophage axis involving LIF and MMP14.
Using synergist ablation as a model of MOV in mouse skeletal muscle, we identified three clusters of macrophages across two time points. To date, macrophage heterogeneity has largely been described as a metric of time from injury. 24 With regard to the metabolic influence, previous in vivo findings from Varga et al., illustrated that changes in macrophage metabolism preceded changes in inflammatory status following injury. 26 Our findings indicate that macrophage heterogeneity early during hypertrophy is largely dictated by the metabolic state as opposed to inflammatory status. On day 4 of MOV, we observed two subclusters of macrophages that demonstrated differential gene expression of inverse glycolytic or oxidative metabolic transcripts. Both subclusters expressed ECM remodeling transcripts that were differentially expressed compared to Sham, including Mmp14 that was increased 22‐fold in 4DMOV macrophages compared to Sham macrophages regardless of subcluster.
MMP14 plays an important role in a variety of remodeling events including myogenesis, angiogenesis, cell migration, and collagen degradation. 10 , 15 , 27 ECM remodeling in response to a hypertrophic stimulus is necessary for permitting myofiber out‐growth through the remodeling of the basement membrane. 28 Following 14 weeks of PRT in humans, we identified a positive association between MMP14 expression and macrophage number. These findings suggest that macrophages may be a primary source for MMP14 expression following PRT. Given the similarity between the macrophage‐associated changes in muscle architecture and the importance of MMP14 in skeletal muscle (satellite cell proliferation/fusion, capillary formation, and enlargement of muscle fiber CSA), it appears that macrophage‐specific MMP14 expression may facilitate skeletal muscle adaptations to PRT. Despite our previously reported changes in ECM remodeling pathways with PRT, 9 PSR staining did not reveal any significant changes to fibrous collagen content following 14 weeks of PRT in MASTERS participants. Additionally, neither macrophages nor MMP14 expression was associated with changes in total fibrous collagen abundance by PSR following 14 weeks of PRT. It is important to note that the age of our subjects may have played an important part in the absence of significant changes to fibrous collagen content by PSR. A recent report has provided evidence that a smaller dynamic protein pool of collagen exists in aged mice (>23 months) compared to young mice (6 months), which researchers attributed to a decline in proteostasis maintenance. 29 With aging, long‐lived proteins such as collagen become more susceptible to damage and protein aggregation which impedes protein turnover by proteases such as MMP14. Additional limitations to PSR staining are that it primarily stains collagens I and III, therefore any changes in the basement membrane, which is primarily collagen IV, would not be detected.
Exercise‐induced muscle cytokines are likely candidates for upregulating macrophage expression of MMP14. LIF has previously been described as a resistance exercise‐induced myokine, although not detectable in plasma following exercise. 11 LIF knockout mice fail to undergo skeletal muscle hypertrophy following 21 days of MOV, which indicates an important paracrine function of LIF in skeletal muscle hypertrophy via mechanisms currently unknown. 13 Our results suggest an important function of LIF is to facilitate remodeling of the ECM through interactions with macrophages. Our in vitro experiments suggest that communication between electrically stimulated myotubes and macrophages promotes collagen degradation via MMP14. The mechanism by which LIF regulates MMP14 expression was first discovered in a HCT116 cell line (human colorectal cancer cell line), where the authors reported an increase in p53 degradation with LIF treatment. 30 In a similar study, IL6 treatment, which is highly homologous to LIF, was used to further describe the mechanism by which p53 degradation enables Sp1 binding to the MMP14 gene promoter in HCT116 cells. 15 Sp1 was previously characterized as the primary transcription factor responsible for MMP14 gene expression in multiple human immortalized cell lines. 31 Here, we show that in macrophages, myotube CM following electrical stimulation promotes Sp1 binding to the Mmp14 promoter. These findings are the first to demonstrate that this mechanism is conserved in macrophages.
Our focus was to identify how changes in muscle macrophages with PRT/MOV influence ECM remodeling. Despite illustrating enhanced type I collagen degradation following E(+) CM treatment of BMDM in vitro, further studies will be needed in vivo using macrophage‐specific Mmp14 knockout mice to assess the physiological impact of macrophage Mmp14 on ECM remodeling. Similarly, one could also generate skeletal muscle‐specific Lif knockout mice to validate our in vitro findings. The underlying role of metabolism in macrophage heterogeneity may also be of interest, especially if impeding shifts in glycolysis or OXPHOS may adversely affect ECM remodeling or muscle growth. The accessibility of monocytes/macrophages and our understanding of how they are recruited to damaged or exercised muscle makes macrophages highly attractive therapeutic drug and gene targets. Deepening our understanding of macrophage function should fuel future research to manipulate and potentially enhance muscle adaptations to resistance exercise and reduce skeletal muscle fibrosis and anabolic resistance associated with aging.
DISCLOSURES
The authors have declared that no conflict of interest exists.
AUTHOR CONTRIBUTIONS
Bailey D. Peck, Kevin A. Murach, Charlotte A. Peterson, Marcas M. Bamman, and R. Grace Walton designed experiments. Bailey D. Peck, Kevin A. Murach, Kate Kosmac, Douglas E. Long, Philip A. Kern, Cory M. Dungan, and Alexander J. Simmons performed experiments. Bailey D. Peck and R. Grace Walton analyzed data. Bailey D. Peck wrote the manuscript and prepared the figures. Charlotte A. Peterson, Marcas M. Bamman, and Kevin A. Murach provided funding support and supervised the study. Charlotte A. Peterson and Kevin A. Murach assisted with data interpretation and manuscript writing. All authors edited and approved the final manuscript.
Supporting information
Fig S1
Fig S2
Fig S3
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
ACKNOWLEDGMENTS
This work was supported by funding from the NIH National Institutes of Arthritis and Musculoskeletal and Skin Diseases (AR060701 to C.A.P and AR071753 to K.A.M), National Institute on Aging (AG046920 to C.A.P, M.M.B, and P.A.K, AG049086 to C.A.P, and AG063994 to K.A.M), National Institute of Diabetes and Digestive and Kidney Diseases (DK119619 to C.A.P), a Paul Glenn Investigator Award (C.A.P) and the University of Kentucky CTSA award (UL1TR001998).
Peck BD, Murach KA, Walton RG, et al. A muscle cell‐macrophage axis involving matrix metalloproteinase 14 facilitates extracellular matrix remodeling with mechanical loading. FASEB J.2022;36:e22155. doi: 10.1096/fj.202100182RR
REFERENCES
- 1. DiPasquale DM, Cheng M, Billich W, et al. Urokinase‐type plasminogen activator and macrophages are required for skeletal muscle hypertrophy in mice. Am J Physiol Cell Physiol. 2007;293(4):C1278‐C1285. [DOI] [PubMed] [Google Scholar]
- 2. Walton RG, Kosmac K, Mula J, et al. Human skeletal muscle macrophages increase following cycle training and are associated with adaptations that may facilitate growth. Sci Rep. 2019;9(1):969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walton RG, Dungan CM, Long DE, et al. Metformin blunts muscle hypertrophy in response to progressive resistance exercise training in older adults: a randomized, double‐blind, placebo‐controlled, multicenter trial: the MASTERS trial. Aging Cell. 2019;18(6):e13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jensen SM, Bechshøft CJL, Heisterberg MF, et al. Macrophage subpopulations and the acute inflammatory response of elderly human skeletal muscle to physiological resistance exercise. Front Physiol. 2020;11(811). doi: 10.3389/fphys.2020.00811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tonkin J, Temmerman L, Sampson RD, et al. Monocyte/macrophage‐derived IGF‐1 orchestrates murine skeletal muscle regeneration and modulates autocrine polarization. Mol Ther. 2015;23(7):1189‐1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Juban G, et al. AMPK activation regulates LTBP4‐dependent TGF‐beta1 secretion by pro‐inflammatory macrophages and controls fibrosis in Duchenne muscular dystrophy. Cell Rep. 2018;25(8):2163‐2176.e6. [DOI] [PubMed] [Google Scholar]
- 7. Chazaud B. Inflammation and skeletal muscle regeneration: leave it to the macrophages! Trends Immunol. 2020;41(6):481‐492. [DOI] [PubMed] [Google Scholar]
- 8. McKee TJ, Perlman G, Morris M, et al. Extracellular matrix composition of connective tissues: a systematic review and meta‐analysis. Sci Rep. 2019;9(1):10542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kulkarni AS, Peck BD, Walton RG, et al. Metformin alters skeletal muscle transcriptome adaptations to resistance training in older adults. Aging. 2020;12(20):19852‐19866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Snyman C, Niesler CU. MMP‐14 in skeletal muscle repair. J Muscle Res Cell Motil. 2015;36(3):215‐225. [DOI] [PubMed] [Google Scholar]
- 11. Broholm C, Mortensen OH, Nielsen S, et al. Exercise induces expression of leukaemia inhibitory factor in human skeletal muscle. J Physiol. 2008;586(8):2195‐2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Broholm C, Pedersen BK. Leukaemia inhibitory factor–an exercise‐induced myokine. Exerc Immunol Rev. 2010;16:77‐85. [PubMed] [Google Scholar]
- 13. Spangenburg EE, Booth FW. Leukemia inhibitory factor restores the hypertrophic response to increased loading in the LIF(−/−) mouse. Cytokine. 2006;34(3‐4):125‐130. [DOI] [PubMed] [Google Scholar]
- 14. Welc SS, Flores I, Wehling‐Henricks M, et al. Targeting a therapeutic LIF transgene to muscle via the immune system ameliorates muscular dystrophy. Nat Commun. 2019;10(1):2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cathcart JM, Banach A, Liu A, et al. Interleukin‐6 increases matrix metalloproteinase‐14 (MMP‐14) levels via down‐regulation of p53 to drive cancer progression. Oncotarget. 2016;7(38):61107‐61120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Long DE, Peck BD, Martz JL, et al. Metformin to augment strength training effective response in seniors (MASTERS): study protocol for a randomized controlled trial. Trials. 2017;18(1):192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murach KA, Peck BD, Policastro RA, et al. Early satellite cell communication creates a permissive environment for long‐term muscle growth. iScience. 2021;24(4):102372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Juhas M, Abutaleb N, Wang JT, et al. Incorporation of macrophages into engineered skeletal muscle enables enhanced muscle regeneration. Nat Biomed Eng. 2018;2(12):942‐954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kosmac K, Peck B, Walton R, et al. Immunohistochemical identification of human skeletal muscle macrophages. Bio Protoc. 2018;8(12). doi: 10.21769/BioProtoc.2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wen Y, Murach KA, Vechetti IJ, et al. MyoVision: software for automated high‐content analysis of skeletal muscle immunohistochemistry. J Appl Physiol. 2017;124:40‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu L, Cheung TH, Charville GW, et al. Isolation of skeletal muscle stem cells by fluorescence‐activated cell sorting. Nat Protoc. 2015;10(10):1612‐1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single‐cell data with Harmony. Nat Methods. 2019;16(12):1289‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Micheli AJ, Laurilliard EJ, Heinke CL, et al. Single‐cell analysis of the muscle stem cell hierarchy identifies heterotypic communication signals involved in skeletal muscle regeneration. Cell Rep. 2020;30(10):3583‐3595.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arnold L, Henry A, Poron F, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. 2007;204(5):1057‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giannakis N, Sansbury BE, Patsalos A, et al. Dynamic changes to lipid mediators support transitions among macrophage subtypes during muscle regeneration. Nat Immunol. 2019;20(5):626‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Varga T, Mounier R, Horvath A, et al. Highly dynamic transcriptional signature of distinct macrophage subsets during sterile inflammation, resolution, and tissue repair. J Immunol. 2016;196(11):4771‐4782. [DOI] [PubMed] [Google Scholar]
- 27. Gramolelli S, Cheng J, Martinez‐Corral I, et al. PROX1 is a transcriptional regulator of MMP14. Sci Rep. 2018;8(1):9531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Q, Joshi SK, Lovett DH, et al. Matrix metalloproteinase‐2 plays a critical role in overload induced skeletal muscle hypertrophy. Muscles Ligaments Tendons J. 2014;4(4):446‐454. [PMC free article] [PubMed] [Google Scholar]
- 29. Abbott CB, Lawrence MM, Kobak KA, et al. A novel stable isotope approach demonstrates surprising degree of age‐related decline in skeletal muscle collagen proteostasis. Function. 2021;2(4):zqab028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu H, et al. LIF negatively regulates tumour‐suppressor p53 through Stat3/ID1/MDM2 in colorectal cancers. Nat Commun. 2014;5:5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lohi J, Lehti K, Valtanen H, et al. Structural analysis and promoter characterization of the human membrane‐type matrix metalloproteinase‐1 (MT1‐MMP) gene. Gene. 2000;242(1‐2):75‐86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
Data Availability Statement
Integration of Human RNA‐seq data sets (GSE157585) and mouse 4DMOV scRNA‐seq (GSE168872) were done using Partek Flow. Raw data sets for human myotube RNA‐seq are deposited in ArrayExpress (E‐MTAB‐10833). Filtered barcoded Sham.h5 data are available upon request. Results are presented as mean ± SEM. Data were analyzed with GraphPad Prism software, via a one‐way repeated measures ANOVA with Sidak's correction for multiple comparisons or a paired or unpaired two‐tailed Student's t‐test. Linear regressions employed the Pearson product‐moment correlation coefficient when the two continuous variables were normally distributed (including all figures depicting regressions). Significance was set at a p < .05. False‐discovery rates were set to Adj‐p < .05 were implemented for all scRNA‐seq data analyzed.