

Abstract
Vascular calcification is a prognostic marker for cardiovascular mortality in chronic kidney disease (CKD) patients. In these patients, magnesium balance is disturbed, mainly due to limited ultrafiltration of this mineral, changes in dietary intake and the use of diuretics. Observational studies in dialysis patients report that a higher blood magnesium concentration is associated with reduced risk to develop vascular calcification. Magnesium prevents osteogenic vascular smooth muscle cell transdifferentiation in in vitro and in vivo models. In addition, recent studies show that magnesium prevents calciprotein particle maturation, which may be the mechanism underlying the anti-calcification properties of magnesium. Magnesium is an essential protective factor in the calcification milieu, which helps to restore the mineral-buffering system that is overwhelmed by phosphate in CKD patients. The recognition that magnesium is a modifier of calciprotein particle maturation and mineralization of the extracellular matrix renders it a promising novel clinical tool to treat vascular calcification in CKD. Consequently, the optimal serum magnesium concentration for patients with CKD may be higher than in the general population.
Keywords: calciprotein particle, chronic kidney disease, mineral homeostasis, phosphate, vascular smooth muscle cells
INTRODUCTION
Chronic kidney disease (CKD) is characterized by severe mineral–bone disturbances including hyperphosphatemia, which stimulates the development of vascular calcification [1]. The presence of vascular calcification is associated with increased cardiovascular mortality risk [2]. Currently there are no treatment options with proven efficacy for patients to prevent or counteract vascular calcification [3]. Over the past decade, a higher serum magnesium (Mg2+) concentration has been associated with lower cardiovascular mortality risk in patients on haemodialysis treatment [4]. In addition, a higher Mg2+ concentration beneficially modifies the association of serum levels of phosphate (Pi) with all-cause mortality in patients treated with dialysis, mitigating risk if Mg2+ is high [5]. Besides these epidemiological association studies, Mg2+ has been demonstrated to reduce vascular calcification in a multitude of experimental models. In this review we provide an overview of the current perspectives on the role of Mg2+ in CKD-induced vascular calcification.
VASCULAR CALCIFICATION IN CKD AND THE ROLE OF MAGNESIUM: OBSERVATIONAL STUDIES
Vascular calcification is a prognostic marker for cardiovascular mortality in CKD patients [6, 7]. The severity of vascular calcification is correlated with, among others, a reduced glomerular filtration rate (GFR) [8]. In dialysis patients, the prevalence of vascular calcifications is >80% [9, 10]. Vascular calcification can be present in both the intimal and the medial layers of arteries. Intimal calcification develops specifically at sites of atheromatous plaques and is typically associated with advanced stages of atherosclerosis [11]. Medial calcification occurs in response to metabolic imbalance of, for instance, minerals and glucose in the larger arteries and generally is associated with ageing, diabetes and CKD [12–14].
Once established, vascular calcification is currently considered to be irreversible [15]. Indeed, there are no confirmed treatment options to regress calcification in arteries of CKD patients. Rather, treatment is directed at preventing the formation of calcification mainly by reducing blood Pi concentrations [16]. The use of calcium (Ca2+)-free Pi binders has been shown to reduce the incidence and progression of vascular calcification, and these are most frequently used in CKD patients to limit vascular calcification development [1]. While Pi binders are successful in lowering blood Pi concentrations, a recent meta-analysis indicated that they do not clearly reduce cardiovascular risk [17]. Studies assessing other medications such as vitamin D3 derivatives, normally used to improve Ca2+ balance, remained inconclusive, as in some cases vascular calcifications worsened rather than improved [18]. In 2011, the ADVANCE trial reported promising effects of low-dose vitamin D3 treatment in combination with the calcimimetic cinacalcet on coronary, aortic and cardiac valve calcification progression [19]. However, this treatment never proceeded to clinical use for this indication, potentially because the benefits seem inferior to the current treatment standards [18]. Cinacalcet is frequently used for the treatment of secondary hyperparathyroidism. Given the limited treatment options, it is of great clinical importance to identify novel modifiable factors that influence the evolution of vascular calcification.
Magnesium and cardiovascular mortality risk
Hypomagnesaemia (serum Mg2+ concentration <0.7 mmol/L) is a well-known risk factor for cardiovascular disease, events and mortality in the patients with higher baseline cardiovascular risk and in patients with dialysis-dependent CKD [4, 20–23]. Besides its blood concentrations, low dietary Mg2+ intake is also associated with all-cause mortality, risk of stroke, heart failure and diabetes mellitus type 2 in the general population [24]. Lower blood Mg2+ concentration is associated with an increased risk of death from heart failure and coronary heart disease, even at concentrations (0.7–0.8 mmol/L) that fall within the reference range for serum Mg2+, which is typically between 0.7 and 1.0 mmol/L [25, 26].
In both CKD and dialysis patients, similar associations exist, as a number of epidemiological studies have revealed an inverse association between serum Mg2+ concentration and (cardiovascular) mortality risk [4, 27, 28]. Observational studies in large groups of dialysis patients reported that a higher serum Mg2+ concentration is associated with a reduced risk of developing vascular calcification [29]. Dialysis patients with a Mg2+ concentration <1.14 mmol/L showed significantly more risk for mortality [30]. In other studies, blood Mg2+ concentrations <1.23 mmol/L [31], 1.15 mmol/L [27] and 1.21 mmol/L [32] were associated with increased cardiovascular and all-cause mortality in dialysis patients. Of note, these concentrations are all above the reference value for serum Mg2+. This suggests that supranormal Mg2+ concentrations beneficially impact survival rate in CKD. Importantly, a large Japanese cohort study reports a U-shaped curve for this association, as a blood Mg2+ concentration >1.27 mmol/L was associated with increased cardiovascular mortality [20]. Together, these observational studies suggest that a blood Mg2+ concentration between 1.14 and 1.27 mmol/L may be optimal and maintaining blood Mg2+ concentrations in this range in CKD and dialysis patients might be of importance. Of note, it may be of importance to determine the optimal Mg2+ concentration on an individual level.
While the relevance of Mg2+ is increasingly recognized in the context of CKD, Mg2+ status is infrequently assessed in clinical practice. Given recent clinical trials that demonstrate a beneficial effect of Mg2+ supplementation on intermediate endpoints like calcification propensity or coronary artery calcification [33, 34], the time seems right to study the effect of Mg2+ supplementation on clinically relevant outcomes, which so far has not been done.
Magnesium homoeostasis in CKD
The kidney is the central organ for maintaining Mg2+ balance, in addition to less pronounced roles for the intestinal tract and bone. Within the kidney, 90% of the filtered load of Mg2+ is reabsorbed paracellular in the proximal tubule and thick ascending limb of Henle’s [35]. Fine-tuning takes place in the distal convoluted tubule (DCT) [36, 37]. Disturbed Mg2+ reabsorption in the DCT inevitably results in Mg2+ wasting, since Mg2+ cannot be reabsorbed downstream. Consequently DCT-targeting diuretics, e.g. thiazides, are associated with hypomagnesaemia [38].
In patients with CKD, Mg2+ balance is disturbed, initially mainly due to limited ultrafiltration of this mineral. However, patients with early-stage CKD have normal blood Mg2+ concentrations due to an effective increase in fractional excretion of Mg2+ [39]. Additionally, the use of loop diuretics in CKD patients indirectly reduces paracellular reabsorption of Mg2+ in the thick ascending limb of Henle’s loop [40]. Type 2 diabetes, often present in CKD patients, has also been associated with hypomagnesaemia and renal Mg2+ wasting [41, 42]. In more severe CKD, blood Mg2+ concentrations tend to increase, especially if the GFR drops to <10 mL/min/1.73 m2 [43]. In dialysis patients, the blood concentration of Mg2+ is dependent on the dialysate Mg2+ concentrations [39]. The mean serum Mg2+ concentration was in the high–normal range of normal blood Mg2+ concentrations (0.98 mmol/L) in a European cohort of haemodialysis patients [4]. However, on an individual level, hypomagnesaemia and hypomagnesaemia may be present depending on the patient's status and disease severity, possibly depending on dietary intake [44].
Hypomagnesaemia is also common in dialysis patients. A relatively low dialysate Mg2+ concentration of 0.5 mmol/L is most frequently used [45]. In 34 Dutch haemodialysis patients, the mean plasma Mg2+ concentration was 0.88 mmol/L prior to dialysis, which declined post-dialysis to a mean value of 0.78 mmol/L when the dialysate Mg2+ was 0.5 mmol/L [46]. The Mg2+ concentration declined in most individuals. Obviously a dialysate concentration of 0.25 mmol/L Mg2+ increases the risk for hypomagnesaemia even more [39]. In CKD, blood Mg2+ concentrations are further affected by the use of medication (e.g. proton pump inhibitors, thiazides and loop diuretics), the prescription of diets low in potassium (food products rich in potassium are generally also rich in Mg2+) and the presence of diabetes [47]. Monitoring and adjusting the blood Mg2+ concentration in CKD patients might be relevant, as hypomagnesaemia is associated with progression to end-stage renal disease in diabetic nephropathy patients [48, 49].
Bone serves as the body’s Mg2+ store. In total, 60% of the total body Mg2+ is embedded at the surface of the hydroxyapatite crystals [50]. In bone, Mg2+ directly contributes to healthy bone growth. In animal models with Mg2+ deficiency, hydroxyapatite crystals grow tighter and more brittle, resulting in less flexible bone that is more prone to fracture [51]. Multiple studies suggest effects of Mg2+ on bone cell function and fate. However, a consensus is lacking, as results have indicated that Mg2+ both promotes and impairs osteoblastogenesis and osteoclastogenesis in different in vitro studies [52–55]. A recent study showed that mild hypomagnesaemia is associated with a lower risk to bone fracture in late-stage CKD patients [56]. However, too highly elevated blood Mg2+ concentrations may compromise bone mineralization, as Mg2+ is known to interfere with hydroxyapatite formation [57, 58]. Triggered by ageing and uraemia, bone abnormalities such as osteoporosis, osteomalacia and other mineralization defects are common in CKD [59]. As such, properly maintaining Mg2+ status may be important for the already compromised health of bones.
PATHOGENESIS OF VASCULAR CALCIFICATION AND THE ROLE OF CALCIPROTEIN PARTICLES
Medial calcification, as observed in CKD patients, was originally thought to be the result of passive precipitation and deposition of accumulating blood Ca2+ and inorganic Pi in the vessel wall. Almost two decades ago, genes and proteins normally restricted to bone tissue were found to be expressed in calcified arteries of CKD patients [60]. This finding led to the premise that vascular calcification is an active cell-orchestrated process by transdifferentiation of vascular smooth muscle cells (VSMCs) into osteoblast-like cells. VMSCs are triggered by the uremic milieu, well established for Pi and indoxyl sulphate, to change their transcriptional repertoire and lose their contractile phenotype [61]. These transformed VSMCs are characterized by the expression of genes such as Runt-related transcription factor 2 (RUNX2), bone morphogenetic proteins (BMP), osteopontin (OPN encoded by SPP1) and osterix (OSX encoded by SP7), as well as higher alkaline phosphatase (ALP) activity [62, 63], thereby resembling osteoblasts. These cells orchestrate the local spread of vascular calcification by increasing extracellular matrix synthesis and releasing Ca2+-rich exosomes [64]. Experimental evidence strongly suggests that these processes are directly initiated at least partially by excess extracellular Pi and its increased cellular uptake [65]. Interestingly, the results of other studies suggest that VSMC transdifferentiation is a consequence rather than a cause of the vascular calcification process. For example, in vitro studies suggest that VSMC transdifferentiation may be induced primarily by formation of calciprotein particles or nanocrystals in the extracellular space [66] and exosome release facilitating Ca–Pi crystal nucleation [64], DNA damage and cellular senescence [67].
Calciprotein particle formation mediate phosphate-induced calcification
For some years it has been established that elevated blood Pi concentrations give rise to colloidal particles in the circulation [68]. In healthy individuals, despite supersaturated concentrations of soluble Ca2+ and Pi in the circulation, the step from soluble Pi towards amorphous Ca–Pi rarely occurs, due to the presence of inhibitors (e.g. fetuin-A, albumin, osteopontin). In CKD, the Pi concentration increases even more and the expression of inhibitory proteins or colloidal components of these particles, such as fetuin A, decreases [69]. Therefore in the circulation of CKD patients, this protective mineral-buffering system is compromised [70]. When this occurs, Pi, Ca2+ and serum proteins aggregate to form amorphous, soluble Ca–Pi particles or primary calciprotein particles (CPP1). Depending on the local milieu, CPP1 mature to crystalline secondary calciprotein particles (CPP2). The maturation time of CPP1–CPP2, which is measured ex vivo in serum samples in the recently developed T50 test, has been proposed as a measure for the calcification propensity in individuals [71].
CPP2 have been identified in the circulation of CKD patients and their presence is associated with aortic stiffness [72–74]. CPP2, in contrast to CPP1, have been shown to directly induce VSMC calcification in cultured VSMCs [75]. Only nanocrystals and CPP2, but not Pi in solution, induced upregulation of osteogenic proteins in VSMCs [66, 76]. Accordingly, vascular calcification can conceptually be prevented by inhibiting hydroxyapatite formation despite persistently high extracellular Pi concentrations in VSMCs [77]. Of note, it remains unknown whether the CPP2 concentrations used in vitro are comparable to concentrations circulating in CKD patients. Obviously the exposure time in vitro is shorter than in in vivo conditions. Altogether, these findings suggest that not Pi as such, but rather the formation of CPP2, is the true culprit in vascular calcification initiation and progression [70, 78]. Studies in animal models and patients are required to further confirm the causal role of CPP in vascular calcification.
VASCULAR CALCIFICATION IN CKD AND THE ROLE OF MAGNESIUM: EXPERIMENTAL STUDIES
Since a higher serum Mg2+ concentration is associated with improved cardiovascular morbidity and mortality in CKD and dialysis, the potentially preventive effect of Mg2+ has been investigated in a multitude of experimental models. In cellular and rodent models, Mg2+ supplementation prevented high Pi-induced vascular calcification. Mg2+ supplementation to Pi and Ca2+– enriched culture medium effectively suppressed calcification in human, bovine and rat VSMCs in vitro [79–81]. Moreover, Mg2+ prevented calcification in rat aortic rings treated with high Pi and in a rat model of CKD [82, 83]. Additionally, vascular calcification in Klotho knock-out mice was abrogated by a high-Mg2+ diet [84]. A note of caution, Mg2+ may increase the risk for osteomalacia in CKD by disturbing bone mineralization when used in too high concentrations or during development [45, 58, 84]. Although longer-term studies are warranted, a 28-day increase (1.0 versus 0.5 mmol/L) in Mg2+ concentration in dialysate did not lead to a change of bone turnover markers [85].
The anti-calcifying properties of Mg2+ are operational at the medial layer of arteries [86, 87]. A substantial body of studies has investigated potential mechanisms underlying these protective properties. Recent insights regarding the impact of Mg2+ on CPP maturation expanded the spectrum of mechanisms that underlie the anti-calcifying properties of Mg2+ (Figure 1).
FIGURE 1.

Overview of the mechanisms by which magnesium prevents vascular calcification. 1 – High Pi concentrations and the absence of circulating inhibitors such as fetuin-A stimulate the formation of CPP1 in the circulation of CKD patients. Subsequently CPP1 transitions into CPP2 due to a lack of circulating crystallization inhibitors. 2 – In vitro studies suggest that CPP2 and Pi induce calcification in VSMCs and stimulate expression of pro-calcification genes such as RUNX2, ALP (ALPL) and osterix (SP7). Simultaneously, contractility genes such as transgelin (SM22α) diminish. 3 – Combined, this cascade results in VSMC transdifferentiation and calcification and loss of VSMC function and contractility. 4 – The resulting calcified VSMCs amplify the calcification process by shedding Ca-loaded exosomes that are engulfed by neighbouring VSMCs. Mg2+ is proposed to interfere with the calcification process on multiple levels. First, Mg2+ inhibits the transition from CPP1 towards CPP2, preventing VSMC calcification. Second, increased Mg2+ entry facilitated by TRPM7 and possibly angiotensin receptor 2 (AT-2) may directly interfere with Pi-mediated VSMC osteoblast-like transdifferentiation. Third, Mg2+ may restore CaSR activity and MGP γ-carboxylation or protein expression.
Influence of magnesium on VSMC transdifferentiation
As discussed in a previous section, VSMC transdifferentiation towards an osteoblast-like cell has often been suggested to be the driving event of vascular calcification, manifestation and progression [88]. Consequently the leading hypothesis has been that Mg2+ prevents vascular calcification primarily by downregulating pathways involved in the transcription of osteogenic genes. A large number of studies have reported that Mg2+ directly downregulates pro-calcification genes and proteins, among others RUNX2, BMP2 and OSX (reviewed in detail in [79, 81, 89–92]). Other studies report that Mg2+ prevents VSMC calcification through restoring activity of the Ca2+-sensing receptor (CaSR), important for matrix Gla protein (MGP) synthesis [93]. An increase in MGP after Mg2+ supplementation has been reported previously [92]. As such, Mg2+ may increase the abundance of calcification inhibitors. Although these results are often interpreted as direct effects of Mg2+ on gene transcription, it is important to note that the experimental setups are often insufficient to distinguish intracellular from extracellular modes of action. In general, these experiments rely on increasing Pi concentrations in the experimental culture media, which will result in CPP formation in the culture medium. Consequently the effects of Mg2+ may be intracellular, or may depend on extracellular inhibition of CPP maturation, preceding osteogenic transdifferentation [76]. In these setups, measurements of osteoblast-like gene or protein expression is therefore insufficient to draw firm conclusions on the mechanisms involved.
Several studies provide more advanced approaches that support an intracellular role of Mg2+ in the prevention of VSMC transdifferentiation. Knock-down or inhibition of the divalent cation channel transient receptor potential melastatin 7 (TRPM7) impaired the preventive effect of Mg2+ on VSMC calcification [94]. Since TRPM7 is the major Mg2+ channel in VSMCs, these findings demonstrate that inhibition of osteogenic transcription depends on intracellular Mg2+ levels. Several studies indicate that TRPM7 is required for the anti-calcifying effects of Mg2+ in VSMCs. In rat VSMCs, 2-aminoethoxydiphenyl borate (2-APB), a non-selective TRPM7 blocker, abrogated the protective effects of Mg2+ in human high Pi–treated VSMCs supplemented with Mg2+ [91]. Similar results were reported by another study using 2-APB in human VSMCs [80]. In addition, treatment of human VSMCs with a small interfering RNA directed at TRPM7 resulted in calcification despite Mg2+ supplementation [92]. Combined, these studies demonstrate the need for intracellular Mg2+ to limit VSMC calcification. Interestingly, other studies have shown that TRPM7 and increased Mg2+ influx may mediate the anti-calcifying properties of angiotensin type 2 [95].
However, the obligate involvement of TRPM7 remains uncertain, as results from our group demonstrated that Mg2+ actually did prevent bovine VSMC calcification even when TRPM7 was blocked using the channel inhibitor 2-APB [77]. While similar concentrations were used, the use of bovine VSMCs rather than human VSMCs might explain these contradictory findings. Importantly, in most studies it was not accurately assessed whether Mg2+ entry had been modified by TRPM7 blocking, as no validated intracellular Mg2+ probes or patch-clamp techniques were used. Other studies may suggest an alternative role for TRPM7 in VSMC calcification. Its activation increased after treating VSMCs with interleukin-18, a cytokine that stimulated VSMC calcification effectively in vitro [96]. Taken together, the impact of intracellular Mg2+ on VSMC gene transcription should be studied in more detail by using validated intracellular Mg2+ probes and more specific methods of TRPM7 blocking.
Influence of magnesium on calciprotein particle formation
In light of recent data demonstrating that CPP2 are important drivers of VSMC transdifferentiation and vascular calcification, the model describing a major role for Mg2+ on intracellular pro-calcifying pathways might be incomplete [66, 75]. The formation of CPP2 may precede transdifferentiation of VSMCs into an osteoblast-like phenotype. Consequently the inhibition of CPP2 maturation by Mg2+ will then result in reduced osteogenic gene expression and maintain VSMCs in their contractile phenotype.
The effect of Mg2+ on hydroxyapatite formation was widely studied in the 1970s. Although not in the context of CKD, the interference on Ca–Pi crystallization by Mg2+ has been established in several experimental studies [97–100]. First, Mg2+ can substitute Ca2+ in hydroxyapatite formation, favouring the formation of Mg2+-containing whitlockite instead [101–103]. Indeed, whitlockite is sometimes observed in calcified arteries of CKD patients and uraemic rats and has been found to be less pathogenic compared with hydroxyapatite [104–107]. Second, Mg2+ may prevent the formation of hydroxyapatite altogether [77]. In aqueous solution, Mg2+ shields amorphous Ca–Pi particles from transition into crystalline mature hydroxyapatite, thus stabilizing the amorphous phase, which is likely to be applicable during CPP2 formation in the serum [97].
The importance of these mechanisms in the context of CKD-induced vascular calcification was recently tested by our group and by others. Interestingly, Mg2+ did not inhibit initial CPP1 formation from supersaturated concentrations of Ca2+ and Pi but prevented maturation into crystalline CPP2 [76, 98, 99, 108]. Once CPP2 was formed, Mg2+ was incapable of preventing CPP2-induced VSMC mineralization and osteogenic protein expression [76]. Therefore these findings suggest that Mg2+-dependent inhibition of CPP2 maturation is instrumental in the prevention of VSMC calcification (Figure 2). The potential strength of the stabilizing effect of Mg2+ after CPP1 formation is illustrated by the observation that nucleation of amorphous Ca–Pi in aqueous solution is delayed by 2 h after the addition of 57 µmol/L Mg2+ [100]. Given that the normal physiological range of blood Mg2+ is 0.7–1.1 mmol/L, even minor differences in Mg2+ availability may greatly affect the formation of CPP2 from Ca–Pi particles in vivo. In patient sera, supplemental Mg2+ from concentrations as low as 0.2 mmol/L (the average Mg2+ concentration of the serum pools was 0.83 mmol/L) dose-dependently reduced serum calcification propensity [71, 76]. Of note, the required concentration of Mg2+ may depend on the exact composition of the calcifying milieu. Indeed, others reported previously that Mg2+ did not inhibit hydroxyapatite formation during VSMC calcification using high Pi concentrations in the medium [109]. Interestingly, a recent study reported that increasing dialysate Mg2+ for 28 days resulted in a 21% reduction of CPP1 and a 68% reduction of CPP2 [85]. These results suggest that Mg2+ not only inhibits the formation of CPP2 formation, but also CPP1, and translates to patients, lowering the overall CPP burden in CKD.
FIGURE 2.

Close-up of calciprotein particle formation and phase transition. Soluble Pi and Ca2+ in combination with fetuin-A and other serum proteins can aggregate into calciprotein monomers and subsequently these monomers cluster in CPP1 [110]. CPP1 are composed of complexes of fetuin-A and other serum proteins and aggregated amorphous Ca–Pi particles. CPP1 are approximately 50 nm in size and kept stable by serum components. While being initially harmless, the disbalance between calcification promotors (Ca2+, Pi) and inhibitors (fetuin-A, albumin, Mg2+ and alkaline pH) in CKD promote further maturation and transition of CPP1 into crystalline CPP2. CPP2 are larger (150–200 nm) and consist of a crystalline hydroxyapatite core in a protein-covered shell. Mg2+ prevents the maturation of CPP1 into CPP2 but not initial CPP1 formation.
In this context, it is important to realize that intracellular transcriptional effects of Mg2+ and extracellular Mg2+ effects on CPP maturation are not mutually exclusive. Both mechanisms may contribute to reduced vascular calcification in CKD.
A CLINICAL PERSPECTIVE OF MODIFYING MAGNESIUM BALANCE
As CPP formation mainly depends on Ca2+, Pi and fetuin-A concentrations, CPP may explain Pi-stimulated vascular calcification in CKD and thus mediate Pi-induced VSMC calcification. Consequently these pathological effects of Pi may be reduced by preventing CPP2 formation by Mg2+. Indeed, many experimental vascular calcification inhibitors were shown to delay CPP2 maturation [71]. The introduction of the calcification propensity test (T50 test), which measures the transition time from CPP1 to CPP2 ex vivo in serum samples, has created an opportunity to quantify the mineral-buffering capacity of CKD patients to withstand vascular calcification (Figure 3) [71]. A higher calcification propensity (T50) is associated with mortality and the presence and severity of vascular calcification in patients with CKD [111, 112]. Factors reducing calcification propensity are of interest as potential treatment options for progressive vascular calcification.
FIGURE 3.
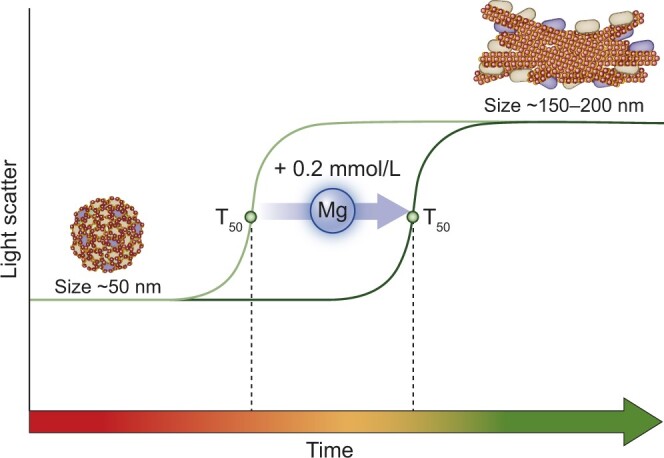
T50 as a measure of calcification propensity and effects of magnesium. The formation of colloidal CPP1 is caused by an imbalance between blood Pi concentrations and calcification inhibitors. The transition time from CPP1 to CPP2, referred to as T50, can be monitored by time-resolved nephelometry and is a measure for the mineral-buffering system of the serum and the calcification propensity. Mg2+ increments of 0.2 mmol/L [76] delay the transition from CPP1 to CPP2, increases T50 and therefore reduces calcification propensity in the serum of CKD patients [71].
The identification of Mg2+ as a targetable factor to modify CPP2 maturation provides an interesting clinical tool for vascular calcification in CKD. Increasing dialysate Mg2+ concentration from 0.5 to 1.0 mmol/L led to an increase in serum Mg2+ concentration of 0.34 mmol/L and improved T50 significantly [113]. In another study, ex vivo addition of Mg2+ to serum samples of pre-transplant CKD patients, dose-dependently improved T50 [76]. In 2019, a clinical trial was published in which MgO was supplemented in 96 pre-dialysis CKD patients, leading to decreased progression of coronary artery calcification, although no effect was seen on thoracic aorta calcification [34]. Now the stage is set for clinical trials in patients with CKD and on dialysis to assess the effectiveness and safety of Mg2+ supplementation in CKD patients on clinically relevant endpoints. However, proper monitoring of efficacy is challenging because establishing differences in clinically relevant endpoints requires long-term follow-up of a substantial number of study participants. It is tempting to assume that, for instance, changes in the T50 antedate changes in these clinical endpoints. While T50 is an informative indicator of calcification propensity in CKD, it is of importance to determine whether Mg2+ supplementation ultimately results in lower calcification scores and its associated clinical events in CKD patients.
CONCLUDING REMARKS
Mg2+ supplementation is a promising treatment option for vascular calcification. Inhibition of CPP2 maturation may be the key molecular mechanism that explains the prevention of vascular calcification by Mg2+, but beneficial effects on gene expression profiles of VSMCs may operate in parallel. As such, Mg2+ is an essential factor in the calcification milieu, which helps to restore the mineral-buffering system overwhelmed by Pi in CKD patients. Consequently the optimal serum Mg2+ concentration for patients with CKD may be higher than in the general population. Future studies are warranted to determine a safe and effective reference range for serum Mg2+ concentrations in CKD patients.
FUNDING
This work was financially supported by grants from the Netherlands Organization for Scientific Research (NWO Veni 016.186.012 and Vici 016.130.668) and the Dutch Kidney Foundation (Kolff 14OKG17, 15OP02 and 16TKI02). The work is co-funded by the PPP Allowance made available by Health∼Holland Top Sector Life Sciences & Health to stimulate public–private partnerships.
CONFLICT OF INTEREST STATEMENT
M.G.V. has received lecture fees and scientific support from Amgen, Vifor Fresenius Medical Care Renal Pharma, Kyowa Kirin, Medice, AbbVie and Fresenius Medical Care. He is an advisor for Amgen, Vifor Fresenius Medical Care Renal Pharma, Kyowa Kirin and Medice and serves on the advisory board of Otsuka. M.G.V. is also a member of the European Renal Association–European Dialysis and Transplantation Association working group on CKD-MBD and a member of the Kidney Disease: Improving Global Outcomes committee on CKD-MBD. All the other authors have declared no competing interests.
REFERENCES
- 1. Schlieper G, Schurgers L, Brandenburg V. et al. Vascular calcification in chronic kidney disease: an update. Nephrol Dial Transplant 2016; 31: 31–39 [DOI] [PubMed] [Google Scholar]
- 2. Blacher J, Guerin AP, Pannier B. et al. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension 2001; 38: 938–942 [DOI] [PubMed] [Google Scholar]
- 3. Wu M, Rementer C, Giachelli CM.. Vascular calcification: an update on mechanisms and challenges in treatment. Calcif Tissue Int 2013; 93: 365–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Roij van Zuijdewijn CLM, Grooteman MPC, Bots ML. et al. Serum magnesium and sudden death in European hemodialysis patients. PLoS One 2015; 10: e0143104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sakaguchi Y, Iwatani H, Hamano T. et al. Magnesium modifies the association between serum phosphate and the risk of progression to end-stage kidney disease in patients with non-diabetic chronic kidney disease. Kidney Int 2015; 88: 833–842 [DOI] [PubMed] [Google Scholar]
- 6. London GM, Guérin AP, Marchais SJ. et al. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 2003; 18: 1731–1740 [DOI] [PubMed] [Google Scholar]
- 7. Sigrist MK, Taal MW, Bungay P. et al. Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin J Am Soc Nephrol 2007; 2: 1241–1248 [DOI] [PubMed] [Google Scholar]
- 8. Budoff MJ, Rader DJ, Reilly MP. et al. Relationship of estimated GFR and coronary artery calcification in the CRIC (Chronic Renal Insufficiency Cohort) study. Am J Kidney Dis 2011; 58: 519–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Braun J, Oldendorf M, Moshage W. et al. Electron beam computed tomography in the evaluation of cardiac calcifications in chronic dialysis patients. Am J Kidney Dis 1996; 27: 394–401 [DOI] [PubMed] [Google Scholar]
- 10. Goodman WG, Goldin J, Kuizon BD. et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med 2000; 342: 1478–1483 [DOI] [PubMed] [Google Scholar]
- 11. Drüeke TB. Arterial intima and media calcification: distinct entities with different pathogenesis or all the same? Clin J Am Soc Nephrol 2008; 3: 1583–1584 [DOI] [PubMed] [Google Scholar]
- 12. Proudfoot D, Shanahan CM, Weissberg PL.. Vascular calcification: new insights into an old problem. J Pathol 1998; 185: 1–3 [DOI] [PubMed] [Google Scholar]
- 13. Everhart JE, Pettitt DJ, Knowler WC. et al. Medial arterial calcification and its association with mortality and complications of diabetes. Diabetologia 1988; 31: 16–23 [DOI] [PubMed] [Google Scholar]
- 14. Chen W, Fitzpatrick J, Monroy-Trujillo JM. et al. Diabetes mellitus modifies the associations of serum magnesium concentration with arterial calcification and stiffness in incident hemodialysis patients. Kidney Int Re 2019; 4: 806–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wyatt CM, Drueke TB.. Vascular calcification in chronic kidney disease: here to stay? Kidney Int 2017; 92: 276–278 [DOI] [PubMed] [Google Scholar]
- 16. O'Neill WC, Lomashvili KA.. Recent progress in the treatment of vascular calcification. Kidney Int 2010; 78: 1232–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ruospo M, Palmer SC, Natale P. Phosphate binders for preventing and treating chronic kidney disease-mineral and bone disorder (CKD-MBD). Cochrane Database Syst Rev2018; 8: CD006023, doi: 10.1002/14651858.CD006023.pub3 [DOI] [PMC free article] [PubMed]
- 18. Lim K, Hamano T, Thadhani R.. Vitamin D and calcimimetics in cardiovascular disease. Semin Nephrol 2018; 38: 251–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Raggi P, Chertow GM, Torres PU. et al. The ADVANCE study: a randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transplant 2011; 26: 1327–1339 [DOI] [PubMed] [Google Scholar]
- 20. Sakaguchi Y, Fujii N, Shoji T. et al. Hypomagnesemia is a significant predictor of cardiovascular and non-cardiovascular mortality in patients undergoing hemodialysis. Kidney Int 2014; 85: 174–181 [DOI] [PubMed] [Google Scholar]
- 21. Cai K, Luo Q, Dai Z. et al. Hypomagnesemia is associated with increased mortality among peritoneal dialysis patients. PLoS One 2016; 11: e0152488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li L, Streja E, Rhee CM. et al. Hypomagnesemia and mortality in incident hemodialysis patients. Am J Kidney Dis 2015; 66: 1047–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guasch-Ferré M, Bulló M, Estruch R. et al. Dietary magnesium intake is inversely associated with mortality in adults at high cardiovascular disease risk. J Nutr 2014; 144: 55–60 [DOI] [PubMed] [Google Scholar]
- 24. Fang X, Wang K, Han D. et al. Dietary magnesium intake and the risk of cardiovascular disease, type 2 diabetes, and all-cause mortality: a dose–response meta-analysis of prospective cohort studies. BMC Med 2016; 14: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lutsey PL, Alonso A, Michos ED, et al. Serum magnesium, phosphorus, and calcium are associated with risk of incident heart failure : the Atherosclerosis Risk in Communities (ARIC). Am J Clin Nutr 2014; 100: 756–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kieboom BCT, Niemeijer MN, Leening MJG. et al. Serum magnesium and the risk of death from coronary heart disease and sudden cardiac death. J Am Heart Assoc 2016; 5: e002707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matias PJ, Azevedo A, Laranjinha I. et al. Lower serum magnesium is associated with cardiovascular risk factors and mortality in haemodialysis patients. Blood Purif 2014; 38: 244–252 [DOI] [PubMed] [Google Scholar]
- 28. Kanbay M, Yilmaz MI, Apetrii M. et al. Relationship between serum magnesium levels and cardiovascular events in chronic kidney disease patients. Am J Nephrol 2012; 36: 228–237 [DOI] [PubMed] [Google Scholar]
- 29. Molnar AO, Biyani M, Hammond I. et al. Lower serum magnesium is associated with vascular calcification in peritoneal dialysis patients: a cross sectional study. BMC Nephrol 2017; 18: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ishimura E, Okuno S, Yamakawa T. et al. Serum magnesium concentration is a significant predictor of mortality in maintenance hemodialysis patients. Magnes Res 2007; 20: 237–244 [PubMed] [Google Scholar]
- 31. Tamura T, Unagami K, Okazaki M. et al. Serum magnesium levels and mortality in Japanese maintenance hemodialysis patients. Blood Purif 2019; 47: 88–94 [DOI] [PubMed] [Google Scholar]
- 32. Yu L, Li H, Wang S-X.. Serum magnesium and mortality in maintenance hemodialysis patients. Blood Purif 2017; 43: 31–36 [DOI] [PubMed] [Google Scholar]
- 33. Bressendorff I, Hansen D, Schou M. et al. Oral magnesium supplementation in chronic kidney disease stages 3 and 4: efficacy, safety, and effect on serum calcification propensity—a prospective randomized double-blinded placebo-controlled clinical trial. Kidney Int Rep 2017; 2: 380–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sakaguchi Y, Hamano T, Obi Y. et al. A randomized trial of magnesium oxide and oral carbon adsorbent for coronary artery calcification in predialysis CKD. J Am Soc Nephrol 2019; 30: 1073–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Baaij JHF, Hoenderop JGJ, Bindels RJM.. Magnesium in man: implications for health and disease. Physiol Rev 2015; 95: 1–46 [DOI] [PubMed] [Google Scholar]
- 36. Blanchard MG, Kittikulsuth W, Nair AV. et al. Regulation of Mg2+ reabsorption and transient receptor potential melastatin type 6 activity by cAMP signaling. J Am Soc Nephrol 2016; 27: 804–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van der Wijst J, Bindels RJM, Hoenderop JGJ.. Mg2+ homeostasis: the balancing act of TRPM6. Curr Opin Nephrol Hypertens 2014; 23: 361–369 [DOI] [PubMed] [Google Scholar]
- 38. Kieboom BCT, Zietse R, Ikram MA. et al. Thiazide but not loop diuretics is associated with hypomagnesaemia in the general population. Pharmacoepidemiol Drug Saf 2018; 27: 1166–1173 [DOI] [PubMed] [Google Scholar]
- 39. Cunningham J, Rodríguez M, Messa P. et al. Magnesium in chronic kidney disease Stages 3 and 4 and in dialysis patients. Clin Kidney J 2012; 5: i39–i59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ryan MP, Devane J, Ryan MF. et al. Effects of diuretics on the renal handling of magnesium. Drugs 1984; 28: 167–181 [DOI] [PubMed] [Google Scholar]
- 41. Kurstjens S, de Baaij JHF, Bouras H. et al. Determinants of hypomagnesemia in patients with type 2 diabetes mellitus. Eur J Endocrinol 2017; 176: 11–19 [DOI] [PubMed] [Google Scholar]
- 42. Kurstjens S, de Baaij JHF, Overmars-Bos C. et al. Increased NEFA levels reduce blood Mg2+ in hypertriacylglycerolaemic states via direct binding of NEFA to Mg2 +. Diabetologia 2019; 62: 311–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Coburn JW, Popovtzer MM, Massry SG. et al. The physicochemical state and renal handling of divalent ions in chronic renal failure. Arch Intern Med 1969; 124: 302–311 [PubMed] [Google Scholar]
- 44. Hruby A, O’Donnell CJ, Jacques PF. et al. Magnesium intake is inversely associated with coronary artery calcification: the Framingham Heart Study. Cardiovasc Imaging 2014; 7: 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Floege J. Magnesium concentration in dialysate: Is higher better? Clin J Am Soc Nephrol 2018; 13: 1309–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leenders NHJ, Van Ittersum FJ, Hoekstra T. et al. Routine hemodialysis induces a decline in plasma magnesium concentration in most patients: a prospective observational cohort study. Sci Rep 2018; 8: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. William JH, Richards K, Danziger J.. Magnesium and drugs commonly used in chronic kidney disease. Adv Chronic Kidney Dis 2018; 25: 267–273 [DOI] [PubMed] [Google Scholar]
- 48. Sakaguchi Y, Shoji T, Hayashi T. et al. Hypomagnesemia in type 2 diabetic nephropathy: a novel predictor of end-stage renal disease. Diabetes Care 2012; 35: 1591–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gommers LMM, Hoenderop JGJ, Bindels RJM. et al. Hypomagnesemia in type 2 diabetes: a vicious circle? Diabetes 2016; 65: 3–13 [DOI] [PubMed] [Google Scholar]
- 50. Alfrey AC, Miller NL, Trow R.. Effect of age and magnesium depletion on bone magnesium pools in rats. J Clin Invest 1974; 54: 1074–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boskey AL, Rimnac CM, Bansal M. et al. Effect of short‐term hypomagnesemia on the chemical and mechanical properties of rat bone. J Orthop Res 1992; 10: 774–783 [DOI] [PubMed] [Google Scholar]
- 52. Wu L, Feyerabend F, Schilling AF. et al. Effects of extracellular magnesium extract on the proliferation and differentiation of human osteoblasts and osteoclasts in coculture. Acta Biomater 2015; 27: 294–304 [DOI] [PubMed] [Google Scholar]
- 53. Mammoli F, Castiglioni S, Parenti S. et al. Magnesium is a key regulator of the balance between osteoclast and osteoblast differentiation in the presence of vitamin D3. Int J Mol Sci 2019; 20: 385–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Leidi M, Dellera F, Mariotti M, Maier JAM.. High magnesium inhibits human osteoblast differentiation in vitro. Magnes Res 2011; 24: 1–6 [DOI] [PubMed] [Google Scholar]
- 55. Lu WC, Pringa E, Chou L.. Effect of magnesium on the osteogenesis of normal human osteoblasts. Magnes Res 2017; 30: 42–52 [DOI] [PubMed] [Google Scholar]
- 56. Sakaguchi Y, Hamano T, Wada A. et al. Magnesium and risk of hip fracture among patients undergoing hemodialysis. J Am Soc Nephrol 2018; 29: 991–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Castiglioni S, Cazzaniga A, Albisetti W. et al. Magnesium and osteoporosis: current state of knowledge and future research directions. Nutrients 2013; 5: 3022–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gonella M, Ballanti PD, Rocca C. et al. Improved bone morphology by normalizing serum magnesium in chronically hemodialyzed patients. Miner Electrolyte Metab 1988; 14: 240–245 [PubMed] [Google Scholar]
- 59. Covic A, Vervloet M, Massy ZA. et al. Bone and mineral disorders in chronic kidney disease: implications for cardiovascular health and ageing in the general population. Lancet Diabetes Endocrinol 2018; 6: 319–331 [DOI] [PubMed] [Google Scholar]
- 60. Shanahan CM, Cary NRB, Salisbury JR. et al. Medial localization of mineralization-regulating proteins in association with Mönckeberg’s sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation 1999; 100: 2168–2176 [DOI] [PubMed] [Google Scholar]
- 61. Bouabdallah J, Zibara K, Issa H. et al. Endothelial cells exposed to phosphate and indoxyl sulphate promote vascular calcification through interleukin-8 secretion. Nephrol Dial Transplant 2019; 34: 1125–1134 [DOI] [PubMed] [Google Scholar]
- 62. Steitz SA, Speer MY, Curinga G. et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res 2001; 89: 1147–1154 [DOI] [PubMed] [Google Scholar]
- 63. Shroff RC, McNair R, Skepper JN. et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 2010; 21: 103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kapustin AN, Chatrou MLL, Drozdov I. et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res 2015; 116: 1312–1323 [DOI] [PubMed] [Google Scholar]
- 65. Crouthamel MH, Lau WL, Leaf EM. et al. Sodium-dependent phosphate cotransporters and phosphate-induced calcification of vascular smooth muscle cells: redundant roles for PiT-1 and PiT-2. Arterioscler Thromb Vasc Biol 2013; 33: 2625–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sage AP, Lu J, Tintut Y. et al. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int 2011; 79: 414–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu Y, Drozdov I, Shroff R. et al. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res 2013; 112: e99–e109 [DOI] [PubMed] [Google Scholar]
- 68. Smith ER, Cai MM, McMahon LP. et al. Serum fetuin-A concentration and fetuin-A-containing calciprotein particles in patients with chronic inflammatory disease and renal failure. Nephrology 2013; 18: 215–221 [DOI] [PubMed] [Google Scholar]
- 69. Moe SM, Reslerova M, Ketteler M. et al. Role of calcification inhibitors in the pathogenesis of vascular calcification in chronic kidney disease (CKD). Kidney Int 2005; 67: 2295–2304 [DOI] [PubMed] [Google Scholar]
- 70. Pasch A, Jahnen-Dechent W, Smith ER.. Phosphate, calcification in blood, and mineral stress: the physiologic blood mineral buffering system and its association with cardiovascular risk. Int J Nephrol 2018; 2018:9182078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pasch A, Farese S, Graber S. et al. Nanoparticle-based test measures overall propensity for calcification in serum. J Am Soc Nephrol 2012; 23: 1744–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Heiss A, Eckert T, Aretz A. et al. Hierarchical role of fetuin-A and acidic serum proteins in the formation and stabilization of calcium phosphate particles. J Biol Chem 2008; 283: 14815–14825 [DOI] [PubMed] [Google Scholar]
- 73. Hamano T, Matsui I, Mikami S. et al. Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J Am Soc Nephrol 2010; 21: 1998–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Smith ER, Ford ML, Tomlinson LA. et al. Serum calcification propensity predicts all-cause mortality in predialysis CKD. J Am Soc Nephrol 2014; 25: 339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Aghagolzadeh P, Bachtler M, Bijarnia R. et al. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor α. Atherosclerosis 2016; 251: 404–411 [DOI] [PubMed] [Google Scholar]
- 76. ter Braake AD, Eelderink C, Zeper LW. et al. Calciprotein particle inhibition explains magnesium-mediated protection against vascular calcification. Nephrol Dial Transplant 2020; 35: 765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. ter Braake AD, Tinnemans PT, Shanahan CM. et al. Magnesium prevents vascular calcification in vitro by inhibition of hydroxyapatite crystal formation. Sci Rep 2018; 8: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vervloet MG. Modifying phosphate toxicity in chronic kidney disease. Toxins (Basel) 2019; 11: 522–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kircelli F, Peter ME, Sevinc Ok E. et al. Magnesium reduces calcification in bovine vascular smooth muscle cells in a dose-dependent manner. Nephrol Dial Transplant 2012; 27: 514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Louvet L, Büchel J, Steppan S. et al. Magnesium prevents phosphate-induced calcification in human aortic vascular smooth muscle cells. Nephrol Dial Transplant 2013; 28: 869–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bai Y, Zhang J, Xu J. et al. Magnesium prevents β-glycerophosphate-induced calcification in rat aortic vascular smooth muscle cells. Biomed Reports 2015; 3: 593–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Diaz-Tocados JM, Peralta-Ramirez A, Rodríguez-Ortiz ME. et al. Dietary magnesium supplementation prevents and reverses vascular and soft tissue calcifications in uremic rats. Kidney Int 2017; 92: 1084–1099 [DOI] [PubMed] [Google Scholar]
- 83. Salem S, Bruck H, Bahlmann FH. et al. Relationship between magnesium and clinical biomarkers on inhibition of vascular calcification. Am J Nephrol 2012; 35: 31–39 [DOI] [PubMed] [Google Scholar]
- 84. Braake AD, Smit AE, Bos C. et al. Magnesium prevents vascular calcification in Klotho deficiency. Kidney Int 2020; 97: 487–501 [DOI] [PubMed] [Google Scholar]
- 85. Bressendorff I, Hansen D, Pasch A. et al. The effect of increasing dialysate magnesium on calciprotein particles, inflammation and bone markers: post hoc analysis from a randomized controlled clinical trial. Nephrol Dial Transplant 2019; 10.1093/ndt/gfz234 [DOI] [PubMed] [Google Scholar]
- 86. Shechter M. Magnesium and cardiovascular system. Magnes Res 2010; 23: 60–72 [DOI] [PubMed] [Google Scholar]
- 87. Shechter M, Sharir M, Labrador MJP. et al. Oral magnesium therapy improves endothelial function in patients with coronary artery disease. Circulation 2000; 102: 2353–2848 [DOI] [PubMed] [Google Scholar]
- 88. Giachelli CM. The emerging role of phosphate in vascular calcification. Kidney Int 2009; 75: 890–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. ter Braake AD, Shanahan CM, de Baaij JHF.. Magnesium counteracts vascular calcification: passive interference or active modulation? Arterioscler Thromb Vasc Biol 2017; 37: 1431–1445 [DOI] [PubMed] [Google Scholar]
- 90. Louvet L, Metzinger L, Büchel J. et al. Magnesium attenuates phosphate-induced deregulation of a microRNA signature and prevents modulation of Smad1 and Osterix during the course of vascular calcification. Biomed Res Int 2016; 2016:7419524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Montezano AC, Zimmerman D, Yusuf H. et al. Vascular smooth muscle cell differentiation to an osteogenic phenotype involves TRPM7 modulation by magnesium. Hypertension 2010; 56: 453–462 [DOI] [PubMed] [Google Scholar]
- 92. De Oca AM, Guerrero F, Martinez-Moreno JM. et al. Magnesium inhibits Wnt/β-catenin activity and reverses the osteogenic transformation of vascular smooth muscle cells. PLoS One 2014; 9: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Alesutan I, Tuffaha R, Auer T. et al. Inhibition of osteo/chondrogenic transformation of vascular smooth muscle cells by MgCl2 via calcium-sensing receptor. J Hypertens 5017; 35523–532 [DOI] [PubMed] [Google Scholar]
- 94. He Y, Yao G, Savoia C, Touyz RM.. Transient receptor potential melastatin 7 ion channels regulate magnesium homeostasis in vascular smooth muscle cells: role of angiotensin II. Circ Res 2005; 96: 207–215 [DOI] [PubMed] [Google Scholar]
- 95. Herencia C, Rodriguez-Ortiz ME, Munoz-Castaneda JR. et al. Angiotensin II prevents calcification in vascular smooth muscle cells by enhancing magnesium influx. Eur J Clin Invest 2015; 45: 1129–1144 [DOI] [PubMed] [Google Scholar]
- 96. Zhang K, Zhang Y, Feng W. et al. Interleukin-18 enhances vascular calcification and osteogenic differentiation of vascular smooth muscle cells through TRPM7 activation. Arterioscler Thromb Vasc Biol 2017; 37: 1933–1943 [DOI] [PubMed] [Google Scholar]
- 97. Boskey AL, Posner AS.. Magnesium stabilization of amorphous calcium phosphate: a kinetic study. Mater Res Bull 1974; 9: 907–916 [Google Scholar]
- 98. Termine JD, Peckauskas RA, Posner AS.. Calcium phosphate formation in vitro. II. Effects of environment on amorphous-crystalline transformation. Arch Biochem Biophys 1970; 140: 318–325 [DOI] [PubMed] [Google Scholar]
- 99. Termine JD, Posner AS.. Calcium phosphate formation in vitro. I. Factors affecting initial phase separation. Arch Biochem Biophys 1970; 140: 307–317 [DOI] [PubMed] [Google Scholar]
- 100. Blumenthal NC, Betts F, Posner AS.. Stabilization of amorphous calcium phosphate by Mg and ATP. Calc Tis Res 1977; 23: 245–250 [DOI] [PubMed] [Google Scholar]
- 101. Tenhuisen KS, Brown PW.. Effects of magnesium on the formation of calcium-deficient hydroxyapatite from CaHPO4 2H2O and Ca4(PO4)2O. Calcif Tissue Int 1996; 4: 538–546 [DOI] [PubMed] [Google Scholar]
- 102. Abbona F, Baronnet A.. A XRD and TEM study on the transformation of amorphous calcium phosphate in the presence of magnesium. J Cryst Growth 1996; 165: 98–105 [Google Scholar]
- 103. Lagier R, Baud C-A.. Magnesium whitlockite, a calcium phosphate crystal of special interest in pathology. Pathol Res Pract 2003; 199: 329–335 [DOI] [PubMed] [Google Scholar]
- 104. Schlieper G, Aretz A, Verberckmoes SC. et al. Ultrastructural analysis of vascular calcifications in uremia. J Am Soc Nephrol 2010; 21: 689–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Verberckmoes SC, Persy V, Behets GJ. et al. Uremia-related vascular calcification: more than apatite deposition. Kidney Int 2007; 71: 298–303 [DOI] [PubMed] [Google Scholar]
- 106. LeGeros RZ, Contiguglia SR, Alfrey AC.. Pathological calcifications associated with uremia – two types of calcium phosphate deposits. Calc Tis Res 1973; 13: 173–185 [DOI] [PubMed] [Google Scholar]
- 107. Ryan LM, Cheung HS, LeGeros RZ. et al. Cellular responses to whitlockite. Calcif Tissue Int 1999; 65: 374–377 [DOI] [PubMed] [Google Scholar]
- 108. Eanes ED, Posner AS.. Kinetics and mechanism of conversion of non-crystalline calcium phosphate to hydroxyapatite. Trans N Y Acad Sci 1965; 28: 233–241 [Google Scholar]
- 109. Louvet L, Bazin D, Büchel J. et al. Characterisation of calcium phosphate crystals on calcified human aortic vascular smooth muscle cells and potential role of magnesium. PLoS One 2015; 10: e0115342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Heiss A, Pipich V, Jahnen-Dechent W. et al. Fetuin-A is a mineral carrier protein: small angle neutron scattering provides new insight on fetuin-A controlled calcification inhibition. Biophys J 2010; 99: 3986–3995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Keyzer CA, de Borst MH, van den Berg E. et al. Calcification propensity and survival among renal transplant recipients. J Am Soc Nephrol 2015; 27: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bundy JD, Cai X, Scialla JJ. et al. Serum calcification propensity and coronary artery calcification among patients with CKD: the CRIC (Chronic Renal Insufficiency Cohort) study. Am J Kidney Dis 2019; 73: 806–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bressendorff I, Hansen D, Schou M. et al. The effect of increasing dialysate magnesium on serum calcification propensity in subjects with end stage kidney disease. Clin J Am Soc Nephrol 2018; 13: 1373–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]