

Abstract
Histone (de)acetylation is important for the regulation of fundamental biological processes such as gene expression and DNA recombination. Distinct classes of histone deacetylases (HDACs) have been identified, but how they are regulated in vivo remains largely unexplored. Here we describe results demonstrating that HDAC4, a member of class II human HDACs, is localized in the cytoplasm and/or the nucleus. Moreover, we have found that HDAC4 interacts with the 14-3-3 family of proteins that are known to bind specifically to conserved phosphoserine-containing motifs. Deletion analyses suggested that S246, S467, and S632 of HDAC4 mediate this interaction. Consistent with this, alanine substitutions of these serine residues abrogated 14-3-3 binding. Although these substitutions had minimal effects on the deacetylase activity of HDAC4, they stimulated its nuclear localization and thus led to enhanced transcriptional repression. These results indicate that 14-3-3 proteins negatively regulate HDAC4 by preventing its nuclear localization and thereby uncover a novel regulatory mechanism for HDACs.
Specific lysine acetylation of histones and nonhistone proteins has been recently recognized as a major mechanism by which eukaryotic transcription is regulated (12, 23, 24, 44, 45, 56, 57). Such acetylation is reversible and dynamic in vivo, and its level is governed by the opposing actions of histone acetyltransferases and histone deacetylases (HDACs). Distinct classes of HDACs have been identified in mammals (21, 36). Class I HDACs (HDAC1, HDAC2, HDAC3, and HDAC8) are homologous to yeast Rpd3 (8, 16, 49, 60, 61). HDAC1 and HDAC2 interact with each other and form the catalytic core of Sin3 and NuRD complexes, both of which play important roles in transcriptional repression and gene silencing (26, 51, 53, 54, 58, 63–65). Various transcriptional repressors recruit these complexes to inhibit transcription (reviewed in references 15, 45, and 56). Class II HDACs (HDAC4, HDAC5, HDAC6, and HDAC7) contain domains significantly similar to the catalytic domain of yeast Hda1 (9, 11, 20, 33, 41, 52, 55). HDAC4, HDAC5, and HDAC7 are homologous, whereas HDAC6 has two Hda1-related catalytic domains and a unique Cys- and His-rich C-terminal domain. HDAC4 and HDAC5 interact with the MEF2 transcription factors (28, 33, 55), and this interaction is regulated (30, 62). Related to this, MITR/HDRP, a protein related to the N-terminal part of HDAC4, HDAC5, and HDAC7, binds to MEF2s and represses transcription (43, 66). Moreover, HDAC4, HDAC5, and HDAC7 were found to interact with the nuclear receptor corepressors SMRT and N-CoR (13, 17, 20). These new findings suggest that like class I HDACs, some class II HDACs are recruited to promoters to inhibit transcription. One interesting but unaddressed question is how the function of HDACs is regulated in vivo.
While HDAC1, HDAC2, and HDAC3 are nuclear, the plant deacetylase HD2 is a nucleolar protein (8, 31). Miska et al. reported that the HDAC4 protein lacking the N-terminal 117 residues is cytoplasmic or nuclear in HeLa cells (33), whereas Fischle et al. found this mutant predominantly nuclear in the same cell line (9). Importantly, this mutant is actively exported to the cytoplasm (33). We found that the same mutant is mainly cytoplasmic in NIH 3T3 cells (M. Vezmar and X. J. Yang, unpublished observation). Very recently, it was reported that HDAC5 and HDAC7 are nuclear in HeLa and CV-1 cells (20, 28). These findings suggest that the subcellular localization of HDAC4 and its homologs may be regulated in a cell context-dependent manner and that controlled subcellular localization may serve as a regulatory mechanism for these HDACs. However, the way by which such regulation is achieved remains entirely unclear.
Emerging evidence indicates that 14-3-3 proteins function as cytoplasmic anchors for some binding partners (1, 38). For example, 14-3-3 proteins bind to and retain phosphorylated CDC25C, a phosphatase important for initiating the G2/M transition during cell cycle progression, in the cytoplasm (39). It has been recently shown that 14-3-3 proteins also regulate the nuclear localization of transcription factors. Upon phosphorylation by the kinase Akt/PKB, the Forkhead transcription factor FKHRL1 interacts with 14-3-3 proteins and is sequestered in the cytoplasm (4). Such regulation may also control the nuclear localization of two other transcription factors related to FKHRL1 (3, 22, 46; reviewed in reference 6). Furthermore, the yeast 14-3-3 protein BMH2 interacts with the transcription factors MSN2 and MSN4 and may regulate their cytoplasmic retention in a TOR kinase-dependent manner (2). Intriguingly, 14-3-3 proteins were found to be part of a HAT1 complex purified from Xenopus oocytes (19). Here we present evidence that 14-3-3 proteins bind to HDAC4 and sequester it in the cytoplasm, suggesting that 14-3-3 proteins negatively regulate HDAC4 and its homologs by excluding them from the nucleus.
MATERIALS AND METHODS
Molecular cloning.
Expression plasmids for HDAC4 and some deletion mutants have been described previously (55). Additional HDAC4 mutants were generated by PCR with the Expand thermostable DNA polymerase (Roche) or by site-directed mutagenesis with single-stranded uracil-containing templates and T7 DNA polymerase. DNA sequencing was performed with T7 Sequenase 2.0 (Amersham Pharmacia Biotech) to confirm the mutations. Green fluorescent protein (GFP) constructs were derived from pEGFP-C2 (Clontech). Luciferase reporters pJLuc, MEF2-E4-Luc, and Gal4-tk-Luc have been described previously (5, 55).
Cytoplasmic and nuclear fractionation.
A previously described procedure was used with modifications (5). Briefly, NIH 3T3 cells (∼106) were washed twice with phosphate-buffered saline (PBS) and lysed in situ using 1 ml of ice-cold hypotonic lysis buffer (20 mM HEPES [pH 7.6], 20% glycerol, 10 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% Triton X-100, 25 mM NaF, 25 mM β-glycerophosphate, 1 mM dithiothreitol, protease inhibitors). After 5 min on ice with occasional shaking, the cell lysate was harvested by scraping and centrifuged for 5 min on a benchtop centrifuge (at 1,300 × g) at 4°C. The supernatant was collected, cleared by high-speed centrifugation (10 min at 16,000 × g) at 4°C, and saved as the cytoplasmic fraction. The nuclear pellet from the low-speed centrifugation was suspended in 0.2 ml of hypotonic lysis buffer containing 0.5 M NaCl and rotated for 20 min at 4°C. After high-speed centrifugation, the supernatant was collected as the nuclear extract.
Immunofluorescence microscopy.
Subconfluent and cycling NIH 3T3, 293, COS1, or SKN (SK-N-SH [American Type Culture Collection]) cells growing on glass coverslips in complete Joklik minimal essential medium (Gibco) were transfected using the Lipofectamine liposome reagent (Gibco). Briefly, 1 μg of Flag-tagged HDAC4 expression construct and 12 μl of Lipofectamine were used to transfect cells on a coverslip. The cells were incubated for 3 h with the plasmid-liposome complex in serum-free medium and washed once with PBS, and complete medium was added. After 15 h, the cells were fixed with PBS–1% paraformaldehyde at room temperature (RT) for 10 min. After being washed once with PBS, the cells were permeabilized with PBS–0.5% Triton X-100 for 5 min at RT. The cells were again washed once with PBS and incubated with the anti-Flag M2 monoclonal antibody (Sigma) for 60 min at RT. They were washed once with PBS and incubated with goat anti-mouse immunoglobulin G conjugated with Alexa 488 (Molecular Probes) for 60 min at RT. They were washed once again with PBS and mounted on glass slides using a glycerol-based mounting medium containing the antifade agent para-phenylenediamine (0.1 mg/ml; Sigma) and 4′,6-diamidino-2-phenylindole (DAPI) (30 μg/ml; Sigma). Labeled cells were visualized using a digital deconvolution epifluorescence microscope (Leica); images were collected using a digital camera containing a 14-bit detector (Princeton Instruments) and further processed with Adobe Photoshop.
Alternatively, NIH 3T3 or 293 cells (2 × 104) were seeded on coverslips in a 12-well plate and transfected with 0.1 μg of a Flag- or GAl4-tagged HDAC4 expression plasmid using 2 to 5 μl of Superfect (Qiagen). Fifteen to 24 h later, the cells were rinsed three times with PBS–1 mM MgCl2–0.1 mM CaCl2 and further processed for immunofluorescence microscopy with the anti-Flag (1:300; Sigma) or anti-Gal4 (RK5C1; Santa Cruz Biotechnology) antibody as described previously (32). For nuclear staining, either DAPI or Hoechst 33258 (20 ng/ml; Sigma) was used.
Live green fluorescence microscopy.
Expression plasmids for GFP fusions were transfected, with SuperFect, into NIH 3T3, 293, or SKN cells cultured in Dulbecco minimal essential medium (Gibco) supplemented with 10% fetal bovine serum and antibiotics. Sixteen hours posttransfection, transfected cells were subjected to live green fluorescence microscopy using a Zeiss Axiovert 135 microscope equipped with a temperature-adjustable platform and linked to a charge-coupled device camera (Princeton Instruments) controlled by a Hewlett-Packard computer running Northern Eclipse (Empix Imaging). Images were taken and exported to a PowerMac computer for further processing with Adobe Photoshop.
To quantify transfected cells with different subcellular localization of GFP fusions, transfected cells with green fluorescence were counted under the fluorescence microscope by eye. For each GFP fusion construct, 50 to 400 cells with green fluorescence were counted per experiment; at least three independent transfection experiments were performed to obtain consistent results.
Protein-protein interaction.
To examine the interaction between HDAC4 and 14-3-3 proteins, the Flag-HDAC4 expression plasmid was cotransfected into 293 cells with or without an expression plasmid for hemagglutinin (HA)-tagged human 14-3-3β. A 3-μg portion of each plasmid was used to transfect 4 × 105 cells (in a 6-cm dish) with 9 μl of SuperFect transfection reagent. Forty-eight hours after transfection, the cells were washed twice with PBS and collected in 0.5 ml of buffer B (20 mM Tris-HCl [pH 8.0], 10% glycerol, 5 mM MgCl2, 0.1% NP-40, protease inhibitors) containing 0.15 or 0.5 M KCl. Cell extracts were prepared for affinity purification on M2 agarose beads (Sigma) or for immunoprecipitation with the mouse anti-HA monoclonal antibody (Babco) and UltraLink immobilized protein A/G beads (Pierce). Beads with bound immunocomplexes were washed four times with buffer B supplemented with 0.15 or 0.5 M KCl, and bound proteins were eluted with Flag peptide (Sigma) or sodium dodecyl sulfate (SDS) sample buffer. Eluted proteins were subsequently resolved by SDS-polyacrylamide gel electrophoresis (10% polyacrylamide) and transferred to nitrocellulose membranes for Western analysis with the anti-Flag or anti-HA antibody. Blots were developed with Supersignal chemiluminescent substrates (Pierce).
To examine the interaction of Flag-HDAC4 with endogenous 14-3-3, Flag-HDAC4 was expressed in and purified from 293 cells as described above. Bound proteins were eluted and subjected to Western analysis with anti-14-3-3 antibodies (K-19 and H-8; Santa Cruz Biotechnology). The interaction of Flag-tagged HDAC4 mutants with 14-3-3 proteins was similarly analyzed.
For interaction between endogenous HDAC4 and 14-3-3 proteins, NIH 3T3 cell extracts (∼1.5 mg in 0.4 ml of buffer B supplemented with 150 mM KCl and 50 mM NaF) were mixed with preimmune IgG or rabbit anti-HDAC4 antibody and incubated at 4°C for 1 h. A 20-μl bed volume of UltraLink Immobilized protein A/G beads was added; after being rotated overnight at 4°C, the beads were washed extensively with buffer B supplemented with 150 mM KCl and 50 mM NaF. Bound immunocomplexes were eluted by boiling in SDS sample buffer and subjected to Western analysis with the rabbit anti-HDAC4 or mouse anti-14-3-3 antibody (H-8).
HDAC assays.
Flag-tagged HDAC4 and mutant proteins were expressed in and purified from 293 or 293T cells as described above. HDAC assays were carried out using [3H]acetyl-histones prepared from HeLa cells as described previously (55).
Reporter gene assays.
Reporter gene assays were performed as described previously, except that transfected cells were lysed for measurement of reporter activities 24 h posttransfection (55).
RESULTS
Subcellular localization of HDAC4.
For examination of the subcellular localization of HDAC4, a rabbit polyclonal antibody was raised. This antibody detected Flag-HDAC4 expressed in and affinity purified from 293 cells (Fig. 1A, lane 1). Western analyses of cytoplasmic and nuclear extracts of NIH 3T3 cells revealed that HDAC4 is mainly in the cytoplasmic fraction (lanes 2 and 3). As expected, anti-14-3-3 and anti-MEF2D antibodies detected 14-3-3 and MEF2D in the cytoplasmic and nuclear fractions, respectively (lanes 5 and 6). These results indicate that in NIH 3T3 cells, endogenous HDAC4 is localized mainly in the cytoplasm.
FIG. 1.

Cytoplasmic localization of HDAC4. (A) Affinity-purified Flag-HDAC4 (lane 1) and cytoplasmic (lanes 2 and 5) and nuclear (lanes 3 and 6) extracts of NIH 3T3 cells were subjected to immunoblotting with the anti-HDAC4 (lanes 1 to 3), anti-14-3-3 (lanes 5 and 6, top), or anti-MEF2D (lanes 5 and 6, bottom) antibody. The amount of extracts was normalized according to cell numbers. The 55-kDa band in lane 2 may not be specific, since it was not reproducibly detected by different bleeds of the anti-HDAC4 antibody. (B) Representative green fluorescence images of NIH 3T3 and 293 cells expressing GFP-HDAC4. (C) Green fluorescence images of two SKN cells (cells a and b) expressing GFP-HDAC4. After initial examination for green fluorescence, LMB (10 ng/ml) was added to the medium and cell b was then analyzed for redistribution of green fluorescence at the indicated times. Under similar conditions, LMB had minimal effects on the pancellular localization of GFP itself (data not shown).
To examine the subcellular localization of HDAC4 in live cells, we performed green fluorescence microscopy. For this, a mammalian vector was constructed to express the fusion protein GFP-HDAC4, with HDAC4 fused to the carboxyl terminus of enhanced GFP. This construct was transfected into NIH 3T3 cells, and live transfected cells were examined for green fluorescence. While GFP itself was pancellular, GFP-HDAC4 was predominantly cytoplasmic in ∼90% of the NIH 3T3 cells transfected (Fig. 1B, left panel, and data not shown). Similarly, unlike GFP, GFP-HDAC4 was cytoplasmic in most 293 cells transfected (middle panel). In a small portion of 293 cells transfected, GFP-HDAC4 was either pancellular or mainly in the nucleus, where it formed dot-like structures (right panel). Compared to NIH 3T3 and 293 cells, more SKN cells (∼25%) expressed GFP-HDAC4 in the nucleus (Fig. 1C and data not shown). Taken together, these results indicate that HDAC4 is localized in the cytoplasm and/or the nucleus in a manner dependent on cellular context.
The distinct subcellular localization of HDAC4 suggests that it may be actively shuttled between the cytoplasm and the nucleus. To address this, we treated transfected SKN cells with leptomycin B (LMB), a specific inhibitor of CRM1-mediated nuclear export (10, 25, 37). As shown in Fig. 1C, LMB elicited rapid nuclear translocation of GFP-HDAC4 in cell b, and after 40 min, GFP-HDAC4 was localized in nuclear dots. LMB treatment of NIH 3T3 and 293 cells also induced nuclear accumulation of GFP-HDAC4 in discrete dots (data not shown). Therefore, like the HDAC4 protein lacking its N-terminal 117 residues (33), full-length HDAC4 is actively exported to the cytoplasm in a CRM1-dependent manner.
HDAC4 interacts with 14-3-3 proteins.
Subcellular compartmentalization of HDAC4 may serve as a regulatory mechanism to control its repression function. We therefore asked how HDAC4 might be retained in the cytoplasm. One possibility is that cytoplasmic anchor proteins are involved. 14-3-3 proteins have been shown to regulate the translocation of FKHRL1 and CDC25C from the nucleus to the cytoplasm (4, 39). 14-3-3 proteins bind to two types of consensus sites: R-(S/Ar)-(+/S)-pS-(L/E/A/M)-P and R-X-(Ar/S)-(+)-pS-(L/E/A/M)-P, where Ar is an aromatic amino acid, pS is phosphoserine, + is a basic amino acid, and X is any amino acid (40, 59). However, atypical 14-3-3 binding sites have also been reported (29). Moreover, 14-3-3 proteins bind to two R-X-R-X-X-pS/T motifs of FKHRL1 (4). With these considerations, we inspected the HDAC4 sequence and found that HDAC4 contains five potential 14-3-3 binding sites: 242-RKTASEP-248, 464-RTQSAP-469, 516-RQPESHP-522, 629-RAQSSP-632, and 703-RGRKATL-709, where the conserved residues are underlined. This observation led us to postulate that HDAC4 may interact with 14-3-3 proteins.
To test this hypothesis, we performed immunoprecipitation. Expression plasmids for Flag-HDAC4 and HA-14-3-3β were transfected into 293 cells, and cell extracts were prepared for affinity purification on anti-Flag M2 agarose or for immunoprecipitation with an anti-HA antibody. As shown in Fig. 2A (top), Flag-HDAC4 was specifically coprecipitated with HA-14-3-3β. Reciprocally, HA-14-3-3β was specifically coprecipitated with Flag-HDAC4 (Fig. 2A, bottom).
FIG. 2.

HDAC4 interacts with 14-3-3. (A) Expression plasmids for Flag-HDAC4 and HA-14-3-3β were cotransfected into 293 cells as indicated. At 48 h after transfection, cell extracts were prepared for affinity purification (AP) on M2 agarose beads (lanes 1 to 4) or immunoprecipitation (IP) with the anti-HA monoclonal antibody (lanes 5 to 8). Bound proteins, eluted with Flag peptide (lanes 1 to 4) or the SDS sample buffer (lanes 5 to 8), were subjected to Western analyses with the anti-Flag (top) or anti-HA antibody (bottom). H, IgG heavy chain; L, light chain. Note that in lanes 1 to 4, no heavy- and light-chain bands are visible because the bound antigens were eluted with Flag peptide from M2 agarose beads, on which the anti-Flag antibody is covalently cross-linked. Whether the bands at the light-chain position in lanes 3 and 4 (bottom) are due to light chains is unclear. (B) NIH 3T3 extracts (lane 1) were subjected to immunoprecipitation with a rabbit preimmune IgG (lane 2) or the rabbit anti-HDAC4 antibody (lane 3) and subsequent Western analysis with the rabbit anti-HDAC4 antibody (top) or a mouse anti-14-3-3 monoclonal antibody (bottom).
We also examined the interaction of endogenous HDAC4 and 14-3-3 proteins by using anti-HDAC4 and anti-14-3-3 antibodies. As shown in Fig. 2B (top), the anti-HDAC4 antibody specifically precipitated endogenous HDAC4. Importantly, the same antibody also precipitated 14-3-3 proteins (Fig. 2B, bottom), further supporting the notion that HDAC4 associates with 14-3-3 proteins.
S246, S467, and S632 of HDAC4 mediate the 14-3-3 binding.
Next we mapped the 14-3-3 binding sites on HDAC4. We first utilized a series of HDAC4 deletion mutants that were already available in our laboratory. Some of these mutants have been described previously (55). These deletion mutants were expressed in 293 cells and affinity purified on anti-Flag M2 agarose, and their ability to copurify 14-3-3 proteins was assessed by immunoblotting. As demonstrated above, endogenous 14-3-3 proteins copurified with Flag-HDAC4 (Fig. 3A, compare lanes 1 and 2). 14-3-3 isoforms have similar properties in binding to their partners (40). These results therefore confirm that HDAC4 physically interacts with 14-3-3 proteins. Like full-length HDAC4 (lanes 1 and 2), mutants hm1 to hm5 coprecipitated 14-3-3 proteins (lanes 3 to 7). This suggests that residues 531 to 1084 of HDAC4 contain a 14-3-3 binding site (Fig. 3B). To test if S632 is essential, we replaced it with alanine to generate the mutant hm6 (Fig. 3B). This mutant was unable to bind to 14-3-3 proteins (Fig. 3A, lane 8), indicating that S632 but not T708 is important for 14-3-3 binding.
FIG. 3.

Mapping of 14-3-3 binding sites. (A) Expression plasmids for HDAC4 and its deletion mutants (all Flag tagged) were transfected into 293 cells, and cell extracts were prepared for affinity purification on M2 agarose. Bound proteins were eluted with the Flag peptide and subjected to Western analyses with the anti-Flag (top) or anti-14-3-3 (bottom) antibody. C (lane 1), control affinity purification using nontransfected cells. For HDAC4 proteins, bands with expected molecular masses are indicated by asterisks. (B) Schematic representation of HDAC4 and its mutants, with their 14-3-3 binding ability indicated at the right. (C) Expression plasmids for HA-tagged hm9 and hm10 were transfected into 293 cells, and cell extracts were prepared for immunoprecipitation with the anti-HA antibody. Immunocomplexes were subjected to immunoblotting with the anti-HA (lanes 1 to 3) or anti-14-3-3 antibody (lanes 4 to 6). H, IgG heavy chain; L, light chain. (D and E) Interaction of Flag-tagged deletion mutants hm11 to hm15 (D) and full-length point mutants (E) with 14-3-3 proteins. The migration difference between hm11 and hm12 may be due to differential phosphorylation. The Flag-tagged HDAC4 proteins were expressed, affinity purified, and analyzed as in panel A.
Unlike hm7, hm8 was able to bind to 14-3-3 proteins (Fig. 3A, lanes 9 and 10), indicating that there are 14-3-3 binding sites between amino acids 208 and 620 of HDAC4 (Fig. 3B). Additional deletion mutants (hm9 to hm11) were analyzed and were all found to bind 14-3-3 proteins (Fig. 3C and D, lanes 1 and 2). This led us to test if S246 of HDAC4 is important for 14-3-3 binding, by replacing S246 with alanine to generate mutant hm12 (Fig. 3B). This mutant was indeed defective in 14-3-3 binding (Fig. 3D, lane 3), indicating that S246 is important for 14-3-3 binding. To address if S467 is required for 14-3-3 binding, mutants hm13 and hm14 were generated (Fig. 3B). Unlike hm13, hm14 was defective in 14-3-3 binding (Fig. 3D, lanes 4 and 5), indicating that S467 is important for 14-3-3 binding. To assess whether S520 is involved in 14-3-3 binding, mutant hm15 was tested (Fig. 3B). This mutant was defective in 14-3-3 binding (Fig. 3D, lane 6). Taken together, these mapping results indicate that S246, S467, and S632 of HDAC4 mediate the binding of 14-3-3 proteins.
For verification of this conclusion and analysis of the functional consequences of 14-3-3 binding, point mutations were introduced at S246, S467, and/or S632 of full-length HDAC4, generating mutants S246A, S467A, S632A, S246/467A, S246/632A, S467/632A, and S246/467/632A. Among these mutants, only S246/467/632A was completely defective in 14-3-3 binding (Fig. 3E and data not shown). These results confirm that S246, S467, and S632 of HDAC4 are all involved in 14-3-3 binding.
14-3-3 binding inhibits nuclear localization of HDAC4.
Next we wished to determine the functional consequences of 14-3-3 binding to HDAC4. The cytoplasmic localization of HDAC4 and its association with 14-3-3 proteins led us to test whether 14-3-3 binding regulates the subcellular localization of HDAC4. To this end, we constructed GFP expression plasmids for the full-length mutants S246A, S467A, S632A, S246/467A, S246/632A, S467/632A, and S246/467/632A. Subcellular localization of these fusion proteins was examined by live-cell fluorescence microscopy. Like the wild-type GFP-HDAC4, the mutants with single mutations were predominantly cytoplasmic in NIH 3T3 cells (Fig. 4A and B). For the mutants with two substitutions, GFP-S246/467A and GFP-S246/632A were nuclear in the majority of transfected cells whereas fewer cells expressed GFP-S467/632A in the nucleus (Fig. 4A and B), suggesting that compared to S467 and S632, S246 plays a more important role in controlling the subcellular localization of HDAC4. The triple mutant GFP-S246/467/632A was nuclear in most transfected cells and formed discrete nuclear dots (Fig. 4A). Similar results were obtained with these mutants in 293 cells (Fig. 4C and data not shown). Since S246, S467, and S632 are important for 14-3-3 binding, these results suggest that 14-3-3 proteins bind to HDAC4 and sequester it in the cytoplasm.
FIG. 4.

Effects of point mutations of S246, S467, and S632 of HDAC4 on its subcellular localization. (A) Representative images of green fluorescence of NIH 3T3 cells expressing HDAC4 and its mutants fused to GFP. (B) Quantitative representation of NIH 3T3 cells expressing HDAC4 or its mutants fused to GFP. Blank bar (C>N), more green fluorescence in the cytoplasm; shaded bar (C=N), fluorescence equally in the cytoplasm and the nucleus; solid bar (N>C), more fluorescence in the nucleus. Average values of three independent experiments are shown with standard deviation indicated by error bars. (C) Representative images of green fluorescence of 293 cells expressing GFP-S246/467/632A.
14-3-3 binding does not affect the deacetylase activity of HDAC4.
The 14-3-3 binding sites were mapped to the N-terminal half of HDAC4, whereas its catalytic domain is located at the C-terminal part. The N-terminal truncations of HDAC4 lead to some activation of its deacetylase activity (9, 55). Moreover, 14-3-3 proteins are known to directly regulate the activity of several enzymes. We thus assessed the effects of 14-3-3 binding on the enzymatic activity of HDAC4. As shown in Fig. 5, the mutant S246/467/632A was almost as active as wild-type HDAC4, suggesting that 14-3-3 binding has minimal effects on the deacetylase activity of HDAC4.
FIG. 5.

Effects of point mutations of S246, S467, and S632 of HDAC4 on its deacetylase activity. (A) Deacetylase activity of HDAC4 and its mutant S246/467/632A. Expression plasmids for Flag-tagged fusion proteins were transfected into 293 cells, and cell extracts were prepared for affinity purification on M2 agarose. Activities of eluted proteins (left) were determined by measuring the release of [3H]acetate from [3H]acetyl-histones. (B) The amount of the eluted proteins was analyzed by immunoblotting with the anti-Flag antibody. The migration position of full-length proteins is indicated by an asterisk.
14-3-3 binding inhibits the repression potential of HDAC4.
Since HDAC4 and its related proteins repress MEF2-dependent transcription (28, 30, 33, 43, 55, 66), we asked whether cytoplasmic retention of HDAC4 indirectly inhibits its repression function. To address this, we conducted reporter gene assays to compare the repression ability of HDAC4 and its mutant S246/467/632A. We first tested MEF2-E4-Luc, which contains a MEF2 consensus site upstream from the adenovirus E4 core promoter driving the luc gene. As shown in Fig. 6A, 50 ng of the HDAC4 construct resulted in reduction of the MEF2C-stimulated reporter activity to the basal level, whereas 10 ng of the mutant construct achieved a similar level of repression. We also tested pJLuc, a Luc reporter driven by the c-Jun promoter (−225/+150) that is known to contain a MEF2 binding site (14). Therefore, compared to wild-type HDAC4, the mutant S246/467/632A was apparently more potent in repressing pJLuc reporter activity (Fig. 6B). To test whether the expression of HDAC4 and its mutant lead to generalized repression, we cotransfected the reporter Gal4-tk-Luc with an expression plasmid for Gal4-VP16. As shown in Fig. 6C, HDAC4 and its triple mutant had minimal effects on the activation mediated by Gal4-VP16, suggesting that expression of HDAC4 and its mutant does not lead to global repression.
FIG. 6.

Repression ability of HDAC4 and its mutant S246/467/632A. (A and B) The reporter (200 ng), MEF2-E4-Luc (A) or pJLuc (B), was transfected into NIH 3T3 cells with a MEF2C expression plasmid (100 ng), an internal control plasmid (CMV-β-Gal; 50 ng), and the expression plasmid for Flag-tagged HDAC4 or S246/467/632A at the indicated amount. The normalized luciferase activity from the transfection without any effector plasmid was arbitrarily set to 1.0. Average values of at least three independent experiments are shown with standard deviation indicated by error bars. (C) A 200-ng sample of the Gal4-tk-Luc reporter was transfected into NIH 3T3 cells with a Gal4-VP16 expression plasmid (5 ng), the internal control plasmid CMV-β-Gal (50 ng), and the expression plasmid for Flag-tagged HDAC4 or S246/467/632A at the indicated amount. The reporter activities were measured as for panels A and B. (D) The Gal4-tk-Luc reporter was transfected into NIH 3T3 cells along with an expression plasmid for Gal4-HDAC4 or Gal4-S246/467/632A. Normalized luciferase activities from transfection with effector plasmids at the indicated amounts were compared with that from the reporter alone to calculate the relative repression. Average values of four independent experiments are shown with standard deviation indicated by error bars.
We also assessed the apparent repression ability of HDAC4 and its mutant by artificially tethering them to a promoter. To do this, HDAC4 and its mutant were expressed as proteins fused to the Gal4 DNA binding domain and tested for the ability to inhibit the reporter activity of Gal4-tk-Luc. As shown in Fig. 6D, Gal4-S246/467/632A was much more potent than Gal4-HDAC4 in repressing Gal4-tk-Luc reporter activity. Indirect-immunofluorescence experiments with an anti-Gal4 antibody revealed that unlike Gal4-HDAC4, Gal4-S246/467/632A was predominantly nuclear in NIH 3T3 cells (data not shown). Taken together, these results support the notion that 14-3-3 proteins sequester HDAC4 away from its targets in the nucleus and thereby indirectly inhibit its repression function.
DISCUSSION
HDAC4 is localized in the cytoplasm and/or the nucleus.
The results presented herein support the notion that HDAC4 is localized in the cytoplasm and/or the nucleus. This is consistent with reports on the subcellular localization of the HDAC4 protein lacking the N-terminal 117 residues (9, 33). An interesting question is why, even for the same cell line, HDAC4 is nuclear in some cells but cytoplasmic in others (Fig. 1) (33). One possibility is that cell cycle progression may affect the subcellular localization. However, we did not find evidence that the subcellular localization of HDAC4 is regulated during the cell cycle (data not shown). Other possibilities include growth conditions, extracellular signaling events, and heterogeneity of cells in the cell lines used. Clearly, these interesting issues merit further investigation. While HDAC4 was evenly distributed in the cytoplasm, it occupied dot-like patterns in the nucleus (Fig. 1 and 4). Such nuclear dots have been observed by others (20, 33), but their physiological significance remains to be established.
The cytoplasmic and nuclear localization of HDAC4 suggests that it may have functions in both compartments. Alternatively, such a subcellular localization may simply serve as a regulatory mechanism for HDAC4. Since HDAC4 is known to be involved in transcriptional regulation (30, 33, 55), its cytoplasmic localization may negatively regulate its function in the nucleus. Indeed, the nuclear localization of HDAC4 is negatively regulated by binding to 14-3-3 proteins (Fig. 2 to 6). This also suggests that by analogy to DNA-binding transcription factors, the control of nuclear localization is an important regulatory mechanism for transcriptional coregulators. This is the case for at least two other coregulators, β-catenin and activated Notch (7, 42).
The distinct subcellular localization of HDAC4 also suggests that it is actively shuttled between the cytoplasm and the nucleus. Consistent with this suggestion, LMB treatment was found to elicit nuclear accumulation of GFP-HDAC4 (Fig. 1C). Since LMB specifically inhibits CRM1 (10, 25, 37), HDAC4 may be actively exported in a CRM1-dependent manner. Using known consensus nuclear import and export sequences (35), we inspected the amino acid sequence of HDAC4 and found that it contains three putative bipartite nuclear localization signals and three potential leucine-rich nuclear export signals. Therefore, HDAC4 possesses putative intrinsic nucleocytoplasmic trafficking signals. It is tempting to speculate that the subcellular localization of HDAC4 is dependent on its nuclear import as well as on its nuclear export (Fig. 7). If its nuclear import dominates, more HDAC4 molecules end up in the nucleus, and if export dominates, the reverse is true. Therefore, factors that alter its nuclear import, export, or both will also affect the subcellular localization of HDAC4.
FIG. 7.
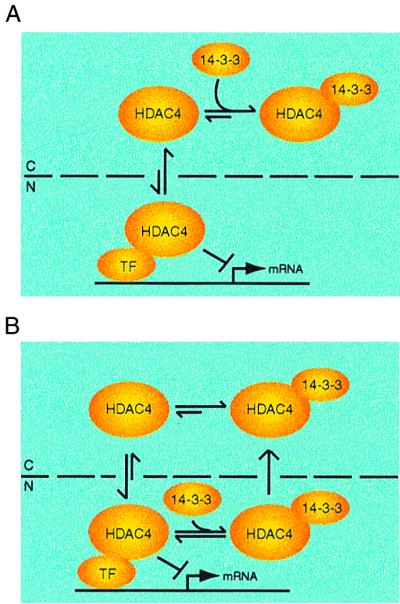
Model depicting possible modes of regulation of HDAC4 by 14-3-3 proteins. HDAC4 is actively shuttled between the cytoplasm (C) and the nucleus (N), and the relative rate of nuclear import and export may determine the subcellular localization. 14-3-3 binding may shift the distribution equilibrium of HDAC4 toward cytoplasmic accumulation by hindering its nuclear import (A) and/or facilitating its nuclear export (B). 14-3-3 proteins have been shown to be subject to active nuclear export (40), so they can interact with HDAC4 in the nucleus (B). Association of HDAC4 with other proteins may also affect its localization. In this study, we have investigated how 14-3-3 proteins regulate the functions of HDAC4. Theoretically, it is also possible that HDAC4 regulates the functions of 14-3-3 proteins such as their ability to regulate the function of their binding partners (1, 4, 19, 38, 39) and to bind to cruciform DNA molecules (19, 50).
Physical association of HDAC4 with 14-3-3 proteins.
Besides its putative nuclear localization and export signals, HDAC4 also contains five putative 14-3-3 binding sites (Fig. 3B). Importantly, we have found that HDAC4 interacts with 14-3-3 proteins (Fig. 2 and 3). Among the five putative 14-3-3 binding sites on HDAC4, only S246, S467, and S632 appeared to mediate the interaction (Fig. 3A to D). Consistent with this, the triple mutant S246/467/632A was completely incapable of binding to 14-3-3 proteins (Fig. 3E). These findings indicate that HDAC4 possesses three functional 14-3-3 binding sites. By contrast, CDC25C contains only one 14-3-3 binding site (39). 14-3-3 proteins exist as homodimers in the cells (40, 59), so one molecule of HDAC4 may bind to two 14-3-3 homodimers with one of their four phosphoserine-binding pockets free. Interestingly, 14-3-3 proteins contain functional nuclear export signals within their binding pockets (40), raising the possibility that 14-3-3 proteins bind to HDAC4 and provide it with functional nuclear export signals (see below).
Regulation of HDAC4 by binding of 14-3-3 proteins.
What is the functional consequence of 14-3-3 binding to HDAC4? The HDAC4 mutant S246/467/632A had deacetylase activity comparable to that of the wild-type protein (Fig. 5), suggesting that 14-3-3 binding does not affect the deacetylase activity of HDAC4. Significantly, unlike the wild-type HDAC4 protein, the triple mutant S246/467/632A was predominantly nuclear (Fig. 4). This is consistent with the finding that this triple mutant was apparently more potent than the wild-type protein in reporter gene assays (Fig. 6D). Therefore, 14-3-3 binding negatively regulates the repression function of HDAC4 by interfering with its nuclear localization. Such a regulatory mode is similar to those reported for CDC25C (39) and Forkhead transcription factors (4, 6) but different from that reported for a homeodomain transcription factor (48).
How does 14-3-3 binding lead to the cytoplasmic accumulation of HDAC4? As discussed above, HDAC4 is actively shuttled between the cytoplasm and the nucleus, and any factors that alter its nuclear import, nuclear export, or both also affect its subcellular localization. We speculate that without 14-3-3 binding, the nuclear import of HDAC4 may prevail and lead to its nuclear localization. Consistent with this speculation, the triple mutant S246/467/632A was incapable of binding to 14-3-3 proteins and was thus predominantly nuclear (Fig. 4). With 14-3-3 binding, the dynamic shuttling of HDAC4 may be shifted toward cytoplasmic accumulation. Therefore, 14-3-3 binding plays a contributing role in determining the subcellular localization of HDAC4. 14-3-3 binding may interfere with the nuclear import of HDAC4. Related to this, there are two putative nuclear localization signals close to the S246 14-3-3 binding site of HDAC4, and 14-3-3 binding to S246 of HDAC4 plays an important role in regulating the subcellular localization of HDAC4 (Fig. 4). Alternatively, association with 14-3-3 proteins may stimulate the nuclear export of HDAC4. Indeed, each 14-3-3 isoform contains a functional nuclear export signal (40). Therefore, we propose that 14-3-3 proteins sequester HDAC4 in the cytoplasm by directly hindering its nuclear import and/or facilitating its nuclear export (Fig. 7). A third possibility is that 14-3-3 proteins simply serve as cytoplasmic anchors for HDAC4. Further studies are needed to distinguish these possibilities.
Once in the nucleus, HDAC4 may initiate the assembly of fully functional repression complexes by association with DNA binding transcription factors such as MEF2s (28, 33, 55) and transcriptional corepressors such as HDAC3 (11) and SMRT/N-CoR (13, 17, 20). 14-3-3 binding to HDAC4 may serve as a switch that controls the assembly of these repression complexes. How is this switch turned on and off? Since 14-3-3 proteins are known phosphoserine-binding adapters (34, 40, 59), they may bind to HDAC4 in a phosphorylation-dependent manner. This is supported by the finding that replacement of S246, S467, and S632 of HDAC4 with the nonphosphorylable residue alanine abolished 14-3-3 binding (Fig. 3). Phosphorylation of these three serine residues may be controlled by known or unknown protein kinases and phosphatases. Consistent with this, we have found that Flag-HDAC4 is heavily phosphorylated in 293 cells (data not shown). How the interaction of HDAC4 with 14-3-3 proteins is regulated by phosphorylation is an interesting question that merits further investigation.
Like HDAC4, HDAC5 and HDAC7 contain putative 14-3-3 binding sites (9, 11, 20, 33, 52, 55), so HDAC5 and HDAC7 may be subject to similar regulation by 14-3-3 proteins. On the other hand, HDAC6 and Hda1 possess no obvious 14-3-3 binding motifs (11, 33, 41, 52, 55). Therefore, 14-3-3 proteins may regulate a subfamily of class II HDACs by affecting their subcellular localization. Interestingly, the subcellular localization of the recently identified NAD-dependent deacetylase SIR2 may be also regulated (18, 27). Furthermore, it has been recently reported that chicken HDAC3 may be subject to active nuclear export (47). Therefore, controlled subcellular compartmentalization may be one general regulatory mechanism for members of different classes of HDACs.
In summary, this study supports the notion that HDAC4 is localized in the cytoplasm and/or the nucleus. Through S246, S467, and S632, HDAC4 interacts with the 14-3-3 family of proteins. Moreover, the binding of 14-3-3 proteins negatively regulates the function of HDAC4 by excluding it from the nucleus. Future experiments on how the association of 14-3-3 proteins with HDAC4 and perhaps its homologs is regulated will shed light on the molecular mechanisms by which deacetylation of acetylated histones and nonhistone proteins is controlled in vivo.
ACKNOWLEDGMENTS
We thank J. Th'ng for advice on isolation of [3H]acetyl-histones, M. Yoshida for LMB, R. Prywes for anti-MEF2D antibody, M. Park and her laboratory members for kind help with fluorescence microscopy, and V. Giguère for helpful discussions.
This work was supported by funds from the National Cancer Institute of Canada (to X.J.Y.). A.H.W. is the recipient of a Canadian Institutes of Health Research (CIHR) doctoral research award. J.W. received support from the Lady Davis Medical Institute, Montreal, Quebec, Canada. X.J.Y. is a CIHR scholar.
A.H.W., M.J.K., and J.W. made equally significant contributions to this work.
ADDENDUM IN PROOF
A similar conclusion about regulation of HDAC4 by 14-3-3 was also recently reported by C. M. Grozinger and S. L. Schreiber (Proc. Natl. Acad. Sci. USA 97:7835–7840, 2000).
REFERENCES
- 1.Aitken A. 14-3-3 and its possible role in co-ordinating multiple signaling pathways. Trends Cell Biol. 1996;6:341–347. doi: 10.1016/0962-8924(96)10029-5. [DOI] [PubMed] [Google Scholar]
- 2.Beck T, Hall M N. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999;402:689–692. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- 3.Biggs W H, Meisenhelder J, Hunter T, Cavenee W K, Arden K C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunet A, Bonni A, Zigmond M J, Lin M Z, Juo P, Hu L S, Anderson M J, Arden K C, Blenis J, Greenberg M E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 5.Champagne N, Bertos N R, Pelletier N, Wang A H, Vezmar M, Yang Y, Heng H H, Yang X J. Identification of a human histone acetyltransferase related to monocytic leukemia zinc finger protein. J Biol Chem. 1999;274:28528–28536. doi: 10.1074/jbc.274.40.28528. [DOI] [PubMed] [Google Scholar]
- 6.Datta S R, Brunet A, Greenberg M E. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 7.Eastman Q, Grosschedl R. Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr Opin Cell Biol. 1999;11:233–240. doi: 10.1016/s0955-0674(99)80031-3. [DOI] [PubMed] [Google Scholar]
- 8.Emiliani S, Fischle W, Van Lint C, Al-Abed Y, Verdin E. Characterization of a human RPD3 ortholog, HDAC3. Proc Natl Acad Sci USA. 1998;95:2795–2800. doi: 10.1073/pnas.95.6.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischle W, Emilian S, Hendzel M J, Nagase T, Nomura N, Voelter W, Verdin E. A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p. J Biol Chem. 1999;274:11713–11720. doi: 10.1074/jbc.274.17.11713. [DOI] [PubMed] [Google Scholar]
- 10.Fornerod M, Ohno M, Yoshida M, Mattaj I W. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 11.Grozinger C M, Hassig C A, Schreiber S L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 13.Guenther M G, Lane W S, Fischle W, Verdin E, Lazar M A, Shiekhattar R. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 2000;14:1048–1057. [PMC free article] [PubMed] [Google Scholar]
- 14.Han T-H, Prywes R. Regulatory role of MEF2D in serum induction of the c-Jun promoter. Mol Cell Biol. 1995;15:2907–2915. doi: 10.1128/mcb.15.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hassig C A, Schreiber S L. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Curr Opin Chem Biol. 1997;1:300–308. doi: 10.1016/s1367-5931(97)80066-x. [DOI] [PubMed] [Google Scholar]
- 16.Hu E, Chen Z, Fredrickson T, Zhu Y, Kirkpatrick R, Zhang G F, Johanson K, Sung C, Liu R, Winkler J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J Biol Chem. 2000;275:15254–15264. doi: 10.1074/jbc.M908988199. [DOI] [PubMed] [Google Scholar]
- 17.Huang E Y, Zhang J, Miska E A, Guenther M G, Kouzarides T, Lazar M A. Nuclear receptor corepressors partner with class II histone deacetylases in a Sin3-independent repression pathway. Genes Dev. 2000;14:45–54. [PMC free article] [PubMed] [Google Scholar]
- 18.Imai S I, Armstrong C M, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–799. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 19.Imhof A, Wolffe A P. Purification and properties of the Xenopus Hat1 acetyltransferase: association with the 14-3-3 proteins in the oocyte nucleus. Biochemistry. 1999;38:13085–13093. doi: 10.1021/bi9912490. [DOI] [PubMed] [Google Scholar]
- 20.Kao H Y, Downes M, Ordentlich P, Evans R M. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 2000;14:55–66. [PMC free article] [PubMed] [Google Scholar]
- 21.Knoepfler P S, Eisenman R N. Sin meets NuRD and other tails of repression. Cell. 1999;99:447–450. doi: 10.1016/s0092-8674(00)81531-7. [DOI] [PubMed] [Google Scholar]
- 22.Kops G J, de Ruiter N D, De Vries-Smits A M, Powell D R, Bos J L, Burgering B M. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;396:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- 23.Kornberg R D, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 24.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner E P, Wolff B, Yoshida M, Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci USA. 1999;96:9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laherty C D, Billin A N, Lavinsky R M, Yochum G S, Bush A C, Sun J M, Mullen T M, Davie J R, Rose D W, Glass C K, Rosenfeld M G, Ayer D E, Eisenman R N. SAP30, a component of the mSin3 corepressor complex involved in N-CoR-mediated repression by specific transcription factors. Mol Cell. 1998;2:33–42. doi: 10.1016/s1097-2765(00)80111-2. [DOI] [PubMed] [Google Scholar]
- 27.Landry J, Sutton A, Tafrov S T, Heller R C, Stebbins J, Pillus L, Sternglanz R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci USA. 2000;97:5801–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemercier C, Verdel A, Galloo B, Curtet S, Brocard M, Khochbin S. mHDA1/HDAC5 histone deacetylase interacts with and represses MEF2A transcriptional activity. J Biol Chem. 2000;275:15594–15599. doi: 10.1074/jbc.M908437199. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y C, Liu Y, Elly C, Yoshida H, Lipkowitz S, Altman A. Serine phosphorylation of Cbl induced by phorbol ester enhances its association with 14-3-3 proteins in T cells via a novel serine-rich 14-3-3-binding motif. J Biol Chem. 1997;272:9979–9985. doi: 10.1074/jbc.272.15.9979. [DOI] [PubMed] [Google Scholar]
- 30.Lu J, McKinsey T A, Nicol R L, Olson E N. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci USA. 2000;8:4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lusser A, Brosch G, Loidl A, Haas H, Loidl P. Identification of maize histone deacetylase HD2 as an acidic nucleolar phosphoprotein. Science. 1997;277:88–91. doi: 10.1126/science.277.5322.88. [DOI] [PubMed] [Google Scholar]
- 32.Maroun C R, Moscatello D K, Naujokas M A, Holgado-Madruga M, Wong A J, Park M. A conserved inositol phospholipid binding site within the pleckstrin homology domain of the Gab1 docking protein is required for epithelial morphogenesis. J Biol Chem. 1999;274:31719–31726. doi: 10.1074/jbc.274.44.31719. [DOI] [PubMed] [Google Scholar]
- 33.Miska E, Karlsson C, Langley E, Nielsen S, Pines J, Kouzarides T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999;18:5099–5107. doi: 10.1093/emboj/18.18.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muslin A J, Tanner J W, Allen P M, Shaw A S. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 35.Nakielny S, Dreyfuss G. Transport of proteins and RNAs in and out of the nucleus. Cell. 1999;99:677–690. doi: 10.1016/s0092-8674(00)81666-9. [DOI] [PubMed] [Google Scholar]
- 36.Ng H H, Bird A. Histone deacetylases: silencers for hire. Trends Biochem Sci. 2000;25:121–126. doi: 10.1016/s0968-0004(00)01551-6. [DOI] [PubMed] [Google Scholar]
- 37.Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278:141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- 38.Pawson T, Scott J D. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 39.Piwnica-Worms H. Cell cycle: fools rush in. Nature. 1999;401:535–537. doi: 10.1038/44029. [DOI] [PubMed] [Google Scholar]
- 40.Rittinger K, Budman J, Xu J, Volinia S, Cantley L C, Smerdon S J, Gamblin S J, Yaffe M B. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol Cell. 1999;4:153–166. doi: 10.1016/s1097-2765(00)80363-9. [DOI] [PubMed] [Google Scholar]
- 41.Rundlett S E, Carmen A A, Kobayashi R, Bavykin S, Turner B M, Grunstein M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc Natl Acad Sci USA. 1996;93:14503–14508. doi: 10.1073/pnas.93.25.14503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schroeter E H, Kisslinger J A, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 43.Sparrow D B, Miska E A, Langley E, Reynaud-Deonauth S, Kotecha S, Towers N, Spohr G, Kouzarides T, Mohun T J. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 1999;18:5085–5098. doi: 10.1093/emboj/18.18.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strahl B D, Allis C D. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 45.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 46.Takaishi H, Konishi H, Matsuzaki H, Ono Y, Shirai Y, Saito N, Kitamura T, Ogawa W, Kasuga M, Kikkawa U, Nishizuka Y. Regulation of nuclear translocation of Forkhead transcription factor AFX by protein kinase B. Proc Natl Acad Sci USA. 1999;96:11836–11841. doi: 10.1073/pnas.96.21.11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takami Y, Nakayama T. N-terminal region, C-terminal region, nuclear export signal, and deacetylase activity of histone deacetylase-3 are essential for the viability of the DT40 chicken cell line. J Biol Chem. 2000;275:16191–16201. doi: 10.1074/jbc.M908066199. [DOI] [PubMed] [Google Scholar]
- 48.Tang S J, Suen T C, McInnes R R, Buchwald M. Association of the TLX-2 homeodomain and 14-3-3 signaling proteins. J Biol Chem. 1998;273:25356–25363. doi: 10.1074/jbc.273.39.25356. [DOI] [PubMed] [Google Scholar]
- 49.Taunton J, Hassig C A, Schreiber S L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 50.Todd A, Cossons N, Aitken A, Price G B, Zannis-Hadjopoulos M. Human cruciform binding protein belongs to the 14-3-3 family. Biochemistry. 1998;37:14317–14325. doi: 10.1021/bi980768k. [DOI] [PubMed] [Google Scholar]
- 51.Tong J K, Hassig C A, Schnitzler G R, Kingston R E, Schreiber S L. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature. 1998;395:917–921. doi: 10.1038/27699. [DOI] [PubMed] [Google Scholar]
- 52.Verdel A, Khochbin S. Identification of a new family of higher eukaryotic histone deacetylases. J Biol Chem. 1999;274:2440–2445. doi: 10.1074/jbc.274.4.2440. [DOI] [PubMed] [Google Scholar]
- 53.Wade P A, Gegonne A, Jones P L, Ballestar E, Aubry F, Wolffe A P. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet. 1999;23:62–66. doi: 10.1038/12664. [DOI] [PubMed] [Google Scholar]
- 54.Wade P A, Jones P L, Vermaak D, Wolffe A P. A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated SNF2 superfamily ATPase. Curr Biol. 1998;8:843–846. doi: 10.1016/s0960-9822(98)70328-8. [DOI] [PubMed] [Google Scholar]
- 55.Wang A H, Bertos N R, Vezmar M, Pelletier N, Crosato M, Heng H H, Th'ng J, Han J, Yang X J. HDAC4, a human histone deacetylase related to yeast HDA1, is a potent transcriptional corepressor. Mol Cell Biol. 1999;19:7816–7827. doi: 10.1128/mcb.19.11.7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolffe A P, Wong J, Pruss D. Activators and repressors: making use of chromatin to regulate transcription. Genes Cells. 1997;2:291–302. doi: 10.1046/j.1365-2443.1997.1260323.x. [DOI] [PubMed] [Google Scholar]
- 57.Workman J L, Kingston R E. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu Rev Biochem. 1998;67:545–579. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- 58.Xue Y, Wong J, Moreno G T, Young M K, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–861. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 59.Yaffe M B, Rittinger K, Volinia S, Caron P R, Aitken A, Leffers H, Gamblin S J, Smerdon S J, Cantley L C. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 1996;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 60.Yang W M, Inouye C, Zeng Y, Bearss D, Seto E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc Natl Acad Sci USA. 1996;93:12845–12850. doi: 10.1073/pnas.93.23.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang W M, Yao Y L, Sun J M, Davie J R, Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J Biol Chem. 1997;272:28001–28007. doi: 10.1074/jbc.272.44.28001. [DOI] [PubMed] [Google Scholar]
- 62.Youn H D, Grozinger C M, Liu J O. Calcium regulates transcriptional repression of myocyte enhancer factor 2 by histone deacetylase 2. J Biol Chem. 2000;275:22563–22567. doi: 10.1074/jbc.C000304200. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y, LeRoy G, Seelig H P, Lane W S, Reinberg D. The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell. 1998;95:279–289. doi: 10.1016/s0092-8674(00)81758-4. [DOI] [PubMed] [Google Scholar]
- 64.Zhang Y, Ng H H, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–1935. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Sun Z W, Iratni R, Erdjument-Bromage H, Tempst P, Hampsey M, Reinberg D. SAP30, a novel protein conserved between human and yeast, is a component of a histone deacetylase complex. Mol Cell. 1998;1:1021–1031. doi: 10.1016/s1097-2765(00)80102-1. [DOI] [PubMed] [Google Scholar]
- 66.Zhou X, Richon V M, Rifkind R A, Marks P A. Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc Natl Acad Sci USA. 2000;97:1056–1061. doi: 10.1073/pnas.97.3.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]