

Abstract
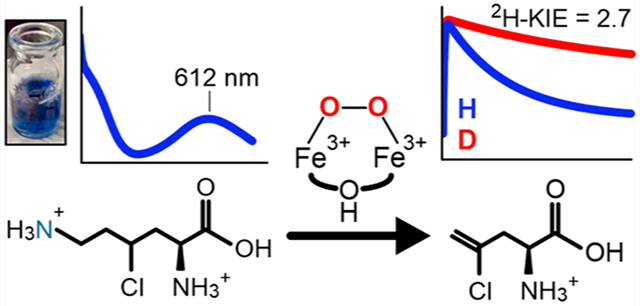
BesC catalyzes the iron- and O2-dependent cleavage of 4-chloro-l-lysine to form 4-chloro-l-allylglycine, formaldehyde, and ammonia. This process is a critical step for a biosynthetic pathway that generates a terminal alkyne amino acid which can be leveraged as a useful bio-orthogonal handle for protein labeling. As a member of an emerging family of diiron enzymes that are typified by their heme oxygenase-like fold and a very similar set of coordinating ligands, recently termed HDOs, BesC performs an unusual type of carbon–carbon cleavage reaction that is a significant departure from reactions catalyzed by canonical dinuclear-iron enzymes. Here, we show that BesC activates O2 in a substrate-gated manner to generate a diferric-peroxo intermediate. Examination of the reactivity of the peroxo intermediate with a series of lysine derivatives demonstrates that BesC initiates this unique reaction trajectory via cleavage of the C4–H bond; this process represents the rate-limiting step in a single turnover reaction. The observed reactivity of BesC represents the first example of a dinuclear-iron enzyme that utilizes a diferric-peroxo intermediate to capably cleave a C–H bond as part of its native function, thus circumventing the formation of a high-valent intermediate more commonly associated with substrate monooxygenations.
Graphical Abstract
INTRODUCTION
Using comparative genomics, a novel biosynthetic pathway for the production of terminal alkyne amino acids was recently elucidated.1 Genetic knockouts and in vitro assembly have revealed a biosynthetic logic whereby a chloride is initially installed at the C4 position of l-lysine by the iron(II)- and α-ketoglutarate (αKG)-dependent halogenase BesD, followed by formation of a terminal alkene that results from a carbon–carbon cleavage reaction catalyzed by the iron-dependent oxidase BesC (Figure 1A). Subsequent elimination of the chloride is performed by the pyridoxal 5′-phosphate (PLP)-dependent acetylenase BesB, generating a terminal-alkyne moiety. Although this pathway is most likely natively used by Streptomyces cattleya for the generation of dipeptide natural products, Chang and co-workers have elegantly shown that the resulting terminal alkyne-containing amino acids can also be leveraged as a unique bio-orthogonal handle for protein labeling through azide–alkyne cycloaddition, or “click”, chemistry.
Figure 1.
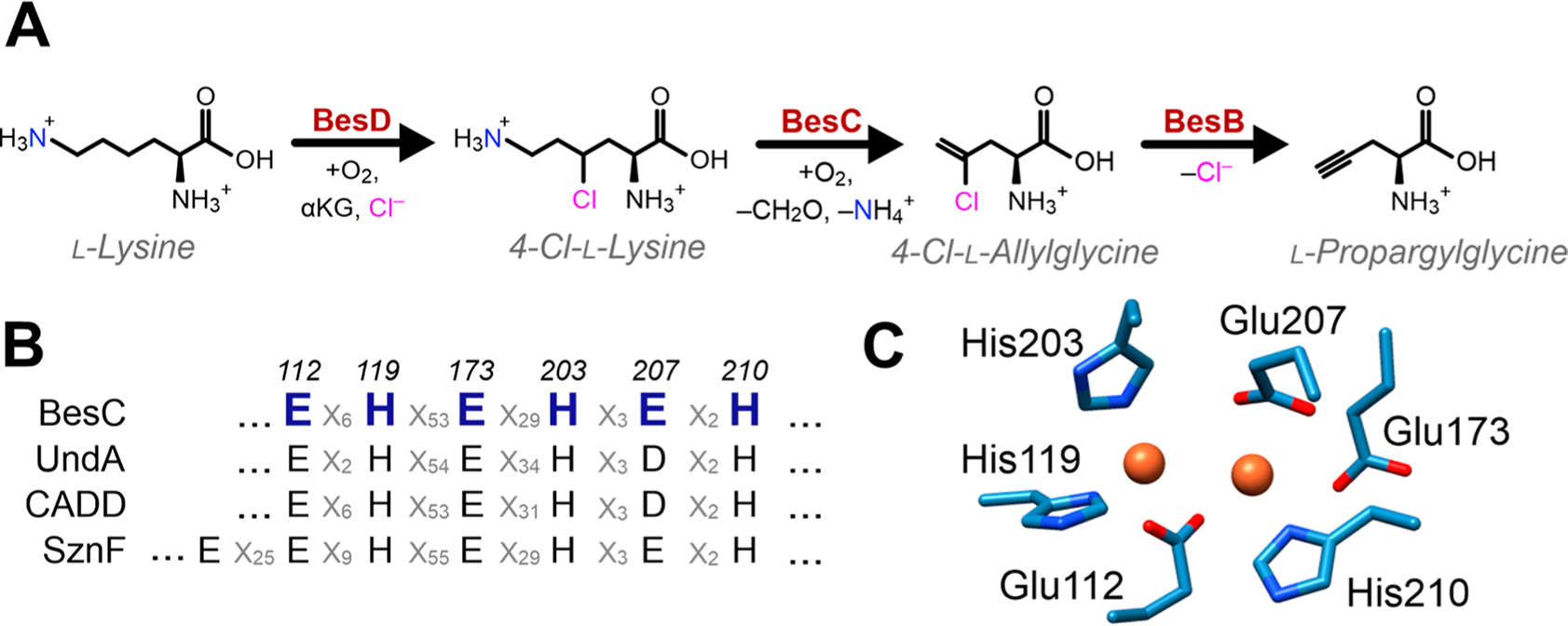
Oxidative C–C cleavage by BesC. (A) The partial biosynthetic pathway for terminal alkyne amino acids from l-lysine in S. cattleya. (B) Sequence alignment demonstrates conservation of the iron-ligating residues of BesC with other selected HDOs. (C) Structural alignment of the diiron site predicted by a homology model of BesC prepared using Swiss-Model using CADD (PDB ID: 1RCW) as a template.
Although the reactions catalyzed by BesD and BesB are more readily rationalized on the basis of relatively well-established Fe(II)/αKG halogenation2,3 and PLP-dependent desaturation,4,5 the carbon–carbon scission of 4-chloro-l-lysine (4-Cl-Lys) by BesC to yield 4-chloro-l-allylglycine (4-Cl-Alg), formaldehyde, and ammonia is poorly understood. Based on sequence similarity, BesC is proposed to be a member of a newly recognized family of dinuclear-iron enzymes that are typified by their heme oxygenase (HO)-like fold (HDOs). Intriguingly, HDOs have been shown to afford a breadth of divergent reactivities for natural product biosynthesis that emanate from a similar diiron core structure. Notably, enzymes from this structural family include the fatty acid decarboxylase UndA,6–8 the N-oxygenase SznF,9–11 and CADD,12 with the latter purportedly producing para-aminobenzoate through cleavage of a protein-derived tyrosine for the ultimate integration into folate.13–15 Intriguingly, each of these proteins is either predicted7,8 or structurally shown11,12 to contain a remarkably similar coordination motif of the diiron cluster that consists of two histidine ligands to one iron, and one histidine ligand and two carboxylates to the second iron (Figure 1B, C). The ligation motif is finalized by bridges consisting of a glutamate and one or more (hydr)oxo ligands. The structure of SznF reveals an additional glutamate ligand to the 2-His Fe that is thought to possibly enable its N-oxygenating activity.11 Notably, a very similar asymmetric ligand set is found in the diiron N-oxygenases CmlI and AurF16,17 even though a 4-helix ferritin (FDO) structural architecture is utilized to house the cluster, rather than 3-helices used by HDOs.
A key mechanistic question that arises is how similar coordination motifs can give rise to such divergent chemical outcomes in HDOs. Examination of the reactions of diferrous SznF and UndA with O2 have demonstrated the presence of a peroxo-diferric intermediate in each enzyme, thus unifying the initial steps of their oxygen activation mechanisms with other diiron enzymes that more commonly perform hydroxylations, radical transfer, or desaturations.18 For SznF, the peroxo species can be readily generated in the absence of a bound substrate, but its decay is appreciably accelerated in the presence of amine substrates, suggesting it may represent the activated-O2 species that initiates catalysis, an attribute shared with the arylamine N-oxygenases CmlI19 and AurF.20 In contrast, the UndA diferric-peroxo (UndA-P) is only detectable in the presence of a bound fatty acid, and has been proposed to transition to a diiron(IV)-oxo intermediate on the basis of isolation of an Fe2(III/IV) species detected upon UndA-P decay,8 analogous to mononuclear decarboxylases that initiate C–H abstraction via a ferryl-intermediate,21,22 and computational grounds.23
To further explore the origins for variations in oxygen-activation strategies and possible branching of the diferric-peroxo intermediate in HDOs between direct reactivity and further activation to a high-valent species, we have investigated the reaction of BesC with dioxygen and a series of substrate analogs. Transient absorption and Mössbauer studies show that diferrous BesC rapidly reacts with O2 in the presence of lysine substrates to form a remarkably long-lived peroxo-diferric species (BesC-P). Through variation of the C4–H bond strength using C4–D and Cl–C4–H lysine analogs, we demonstrate that BesC likely initiates C–C cleavage through direct substrate C–H abstraction by this peroxo species. The observed reactivity of BesC-P represents the first known example of an enzymatic diferric-peroxo intermediate that capably cleaves relatively strong C–H bonds as part of its native reaction coordinate and rationalizes the need for halogen installation to promote efficient C–C cleavage.
RESULTS
Substrate-Triggered Formation of a Diiron Peroxo Species.
As found for other members of the HDO family,7–10 BesC contained very little iron bound (<0.05 mol equiv per enzyme) to the enzyme following affinity chromatography (Figure S1). Following anaerobic incubation with 2 equiv of Fe(II) and exposure to O2, the substrate-free enzyme oxidized over the course of 10 min in a slow, monophasic manner, as monitored by a gradual increase in absorbance at ~340 nm (Figure 2A). To investigate the BesC reaction, 4-Cl-Lys, the presumptive native substrate, was prepared. The enzymatic generation and purification of 4-Cl-Lys using the BesD halogenase is described in the SI, and characterization is shown in Figures S2 to S10. Upon the addition of 4-Cl-Lys to the ferrous enzyme and subsequent mixing with O2, a blue species with absorption maxima at 320 and 612 nm rapidly formed within seconds (Figure 2B). The absorption features of this species (ε612 ≈ 2400 M−1 cm−1) strongly resemble μ-peroxodiiron(III) intermediates that are found in several diiron enzymes and biomimetic complexes that typically exhibit peroxo-to-Fe(III) charge transfer bands with λmax values that range from 500 to 720 nm (see Table S1 for a compilation). The decay process proceeds slowly over 100 s without the accumulation of any other distinguishable intermediates in the visible region.
Figure 2.
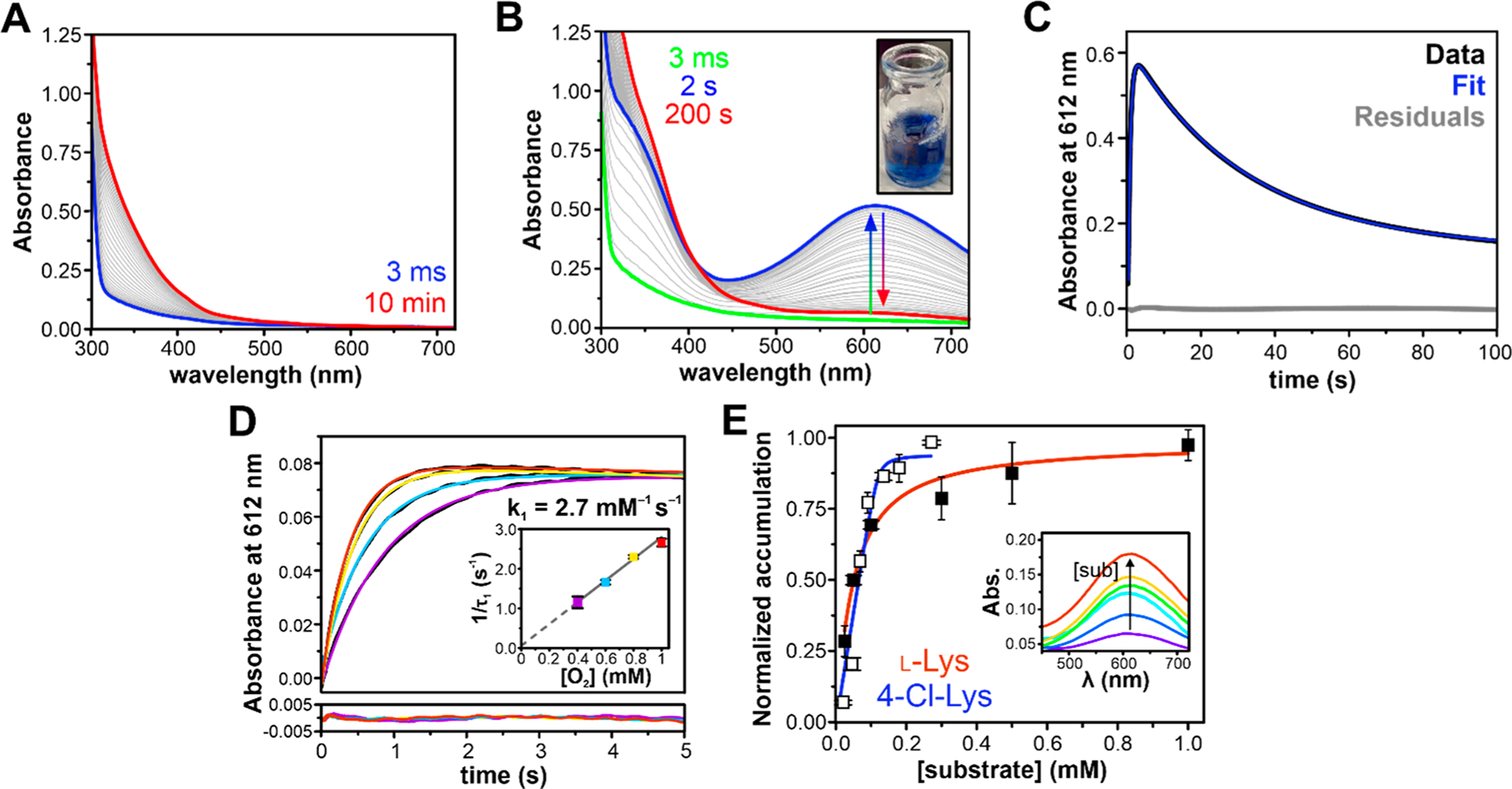
Substrate-triggered formation of an oxygenated intermediate in BesC. (A) Rapid mixing of diferrous BesC (300 μM postmix concentration) with O2-saturated buffer (1 mM postmix) in the absence of substrate results in slow autoxidation. (B) Substrate-bound BesC (300 μM BesC, 1.5 mM 4-Cl-Lys postmix) activates oxygen to produce a blue 612 nm chromophore, shown at a 1 mM O2 postmix concentration in the inset. (C) The single-wavelength time-course measured at 612 nm can be fit with a two-summed exponential expression showing single formation and decay phases. (D) The formation rate of BesC-P is linearly dependent on [O2], measured at a postmix O2 concentrations of 1.0 mM (red), 0.8 mM (yellow), 0.6 mM (blue), and 0.4 mM (purple) and a BesC concentration of 50 μM. (E) Accumulation of BesC-P shows a hyperbolic dependence on substrate concentration and is used to determine the relative efficiencies for l-(Cl)-Lys binding and O2 activation. Postmixing concentration of protein was 100 μM, and substrate concentrations used were 25, 50, 100, 300, 500, and 1000 μM for Lys (filled squares) and 25, 50, 75, 100, 125, 175, and 250 μM for 4-Cl-Lys (open squares). Inset: Representative PDA spectra of maximum BesC-P accumulation at different concentrations of 4-Cl-Lys, with warmer colors indicating higher concentrations. All experiments were performed at 4 °C.
The full time-course for the 612 nm absorbing intermediate, as generated with excess O2 and 4-Cl-Lys, could be adequately fit to a two-exponential expression, revealing reciprocal relaxation times of 1.6 ± 0.3 s−1 and 0.030 ± 0.001 s−1 using a postmix concentration of 1 mM O2 at 4 °C (Figure 2C). The kinetic behavior of the intermediate was further probed with varying O2 concentrations under pseudo-first-order conditions, enabling assignment of the two kinetic phases. Only the fast reciprocal relaxation time (designated here as 1/t1) is dependent on oxygen concentration, and the 1/t1 versus [O2] plot reveals a zero y-intercept (Figure 2D, Figure S11). This implies that, in the presence of bound 4-Cl-Lys, the 612 nm intermediate is formed essentially irreversibly, as observed for several synthetic peroxo-adducts (e.g., refs 24 and 25). The slope of the plot indicates a bimolecular rate constant for O2-binding ~2.7 mM−1 s−1.
Multiple turnover experiments using ascorbate as a redox donor1 suggested that both 4-Cl-Lys and l-Lys can serve as competent substrates for BesC and that both are cleaved to form a terminal alkene. We observed that l-Lys also enables efficient formation of the blue chromophore. A comparison of the 612 nm formation time-course for both substrates in the presence of 5 mol equiv of substrate is shown in Figure S12A. In the case of l-Lys, the 612 nm intermediate forms an order of magnitude more slowly, maximizing over 20 s rather than <2 s for 4-Cl-Lys. Accordingly, the formation kinetics for the intermediate in the presence of l-Lys are significantly more complex and required fitting to a two-summed exponential with only one phase that is dependent on O2 concentration and a second phase that is O2 independent (Figure S12B, C).
The triggering capacity for both substrates was further interrogated through examination of the amplitude of the 612 nm species that is maximally formed versus substrate concentration. Given that substrate binding is obligatory for intermediate formation, that the rates for formation far exceed those for decomposition (>2 orders of magnitude), and that the decay kinetics are independent of substrate concentration (Figure S13), this approach provides a convenient handle to probe for differences in apparent substrate-binding parameters. The accumulation of the blue intermediate (Figure 2E) displayed a hyperbolic dependence on substrate concentration and could be fit by nonlinear Morrison fitting procedures that are appropriate for tight-binding substrates to yield apparent substrate affinities of 1 and 40 μM for 4-Cl-Lys and l-Lys, respectively.
Mössbauer Characterization of the BesC Reaction Coordinate.
We used Mössbauer spectroscopy to interrogate how substrates potentially prime the diiron cluster to enable oxygen activation and to further probe the nature of the 612 nm intermediate. The spectrum of diferrous BesC in the absence of substrate showed a broad quadrupole doublet (Figure 3A). The spectral simulation requires two partially overlapping quadrupole doublets with parameters of typical high-spin ferrous species (δ ≈ 1.25 mm/s, ΔEQ = 2.0–3.0 mm/s; see Table 1). The addition of 4-Cl-Lys clearly perturbs the Mössbauer spectrum of diferrous BesC, suggesting 4-Cl-Lys interacts with the diferrous center (Figure 3B). Although a spectral simulation including two ferrous doublets can reasonably reproduce the experimental data, more rigorous simulation requires the inclusion of three ferrous sites with a nearly equal ratio (Tables 1, S2 and Figure S14), which may suggest that the addition of 4-Cl-Lys induces further structural inhomogeneity on the diferrous center. The addition of l-Lys to diferrous BesC also perturbs the Mössbauer spectrum and results in a spectrum that is clearly different from those of substrate-free and 4-Cl-Lys-bound diferrous BesC (Table S3), implying that the binding of l-Lys to the diferrous center is slightly different from that of 4-Cl-Lys. A comparison of diferrous BesC in the presence and absence of these derivatives is shown in Figure S14. The spectral perturbations observed for diferrous BesC upon substrate binding are remarkably reminiscent of those observed upon fatty acid binding to diferrous UndA,7,8 which also requires a bound substrate to enable efficient O2 activation. Notably, this behavior deviates from N-oxygenases that do not exhibit substrate-triggering requirements (Table S4).
Figure 3.
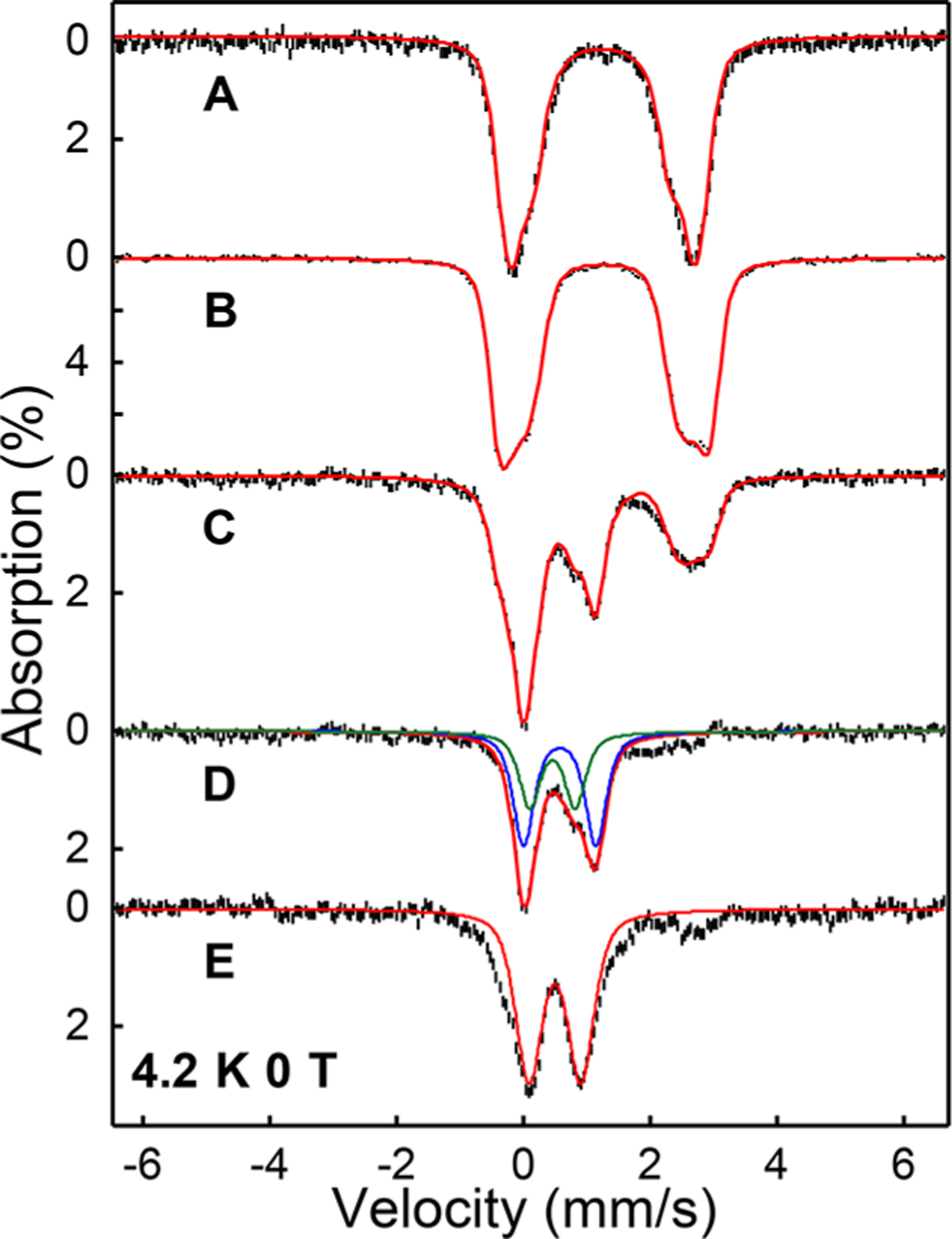
Mössbauer spectroscopy of BesC-P. (A) Zero-field, 4.2 K Mössbauer spectra of diferrous BesC, (B) 4-Cl-Lys-bound diferrous BesC, (C) the oxygenated 4-Cl-Lys-bound diferrous BesC frozen at 1 min, (D) the difference spectrum generated by subtracting 40% (B) from (C), and (E) fully decayed BesC-P. The red solid lines are the overall spectral simulations; the simulation parameters are listed in Table 1 and Table S5. For (D), the blue and the green lines represent the spectral simulation of BesC-P (Species I) and the initial product complex (Species II), respectively.
Table 1.
Parameters Used for the Fitting of the Diiron Clusters of BesC in the Mössbauer Spectra Shown in Figure 3a
| Site | δ (mm/s) | ΔEQ (mm/s) | Γb (mm/s) | Spectral area (%) | |
|---|---|---|---|---|---|
| Diferrous | 1 | 1.25(1) | 2.97(3) | −0.45 | 60(2) |
| 2 | 1.24(1) | 2.15(3) | −0.50 | 40(2) | |
| Diferrous (4-Cl-Lys) | 1 | 1.26(1) | 2.26(3) | −0.45 | 35(2) |
| 2 | 1.26(1) | 2.83(3) | −0.42 | 28(2) | |
| 3 | 1.28(1) | 3.34(3) | −0.34 | 37(2) | |
| BesC-P (4-Cl-Lys) Species I | 1 | 0.56(1) | 1.15(3) | 0.36 | 33(2) |
| Species II | 1 | 0.46(1) | 0.68(3) | 0.45 | 18(2) |
| Decayed BesC-P | 1 | 0.50(1) | 0.82(3) | 0.5 | 45(2) |
The numbers in the parentheses represent the uncertainties of the simulation parameters.
The negative line width represents a Voigt line shape function with a convolution of 50% Gaussian and 50% Lorentzian.
Upon exposure of 4-Cl-Lys-bound BesC to O2, the intensity of the three quadrupole doublets associated with the reactant diferrous complex decreased, and two new species (denoted as Species I and II) represented by two distinct quadrupole doublets concomitantly formed at the earliest freezing time (Figure 3C, 3D). Species I with Mössbauer parameters of δ = 0.57 mm/s and ΔEQ = 1.15 mm/s was also formed upon exposure of the l-Lys reactant complex with O2 (Figure S15 and Table S3), both of which have parameters that are similar to synthetic and enzymatic μ-peroxodiiron(III) complexes, including those trapped in UndA8 and SznF.10 Thus, we assign Species I as BesC-P. The temperature- and magnetic-field-dependence of the BesC-P signal allows for determination of the exchange coupling constant of J ≈ 35 cm−1 (Figure S20), suggesting the two Fe(III) centers are antiferromagnetically coupled and similar to many reported peroxo-diferric intermediates (Table S1). The decay of the BesC-P quadrupole doublet closely approximates the kinetics observed at 612 nm, further confirming the identity of the blue species (Figure S16, Tables S5 and S6).
Species II, with Mössbauer parameters of ~0.46 mm/s and ΔEQ = 0.68 mm/s, was only identified in the reaction of 4-Cl-Lys-bound BesC with O2. Based on the Mössbauer spectra of samples frozen at different reaction times, Species I (BesC-P) and II appear sequentially, with the decay of BesC-P leading to further accumulation of Species II up to a reaction time of ~3 min (Figure S17 and Table S5). Thus, Species II is most likely an intermediate directly downstream of the diferric-peroxo species, and we currently assign it as an initial product complex. We further reason that the inability to observe a similar product complex in the BesC reaction with l-Lys (Figure S17 and Table S6) may result from significant reaction uncoupling in l-Lys metabolism and the slower kinetics for BesC-P decay (vide infra) that render accumulation of a downstream intermediate less probable.
The Mössbauer parameters of Species II clearly indicate that it is a ferric species. However, its magnetic behavior suggests it is not a conventional diferric species with an S = 0 ground spin state. Instead, Species II significantly magnetizes even under a small applied magnetic field (450 G) (Figure S18), which strongly indicates that it exhibits an integer spin ground state (S > 0). Due to the relatively low accumulation of this species, we cannot yet reliably determine its spin state and the electronic structure under the current experimental conditions. Finally, the spectra of samples frozen at prolonged reaction times demonstrate the disappearance of the diferric-peroxo signal and formation of a broad quadrupole doublet with Mössbauer parameters of δ = 0.5 mm/s and ΔEQ = 0.8 mm/s, which could be assigned to the final ferric species (Figure 3E, Table 1). In addition, the spectra collected under high applied magnetic fields suggest that as much as ~60% of this new species is mononuclear in nature, suggesting cluster decomposition under these conditions (Figure S19). We address the potential ramifications of this cluster instability further in the Discussion.
Evidence that Peroxo BesC Mediates C–C Cleavage via Substrate C–H Abstraction.
Single-turnover reactions of BesC with 4-Cl-Lys as monitored by LC-MS demonstrated stoichiometric metabolism of 4-Cl-Lys (Figure 4A, Figure S21). The detected products had m/z = 150.1 and 152.1 in a ~3:1 ratio, as expected for 4-Cl-Alg with the typical Cl isotopic ratio (35Cl/37Cl ≈ 75/25). No such product was observed for reactions lacking BesC nor were any other product peaks observed. However, due to the tendency of 4-Cl-Lys to be nonenzymatically hydrolyzed upon prolonged incubation in aqueous conditions, trace amounts of 4-OH-Lys were consistently detected in LC-MS chromatograms. However, this side-product was not further metabolized by BesC nor did it cause BesC-P to decay more rapidly (Figures S21–23). Thus, we attribute the measured decay kinetics of BesC-P (kdecay = 0.030 s−1) solely to producing 4-Cl-Alg, establishing its role as a catalytically competent intermediate that is involved in carbon–carbon cleavage. In reacting BesC with l-Lys, the longevity of BesC-P was significantly prolonged as evidenced by the persistence of the blue chromophore. Measuring the full decay time-course produced a half-life of 18 ± 2 min at 4 °C (Figure 4B), representing an ~50-fold deceleration relative to 4-Cl-Lys. The sluggish reactivity toward l-Lys was also reiterated under multiple turnover conditions (Figure S24).
Figure 4.
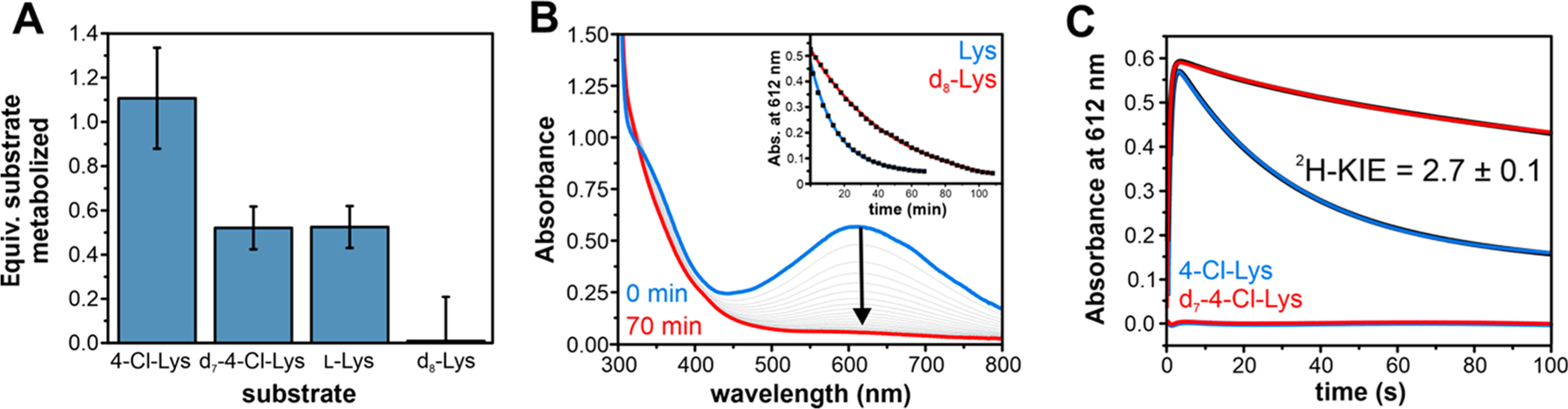
Reactivity of BesC-P with Lys derivatives. (A) Substrate consumption in single-turnover reactions of BesC with 4-Cl-Lys, l-Lys, and their perdeuterated analogs. (B and inset) UV–visible spectra show that BesC-P decay with l-Lys occurs slowly and can be fit to a single exponential decay phase with an apparent 2H KIE. (C) BesC-P decay with 4-Cl-Lys and its perdeuterated analog as monitored at 612 nm. The raw data is shown in black, and the fits to a double-exponential expression are shown in blue (protiated) and red (deuterated). The residuals are shown in the corresponding color. Protein concentration used in (B) and (C) was 300 μM with 5 mol equiv substrate.
The large difference in the decay rate constants for BesC-P between 4-Cl-Lys and l-Lys suggests that C–C cleavage could proceed through activation (e.g., hydrogen atom transfer (HAT)) at the substrate C4 position. Cl installation could be envisaged to enhance this process by lowering the C4–H bond dissociation energy (BDE).26 An alternative possibility is that the Cl-group instead promotes the transition to another species, such as a high-valent intermediate that rapidly reacts with the substrate but does not appreciably accumulate. To delineate between these two scenarios, we examined the reaction of perdeuterated 4-Cl-Lys (3,3,4,5,5,6,6-d7-4-Cl-Lys) with BesC. Should BesC-P proceed to a subsequent intermediate that performs HAT, no isotope effect should be observed for peroxo decay. Conversely, if BesC-P performs HAT directly, substrate deuteration would be anticipated to slow its decay kinetics. In the reaction of BesC with deuterated 4-Cl-Lys, no new optically detectable intermediates were observed (Figure S25). The reactivity of BesC-P with the deuterated substrate displayed a decay rate constant kobs = 0.011 ± 0.001 s−1, giving rise to an apparent 2H kinetic isotope effect (2H-KIE) of 2.7 ± 0.1 (Figure 4C, and Table S7). Considering the uncoupling observed for BesC-P metabolism of d7-4-Cl-Lys, described below, a 2H-KIE of ~2.7 represents a lower bounds and values that correct for uncoupling may be as high as kH/kD ≈ 5 (see SI for further discussion). A significant 2H-KIE was also observed under multiple-turnover conditions using ascorbate as a surrogate redox donor, implicating C–H abstraction as a rate-determining process in the overall reaction of BesC (Figure S26). The reaction of BesC with 3,3,4,4,5,5,6,6-d8-l-Lys under single-turnover conditions was also examined and compared to the protiated substrate. BesC-P decayed more slowly with perdeuterated l-Lys (t1/2 = 42 ± 6 min at 4 °C, Figure 4B, inset), yielding a very similar apparent 2H-KIE ≈ 2.7. However, given the highly uncoupled nature of d8-Lys metabolism, this apparent 2H-KIE for (d8) l-Lys may largely originate from a difference in the rate constants for productive metabolism and uncoupling that leads to cluster decomposition.
Further evidence for a direct reaction of BesC-P for C–C scission is provided through examination of the coupling parameters (substrate consumed per peroxo, Figure 4A) across the substrate series. Although the consumption of BesC-P is completely coupled to product formation for 4-Cl-Lys, quantitation of the substrate remaining from the reactions with perdeuterated d7-4-Cl-Lys revealed that ~50% of the substrate is unmetabolized. LC-MS analysis shows that the metabolized substrate exclusively formed the typical cleavage product, demonstrating that the 2H-KIE does not stem from a significant change in reaction mechanism (Figure S27). Although the origin of this uncoupling is unclear, a lack of appreciable H2O2 detected from reactions alludes to either an oxidase shunt pathway27 or the cluster decomposition as detected by Mössbauer. Irrespective of the precise origin, this behavior implies there is a parallel pathway for BesC-P decomposition that directly competes with productive catalysis. As the rate of uncoupling should be insensitive to substrate isotopic composition, this observation is best rationalized by deceleration of a productive pathway (by raising the substrate BDE) that favors the uncoupling pathway. This trend is reinforced in an examination of l-Lys metabolism and comparison with the deuterated isotopologue. Analysis of single turnover reactions (Figure 4A, Figure S21) revealed only ~0.5 equiv of l-Lys were consumed, compared to <2% for the deuterated substrate. Alongside the product l-Alg, LC-MS revealed that l-Lys cleavage was accompanied by an additional (~20%) Δm/z + 16 coproduct, consistent with the formation of OH-Lys. Isotope tracing experiments (with 18O2 or H218O) revealed that the introduced oxygen was completely incorporated from solvent and not from O2 (Figure 5). Possible mechanistic implications of this side-product are described in the Discussion.
Figure 5.
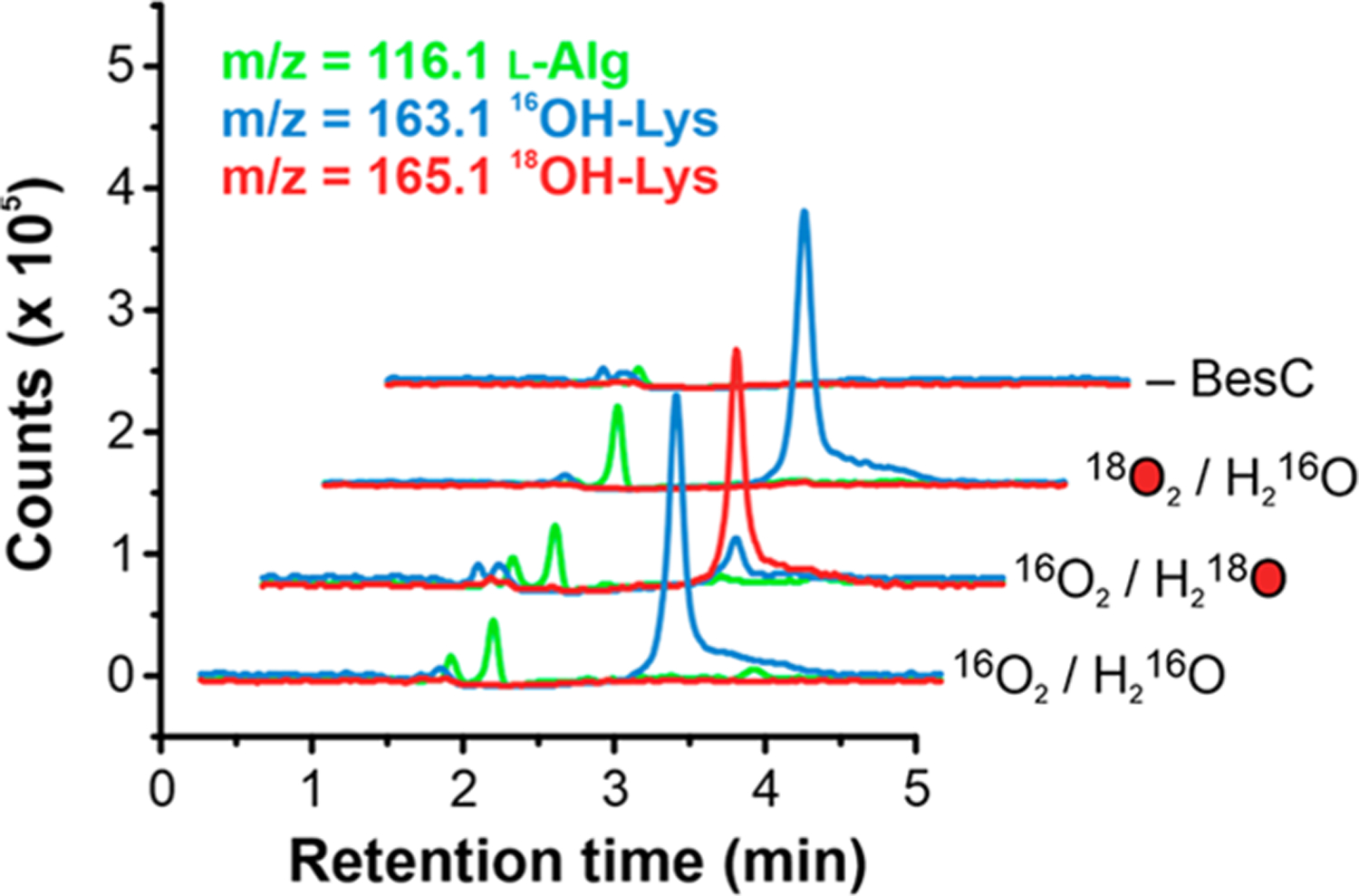
LC-MS extracted ion chromatograms of reactions of BesC with l-Lys and labeled 18O2 or H218O and a negative control where BesC was omitted.
DISCUSSION
The spectroscopic and kinetic characterization shown here demonstrates that BesC forms a coupled diiron center that can react with dioxygen, authenticating its assignment as an HDO. The substrate-dependent O2 activation strategy utilized by BesC has long been appreciated for various iron-enzyme classes, including several diiron enzymes,8,28 to prevent errant side-reactions including self-oxidation and the generation of reactive oxygen species. The HDOs characterized thus far have been shown to regulate O2 activation in either a substrate-dependent (UndA8 and BesC) or unregulated manner (SznF), both of which differ from multicomponent diiron oxygenases/oxidases that can often require auxiliary proteins (e.g., effector29,30 or redox partners31) to elicit structural changes at the diiron site to enable catalysis. Although triggering in other HDOs can be rationalized on the basis of provision of an additional carboxylate ligand to the cluster, from either the substrate (UndA8) or protein (SznF11), the structural origins for substrate-regulated activation in BesC appear to be more complex. A preliminary screen of potential substrate analogs, using BesC-P formation as an optical guide, suggests that neither the substrate carboxylate alone nor the C2 amino group is sufficient for triggering and that the presence of an amine-containing side chain is not a strict requirement (Table S8). Taken together with the role that the Cl-group has for directing efficient binding and for modulating the kinetics of BesC-P formation, more complex structural underpinnings likely prime the cluster for rapid O2 binding.
The influence of the substrate C4–H bond dissociation energy (BDE) on BesC-P decay is shown in Figure 6. The evident correlation, along with the influence of substrate deuteration on the ratio of productive coupling, is best rationalized through a mechanism involving direct electrophilic attack of C4–H by BesC-P, as shown in Figure 7. A peroxo-based HAT mechanism is further supported by (i) the overall sluggish decay of the intermediate; (ii) a 2H KIE within the semiclassical limit32,33 (diiron Fe(IV)-oxo intermediates often exhibit significantly larger 2H KIEs due to significant tunneling contributions34); and (iii) the narrow window of substrate BDEs that can be efficiently metabolized. These characteristics disfavor a mechanism involving the formation of an undetected high-valent species on several grounds. First, the observed 2H-KIE for BesC-P decay would necessitate the quasi-reversible formation of a presumptive high-valent intermediate, a scenario that is unlikely for a process that would involve O–O bond cleavage.35 Furthermore, high-valent species do not typically exhibit such strict limitations for substrate C–H BDE and are known to readily oxidize highly challenging substrates. Rather, the features observed here for BesC-P reactivity are similarly shared in the reaction of methane monooxygenase peroxo (MMO-P) with non-native substrates, and in particular the upper bounds of accessible C–H BDEs ≈ 96 kcal/mol.36,37
Figure 6.
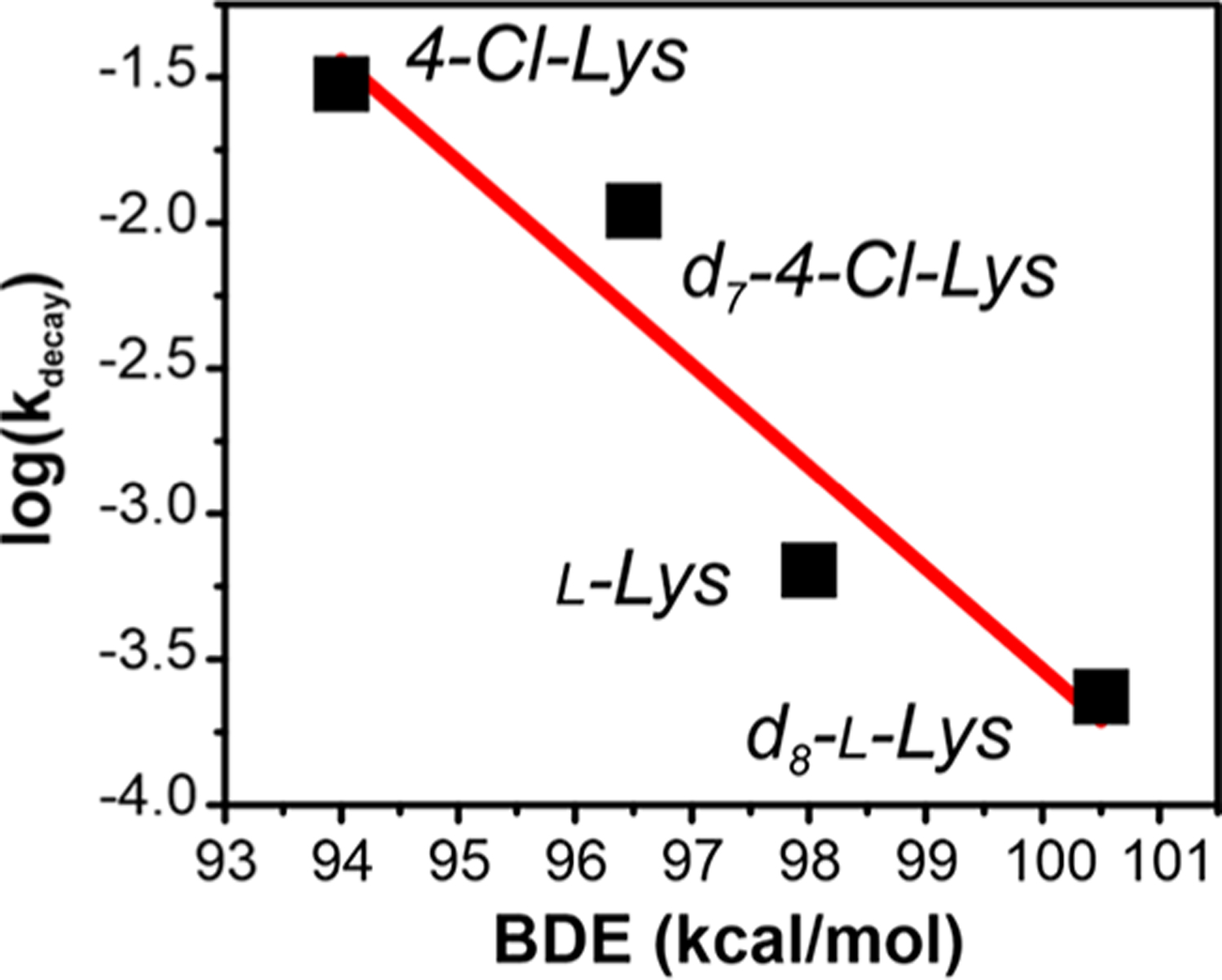
Correlation of the apparent BesC-P decay rates for various lysine derivatives plotted against the estimated substrate C4–H BDE.26
Figure 7.
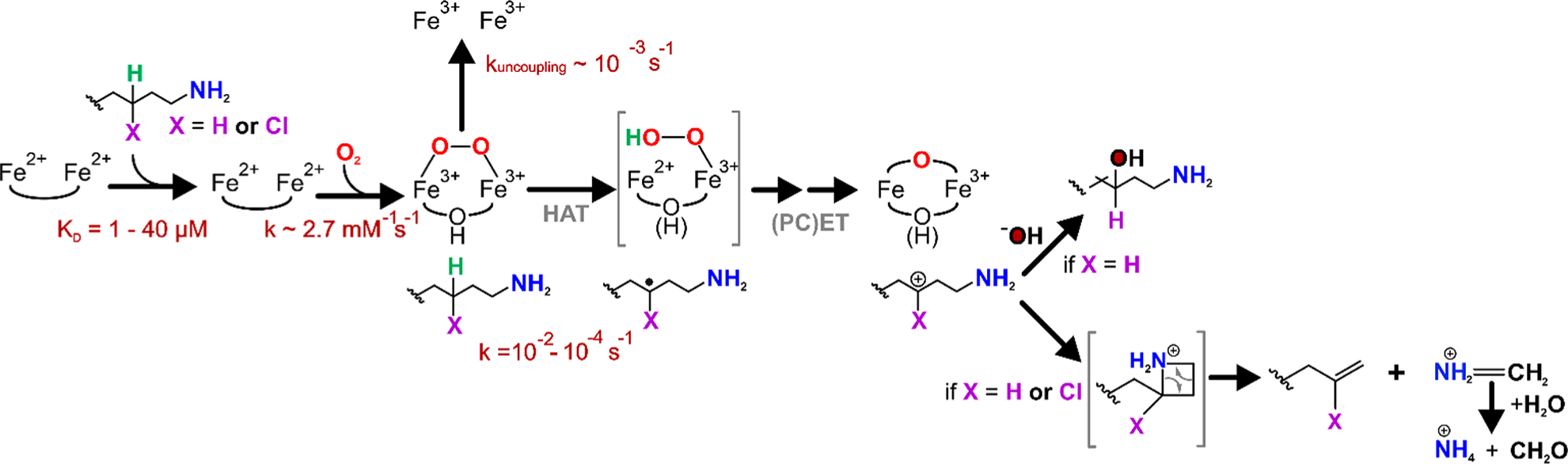
Proposed mechanism for oxidative C–C cleavage by BesC. Substrate-triggered activation of O2 yields the diferric-peroxo intermediate BesC-P, which branches between C4–H cleavage and uncoupling depending on the identity of X (H or Cl). HAT by BesC-P may be followed by an iron(II/III)-hydroperoxo species and subsequent iron(III/IV)-oxo/hydroxo species (bracketed because these have not been directly observed). (PC)ET then yields the carbocation intermediate. At this point, the reaction may proceed through a cyclic intermediate before cleaving to yield (4-chloro)-l-allylglycine and methylene imine. The latter may be nonenzymatically hydrolyzed to ammonium and formaldehyde. An alternative fate of the carbocation intermediate is quenching by a solvent-derived hydroxide, observed when X = H.
Although peroxo-diiron intermediates have been implicated in electrophilic reactions such as N-oxygenations and aromatic substitutions, their reactivity is most often associated with variations in O2 coordination geometry, leading to distinctive optical (e.g., λmax ≤ 500 nm or ε ≤ 500 M−1 cm−1) and Mössbauer spectroscopic properties (ΔEQ < 0.7 mm/s).19,28,38,39 On the contrary, the spectroscopic properties of BesC-P are strikingly similar to synthetic complexes, MMO-P, and, in particular, the enzyme deoxyhypusine hydroxylase (DOHH). DOHH-P has been structurally validated by rR, XAS, and crystallography as containing a cis-μ−1,2-peroxo adduct.40,41 The antiferromagnetic coupling constant of BesC-P (J ≈ 35 cm−1, Figure S20) suggests that exchange is dominated by the peroxide-unit and, like DOHH, may similarly contain a hydroxo single-atom bridge. Such an arrangement would promote the stability of BesC-P and thwart O–O bond cleavage to form a diferryl species.42–45 Following C–H abstraction to form a substrate radical, there are several potential pathways that could lead to C–C fragmentation. Depicted in Figure 7 is a subsequent proton-coupled electron transfer to produce a carbocation intermediate, although a radical fragmentation pathway could also be envisioned. However, the presence of the OH-Lys product, with the integrated alcohol deriving exclusively from solvent rather than O2, suggests that a cationic pathway may indeed be operative.
Bioinformatic analysis further reiterates the necessity of the Cl group for efficient C–C cleavage by BesC (Figure S28). A search for BesC orthologs and examination of the associated genome neighborhood network revealed ~20 biosynthetic gene clusters (BGCs). The presence of a halogenase was universally conserved even though, in many cases, the BesB ortholog was absent. This observation highlights that substrate chlorination is necessary not only for alkyne formation but specifically for alkene formation.
The instability of the diiron cluster upon exposure to O2 appears to reflect a common property for some HDOs11 and is similarly observed for BesC via Mössbauer spectroscopy (Figure S19). However, it should be noted that BesC-P can be readily recycled in multiple rounds of reduction and reoxygenation and capably performs multiple turnovers in the presence of the excess ascorbate (Figures S24, S29). The 2H-KIE that is also observed in the multiple turnover system (Figure S26) implies that either rapid reduction occurs prior to dissociation of the iron or cluster reassembly happens at rates that far exceed those observed for HAT.
Given the similarity of UndA and BesC in terms of overall structure, coordination motif, and olefin-installation function, one would envision that the reactions of the two HDOs would proceed in a very similar fashion. The nature of the C–H abstracting species in UndA has yet to be unambiguously identified, though a computational study has suggested that a high-valent intermediate is needed.23 Thus, there are striking differences in the stabilities of the peroxo-diferric species between BesC and UndA, which differ by more than 3 orders of magnitude. This could arise from as-of-yet determined facets of the secondary coordination sphere or structural differences of the peroxo unit itself.
CONCLUSION
Our results demonstrate that BesC utilizes a coupled dinuclear iron cofactor to perform the oxidative carbon–carbon bond cleavage of 4-chloro-l-lysine to generate 4-chloro-l-allylglycine. Mössbauer and optical spectroscopy confirms its position as a new member of the emerging and expanding heme-oxygenase-like subfamily of diiron oxidases (HDOs) that activate dioxygen for diverse transformations.46 By analogy to the C–C-cleaving HDO UndA, it would be tempting to speculate that BesC would activate dioxygen to ultimately generate a high-valent intermediate to mediate this transformation. Although BesC indeed activates O2 to form a diferric-peroxo intermediate (BesC-P), the decay kinetics for this species are clearly modulated by the strength of the C4–H bond, a scenario that is best rationalized by a mechanism in which the diferric-peroxo, rather than a high-valent intermediate, directly performs HAT to initiate C–C bond cleavage. This represents an unprecedented reaction trajectory for diferric-peroxo intermediates, thus broadening the repertoire of catalytic mechanisms accessible to HDOs and, more generally, diiron enzymes.
EXPERIMENTAL SECTION
Experimental materials and methods are detailed in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grants GM135315 (to T.M.M.), GM125924 (to Y.G.), and GM127588 (to W.-c.C.). The authors thank Profs. John D. Lipscomb and Lawrence Que and Drs. Rahul Banerjee and Melanie Rogers for helpful discussions.
ABBREVIATIONS
- HDO
heme oxygenase-like diiron oxidase
- HAT
hydrogen atom transfer
- PCET
proton-coupled electron transfer
- Alg
allylglycine
- BDE
bond dissociation energy
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c11109.
Detailed experimental procedures, characterization of all compounds, kinetic and Mössbauer fitting parameters, Figures S1–S29, and Tables S1–S8. (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c11109
Contributor Information
Olivia M. Manley, Department of Molecular and Structural Biochemistry, North Carolina State University, Raleigh, North Carolina 27695, United States
Haoyu Tang, Department of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, United States.
Shan Xue, Department of Chemistry, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, United States.
Yisong Guo, Department of Chemistry, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, United States.
Wei-chen Chang, Department of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, United States.
Thomas M. Makris, Department of Molecular and Structural Biochemistry, North Carolina State University, Raleigh, North Carolina 27695, United States; Department of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, United States.
REFERENCES
- (1).Marchand J; Neugebauer M; Ing M; Lin C-I; Pelton J; Chang M Discovery of a pathway for terminal-alkyne amino acid biosynthesis. Nature 2019, 567 (7748), 420–424. [DOI] [PubMed] [Google Scholar]
- (2).Galonic DP; Barr EW; Walsh CT; Bollinger JM; Krebs C Two interconverting Fe(IV) intermediates in aliphatic chlorination by the halogenase CytC3. Nat. Chem. Biol. 2007, 3 (2), 113–116. [DOI] [PubMed] [Google Scholar]
- (3).Vaillancourt FH; Yin J; Walsh CT SyrB2 in syringomycin E biosynthesis is a nonherne FeII alpha-ketoglutarate- and O2-dependent halogenase. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (29), 10111–10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brzovic P; Holbrook EL; Greene RC; Dunn MF Reaction mechanism of Escherichia coli cystathionine γ-synthase: direct evidence for a pyridoxamine derivative of vinylgloxylate as a key intermediate in pyridoxal phosphate dependent γ-elimination and γ-replacement reactions. Biochemistry 1990, 29 (2), 442–451. [DOI] [PubMed] [Google Scholar]
- (5).Walsh CT; Wencewicz TA Flavoenzymes: Versatile catalysts in biosynthetic pathways. Nat. Prod. Rep. 2013, 30 (1), 175–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rui Z; Li X; Zhu X; Liu J; Domigan B; Barr I; Cate JH; Zhang W Microbial biosynthesis of medium-chain 1-alkenes by a nonheme iron oxidase. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (51), 18237–18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Manley OM; Fan R; Guo Y; Makris TM Oxidative Decarboxylase UndA Utilizes a Dinuclear Iron Cofactor. J. Am. Chem. Soc. 2019, 141 (22), 8684–8688. [DOI] [PubMed] [Google Scholar]
- (8).Zhang B; Rajakovich LJ; Van Cura D; Blaesi EJ; Mitchell AJ; Tysoe CR; Zhu X; Streit BR; Rui Z; Zhang W; Boal AK; Krebs C; Bollinger JM Substrate-triggered Formation of a Peroxo-Fe2(III/III) Intermediate during Fatty Acid Decarboxylation by UndA. J. Am. Chem. Soc. 2019, 141 (37), 14510–14514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ng TL; Rohac R; Mitchell AJ; Boal AK; Balskus EP An N-nitrosating metalloenzyme constructs the pharmacophore of streptozotocin. Nature 2019, 566 (7742), 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).McBride M; Sil D; Ng TL; Crooke AM; Kenney GE; Tysoe CR; Zhang B; Balskus EP; Boal AK; Krebs C A peroxodiiron(III) intermediate mediating both N-hydroxylation steps in biosynthesis of the N-nitrosourea pharmacophore of streptozotocin by SznF. J. Am. Chem. Soc. 2020, 142 (27), 11818–11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).McBride MJ; Pope SR; Hu K; Okafor CD; Balskus EP; Bollinger JM; Boal AK Structure and assembly of the diiron cofactor in the heme-oxygenase-like domain of the N-nitrosourea-producing enzyme SznF. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (4), No. e2015931118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Schwarzenbacher R; Stenner-Liewen F; Liewen H; Robinson H; Yuan H; Bossy-Wetzel E; Reed JC; Liddington RC Structure of the Chlamydia protein CADD reveals a redox enzyme that modulates host cell apoptosis. J. Biol. Chem. 2004, 279 (28), 29320–29324. [DOI] [PubMed] [Google Scholar]
- (13).Macias-Orihuela Y; Cast T; Crawford I; Brandecker KJ; Thiaville JJ; Murzin AG; de Crécy-Lagard V; White RH; Allen KD An unusual route for p-aminobenzoate biosynthesis in Chlamydia trachomatis involves a probable self-sacrificing diiron oxygenase. J. Bacteriol. 2020, 202 (20), No. e00319–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Adams NE; Thiaville JJ; Proestos J; Juárez-Vázquez AL; McCoy AJ; Barona-Gómez F; Iwata-Reuyl D; de Crécy-Lagard V; Maurelli AT Promiscuous and adaptable enzymes fill “holes” in the tetrahydrofolate pathway in Chlamydia species . MBio 2014, 5 (4), No. e01378–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Satoh Y; Kuratsu M; Kobayashi D; Dairi T New gene responsible for para-aminobenzoate biosynthesis. J. Biosci. Bioeng. 2014, 117 (2), 178–183. [DOI] [PubMed] [Google Scholar]
- (16).Knoot CJ; Kovaleva EG; Lipscomb JD Crystal structure of CmlI, the arylamine oxygenase from the chloramphenicol biosynthetic pathway. JBIC, J. Biol. Inorg. Chem. 2016, 21 (5–6), 589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Choi YS; Zhang H; Brunzelle JS; Nair SK; Zhao H In vitro reconstitution and crystal structure of p-aminobenzoate N-oxygenase (AurF) involved in aureothin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (19), 6858–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jasniewski AJ; Que L Jr Dioxygen activation by nonheme diiron enzymes: Diverse dioxygen adducts, high-valent intermediates, and related model complexes. Chem. Rev. 2018, 118 (5), 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Makris TM; Vu VV; Meier KK; Komor AJ; Rivard BS; Munck E; Que L Jr.; Lipscomb JD An unusual peroxo intermediate of the arylamine oxygenase of the chloramphenicol biosynthetic pathway. J. Am. Chem. Soc. 2015, 137 (4), 1608–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Li N; Korboukh VK; Krebs C; Bollinger JM Four-electron oxidation of p-hydroxylaminobenzoate to p-nitrobenzoate by a peroxodiferric complex in AurF from Streptomyces thioluteus. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (36), 15722–15727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Grant JL; Hsieh CH; Makris TM Decarboxylation of fatty acids to terminal alkenes by cytochrome P450 compound I. J. Am. Chem. Soc. 2015, 137 (15), 4940–4943. [DOI] [PubMed] [Google Scholar]
- (22).Yu C-P; Tang Y; Cha L; Milikisiyants S; Smirnova TI; Smirnov AI; Guo Y; Chang W.-c. Elucidating reaction pathway of decarboxylation-assisted olefination catalyzed by a mononuclear non-heme iron enzyme. J. Am. Chem. Soc. 2018, 140 (45), 15190–15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lin Y-T; Stańczak A; Manchev Y; Straganz G; de Visser S Can a mononuclear iron(III)-superoxo active site catalyze the decarboxylation of dodecanoic acid in UndA to produce biofuels? Chem.—Eur. J 2020, 26 (10), 2233–2242. [DOI] [PubMed] [Google Scholar]
- (24).Feig AL; Becker M; Schindler S; van Eldik R; Lippard SJ Mechanistic studies of the formation and decay of diiron(III) peroxo complexes in the reaction of diiron(II) precursors with dioxygen. Inorg. Chem. 1996, 35 (9), 2590–2601. [DOI] [PubMed] [Google Scholar]
- (25).Kryatov SV; Rybak-Akimova EV; MacMurdo VL; Que L A mechanistic study of the reaction between a diiron(II) complex [Fe-2(II)(mu-OH)(2)(6-Me-3-TPA)(2)](2+) and O-2 to form a diiron(III) peroxo complex. Inorg. Chem. 2001, 40 (10), 2220–2228. [DOI] [PubMed] [Google Scholar]
- (26).Luo Y-R Comprehensive handbook of chemical bond energies; CRC press: 2007. [Google Scholar]
- (27).Broadwater JA; Ai J; Loehr TM; Sanders-Loehr J; Fox BG Peroxodiferric intermediate of stearoyl-acyl carrier protein Δ9 desaturase: oxidase reactivity during single turnover and implications for the mechanism of desaturation. Biochemistry 1998, 37 (42), 14664–14671. [DOI] [PubMed] [Google Scholar]
- (28).Pandelia ME; Li N; Nørgaard H; Warui DM; Rajakovich LJ; Chang W.-c.; Booker SJ; Krebs C; Bollinger JM Jr Substrate-triggered addition of dioxygen to the diferrous cofactor of aldehyde-deformylating oxygenase to form a diferric-peroxide intermediate. J. Am. Chem. Soc. 2013, 135 (42), 15801–15812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Liu Y; Nesheim JC; Lee S-K; Lipscomb JD Gating effects of component B on oxygen activation by the methane monooxygenase hydroxylase component. J. Biol. Chem. 1995, 270 (42), 24662–24665. [DOI] [PubMed] [Google Scholar]
- (30).Lountos GT; Mitchell KH; Studts JM; Fox BG; Orville AM Crystal structures and functional studies of T4moD, the toluene 4-monooxygenase catalytic effector protein. Biochemistry 2005, 44 (19), 7131–7142. [DOI] [PubMed] [Google Scholar]
- (31).Broadwater JA; Achim C; Münck E; Fox BG Mössbauer studies of the formation and reactivity of a quasi-stable peroxo intermediate of stearoyl-acyl carrier protein Δ9-desaturase. Biochemistry 1999, 38 (38), 12197–12204. [DOI] [PubMed] [Google Scholar]
- (32).Decker A; Chow MS; Kemsley JN; Lehnert N; Solomon EI Direct hydrogen-atom abstraction by activated bleomycin: an experimental and computational study. J. Am. Chem. Soc. 2006, 128 (14), 4719–33. [DOI] [PubMed] [Google Scholar]
- (33).Zhu W; Jang S; Xiong J; Ezhov R; Li XX; Kim T; Seo MS; Lee YM; Pushkar Y; Sarangi R; Guo Y; Nam W A Mononuclear Non-heme Iron(III)-Peroxo Complex with an Unprecedented High O-O Stretch and Electrophilic Reactivity. J. Am. Chem. Soc. 2021, 143 (38), 15556–15561. [DOI] [PubMed] [Google Scholar]
- (34).Nesheim JC; Lipscomb JD Large kinetic isotope effects in methane oxidation catalyzed by methane monooxygenase: evidence for C-H bond cleavage in a reaction cycle intermediate. Biochemistry 1996, 35 (31), 10240–7. [DOI] [PubMed] [Google Scholar]
- (35).Banerjee R; Proshlyakov Y; Lipscomb JD; Proshlyakov DA Structure of the key species in the enzymatic oxidation of methane to methanol. Nature 2015, 518 (7539), 431–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tinberg CE; Lippard SJ Oxidation reactions performed by soluble methane monooxygenase hydroxylase intermediates Hperoxo and Q proceed by distinct mechanisms. Biochemistry 2010, 49 (36), 7902–7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Beauvais LG; Lippard SJ Reactions of the peroxo intermediate of soluble methane monooxygenase hydroxylase with ethers. J. Am. Chem. Soc. 2005, 127 (20), 7370–7378. [DOI] [PubMed] [Google Scholar]
- (38).Jasniewski AJ; Komor AJ; Lipscomb JD; Que L Jr Unprecedented (μ−1,1-peroxo) diferric structure for the ambiphilic orange peroxo intermediate of the nonheme N-oxygenase CmlI. J. Am. Chem. Soc. 2017, 139 (30), 10472–10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Murray LJ; Naik SG; Ortillo DO; García-Serres R; Lee JK; Huynh BH; Lippard SJ Characterization of the arene-oxidizing intermediate in ToMOH as a diiron(III) species. J. Am. Chem. Soc. 2007, 129 (46), 14500–14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Vu VV; Emerson JP; Martinho M; Kim YS; Münck E; Park MH; Que L Human deoxyhypusine hydroxylase, an enzyme involved in regulating cell growth, activates O2 with a nonheme diiron center. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (35), 14814–14819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Han Z; Sakai N; Böttger LH; Klinke S; Hauber J; Trautwein AX; Hilgenfeld R Crystal structure of the peroxodiiron(III) intermediate of deoxyhypusine hydroxylase, an oxygenase involved in hypusination. Structure 2015, 23 (5), 882–892. [DOI] [PubMed] [Google Scholar]
- (42).Dong Y; Yan S; Young VG Jr; Que L Jr Crystal Structure Analysis of a Synthetic Non-Heme Diiron-O2 Adduct: Insight into the Mechanism of Oxygen Activation. Angew. Chem., Int. Ed. 1996, 35 (6), 618–620. [Google Scholar]
- (43).Fiedler AT; Shan X; Mehn MP; Kaizer J; Torelli S; Frisch JR; Kodera M; Que L Jr. Lawrence, Spectroscopic and computational studies of (μ-oxo)(μ-1,2-peroxo)diiron(III) complexes of relevance to nonheme diiron oxygenase intermediates. J. Phys. Chem. A 2008, 112 (50), 13037–13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhang X; Furutachi H; Fujinami S; Nagatomo S; Maeda Y; Watanabe Y; Kitagawa T; Suzuki M Structural and spectroscopic characterization of (μ-hydroxo or μ-oxo)(μ-peroxo) diiron(III) complexes: models for peroxo intermediates of non-heme diiron proteins. J. Am. Chem. Soc. 2005, 127 (3), 826–827. [DOI] [PubMed] [Google Scholar]
- (45).Cranswick MA; Meier KK; Shan X; Stubna A; Kaizer J; Mehn MP; Münck E; Que L Jr Protonation of a peroxodiiron (III) complex and conversion to a diiron (III/IV) intermediate: implications for proton-assisted O-O bond cleavage in nonheme diiron enzymes. Inorg. Chem. 2012, 51 (19), 10417–10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Patteson JB; Putz AT; Tao L; Simke WC; Bryant LH 3rd; Britt RD; Li B Biosynthesis of fluopsin C, a copper-containing antibiotic from Pseudomonas aeruginosa. Science 2021, 374 (6570), 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.