

Abstract
Protein kinase D3 (PKD3) is upregulated in triple-negative breast cancer (TNBC) and associated with cell proliferation and metastasis development but its precise pro-oncogenic function is unknown. Here we show that PKD3 is required for the maintenance of the TNBC stem cell population. Depletion of PKD3 in MDA-MB-231 cells reduced the cancer stem cell frequency in vitro and tumor initiation potential in vivo. We further provide evidence that the RhoGEF GEF-H1 is upstream of PKD3 activation in TNBC stem cells. Most importantly, pharmacological PKD inhibition in combination with paclitaxel synergistically decreased oncosphere and colony formation efficiency in vitro and tumor recurrence in vivo. Based on our results we propose that targeting the GEF-H1/PKD3 signaling pathway in combination with chemotherapy might provide an effective therapeutic option for TNBC.
Keywords: cancer stem cells, TNBC, PKD3, paclitaxel, ALDH
Introduction
Despite improved early detection and the development of novel therapeutics, triple-negative breast cancer (TNBC) still is associated with a high mortality rate. TNBC accounts for 15–20% of all breast carcinomas and is characterized by the lack of estrogen and progesterone receptors (ER/PR) and HER2 overexpression1. Compared to HER2 and ER/PR positive breast cancers TNBC is associated with an increased risk for metastasis and lower 5-year survival2–4. TNBC treatment is mainly based on chemotherapeutics such as anthracyclines and taxanes, however, many TNBCs rapidly develop resistance to these drugs5. Notably, after the treatment with taxanes an expansion of CD44+/CD24- and aldehyde dehydrogenase 1 (ALDH)-positive TNBC cells was observed6–8. Expression of these markers is characteristic of cancer stem cells (CSC), a small subpopulation of undifferentiated cells within the tumor mass. The presence of CSC is linked to early metastases, faster tumor recurrence, and worse overall survival9–12. Moreover, apart from infinite proliferation and self-renewal, CSCs have the capability to initiate tumor formation13. Understanding the mechanisms that underlie the self-renewal behaviour and survival of CSCs is thus critical for developing novel therapeutic strategies that also target CSCs.
The protein kinase D (PKD) family comprising PKD1, 2 and 3 is best known for controlling vesicle fission at the Golgi complex and actin remodeling during cell migration (reviewed in 14, 15). In normal breast tissue, PKD1 is the predominant isoform but its gene expression is suppressed during cancer progression by epigenetic silencing16. In TNBC, an isoform switch towards high PKD3 expression occurs, resulting from the lack of ER, a repressor of PKD3 gene expression17, 18. Elevated PKD3 levels are associated with increased proliferation and cell motility, metastatic progression, as well as poor prognosis, supporting a pro-oncogenic role for PKD3 in TNBC17, 19, 20. However, molecular pathways controlling the activity of PKD3 and its precise pro-oncogenic function in TNBC have remained elusive.
By targeted cell surface screening, we have discovered a novel function for PKD3 in the maintenance of CSCs. We show that the loss of PKD3 in TNBC cell lines reduced the stem cell frequency in vitro and decreased the tumor initiation potential of implanted MDA-MB-231 cells in vivo. We further provide evidence that the function of PKD3 in the maintenance of CSCs requires upstream activation by the Rho guanine nucleotide exchange factor 2 (GEF-H1). Significantly, the combinatorial treatment with the pharmacological PKD inhibitor CRT0066101 and the chemotherapeutic paclitaxel was superior in reducing oncosphere formation in vitro and tumor recurrence in vivo when compared to monotherapy. Our results thus demonstrate the importance of PKD3-mediated CSC regulation and provide a rationale for targeting the GEF-H1/PKD3 signaling pathway to eliminate the tumor initiating cell population in TNBC.
Material and Methods
Cell Culture and Cell Line Authentication
MDA-MB-231 cells (CLS, RRID:CVCL_0062) and MDA-MB-231-based knockdown cell lines (shNon_CTRL, shPKD3_1, shPKD3_2; 17) were cultured in DMEM low glucose. MDA-MB-436 cells (Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology, Stuttgart, RRID:CVCL_0623) were cultured in DMEM high glucose. MDA-MB-468 (CLS, RRID:CVCL_0419), BT-549 (CLS, RRID:CVCL_1092), HCC1806 (ATCC, RRID:CVCL_1258), SKBR3 (CLS, RRID:CVCL_0033), T47D (Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology, Stuttgart, RRID:CVCL_055) and MCF7 (Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology, Stuttgart, RRID:CVCL_0031) cells were cultured in RPMI-1640. All media were supplemented with 10 % fetal bovine serum (PAA Laboratories). MCF10A_EcoR_PKD3WT-EGFP (generated in this study) and MCF10A cells (Department of Biomedicine, University of Basel, RRID:CVCL_0598) and were cultured in DMEM/F12 GlutaMAX supplemented with 5 % horse serum, 20 ng/ml EGF, 0.5 µg/ml hydrocortisone, 100 ng/ml cholera toxin and 10 µg/ml insulin. All cell lines were cultured at 37°C in a humidified chamber with 5 % CO2, tested negative for Mycoplasma (Lonza, LT07–318) and were authenticated within the last three years by SNP profiling (Multiplexion GmbH).
Cell Surface Protein Screen
LEGENDScreen kit was used according to manufacturer’s instructions. In brief, cells were detached with accutase and seeded (3×104 cells) into 96-well plates containing the pre-diluted PE-labelled antibodies. After 40 min of incubation at 4°C, cells were fixed for 10 min at RT,.washed and analyzed by flow cytometry using MACSQuant® Analyzer 10 (Miltenyi Biotec). Data were evaluated using FlowJo. Relative mean fluorescence intensities (rel. MFI) were calculated as follows: rel. MFI = [MFIsample – (MFIisotype – MFIcells)] / MFIcells. Rel MFI values < 1.5 were not considered. After normalizing to shNon_CTRL, surface proteins in the shPKD3_1 cells with values ≤ 0.9 were considered to be downregulated, those with values ≥ 1.1 were considered to be upregulated.
Sphere Formation Assays
For extreme limiting dilution assays (ELDA), cells were trypsinized, singularized using a 27G needle and 1, 10 or 100 cells were seeded onto Poly(2-hydroxyethyl methacrylate)-(pHEMA)-treated 96-well plates, respectively. Cells were cultured in 300 µl sphere formation medium containing DMEM/F12 GlutaMAX supplemented with 10 µg/ml insulin, 20 ng/ml EGF, 1 µg/ml hydrocortisone and 1x B27-supplement. 20 technical replicates per condition were cultured for 10 days. Only wells positive for spheres, no matter the actual number, were regarded as a positive. Multiple group comparison and estimated stem cell frequency was analyzed using the ELDA software21.
For primary sphere formation assays, cells were trypsinized, singularized using a 27G needle, seeded onto pHEMA-treated plates in sphere formation medium and cultured for 5 days. For secondary sphere formation assays, primary spheres were collected, trypsinized, singularized and cultured as described above. shNon, shPKD3_1, shPKD3_2, MDA-MB-231, MDA-MB-468, MDA-MB-436, HCC1806 and MCF7 cells were seeded onto 12-well plates (primary assay: 3×103 cells; secondary assay: 1×103 cells) in 1.5 ml sphere formation medium. BT-549 cells were seeded onto 6-well plates (primary/ secondary assay: 5×103 cells) in 3 ml sphere formation medium. MCF10A_EcoR_PKD3WT-EGFP cells were pre-treated with doxycycline (Dox) for 24 h before the cells were singularized and seeded (5×103 cells) onto 6-well plates in 3 ml sphere formation medium. Subsequently, the sphere formation medium was supplemented with Dox and cells were cultured for 5 days. Spheres were imaged and the sphere area was analyzed using ImageJ. Only spheres larger than 2,500 µm2 were taken into consideration. Data are presented as sphere area (mm2) or sphere formation efficiency (SFE; spheres formed per 1000 cells seeded).
ALDH activity assay
For the ALDH assay, 1×105 singularized cells were diluted in 100 µl Assay Buffer containing 1.5 µl of activated ALDEFLUOR Reagent. Subsequently, 50 µl were removed and mixed with 1.5 µl DEAB Reagent (control). Control and test samples were incubated at 37°C for 30 min, followed by flow cytometry analysis using MACSQuant Analyzer 10 (Miltenyi Biotec). Data were evaluated using FlowJo. DEAB treated samples served as internal controls.
Orthotopic Tumor Models
Animal experiments were approved by state authorities and carried out according to federal guidelines. For the ELDA, 8-week-old female SCID mice were injected with 5×105 or 5×104 MDA-MB-231 shNon_CTRL or shPKD3_1 cells in 100 µl PBS into the right fat pad of the 4th nipple. Tumor growth was analyzed by caliper measurements. Mice were sacrificed after 8 weeks. Multiple group comparison and estimated stem cell frequency was analyzed using the ELDA software21. Tumors were imaged, dissociated according to the manufacturer’s instructions and processed for oncosphere formation assays and ALDH analysis.
For combined paclitaxel and CRT0066101 treatment, 8-week-old female SCID mice were injected with 2×106 MDA-MB-231 cells in 100 µl PBS into the right fat pad of the 4th nipple. Tumor growth was analyzed by caliper measurements ((length x width2)/2). After the tumors had reached 100 mm3 mice were assigned to control and 3 treatment groups (n=7 mice per group). During the following treatment-phase (21 days), mice were treated with either (a) CRT0066101 (peroral, 80 mg/kg diluted in a 5 % dextrose saline solution), (b) paclitaxel (intraperitoneal injection, 10 mg/kg body weight diluted in a 10 % polysorbate PBS solution), (c) CRT0066101 in combination with paclitaxel or (d) the combination of the respective carriers (control). Paclitaxel was injected at day 23, 30, 37 and 44. CRT0066101 was given once daily from day 23 to day 43. After the treatment-phase, tumor growth was monitored for 20 additional days.
Statistical analysis
Data are presented as mean ± SEM. Significance between multiple groups was determined by one-way or two-way ANOVA and Bonferroni’s test for multiple comparison. Significance between two groups was determined by t-test. Data were analyzed using GraphPad Prism 7. P-values: P ≤ 0.05 (*), P ≤ 0.01 (**), P ≤ 0.001 (***), P ≤ 0.0001 (****). Combination index (CI) values were calculated using Webb’s fractional product method. CI<1 indicates synergism, 1≤CI≤1.09 addition and CI>1.09 antagonism.
Additional material and methods can be found in the supplementary materials and methods.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
PKD3 depletion decreases CSC-like properties in MDA-MB-231 cells
It is well established that PKD3 controls vesicular traffic to the plasma membrane thereby modulating the expression of cell surface proteins22. To analyze how PKD3 controls the cell surface phenotype of breast cancer cells, we used the TNBC cell line MDA-MB-231 stably depleted of PKD3 (shPKD3_1)17 and performed a flow cytometry-based screening of 332 molecules (Figure 1A). MDA-MB-231 cells stably expressing a scrambled shRNA were used as control (shNon_CTRL) (Figure S1A-B). 102 cell surfaces proteins were above the detection threshold (see Table S1). In the PKD3 deficient cells, 68 proteins displayed dysregulated surface levels with respect to the control (24 upregulated and 44 downregulated). Several of the proteins with decreased cell surface expression have been associated with the maintenance of breast CSCs, such as CD44, CD184, CD61, NOTCH2, NOTCH4, and CD3046, 7, 23–27 (Figure 1B, Figure S1C-H, Table S1). This prompted us to measure the stem cell frequency of the stable MDA-MB-231 cell lines by performing extreme limiting dilution assays (ELDA) (Figure 1C). Indeed, the estimated stem cell frequency was significantly decreased in PKD3 depleted cells when compared to the stem cell frequency of the control cells (Figure 1D). We then analyzed the gene expression of a panel of breast cancer stemness markers including SOX2, OCT3/4, NANOG, NOTCH4, CD44, SNAI1 and ALDH1 in monolayer (2D)-cultured shPKD3_1 and shNon_CTRL cells11, 28. Compared to the control cells, we observed a significant downregulation of the corresponding genes in shPKD3_1 cells (Figure 1E). Enhanced ALDH activity is considered a hallmark of breast CSCs29, 30. When comparing MDA-MB-231 shPKD3_1 cells with shNon_CTRL cells, we observed that PKD3 depletion caused a significant decrease in ALDH activity as measured by aldefluor assay (Figure 1F), which further substantiated a potential role of the kinase in stem cell regulation. Finally, we investigated the formation of primary and secondary oncospheres as a measure of CSC activity31. Here, the depletion of PKD3 caused an up to 70% decrease in primary and s econdary sphere forming efficiency (SFE) (Figure 1G) in comparison to the control. Remarkably, the knockdown of PKD3 neither changed the size of the oncospheres (Figure 1H), nor did it induce cell death by anoikis, excluding anti-proliferative and pro-apoptotic effects (Figure S1I-J).
Figure 1: Loss of PKD3 reduces stemness in MDA-MB-231 cells.

(A) Cell surface screen workflow. (B) Flow cytometry-based surface protein screening. Data are presented as relative mean fluorescence intensity (rel. MFI) normalized to shNon_CTRL, n=2. (C) ELDA, analysis of 20 wells of a 96-well plate per condition. Only sphere-positive wells were considered. Data are presented as mean ± SEM, n=3. Statistical comparison by ELDA multiple group analysis. (D) ELDA-based estimated stem cell frequency. Data are presented as mean estimated stem cell frequency normalized to shNon_CTRL, n=3. Statistical comparison by t-test. (E) qPCR analysis of stemness markers. Data are presented as mean mRNA expression normalized to shNon_CTRL, n=4. Statistical comparison by t-test. (F) Left: flow cytometry-based ALDH activity analysis. Data are presented as mean ALDHhigh population normalized to shNon_CTRL, n=4. Statistical comparison by t-test. Right: Dot plot of ALDH measurements. (G) Mean SFE of primary and secondary oncosphere assays, n=4. Statistical comparison by one-way ANOVA and Bonferroni-test. (H) Left: mean sphere area of primary and secondary oncospheres, n=4. Right: representative oncosphere pictures. Scale bar: 200 µm.
In an independent MDA-MB-231 PKD3 knockdown cell line, shPKD3_217, we could confirm the significant downregulation of stemness-related marker gene expression (Figure S1K), ALDH activity (Figure S1L) and oncosphere forming efficiency (Figure S1M-N) upon loss of PKD3 expression. Activation of YAP (yes-associated protein)/YAZ (transcriptional co-activator with PDZ-binding motif) and Notch signaling are required to sustain self-renewal and tumor-initiation capacities in breast CSCs 27, 32. Indeed, in both PKD3 knockdown cell lines, decreased expression of the YAP/TAZ and Notch target genes CTGF and HES1/HEY1, respectively, indicated impaired signaling through these pathways (Figure S1O). We further observed downregulation of ABCG2, which contributes to drug resistance in breast CSCs (Figure S1O)8 and the bona fide EMT marker gene SLUG (Figure S1O). These results were confirmed in BT549 cells in which depletion of PKD3 significantly increased CDH1 levels while SNAI1 and SLUG levels were reduced (Fig. S1P). Thus, our results univocally show that MDA-MB-231 cells require PKD3 for CSC maintenance in vitro.
PKD3 knockdown decreases the tumor initiation potential in vivo.
To prove that PKD3 is also important for maintaining the tumor initiation potential in vivo, we implanted four different concentrations of MDA-MB-231 shPKD3_1 and shNon_CTRL cells into the 4th mammary fat pad of immunocompromised mice (Figure 2A). Strikingly, shPKD3_1 cells showed a strongly diminished tumor initiation potential compared to shNon_CTRL cells (Figure 2B-C). In line with the reduced tumor initiation potential, the estimated stem cell frequency of PKD3 deficient tumor cells was significantly lower (Figure 2C). Apart from the lowest cell concentration injected, shNon_CTRL and shPKD3_1 tumors did not differ in size (Figure 2D). We next singularized the tumor cells, analyzed ALDH activity (Figure 2E) and primary as well as secondary oncosphere formation (Figure 2F-G). In agreement with the decreased stem cell frequency, ALDH activity and SFE were significantly reduced in PKD3 deficient MDA-MB-231 tumor cells. Our data thus confirm a role for PKD3 in promoting stemness and tumor-initiating capacity of MDA-MB-231 cells.
Figure 2: In vivo tumor initiation potential is decreased in PKD3-depleted TNBC cells.
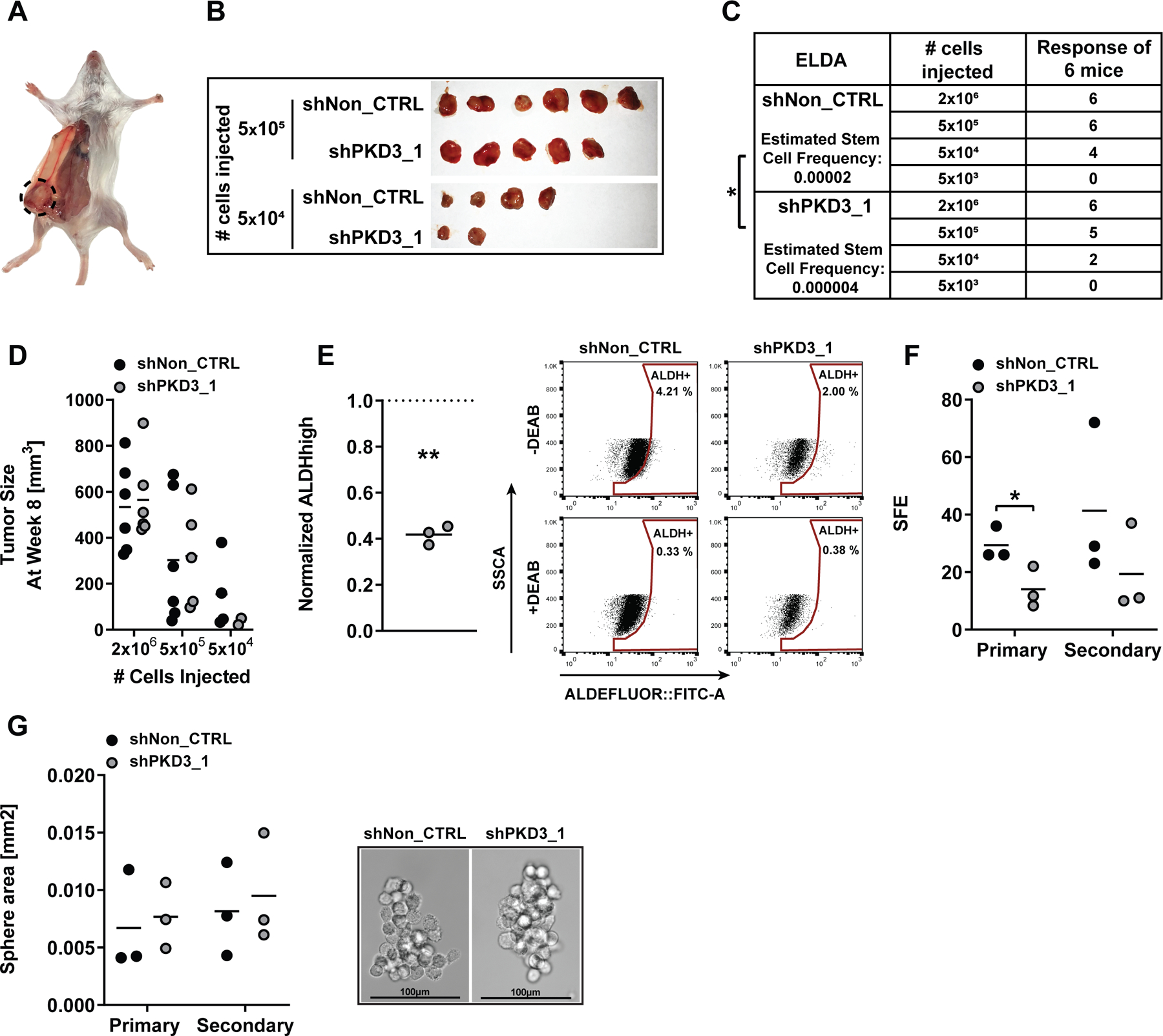
(A) Representative picture of orthotopic tumor formation. (B) Tumors of mice injected with 5×105 and 5×104 MDA-MB-231 cells expressing the indicated shRNAs. (C) ELDA, 6 mice per condition. Data are presented as tumor-bearing mice per injected cell concentration and estimated stem cell frequency. Statistical comparison by ELDA multiple group analysis. (D) Tumor size at week 8, n=2–6. Statistical comparison by one-way ANOVA and Bonferroni-test. (E) Left: flow cytometry-based ALDH activity analysis of singularized tumor cells. Data are presented as mean ALDHhigh population of shPKD3_1 normalized to shNon_CTRL, n=3. Statistical comparison by t-test. Right: Dot plot of ALDH measurements. (F) Mean SFE of primary and secondary oncosphere assays, n=3. Statistical comparison by t-test. (G) Left: mean sphere area of primary and secondary oncospheres, n=3. Right: representative oncosphere pictures. Scale bar: 100 µm.
PKD3-mediated TNBC stem cell regulation is dependent on GEF-H1.
We recently reported that GEF-H1 is an upstream activator of PKD at the Golgi complex in HeLa cells33 (Figure 3A). Therefore, we analyzed whether GEF-H1 is required for PKD3 to promote TNBC cell stemness. To drive Rho-dependent PKD activation we treated MDA-MB-231 cells with nocodazole33 and detected PKD activity by activation loop phosphorylation (pS744/748), which is conserved in all PKD isoforms. Treatment with PMA, a potent inducer of PKD activity14, served as a positive control. Nocodazole strongly increased PKD activity and this was fully abrogated by PKD3 depletion despite the presence of PKD2. Importantly, nocodazole failed to induce PKD activity in the absence of GEF-H1, proving that PKD3, but not PKD2, is activated by GEF-H1 in MDA-MB-231 cells (Figure 3B).
Figure 3: GEF-H1-mediated activation of PKD3 is crucial for mammosphere formation.

(A) Western blot of MDA-MB-231 cells treated with nocodazole (Noc) (5 µg/ml, 1 h), phorbol 12-myristate 13-acetate (PMA) (1 µM, 15 min) or DMSO. Before treatment, MDA-MB-231 cells were transiently transfected with smartpool (sp) siRNAs as indicated. Membranes were probed with specific antibodies as indicated. α-tubulin was used as loading control. (B) Western blot of TNBC and non-TNBC cell lines. Membranes were probed with specific antibodies as indicated. GAPDH was used as loading control. (C) GEF-H1 mRNA expression levels within PKD3 high and low expression groups in tumor samples from the TCGA-BRCA project. The samples were divided according to PKD3 expression by tertile separation, where “PKD3 low” is represented by the lower tertile (quantile < 0.33, n = 403) and “PKD3 high” by the upper tertile (quantile > 0.67, n = 312), normal tissue samples excluded. GEF-H1 expression is visualized as log2[GEF-H1 expression (FPKM) + 0.01]. (D-E) Western blot of (D) MDA-MB-231 and (E) MDA-MB-468 cells, comparing monolayer-cultured cells (2D) with oncospheres (3D). Immunoblotting was conducted, and membranes were probed with specific antibodies as indicated. GAPDH was used as loading control. (F-G) Western blot of (F) MDA-MB-231 or (G) MDA-MB-468 oncospheres. 24 h prior to seeding, MDA-MB-231 or MDA-MB-468 cells were transiently transfected with smartpool (sp) siRNAs as indicated. Membranes were probed with specific antibodies as indicated. GAPDH was used as loading control. (H) Quantification of data shown in (E-F), using densitometry analysis. Data are presented as mean line density of spGEF-H1 normalized to spNon, n=2–3. Statistical comparison by t-test. (I) Workflow: Oncosphere formation assay of transfected cells. (J-K) Left: Mean SFE of primary oncosphere assays of (J) MDA-MB-231 and (K) MDA-MB-468 cells. 24 h prior to seeding, MDA-MB-231 and MDA-MB-468 cells were transiently transfected with smartpool (sp) siRNAs as indicated, n=3–4. Statistical comparison by one-way ANOVA and Bonferroni-test. Right: representative oncosphere pictures. Bar represents 200 µm.
We next analyzed the correlation of GEF-H1 and PKD3 expression in a panel of breast cancer cell lines. In general, TNBC cell lines with elevated PKD3 expression19 also expressed higher GEF-H1 levels whereas low PKD3 and GEF-H1 expression was observed in the non-TNBC cell lines (Figure 3C). To verify these findings in clinical patient data, we analyzed PKD3 and GEF-H1 mRNA levels in human breast cancers from the TCGA database. In accordance with the cell line data, significantly higher GEF-H1 expression was observed in the tumor samples belonging to the high PKD3 expression group (Figure 3D). We next addressed the functional relevance of GEF-H1 and PKD3 expression in primary oncospheres (3D) obtained from two independent TNBC cell lines (Figure 3E-F). In both MDA-MB-231 and MDA-MB-468 cell lines, GEF-H1 as well as PKD3 expression was upregulated in oncospheres compared to the monolayer culture (2D). To validate a potential functional link between GEF-H1 and PKD3 in CSCs, we performed a transient GEF-H1 knockdown using two independent siRNAs prior to seeding of the cells into the primary oncosphere assay and then analyzed PKD3 activity. Remarkably, the depletion of GEF-H1 in MDA-MB-231 and MDA-MB-468 cells strongly reduced the activation loop phosphorylation of PKD3 and phosphorylation of its downstream target S6K119 in oncospheres (Figure 3G-H, Figure S2I). In line with these results, we observed that the transient knockdown of PKD3 or GEF-H1 (Figure 3I) significantly decreased SFE in the TNBC cell lines MDA-MB-231, MDA-MB-468, MDA-MB-436, BT-549 and HCC1806 but not in the luminal A breast cancer cell line MCF7 (Figure 3J-K, Figure S2A-H, J). Our data thus show that GEF-H1 activates PKD3 to promote stem cell maintenance.
PKD3 overexpression increases CSC-like properties
We next explored whether ectopic expression of PKD3 is sufficient to drive oncosphere formation in non-tumorigenic breast epithelial MCF10A cells. Therefore, we generated a doxycycline (Dox)-inducible PKD3 (PKD3-EGFP) MCF10A cell line (Figure 4A) and analyzed stem cell activity and marker gene expression. Indeed, the induction of PKD3-EGFP expression in MCF10A cells significantly increased SFE (Figure 4B), without affecting the sphere area (Figure 4C). Additionally, PKD3-EGFP increased the mRNA expression of stemness markers, such as SOX2 and OCT3/428, 34 (Figure 4D). To verify that GEF-H1 is required for PKD3-mediated stem cell maintenance also in the MCF10A model, we depleted GEF-H1 prior to inducing PKD3-EGFP expression and subjected the cells to a sphere formation assay. Strikingly, the depletion of GEF-H1 completely blocked the increase in sphere formation mediated by PKD3 (Figure 4E-F), indicating that PKD3-mediated oncosphere formation is strictly dependent on GEF-H1.
Figure 4: PKD3 overexpression-induced sphere formation is dependent on GEF-H1.
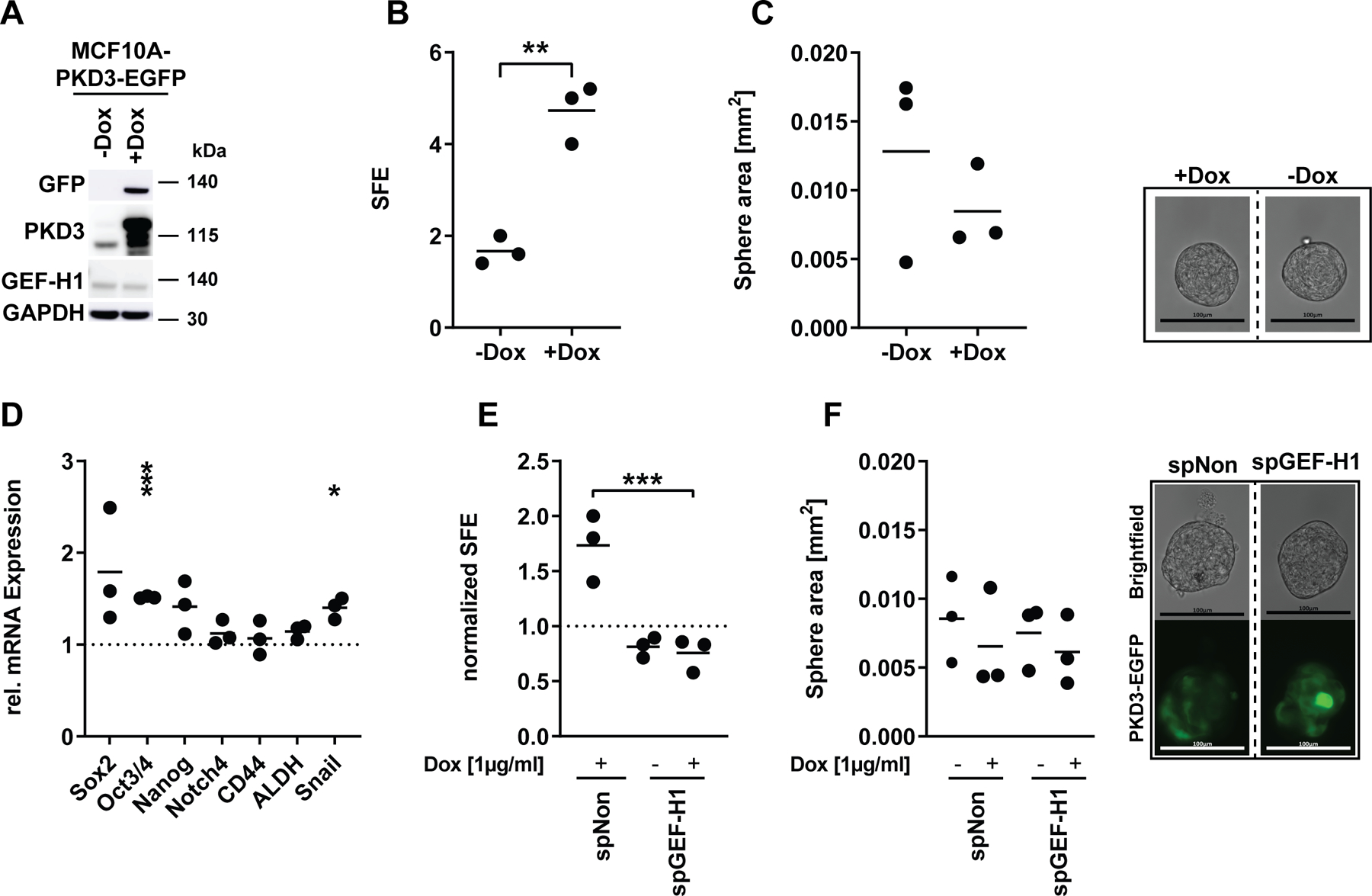
(A) Western blot of MCF10A-EcoR-PKD3WT cells. Cells were treated with doxycycline (+Dox) for 48 h. Water served as control (-Dox). Membranes were probed with specific antibodies as indicated. GAPDH was used as loading control. (B) Mean SFE of primary oncosphere formation assays of MCF10A-EcoR-PKD3WT-EGFP cells. 24 h prior to and directly after seeding cells into the oncosphere assay the medium was supplemented with Dox and spheres were grown for 5 days, n=3. Statistical comparison by t-test. (C) Left: mean sphere area of primary oncospheres, n=3. Statistical comparison by t-test. Right: representative oncosphere pictures. Bar represents 100 µm. (D) qPCR analysis of stemness markers. Data are presented as mean mRNA expression of Dox-treated cells normalized to untreated control, n=3. Statistical comparison by t-test. (E-F) 48 h prior to seeding MCF10A-EcoR-PKD3WT-EGFP cells were transiently transfected with smartpool (sp) siRNAs as indicated. 24h prior to and directly after seeding into the oncosphere assay, cells were treated with Dox. (E) Mean SFE of primary oncosphere assay. Data were normalized to untreated spNon, n=3. Statistical comparison by one-way ANOVA and Bonferroni-test. (F) Left: mean sphere area of primary spheres, n=3. Right: representative pictures of transiently transfected and Dox-treated (continuously for 6 days) MCF10A-PKD3-EGFP cells after 5 days in the oncosphere formation assay. Bar represents 100 µm.
Combined paclitaxel treatment and PKD3 inhibition synergistically decreases TNBC stem cell-mediated oncosphere and colony formation
Loss-of-function by stable or transient knockdown of PKD3 in TNBC cell lines revealed a crucial role for the kinase in CSC maintenance. We thus explored whether pharmacological inhibition of kinase activity using the selective pan-PKD inhibitor CRT006610117 would mimic the loss of PKD3 gene expression. First, we tested the efficacy of the inhibitor in oncosphere formation assays. Equally to PKD3 depletion, CRT0066101 treatment of MDA-MB-231, MDA-MB-468, MDA-MB-436 and BT-549 TNBC cells significantly reduced the number of primary and secondary oncospheres without affecting the respective sphere area (Figure 5A-B, Figure S3A-D). The chemotherapeutic paclitaxel is widely used in the clinical setting35. We therefore asked whether PKD inhibition sensitized TNBC cells to paclitaxel treatment. Firstly, we tested the response behavior of MDA-MB-231 and MDA-MB-468 cells to the combination of paclitaxel and CRT0066101 in a 3D viability assay. Especially low, clinically relevant paclitaxel concentrations36 as well as low micromolar CRT0066101 concentrations showed synergistic or additive effects (Figure S3E-F). In line with these results, we further observed that shPKD3_1 cells were significantly more sensitive when treated with increasing concentrations of paclitaxel compared to shNon_CTRL cells (Figure S3G). Next, we addressed potential synergistic effects of the combinatorial treatment on TNBC stem cells and analyzed ALDH activity as well as sphere forming efficiency. We first performed a primary oncosphere formation assay to enrich for TNBC stem cells (Figure 5C). These oncospheres were singularized, reseeded into a secondary oncosphere formation assay and immediately treated with either CRT0066101, paclitaxel or the combination of both. Both single and combination treatments reduced ALDH activity in MDA-MB-231 and MDA-MB-468 cells (Figure 5D-E). Regarding SFE, the combination was highly synergistic in MDA-MB-231, MDA-MB-468, MDA-MB-436, BT-549 and HCC1806 TNBC cell lines (Figure 5F-G, Figure S4). Because clonogenic activity is a sensitive marker of undifferentiated CSCs37 we tested the combination treatment of CRT0066101 and paclitaxel in a colony formation assay. In line with our previous results, the combinatorial treatment synergistically decreased the clonogenic activity in MDA-MB-231, MDA-MB-468, MDA-MB-436 and BT-549 TNBC cells (Figure 6A-B, Figure S5). Thus, combining paclitaxel with CRT0066101 was superior to the single treatments and synergistically reduced the prevalence of CSCs in these TNBC cell lines.
Figure 5: The combination of paclitaxel and CRT0066101 synergistically reduces sphere formation in vitro.
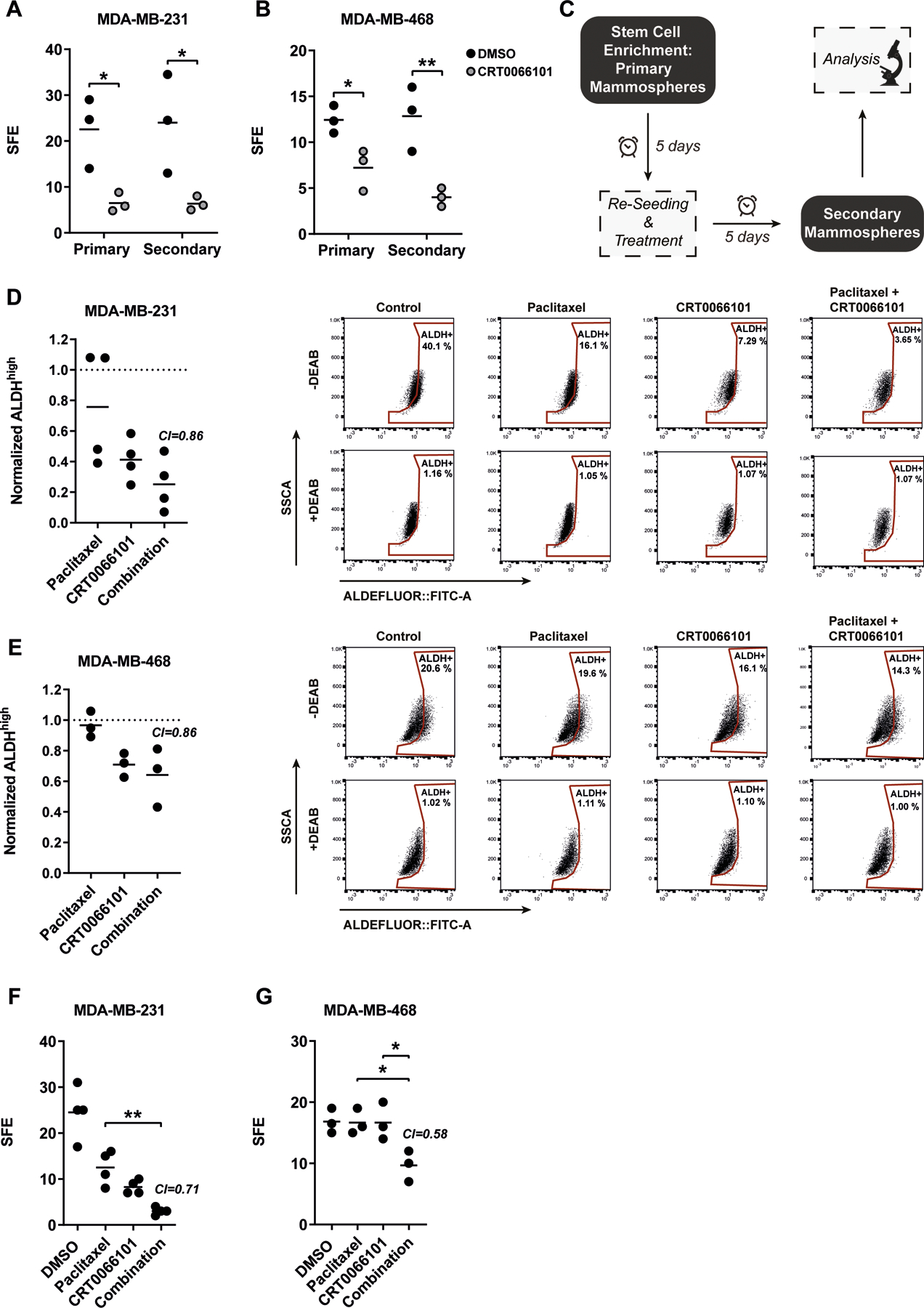
(A-B) Counting of primary and secondary oncospheres of (A) MDA-MB-231 or (B) MDA-MB-468 cells. Cells were treated with 1 µM CRT0066101 directly after seeding into the respective oncosphere assay. Data is presented as mean sphere formation efficiency (SFE), n=3. Statistical comparison by one-way ANOVA and Bonferroni-test. (C) Workflow of paclitaxel and CRT0066101 oncosphere treatment. (D-E) Left panel: Flow cytometry-based stemness analysis of (D) MDA-MB-231 or (E) MDA-MB-468 cells via ALDEFLUOR™. Directly after seeding into the secondary oncosphere assay, cells were treated with paclitaxel (5 nM) or CRT0066101 (MDA-MB-231: 1 µM; MDA-MB-468: 0.5 µM), or in combination. DMSO served as control. Data is presented as mean ALDHhigh population normalized to DMSO control, n=3–4. Statistical comparison by one-way ANOVA and Bonferroni-test. Combination index (CI) values were calculated via Webb’s fractional product method using the respective mean of % ALDHhigh populations. CI<1 indicates synergism. Right panels: Dot plot of ALDEFLUOR measurements. DEAB-treated samples served as internal control. (F-G) Counting of secondary oncospheres of (F) MDA-MB-231 or (G) MDA-MB-468 cells. Directly after seeding into the secondary oncosphere assay, cells were treated with paclitaxel (5 nM) or CRT0066101 (MDA-MB-231: 1 µM; MDA-MB-468: 0.5 µM), or in combination. DMSO served as control. Data is presented as mean sphere formation efficiency (SFE), n=3–4. Statistical comparison by one-way ANOVA and Bonferroni-test. Combination index (CI) values were calculated using Webb’s fractional product method. CI<1 indicates synergism
Figure 6: The combination of paclitaxel and CRT0066101 synergistically reduces colony formation in vitro as well as tumor recurrence in vivo.
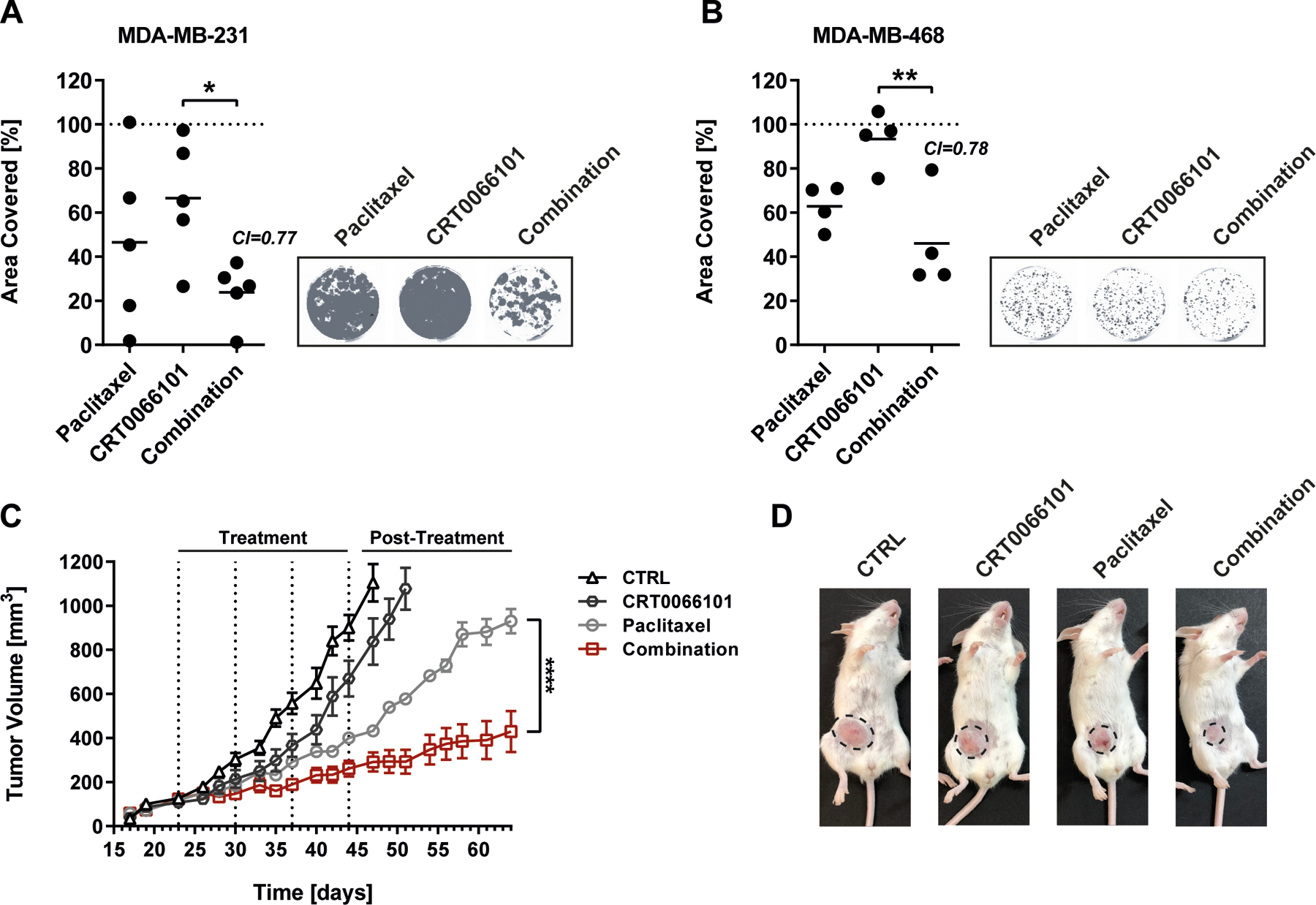
(A-B) Left panels: colony formation of (H) MDA-MB-231 or (I) MDA-MB-468 cells. 24 h after seeding into 6-well culture dishes, MDA-MB-231 or MDA-MB-468 cells were treated for 48 h with paclitaxel (1 nM) or CRT0066101 (MDA-MB-231: 1 µM; MDA-MB-468: 0.1 µM), or the combination of both. DMSO served as control. Afterwards, cells were further cultured for 2 weeks and analyzed using the Odyssey imaging system. Data is presented as mean area covered. DMSO control was set to 100 %, n=4–5. Statistical comparison by one-way ANOVA and Bonferroni-test. Combination index (CI) values were calculated using Webb’s fractional product method. CI<1 indicates synergism. Right panels: Representative pictures of the respective colony formation assays. (C) 8-week-old female SCID mice were injected with 2×106 MDA-MB-231 cells into the right fat pad of the 4th nipple. After the tumors had reached 100 mm3 mice were treated with either CRT0066101, paclitaxel or the combination of both. The combination of the respective carriers served as control. Data is presented as tumor volume (mm3), mean ± SEM, n=7. Statistical comparison by two-way ANOVA and Bonferroni-test. (D) Representative pictures of animals from the respective treatment groups. Tumors are indicated by dotted circles.
The combination of CRT0066101 and paclitaxel is superior in decreasing tumor recurrence in vivo.
Finally, we used an orthotopic xenograft mouse model to investigate the growth and recurrence of MDA-MB-231 tumors after treatment with paclitaxel, CRT0066101 or the combination of both. The cells were transplanted into SCID mice and allowed to form tumors with a size of ~100 mm3. Compared to the control (vehicle only) CRT0066101 had only little effect on tumor growth. Accordingly, tumor-bearing mice belonging to the control and CRT0066101 groups had to be removed from the study shortly after the end of the treatment period. By contrast, paclitaxel caused an initial inhibition of tumor growth, which was further enhanced by CRT0066101. Upon therapy termination, paclitaxel-treated tumors displayed a strong re‐growth. More importantly, in the post-treatment period, animals that had received the combination treatment showed a significant reduction in tumor re-growth compared to paclitaxel treated animals (Figure 6C-D), providing support for the superior targeting of drug-resistant cancer cell subpopulations by paclitaxel plus CRT0066101.
Discussion
PKD3 has been associated with TNBC progression17, 19 but so far, a specific role for PKD3 in CSC stemness has not been described. In this study, we have defined a critical role for PKD3 in the maintenance and propagation of TNBC stem cells in vitro and in vivo. The PKD inhibitor CRT0066101 demonstrated strong efficacy in eliminating TNBC stem cells in vitro. More importantly, CRT0066101 synergistically increased the response to paclitaxel in vitro and in vivo. These findings are of high clinical relevance as taxanes have shown to increase the TNBC stem cell population6, 7, 38. Thus, inhibiting PKD3 in combination with paclitaxel has a dual effect by targeting differentiated cancer cells and at the same time eradicating the cell population responsible for chemoresistance and tumor recurrence.
The role of PKD3 in cancer stemness was specific for TNBC cell lines as in luminal A MCF7 cells with only marginal PKD3 expression depletion of the kinase did not affect oncosphere formation (Figure S2H). Our findings further support the postulated isoform switch from PKD1 to PKD3 in breast tumor progression. While PKD1 is a negative regulator of cell motility and invasion and contributes to the maintenance of the epithelial phenotype of breast cells, PKD3 drives cell proliferation and invasion17 and, as shown in this study, CSC maintenance.
Taxanes were shown to increase the CSC population in patients with invasive breast cancer6, 7, 9. Moreover, paclitaxel increased the ALDH-positive population in MDA-MB-231 cells by enhancing SOX2, ABCG2 and TWIST1 expression, unraveling an interconnected pluripotency-chemoresistance-EMT (epithelial to mesenchymal transition) axis8. EMT is linked to breast CSC regulation, and exposing TNBC cells to the EMT inducer transforming growth factor beta (TGF-β) increased oncosphere formation and the CD44+/CD24- stem cell signature39. Interestingly, PKD3 has also been connected with EMT, as treating MDA-MB-231 cells with CRT0066101 reduced the expression of the EMT transcription factor SNAI140. Strikingly, PKD3 depletion in MDA-MB-231 cells decreased SOX2, ABCG2, SLUG and SNAI1 expression (Figure 1E, Figure S2K, O) and increased CDH1 expression in BT549 cells (Figure S2P). PKD3 inhibition thus likely counteracts the paclitaxel-induced upregulation of EMT genes. Breaking up the pluripotency-chemoresistance-EMT axis might contribute to the strong response to combined CRT0066101 and paclitaxel treatment in vivo.
We further identified GEF-H1 to be upstream of PKD3 activity in oncosphere formation, in support of the proposed contribution of GEF-H1/Rho signaling to breast cancer metastasis41. But how does GEF-H1/PKD3 signaling contribute to the maintenance of TNBC stem cells? Recently, we showed that a GEF-H1-RhoA-PLCε-PKD pathway regulates the fission of exocytic vesicles at the Golgi complex33. The changes in surface protein expression upon PKD3 depletion could thus stem from deregulated exocytosis. Various secreted factors derived from the bulk tumor and CSC population, acting in paracrine and autocrine manners, are required for the CSC survival. For example, in basal-like breast cancer the mesenchymal-like tumor cells were reported to produce WNT2, CXCL12, and IL6, which drive the self-renewal of the CSCs42. Furthermore, TGF-β together with autocrine canonical and non-canonical WNT signaling controls migratory and self-renewal ability of breast CSCs by promoting the expression of EMT-associated transcription factors43. By promoting the secretion of paracrine and/or autocrine survival factors PKD3 signaling might positively regulate the expression of EMT-associated transcription factors to maintain the CSC state. In support of our hypothesis, Zeng and co-workers reported that the mesenchymal stem cell marker CD146 promotes EMT through RhoA-induced upregulation of SLUG thereby contributing to the maintenance of CSC-like cells in breast cancer44. Our data further indicate that PKD3 depletion reduces YAP/TAZ signaling (Figure S1O), congruent with the reported regulation of YAP/TAZ signaling by PKD in pancreatic cancer cells45. EMT induces and requires YAP/TAZ for triggering breast cancer stemness and metastasis32 and vice versa, YAP/TAZ are active inducers of EMT46, 47. This suggests that several positive and negative feedback loops are in place to sustain the maintenance of CSCs.
TNBC patients have a high probability of tumor recurrence within the first five years after the end of treatment, which is largely driven by CSC activity30, 48, 49. The development of new therapeutic strategies targeting TNBC stem cells has thus been intensified over the last years50. For example, the kinase inhibitor Dasatinib and TGF-β pathway inhibition, respectively, were shown to enhance the responsiveness to paclitaxel treatment in preclinical models of TNBC51. Thus, these studies and our results demonstrate that combining paclitaxel with therapeutic antibodies or small molecule inhibitors such as CRT0066101 has great clinical potential by targeting more effectively the TNBC bulk tumor and stem cell populations.
Supplementary Material
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-αTubulin | Cell Signaling Technology | Cat# 3873, RRID:AB_1904178 |
| Anti-GAPDH | Cell Signaling Technology | Cat# 2118, RRID:AB_561053 |
| Anti-GEF-H1 | Cell Signaling Technology | Cat# 4076, RRID:AB_2060032 |
| Anti-GFP | Roche | Cat# 11814460001, RRID:AB_390913 |
| Anti-Phospho-(Ser744/748)-PKD | Cell Signaling Technology | Cat# 2054, RRID:AB_2172539 |
| Anti-PKD3 | Cell Signaling Technology | Cat# 5655, RRID:AB_10695917 |
| Anti-p70 S6 Kinase | Cell Signaling Technology | Cat# 2708, RRID:AB_390722 |
| Anti-Phospho-(Thr389)-p70 S6 Kinase | Cell Signaling Technology | Cat# 9205, RRID:AB_330944 |
| Bacterial and Virus Strains | ||
| Platinum-A (Plat-A) Retroviral Packaging Cell Line | Cell Biolabs | RV-101 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cholera toxin | Merck | Cat# C8052 |
| CRT0066101 (cell culture) | Tocris | Cat# 4975 |
| CRT0066101 (in vivo) | Selleckchem | Cat# S8366 |
| Crystal violet | Merck | Cat# C0775 |
| Dextrose | Merck | Cat# D9434 |
| Doxicycline | Calbiochem | Cat# 324385 |
| EGF | R&D Systems | Cat# 236-EG |
| G418 | Carl Roth | Cat# CP11.1 |
| Hydrocortisone | Merck | Cat# 3867 |
| Insulin | Merck | Cat# I5500 |
| Lipofectamine™ RNAiMAX | Thermo Fisher Scientific | Cat# 13778030 |
| Paclitaxel (cell culture) | Merck | Cat# T7402 |
| Paclitaxel (in vivo) | Fresenius Kabi | 6 mg/ml infusion solution |
| Paraformaldehyde | Merck | Cat# P6148 |
| Poly(2-hydroxyethyl methacrylate) (pHEMA) | Merck | Cat# P3932 |
| Polysorbate | Roth | Cat# 9139.1 |
| Critical Commercial Assays | ||
| ALDEFLUOR | STEMCELL Technologies | Cat# 01700 |
| LegendScreen (human) | BioLegend | Cat# 700007 |
| NucleoSpin® miRNA | Macherey-Nagel | Cat# 740971 |
| Power SYBR® Green RNA-to-CTTM 1-Step kit | Thermo Fisher Scientific | Cat# 4389986 |
| Tumor dissociation kit | Miltenyi Biotec | Cat# 130–096-730 |
| CellTiter-Glo | Promega | Cat# G7570 |
| Organisms/Strains | ||
| SCID Mice | Charles River Laboratories | IMSR Cat# CRL:236, RRID:IMSR_CRL:236 |
| Oligonucleotides (qPCR; F: Forward; R: Reverse) | ||
| ABCG2 F: CAGGTGGAGGCAAATCTTCGT; R: TCCAGACACACCACGGATAAA |
Microsynth | - |
| Actin | Qiagen | Cat# HS_ACTB_2_SG |
| ALDH1A1 F: GCACGCCAGACTTACCTGTC R: CCTCCTCAGTTGCAGGATTAAAG |
Microsynth | - |
| CD44 F: CTGCCGCTTTGCAGGTGTA R: CATTGTGGGCAAGGTGCTATT |
Microsynth | - |
| HES1 F: TGAAGAAAGATAGCTCGCGG R: GGTACTTCCCCAGCACACTT |
Microsynth | - |
| HEY1 F: TGGATCACCTGAAAATGCTG R: TTGTTGAGATGCGAAACCAG |
Microsynth | - |
| NANOG F: CAGAAGGCCTCAGCACCTAC R: CTGTTCCAGGCCTGATTGTT |
Microsynth | - |
| NOTCH4 F: AACTCCTCCCCAGGAATCTG R: CCTCCATCCAGCAGAGGTT |
Microsynth | - |
| OCT3/4 F: GAAGCTGGAGAAGGAGAAGCTG R: CAAGGGCCGCAGCTTACACATGTTC |
Microsynth | - |
| PKD3 | Qiagen | Cat# HS_PRKD3_1_SG |
| SNAIL F: CACTATGCCGCGCTCTTTC R: GCTGGAAGGTAAACTCTGGATTAGA |
Microsynth | - |
| SOX2 F: AGTCTCCAAGCGACGAAAAA R: GGAAAGTTGGGATCGAACAA |
Microsynth | - |
| SNAIL2 F: CTGGTCAAGAAGCATTTCAACGCC R: ATGGGTTACCGGAGAGAGGAGAAA |
Microsynth | - |
| Oligonucleotides (siRNA) | ||
| spNon: ON-Target plus non-targeting control pool | Dharmacon | Cat# D-001810–10 |
| spGEF-H1: ON-Target plus SMARTpool human ARHGEF2 | Dharmacon | Cat# L-009883 |
| GEF-H1 SS#2: Silencer® Select human ARHGEF2 | ThermoFisher Scientific | Cat# 4392420 |
| spPKD3: ON-Target plus SMARTpool human PRKD3 | Dharmacon | Cat# L-005029 |
| PKD3 SS#2: Silencer® Select human PRKD3 |
ThermoFisher Scientific | Cat# 4390824 |
| Recombinant DNA | ||
| pWPXLd-rtTA3-IRES-EcoR-PGK-Puro | IBTB, University of Stuttgart | - |
| pCMV-Gag-Pol | Cell Biolabs | Cat# RV-111 |
| TRE3G-PGK-NEO retroviral backbone | IBTB, University of Stuttgart | - |
| Software and Algorithms | ||
| FlowJo | FlowJo | RRID:SCR_008520 |
| GraphPad Prism | GraphPad | RRID:SCR_002798 |
| ImageJ | ImageJ | RRID:SCR_003070 |
| Other | ||
| Accutase | Thermo Fisher Scientific | Cat# 00–4555-56 |
| B27 Supplement | Thermo Fisher Scientific | Cat# 17504–044 |
| DMEM high glucose | Thermo Fisher Scientific | Cat# 11965092 |
| DMEM low glucose | Thermo Fisher Scientific | Cat# 31885023 |
| DMEM/F12 GlutaMAX | Thermo Fisher Scientific | Cat# 31331028 |
| Horse serum | Thermo Fisher Scientific | Cat# 16050122 |
| NuPage Novex 4–12% Bis-Tris gels | Thermo Fisher Scientific | Cat# NP0336 |
| RPMI-1640 | Thermo Fisher Scientific | Cat# 11875093 |
Novelty and Impact.
Cancer stem cells are endowed with tumor-initiating capacity and associated with metastasis development and poor survival. We identify the serine-threonine kinase PKD3 to be activated downstream of GEF-H1 in TNBC stem cells and to be crucial for stem cell maintenance. Importantly, the PKD inhibitor CRT0066101 and paclitaxel synergistically decreased TNBC stem cell prevalence and tumor recurrence in vivo. Our results thus provide a rationale for targeting PKD3 to eliminate the tumor-initiating cell population in TNBC.
Acknowledgements
This project was supported by the Deutsche Krebshilfe (grant 70111941 to M.A.O. and A. H). P.S. was supported by the NIH grants CA184527 and CA200572.
Abbreviations:
- PKD
Protein Kinase D
- CSC
cancer stem cells
- TNBC
triple negative breast cancer
- ALDH
aldehyde dehydrogenase 1
Footnotes
Conflict of interest
The authors have no conflict of interest to declare.
References
- 1.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, et al. Molecular portraits of human breast tumours. Nature 2000;406: 747–52. [DOI] [PubMed] [Google Scholar]
- 2.Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2008;26: 1275–81. [DOI] [PubMed] [Google Scholar]
- 3.Smid M, Wang Y, Zhang Y, Sieuwerts AM, Yu J, Klijn JG, Foekens JA, Martens JW. Subtypes of breast cancer show preferential site of relapse. Cancer research 2008;68: 3108–14. [DOI] [PubMed] [Google Scholar]
- 4.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010;363: 1938–48. [DOI] [PubMed] [Google Scholar]
- 5.von Minckwitz G, Schneeweiss A, Loibl S, Salat C, Denkert C, Rezai M, Blohmer JU, Jackisch C, Paepke S, Gerber B, Zahm DM, Kummel S, et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. The Lancet Oncology 2014;15: 747–56. [DOI] [PubMed] [Google Scholar]
- 6.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America 2009;106: 13820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008;100: 672–9. [DOI] [PubMed] [Google Scholar]
- 8.Mukherjee P, Gupta A, Chattopadhyay D, Chatterji U. Modulation of SOX2 expression delineates an end-point for paclitaxel-effectiveness in breast cancer stem cells. Scientific reports 2017;7: 9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin Y, Zhong Y, Guan H, Zhang X, Sun Q. CD44+/CD24- phenotype contributes to malignant relapse following surgical resection and chemotherapy in patients with invasive ductal carcinoma. J Exp Clin Cancer Res 2012;31: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adamczyk A, Niemiec JA, Ambicka A, Mucha-Malecka A, Mitus J, Rys J. CD44/CD24 as potential prognostic markers in node-positive invasive ductal breast cancer patients treated with adjuvant chemotherapy. J Mol Histol 2014;45: 35–45. [DOI] [PubMed] [Google Scholar]
- 11.Ma F, Li H, Li Y, Ding X, Wang H, Fan Y, Lin C, Qian H, Xu B. Aldehyde dehydrogenase 1 (ALDH1) expression is an independent prognostic factor in triple negative breast cancer (TNBC). Medicine (Baltimore) 2017;96: e6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abraham BK, Fritz P, McClellan M, Hauptvogel P, Athelogou M, Brauch H. Prevalence of CD44+/CD24-/low cells in breast cancer may not be associated with clinical outcome but may favor distant metastasis. Clinical cancer research : an official journal of the American Association for Cancer Research 2005;11: 1154–9. [PubMed] [Google Scholar]
- 13.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America 2003;100: 3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu Y, Rubin CS. Protein kinase D: coupling extracellular stimuli to the regulation of cell physiology. EMBO reports 2011;12: 785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olayioye MA, Barisic S, Hausser A. Multi-level control of actin dynamics by protein kinase D. Cellular signalling 2013;25: 1739–47. [DOI] [PubMed] [Google Scholar]
- 16.Borges S, Doppler H, Perez EA, Andorfer CA, Sun Z, Anastasiadis PZ, Thompson E, Geiger XJ, Storz P. Pharmacologic reversion of epigenetic silencing of the PRKD1 promoter blocks breast tumor cell invasion and metastasis. Breast cancer research : BCR 2013;15: R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borges S, Perez EA, Thompson EA, Radisky DC, Geiger XJ, Storz P. Effective Targeting of Estrogen Receptor-Negative Breast Cancers with the Protein Kinase D Inhibitor CRT0066101. Mol Cancer Ther 2015;14: 1306–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao Q, McKenzie R, Gan H, Tang H. Protein kinases D2 and D3 are novel growth regulators in HCC1806 triple-negative breast cancer cells. Anticancer Res 2013;33: 393–9. [PubMed] [Google Scholar]
- 19.Huck B, Duss S, Hausser A, Olayioye MA. Elevated protein kinase D3 (PKD3) expression supports proliferation of triple-negative breast cancer cells and contributes to mTORC1-S6K1 pathway activation. The Journal of biological chemistry 2014;289: 3138–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huck B, Kemkemer R, Franz-Wachtel M, Macek B, Hausser A, Olayioye MA. GIT1 phosphorylation on serine 46 by PKD3 regulates paxillin trafficking and cellular protrusive activity. The Journal of biological chemistry 2012;287: 34604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 2009;347: 70–8. [DOI] [PubMed] [Google Scholar]
- 22.Bossard C, Bresson D, Polishchuk RS, Malhotra V. Dimeric PKD regulates membrane fission to form transport carriers at the TGN. J Cell Biol 2007;179: 1123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dubrovska A, Hartung A, Bouchez LC, Walker JR, Reddy VA, Cho CY, Schultz PG. CXCR4 activation maintains a stem cell population in tamoxifen-resistant breast cancer cells through AhR signalling. British journal of cancer 2012;107: 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaillant F, Asselin-Labat ML, Shackleton M, Forrest NC, Lindeman GJ, Visvader JE. The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer research 2008;68: 7711–7. [DOI] [PubMed] [Google Scholar]
- 25.Glinka Y, Mohammed N, Subramaniam V, Jothy S, Prud’homme GJ. Neuropilin-1 is expressed by breast cancer stem-like cells and is linked to NF-kappaB activation and tumor sphere formation. Biochemical and biophysical research communications 2012;425: 775–80. [DOI] [PubMed] [Google Scholar]
- 26.Yen WC, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B, Henner WR, Meisner R, Sato A, Shah J, Tang T, Wallace B, et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21: 2084–95. [DOI] [PubMed] [Google Scholar]
- 27.Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer research 2010;70: 709–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R, Martin AG. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 2012;31: 1354–65. [DOI] [PubMed] [Google Scholar]
- 29.Iriondo O, Rabano M, Domenici G, Carlevaris O, Lopez-Ruiz JA, Zabalza I, Berra E, Vivanco M. Distinct breast cancer stem/progenitor cell populations require either HIF1alpha or loss of PHD3 to expand under hypoxic conditions. Oncotarget 2015;6: 31721–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell 2007;1: 555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cioce M, Gherardi S, Viglietto G, Strano S, Blandino G, Muti P, Ciliberto G. Mammosphere-forming cells from breast cancer cell lines as a tool for the identification of CSC-like- and early progenitor-targeting drugs. Cell Cycle 2010;9: 2878–87. [PubMed] [Google Scholar]
- 32.Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, Daidone MG, Dupont S, et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011;147: 759–72. [DOI] [PubMed] [Google Scholar]
- 33.Eisler SA, Curado F, Link G, Schulz S, Noack M, Steinke M, Olayioye MA, Hausser A. A Rho signaling network links microtubules to PKD controlled carrier transport to focal adhesions. eLife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu T, Liu S, Breiter DR, Wang F, Tang Y, Sun S. Octamer 4 small interfering RNA results in cancer stem cell-like cell apoptosis. Cancer research 2008;68: 6533–40. [DOI] [PubMed] [Google Scholar]
- 35.McAndrew N, DeMichele A. Neoadjuvant Chemotherapy Considerations in Triple-Negative Breast Cancer. J Target Ther Cancer 2018;7: 52–69. [PMC free article] [PubMed] [Google Scholar]
- 36.Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME, Weaver BA. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med 2014;6: 229ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajendran V, Jain MV. In Vitro Tumorigenic Assay: Colony Forming Assay for Cancer Stem Cells. Methods Mol Biol 2018;1692: 89–95. [DOI] [PubMed] [Google Scholar]
- 38.Gomez-Miragaya J, Palafox M, Pare L, Yoldi G, Ferrer I, Vila S, Galvan P, Pellegrini P, Perez-Montoyo H, Igea A, Munoz P, Esteller M, et al. Resistance to Taxanes in Triple-Negative Breast Cancer Associates with the Dynamics of a CD49f+ Tumor-Initiating Population. Stem cell reports 2017;8: 1392–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133: 704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Durand N, Borges S, Storz P. Protein Kinase D Enzymes as Regulators of EMT and Cancer Cell Invasion. J Clin Med 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao YC, Ruan JW, Lua I, Li MH, Chen WL, Wang JR, Kao RH, Chen JH. Overexpressed hPTTG1 promotes breast cancer cell invasion and metastasis by regulating GEF-H1/RhoA signalling. Oncogene 2012;31: 3086–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang M, Tsimelzon A, Chang CH, Fan C, Wolff A, Perou CM, Hilsenbeck SG, Rosen JM. Intratumoral heterogeneity in a Trp53-null mouse model of human breast cancer. Cancer discovery 2015;5: 520–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, Weinberg RA. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011;145: 926–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng Q, Li W, Lu D, Wu Z, Duan H, Luo Y, Feng J, Yang D, Fu L, Yan X. CD146, an epithelial-mesenchymal transition inducer, is associated with triple-negative breast cancer. Proceedings of the National Academy of Sciences of the United States of America 2012;109: 1127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Sinnett-Smith J, Stevens JV, Young SH, Rozengurt E. Biphasic Regulation of Yes-associated Protein (YAP) Cellular Localization, Phosphorylation, and Activity by G Protein-coupled Receptor Agonists in Intestinal Epithelial Cells: A NOVEL ROLE FOR PROTEIN KINASE D (PKD). The Journal of biological chemistry 2016;291: 17988–8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shao D, Zhai P, Del Re DP, Sciarretta S, Yabuta N, Nojima H, Lim DS, Pan D, Sadoshima J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nature communications 2014;5: 3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes & development 2007;21: 2747–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stratford AL, Reipas K, Maxwell C, Dunn SE. Targeting tumour-initiating cells to improve the cure rates for triple-negative breast cancer. Expert reviews in molecular medicine 2010;12: e22. [DOI] [PubMed] [Google Scholar]
- 49.Rangel MC, Bertolette D, Castro NP, Klauzinska M, Cuttitta F, Salomon DS. Developmental signaling pathways regulating mammary stem cells and contributing to the etiology of triple-negative breast cancer. Breast cancer research and treatment 2016;156: 211–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tian J, Raffa FA, Dai M, Moamer A, Khadang B, Hachim IY, Bakdounes K, Ali S, Jean-Claude B, Lebrun J-J. Dasatinib sensitises triple negative breast cancer cells to chemotherapy by targeting breast cancer stem cells. British journal of cancer 2018;119: 1495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhola NE, Balko JM, Dugger TC, Kuba MG, Sanchez V, Sanders M, Stanford J, Cook RS, Arteaga CL. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest 2013;123: 1348–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.