

Abstract
Rift Valley fever virus (RVFV) is an arbovirus that was first reported in the Rift Valley of Kenya which causes significant disease in humans and livestock. RVFV is a tri-segmented, negative-sense RNA virus consisting of a L, M, and S segments with the M segment encoding the glycoproteins Gn and Gc. Host factors that interact with Gn are largely unknown. To this end, two viruses containing an epitope tag (V5) on the Gn protein in position 105 or 229 (V5Gn105 and V5Gn229) were generated using the RVFV MP-12 vaccine strain as a backbone. The V5-tag insertion minimally impacted Gn functionality as measured by replication kinetics, Gn localization, and antibody neutralization assays. A proteomics-based approach was used to identify novel Gn-binding host proteins, including the E3 ubiquitin-protein ligase, UBR4. Depletion of UBR4 resulted in a significant decrease in RVFV titers and a reduction in viral RNA production.
Keywords: Rift Valley fever virus, glycoproteins, UBR4, Gn
Introduction
Rift Valley fever virus (RVFV) is an arthropod-borne virus (arbovirus) that can infect both humans and ruminants, such as sheep, cattle, goats, and camels (Bird and Nichol, 2012; Bouloy and Weber, 2010; Ikegami, 2012; Kroeker et al., 2020; Paweska, 2015; Weaver and Reisen, 2010). Infection of ruminants can result in abortions (80-100% for sheep and cattle) regardless of pregnancy stage. High rates of mortality have been observed in young, infected animals. Humans infection can result in a wide variety of clinical symptoms ranging from mild febrile illness to hepatitis, retinitis, delayed-onset of encephalitis, or in more severe cases, hemorrhagic fever. The case fatality rate is estimated to be between 0.5-2% overall, although individuals with severe complications (jaundice, neurological or hemorrhagic manifestations) are at higher risk for a fatal outcome. While outbreaks have been limited to the African continent and Arabian Peninsula, the increase in travel and commerce, expansion of the vector range, as well as the presence of domestic mosquito species and vertebrate animals that can support RVFV replication have caused increased concerns about the introduction into the US (Gaudreault et al., 2015; Linthicum et al., 2016; Rolin et al., 2013; Wilson et al., 2018). Furthermore, there are no FDA-approved prophylactic therapies or vaccines currently available for human use.
RVFV is from the Phlebovirus genus in the Phenuiviridae family and is characterized by an enveloped virus particle (~100nm size) that contains a tripartite, negative stranded RNA genome (Adams et al., 2017; Elliott, 2014; Elliott and Brennan, 2014). Based on their size, the three segments are termed large (L), medium (M), and small (S). The L segment encodes for the RNA-dependent RNA polymerase (RdRp) L protein, while the S segment encodes for the nucleocapsid protein, NP, and the virulence factor, NSs. The M segment encodes for several polyprotein precursors which upon proteolytic processing, produce the mature proteins including NSm, envelope glycoproteins Gn and Gc, as well as an NSm-Gn fusion protein termed 78kD (Collett et al., 1985).
Gn and Gc are type I transmembrane proteins and facilitate viral entry, fusion, and assembly (Spiegel et al., 2016). During attachment, Gn recognizes host cell receptors, like dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN). The interaction of the DC-SIGN receptor and Gn subsequently triggers viral uptake (Lozach et al., 2011). DC-SIGN is just one of the three host cell receptors identified that Gn interacts with to facilitate viral attachment (Spiegel et al., 2016). Gc has been designated as a class II membrane fusion protein that is responsible for the fusion of the viral envelope with the endosomal membrane (Garry and Garry, 2004; Vaney and Rey, 2011). After genome replication, the precursor glycoprotein Gn/Gc is relocated to the ER, subsequently cleaved by a host protease, and Gn and Gc are glycosylated in the ER lumen before viral assembly and egress.
RVFV assembly occurs at the Golgi complex where particles bud into the cisternae and egress through the trans-Golgi network (Hornak et al., 2016). The Gn cytosolic tail contains the genetic determinants required for targeting the glycoprotein heterodimers from the ER to the Golgi, as well as binding to the L polymerase and ribonucleoprotein (RNP) complex (Carnec et al., 2014). Several studies have focused on the co-packaging of the RVFV genomic segments which has indicated that S and M segments are packaged at a higher ratio when compared to L. Although this process is nonselective with only a small percentage of virions incorporating all three segments, there are host specific fluctuations that occur (Bermúdez-Méndez et al., 2021; Hornak et al., 2016; Wichgers Schreur et al., 2018; Wichgers Schreur and Kortekaas, 2016). A three-dimensional reconstruction of the RVFV viral particle detailing the arrangement of the capsomers comprised of Gn/Gc oligomers that resemble a cylindrical hollow spike on the particle surface has been completed (Freiberg et al., 2008; Huiskonen et al., 2009; Sherman et al., 2009). Furthermore, both antigenic and glycosylation sites on Gn/Gc have been mapped (Kakach et al., 1989; Kakach et al., 1988; Keegan and Collett, 1986; Phoenix et al., 2016). The importance of the 78kD/NSm-Gn fusion protein for assembly has also been examined. While this fusion protein is dispensable for in vitro propagation of the virus, it is required for dissemination within the arthropod vector (Crabtree et al., 2012; Kreher et al., 2014) and produced in larger quantities in invertebrate versus mammalian cells (Weingartl et al., 2014).
A complete picture of host factors required for RVFV entry and assembly, specifically as it relates to Gn protein interactions, is still unknown. In the interest of providing a means to isolate Gn-associated partners at different points within the RVFV lifecycle, we engineered a recombinant RVFV virus containing a V5 epitope tag within the ectodomain of Gn. The impact of the epitope tag insertion on Gn functionality and suitability as a means of isolating Gn interacting partners was examined.
Proteomic analysis of Gn immunoprecipitated complexes identified Ubiquitin Protein Ligase E3 Component N-Recognin 4, UBR4, as a Gn interacting protein. UBR4 is a human E3 ubiquitin ligase. It recruits a ubiquitin conjugated E2 through its association with the target protein (Kim et al., 2018; Tasaki et al., 2005). UBR4 has been shown to impact several key cellular functions, such as proteasomal degradation, secretion of macrovesicles, and autophagy (Kim et al., 2018; Rinschen et al., 2016). UBR4 is of importance because it has already been shown to significantly affect a myriad of other viruses including Influenza A virus (IAV), human papillomavirus (HPV), and dengue virus (DENV) (Huh et al., 2005; Morrison et al., 2013; Tripathi et al., 2015). The interaction of UBR4 with the M2 protein of IAV allows for efficient budding from the plasma membrane. The absence of UBR4 results in altered trafficking of viral proteins, where they are no longer brought to the plasma membrane and instead targeted to autophagosomes for degradation (Tripathi et al., 2015). Conversely, in DENV-infected cells, the interaction between UBR4 and the viral NS5 protein allows for the degradation of STAT2, subsequently decreasing interferon signaling (Morrison et al., 2013). This study aims to characterize two novel tagged virus and use them to determine the impact UBR4 has on RVFV replication.
Materials and Methods
Cell Culture.
Vero cells (ATCC) and the UBR4 +/+ (wild-type, WT) and UBR4 −/− (knockout, KO) HEK 293Ts (Tripathi et al., 2015) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat inactivated fetal bovine serum (FBS) and 1% L-glutamine. Huh-7 cells were maintained in DMEM containing 10% FBS, 1% nonessential amino acids (NEAA), 1% L-glutamine, and 1% sodium pyruvate. Human small airway epithelial cells (HSAECs) were cultured in Ham’s F12 modified media supplemented with 10% FBS, 1% NEAA, 1% L-glutamine, 1% sodium pyruvate, and 0.1% β-mercaptoethanol (Popova et al., 2010). Cells were maintained in a humidified environment at 37°C and 5% CO2. Mosquito C6/36 cells (Aedes albopictus; ATCC) and CxQx (Culex quinquefasciatus) were cultured (29°C, 5%CO2) in Eagle’s Minimum Essential Medium (EMEM) containing 10% FBS as previously described (Hernandez and Brown, 2010).
Cloning of V5 insertion within RVFV Gn.
The V5 epitope (amino acid (aa) sequence-GKPIPNPLLGLDST) derived from the RNA polymerase α subunit of the simian parainfluenza virus type 5 was cloned into the Gn coding region within the pProT7-M(+) plasmid (RVFV MP12 strain). The V5 insertion sites were at Gn aa 105 and 229 (M polyprotein aa 258 and 382, respectively). The two amino acids before each insertion site was duplicated after the V5 sequence. All plasmid constructs were verified by restriction enzyme digestion and sequencing. Plasmid and primer sequences are available upon request.
Viral stocks.
Recombinant RVFV MP12 and ZH548 viruses were rescued and tittered as previously described (Baer and Kehn-Hall, 2014; Benedict et al., 2015). Briefly, transfection of BSR-T7/5 cells with the following plasmids was performed as previously described: pProT7-M(+), pProT7-L(+), pProT7-S(+), pT7-IRES-vN, pT7-IRES-vL, and pCAGGS-vG (Cyr et al., 2015). For V5-tagged Gn rMP12 viruses, pProT7-M(+)_V5Gn105 and pProT7-M(+)_V5Gn229 were utilized to generate MP12-V5Gn105 and MP12-V5Gn229 viruses, respectively. A passaged (P)1 viral stock was generated for infections. Additionally, the parental rMP12 containing a V5-tag inserted between amino acid position 1852 and 1853 of the viral L protein, termed V5L, was also used in this study (Baer et al., 2016). Venezuelan equine encephalitis virus (VEEV) TC-83 viral stocks were produced from electroporation of in vitro transcribed viral RNA generated from the pTC83 plasmid as described previously (Lundberg et al., 2016). Zika virus (ZIKV) MR766 (Cat# NR-50065) was obtained from BEI Resources.
Viral kinetic analysis.
Viral kinetics were measured by either infectious virus titers or percentage of RVFV positive cells. HSAECs, Huh-7, C6/36 and the UBR4 WT and KO HEK 293T cells were infected a MOI of 0.1. Extracellular media supernatants were collected at various times post-infection and infectious viral titers were determined by plaque assay on Vero cells (Baer and Kehn-Hall, 2014). An intracellular infectivity assay was performed as previously described (Benedict et al., 2015). Briefly, the WT and KO UBR4 293T cells were infected at a MOI of 0.1 with MP12. At 8 hours post infection (hpi) supernatants were collected for extracellular plaque assay. Cells were washed with PBS, collected in DMEM, and lysed using multiple freeze thaw cycles (using a dry ice-ethanol bath and 37°C water bath. Cellular lysates (containing infectious intracellular virions) were centrifuged and the supernatant was used for the intracellular plaque assay. For flow cytometry analysis, Huh-7 and C6/36 cells were trypsinized at various time points after infection (MOI 0.1) and fixed in 4% paraformaldehyde. Cells were permeabilized and then probed for RVFV nucleoprotein (NP; clone 1D8 [1:1000], BEI Resources, Cat# NR-43188). The percentage of RVFV NP positive cells from 10,000 cells was determined using EMD Millipore Guava® easyCyte HT Sampling Flow Cytometer.
Gn antibody neutralization.
Vero cells were seeded at 2 × 105 cells/well in 12-well plates. Media containing Gn antibody (clone 4D4, BEI Resources, Cat# NR-43190) at the two-fold dilutions (1:200, 400, 800, 1600, 3200, 6400, 12800, and 25600) or NP mouse antibody at 1:200 dilution was added first to Vero cells. Next RVFV inoculum, at a MOI 0.1, was added to the well. In the case of no antibody incubation, fresh complete DMEM was added to a final well volume of 1mL. After 24 hpi, supernatant was removed, cells were washed three times with phosphate buffered saline (PBS), and incubated for an additional 24h. RVFV positive cells, as indicated by RVFV NP staining, was determined by flow cytometry.
Immunofluorescence.
HSAECs were seeded on poly-L-lysine coated glass coverslips (Neuvitro, Cat# GG-22-1,5-PDL) in 6-well plates at 5 × 105 cells/well seeding density. Cells were infected (MOI 1) with MP12, V5Gn105 or V5Gn229 viruses. At 24 hpi, cells were fixed with 4% paraformaldehyde and permeabilized as previously described (Pinkham et al., 2016). Cells were then probed for RVFV Gn (clone 4D4, 1:5000 BEI Resources), RVFV Gc (clone 4B6, 1:5000, BEI Resources, Cat# NR-43184), TGN46 (AbD Serotec, Cat# AHP500G), and calnexin (Santa Cruz, Cat# sc11397) expression. The following secondary antibodies (1:500) were used to visualize the primary antibodies, Alexa Fluor 488 anti-mouse (Thermo Fisher Scientific, Cat# A-11001), Alexa Fluor 568 anti-rabbit (Thermo Fisher Scientific, Cat# A-11011), and Alexa Fluor 568 anti-sheep (Thermo Fisher Scientific, Cat# A-21099). Cells were counterstained with DAPI before mounting coverslips on glass slides. Slides were imaged using the Nikon Eclipse TE 2000-U microscope with a 60x oil immersion lens. Five images were taken per sample, with one representative shown.
Western blotting.
Cells were harvested at the indicated timepoint by one wash with PBS (without Ca2+ or Mg2+) and lysed directly in blue lysis buffer [1.25:1 ratio of 2x Novex® Tris-Glycine SDS Sample Buffer (Thermo Fisher Scientific, Cat# LC2676) and Tissue Protein Extraction Reagent (T-PER; Thermo Fisher Scientific, Cat# 78510), 2.0mM EDTA pH 8.0, 0.8mM NaF, 160μM Na3VO4, 26mM dithiothreitol (DTT), and EDTA-free Complete Protease Inhibitor Cocktail tablet (Sigma Aldrich, Cat# 11873580001)]. One tenth of the total volume was loaded on 3-12% Bis-Tris glycine gels (Thermo Fisher Scientific, Cat# NP0321) or, in the case of UBR4 protein, 3-8% Tris Acetate gels (Thermo Fisher Scientific, Cat# EA0375). After separation, proteins were transferred to PVDF membrane and blots were probed with antibodies against β-actin (Abcam, Cat# ab49900, 1:20,000), UBR4 (Abcam, Cat# ab86738, 1:1000), V5-tag (Biorad, Cat# MCA1360, 1:500), RVFV Gn (clone 4D4, 1:20,000) and NP (clone 1D8, 1:500). The blots were visualized by chemiluminescence using SuperSignal West Femto Maximum Sensitivity Substrate kit (Thermo Fisher Scientific, Cat# 34095) and the BioRad Molecular Imager ChemiDoc or ChemiDoc XRS systems.
Immunoprecipitation.
For immunoprecipitation, Huh-7 cells or HSAECs were mock-infected (complete media alone) or infected with MP12, V5Gn105, V5Gn229, or LV5 (MOI of 1). At 24 hpi, cells were lysed directly in clear lysis buffer (50mM Tris-HCl pH 7.5, 120mM NaCl, 5mM EDTA, 50mM NaF, 0.2mM Na3VO4, 0.5% NP-40, and EDTA-free Complete Protease Inhibitor Cocktail tablet) and protein concentration determined. Clarified cell lysates (2 mg) were mixed overnight at 4°C with 10-20 μg of either Gn (clone 4D4), V5 (BioRad), or HA (Cell Signaling, Cat# 3724) antibodies. The next morning, Protein G conjugated Dynabeads (Thermo Fisher Scientific, Cat# 10004D) were added. After 2 h, antibody-bound complexes were washed three times with TNE150 with 0.1% NP40 (50mM Tris pH 7.5, 50mM NaCl, 1mM EDTA, 0.1% NP-40, and EDTA-free Complete Protease Inhibitor Cocktail tablet) and once with TNE50 with 0.1% NP40 (50mM Tris pH 7.5, 150mM NaCl, 1mM EDTA, 0.1% NP-40, and EDTA-free Complete Protease Inhibitor Cocktail tablet). Complexes were eluted from the beads by boiling for 10 min in blue lysis buffer. As input controls, 50ug of total cell lysates were used. Proteins were separated by SDS-PAGE and blots probed for RVFV Gn, V5 epitope tag, and UBR4 as described above.
MS/MS analysis.
LC-MS/MS analysis was performed using a Thermo-LTQ mass spectrometer. Immunoprecipitated samples were lysed in 8M urea, reduced using DTT and acetylated with iodoacetamide. The reduced and alkylated proteins were trypsin digested (Promega, Cat# V5113) for 24 hours at 37°C. The trypsinized peptides were eluted using ZipTip purification (Millipore, Cat# ZTC18S) and analysis of the peptides was performed by LTQ-tandem MS/MS equipped with a reverse-phase liquid chromatography nanospray (Thermo Fisher Scientific). The peptides from the injected samples were eluted after washing using a 90 minute linear gradient from 0 to 40% acetonitrile and an additional step of 80% acetonitrile (all in 0.1% formic acid) for 5 minutes. The instrument was operated in a data-dependent mode and tandem mass spectra were matched against the National Center for Biotechnology Information human database by Sequest Bioworks software (Thermo Fisher Scientific) using full tryptic cleavage constraints and static cysteine alkylation by iodoacetamide.
siRNA Transfection.
Huh-7 cells were seeded at 3.5x105 cell/well in 6-well plates. Following the manufacturer’s instructions, cells were transfected using DharmaFECT 1 Transfection Reagent (Horizon Discovery, Cat # T-2001-02) with 50nM nontargeting siRNA (siNTC; Dharmacon cat #: D-001810-01-05), 50 nM siRNA against UBR4 (siUBR4; Dharmacon, cat #: L-014021-01-0005), or no siRNA (Mock control). Transfection media was replaced after 24-48 hours and cells were infected with MP12 (MOI 0.1) at 72 hours post transfection. Both extracellular media supernatants and protein lysates were harvested at 8 and 24 hpi. Viral titers were analyzed by plaque assay, while protein expression was analyzed by western blot.
Cell viability assay.
The HEK 293T KO and WT UBR4 cells were cultured and infected at various MOIs with MP12. At 16 hpi cell viability was measured using CellTiter-Glo Cell Luminescent Viability Assay (Promega, Cat# G7570) according to manufacturer’s protocol. Briefly, an equal volume of room temperature media and CellTiter-Glo reagent was added to the cells and mixed thoroughly. The plate was then incubated on a rocker for 10 minutes at room temperature. The luminescence was detected using the Promega GloMax® Discover Microplate Reader.
UBR4 gene knock-down in mosquito cells.
A pair of primers with 5’ & 3’ flanking T7 promoter sequences (UBR4 F & UBR4 R; see Suppl. Table 1) was designed to amplify a fragment of 455 bp size from the Culex UBR4 gene (Table 1). This fragment was obtained from gDNA extracted from Cx.quinquefaciatus (CxQx) cells. The amplified fragment was further purified using a PCR purification kit (Qiagen, Germany). The purified fragment (200 ng) was in vitro transcribed using the T7 Megascript kit (Ambion, USA) according to the manufacturer’s instructions. Afterwards, the RNA was purified using the Mega-clear transcription clean up kit (Invitrogen, USA) and stored at −80°C until further use. dsRNA targeting the green fluorescent protein (GFP) ORF was used as a negative control and also for assessing the toxic effect of dsRNAs in CxQx cells. A pair of primers (GFP F+ GFP R; see Suppl. Table 1) was designed to amplify a 495 bp segment of the GFP ORF from the pCAG-GFP plasmid (Addgene, USA) and dsRNA was prepared following the same protocol as described above. CxQx (Culex quinquefasciatus) cells were plated at the density of 8 x104 cells/well in a 24 well plate. Forty-eight hours post plating, cells were transfected with 100 ng of dsRNA specific for the GFP or UBR4 gene per well using Lipofectamine 2000 (Cat # 11668027, Invitrogen, USA). Twenty-four hours post transfection, the cells were infected with the RVFV vaccine strain MP-12 (MOI 0.01), and the supernatants were collected at 24 hpi. The virus titer in the supernatant was determined by plaque assay on Vero-MARU cells in 24 well plates.
Table 1. LC-MS/MS analysis of V5-tagged Gn interacting proteins.
List including XC Score, size, and # of peptides identified for relevant proteins.
| Gene Ontology | UniProt # | Protein Symbol | Full Name | Score XC | MW (kDa) | Peptides Identified |
|---|---|---|---|---|---|---|
| Cytoskeleton | P63261 | ACTG1 | Actin, gamma 1 propeptide | 60.31 | 41.8 | 9 |
| P63267 | ACTG2 | Actin, gamma 2 propeptide | 90.24 | 41.8 | 14 | |
| P35579 | MYH9 | Myosin, heavy polypeptide 9, non-muscle | 80.33 | 226.4 | 11 | |
| P19105 | MYL12A | Myosin, light chain 12A, regulatory,non-sarcomeric | 60.25 | 19.8 | 15 | |
| P60660 | MYL6 | Myosin, light chain 6, alkali, smooth muscle and non-muscle isoform 1 | 60.22 | 16.9 | 7 | |
| P35580 | MYH10 | Myosin, heavy polypeptide 10, non-muscle | 30.19 | 228.9 | 4 | |
| P67936 | TPM4 | Tropomyosin 4 isoform 2 | 80.28 | 28.5 | 18 | |
| P14649 | MYL6B | Myosin alkali light chain 6B | 20.14 | 22.7 | 2 | |
| P09493 | TPM1 | Tropomyosin 1 alpha chain isoform 5 | 20.2 | 32.8 | 2 | |
|
| ||||||
| Trafficking | Q00610 | CLTC | Clathrin heavy chain 1 | 20.19 | 191.5 | 2 |
| P55735 | SEC13 | SEC13 protein isoform 1 | 20.21 | 35.5 | 2 | |
|
| ||||||
| Protein Folding/Stability | Q5T4S7 | UBR4 | Ubiquitin Protein Ligase E3 Component N-Recognin 4 | 10.15 | 573.5 | 7 |
| P11021 | HSPA5 | Heat shock 70kDa protein 5 | 20.14 | 72.3 | 3 | |
|
| ||||||
| Viral | P21401 | GP_RVFVZ | ZH548/MP12 strain M polyprotein | 60.24 | 130.7 | 7 |
Quantitative RT-PCR.
Infected HEK 293T KO and WT UBR4 cells were lysed using Trizol (Thermo Fisher Scientific, Cat# 15596026) to analyze intracellular RNA. Intracellular RNA was isolated using the Direct-zol RNA Miniprep (Zymo Research, Cat # R2051). All samples were normalized to 10ng/ul of RNA using Nanodrop™ One prior to PCR analysis. qRT-PCR was performed using the RNA UltraSense One-Step Quantitative RT-PCR system (Thermo Fisher Scientific, Cat#11732-927) with primers and probe targeting the viral M segment (Forward AAAGGAACAATGGACTCTGGTCA, Reverse CACTTCTTACTACCATGTCCTCCAAT, and Probe 6FAM-AAAGCTTTGATATCTCTCAGTGCCCCAA-TAMRA).
Entry assay.
HEK 293T WT and UBR4 KO cells were cultured in 6 well plates and pretreated with either 12 mM NH4Cl dissolved in complete growth media or complete growth media alone for 1 h. Cells were infected with the RVFV MP-12 (MOI 10) for 1h at 37°C. Cells were washed with PBS and 0 hpi samples were collected using Trizol. The corresponding treatment was returned to the 2 hpi samples and were subsequently collected at 2 hpi in Trizol. Intracellular RNA was isolated using the Direct-zol RNA Miniprep. RT-qPCR was performed using the RNA UltraSense One-Step Quantitative RT-PCR system with primers and probe targeting the viral M segment. Samples were compared to a standard curve. Fold change was determined by comparison to WT control.
Results
Insertion of a V5-tag into RVFV Gn does not alter RVFV replication
Of the two RVFV glycoproteins, Gn mediates many of the interactions necessary for targeting viral proteins to the Golgi complex, RNP encapsidation, and receptor binding. Thus, we focused on providing a means to isolate Gn-associated complexes. There are several RVFV Gn antibodies available, however, the use of isotype control antibodies as a negative control can be problematic with a high degree of nonspecific interactions identified. To avoid these issues, we first generated a recombinant RVFV clone that contains a foreign epitope tag within Gn. At the time of creation, there was no crystallized structure of the RVFV Gn glycoprotein available. However, the model formulated by Rusu et al. was able to create a molecular model based on the 3D structure of the RVFV virion and homologous fold recognition analysis (Rusu et al., 2012). From this analysis, the ectodomain of Gn formed most of the protruding capsomer (Huiskonen et al., 2009; Sherman et al., 2009). Based on this model, two solvent exposed areas on Gn that were adjacent to previously mapped neutralizing epitopes (aa. 105 (epitope I) and 229 (epitope II); (Besselaar and Blackburn, 1991)) were utilized for V5 epitope tag insertion (Figure 1A). Recombinant RVFV MP12 containing the V5 insertions, termed hereafter V5Gn105 and V5Gn229 were rescued with well-defined plaques and with similar morphology to the parental RVFV MP12.
Figure 1. Recombinant MP12 viruses containing V5-tagged Gn glycoproteins.
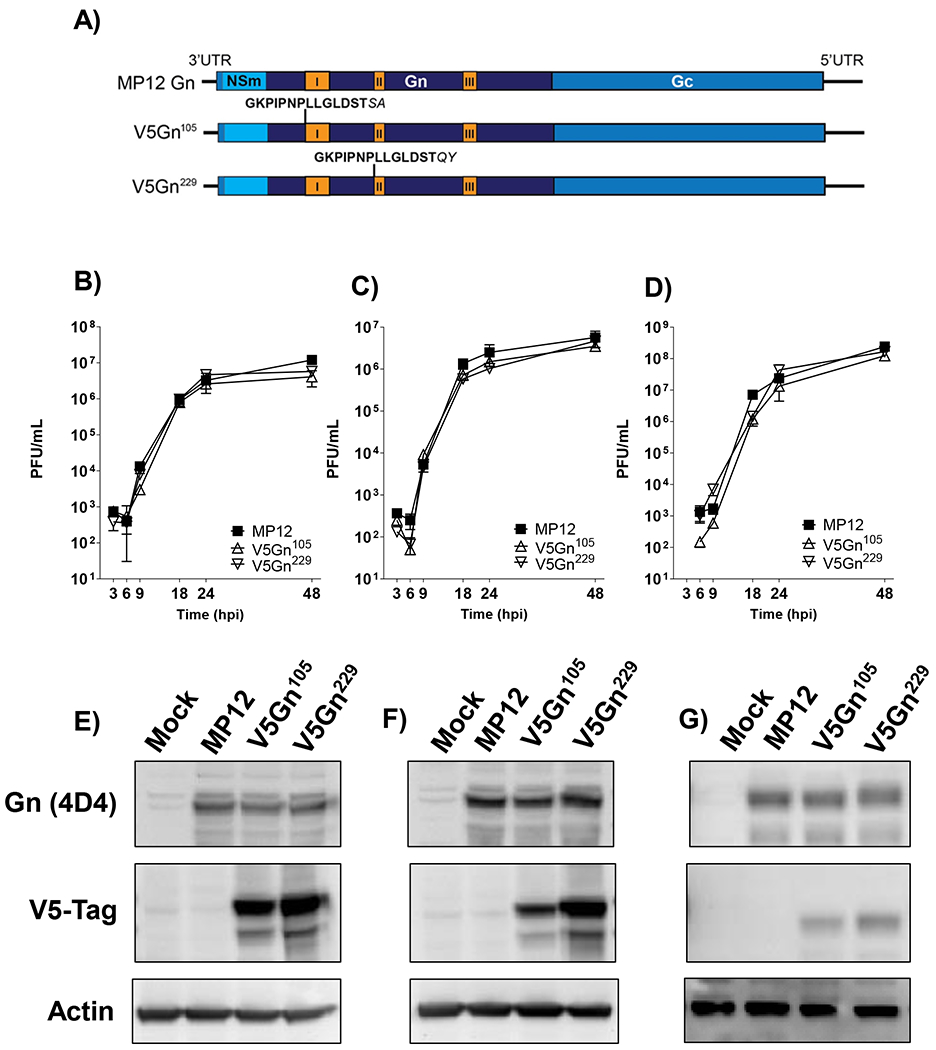
A) Schematic depicting RVFV M segment containing insertion of the V5 epitope tag (bold) within Gn at positions 105 and 229 (Gn amino acid numbering). The two amino acids prior to the insertion site were duplicated (italicized) after the V5 sequence. Mapped Gn antigenic epitopes are indicated in orange and labelled I, II, and III (Besselaar and Blackburn, 1991). B-D) Viral kinetics of MP12 viruses in human and mosquito cells. HSAECs (B), Huh-7 (C), and C6/36 (D) cells were infected at a MOI 0.1 with either untagged parental MP12 virus or the recombinant MP12 viruses containing V5-tagged Gn proteins (V5Gn105 and V5Gn229). Infectious viral titers were determined by plaque assay. Both mean and standard deviations are shown for three biological replicates. HSEACs (E), Huh-7 (F), and C6/36 (G) cells were infected at a MOI 1 with either parental MP12 virus, V5Gn105 or V5Gn229. At 24 hpi, cell lysates were harvested and levels of untagged Gn, V5-tagged Gn, and actin were determined by western blot.
To assess whether the V5-tag impacted Gn functionality due to disruption of Gn structure or protein interactions, a number of complementary assays were performed. First, viral kinetics were analyzed in both mammalian cell lines, HSAECs and Huh-7 cells, as well as the mosquito cell line, C6/36. HSAECs were chosen as a model of inhalational exposure to RVFV, while hepatocytes are a primary site for RVFV replication in vivo (Faburay et al., 2016; Paweska, 2015; Smith et al., 2010; Wilson et al., 2016). The derived A. albopictus cell line (C6/36) has been shown to yield high levels of RVFV replication in vitro. Infectious virus titers for the V5-tagged viruses were comparable to the MP12 parental strain in all three cell types (Figure 1B–D). Additionally, by 24 hpi, the levels of Gn protein expression were also similar between all three viruses (Figure 1E–G). When utilizing the V5 antibody, both the mature Gn, as well as, a truncated Gn protein was detectible via western blot analysis.
The cellular localization of Gn/Gc glycoproteins was then examined by confocal microscopy (Figure 2). Although the site of virus assembly is the Golgi complex, both glycoproteins are initially synthesized within the ER. If the tag insertion disrupted the native Gn structure, then localization of Gn, as well Gc, may be affected. Utilizing glycoprotein specific antibodies, all three viruses demonstrated only a small portion of both Gn and Gc staining overlap with the ER marker, calnexin (Figure 2A, B). Conversely, a larger overlap of Gn with Golgi marker, TGN46 (Figure 2C, D) was observed. Gc also demonstrated some overlap with the Golgi, but it to a lesser extent when compared to Gn. The pattern of localization observed between the parental and V5-tagged viruses was similar, suggesting that localization was not perturbed due to the V5-tag.
Figure 2. Cellular localization of Gn and Gc proteins.
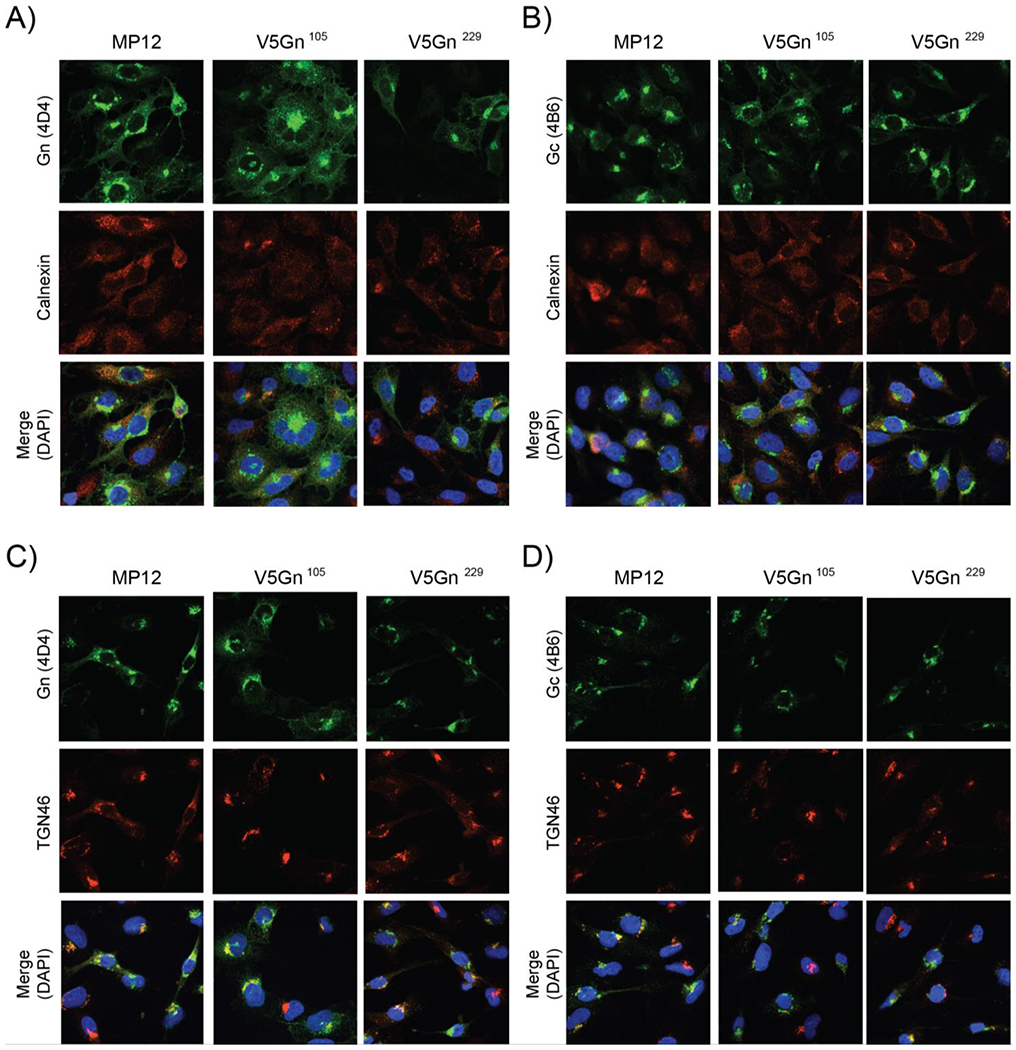
HSAECs cells were infected with either the parental MP12 or V5-tagged viruses at a MOI 1. At 24 hpi, cells were then fixed and analyzed by immunofluorescence for Gn (panels A, C) or Gc (B, D) protein localization. Calnexin (A, B), TGN46 (C, D), and DAPI staining were utilized as controls for the endoplasmic reticulum (ER), Golgi, and the nucleus, respectively.
To examine whether viral spread was altered, the percentage of RVFV infected cells was examined in both Huh-7 and C6/36 cells (Figure 3A and B, respectively). While there was no difference in the percent of cells infected between the V5-tagged viruses and MP12 in Huh-7 cells, there was a slight lag in the C6/36 cells starting at 18 hpi. However, by 48 hpi, the average percentage of RVFV positive cells was 79% for the parental virus and 71% for both V5-tagged viruses. Additionally, neutralization with a titrated Gn antibody was performed to determine whether insertion of the V5-tag disrupted native Gn/Gc conformation in the viral particles produced (Figure 3C). If the glycoprotein arrangement on the particles was severely altered, then a shift in neutralization curves relative to the parental virus would be expected to occur. Shifts to the left and right would indicate more or less neutralization sensitivity, respectively. RVFV nucleoprotein antibody was utilized as a negative control and at the highest concentration did not neutralize any of the RVFV viruses tested. While both V5-tagged viruses did have increased sensitivity, neutralization of the V5Gn229 virus was similar to the parental MP12 virus.
Figure 3. Impact of Gn V5 insertion on viral spread and neutralization.
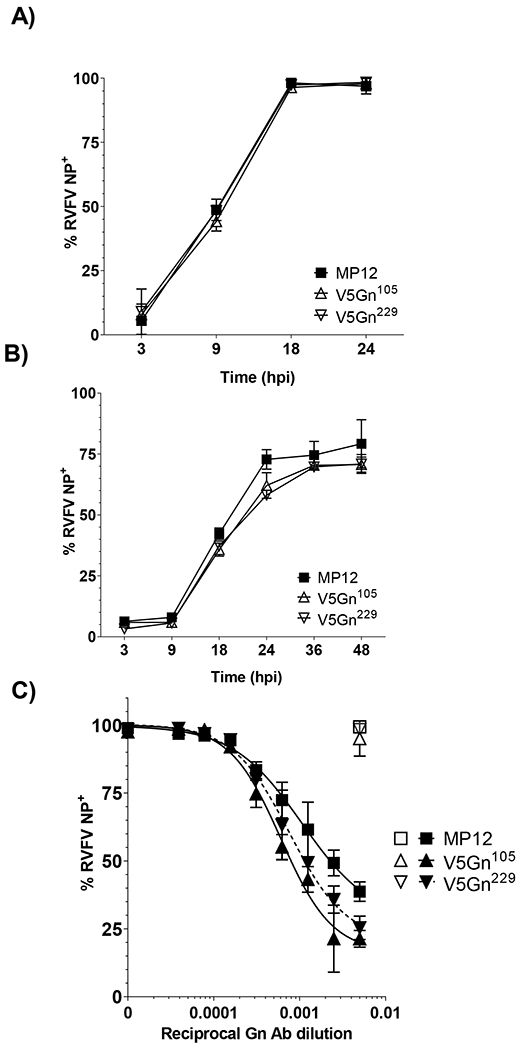
A) Huh-7 and B) mosquito C6/36 cells were infected at a MOI 0.1 with parental MP12 or V5-tagged viruses. Viral spread over time was assessed by RVFV NP staining and flow cytometry. Both mean and standard deviations are plotted for three biological replicates. C) RVFV virus inoculum (MOI 0.1) was mixed with increasing 2-fold dilutions of Gn antibody and incubated on Vero cells for 24h. After 48 hpi, cells were harvested and the percentage of RVFV positive cells were determined by flow cytometry. Filled symbols represent data from virus inoculum mixed with Gn antibody. Open symbols represent data from virus inoculum mixed with NP antibody at 1:200 dilution. Data is graphed as a percentage relative to no antibody control. Both mean and standard deviations are plotted for three biological replicates and non-linear regression analysis was performed. The EC50 for MP12 is a 935-fold dilution, while the EC50 for V5Gn105 and V5Gn229 is a 1649-fold and a 1314-fold dilution, respectively.
Mass spectrometry yields host-binding partners of RVFV Gn
As our goal was to utilize these tagged viruses to identify potential Gn protein interacting partners, we compared the level of Gn protein immunoprecipitated by the V5-tag or Gn-specific antibody. Huh-7 cells were mock-infected (i.e. media alone) or infected with MP12, V5Gn105, or V5Gn229 viruses, then immunoprecipitation was performed on the collected lysates (Figure 4A). While higher levels of Gn protein was isolated with the Gn (clone 4D4) antibody, there was a high degree of background present. The amount of Gn isolated was similar between MP12 constructs suggesting that insertion of the V5 epitope tags at amino acid positions 105 or 229 did not negatively impact the antigenic epitope II structure. The V5 antibody isolated only those Gn proteins that contained the V5 epitope. It was observed that of the two tagged viruses, slightly more V5Gn229 protein than V5Gn105 was collected. In conclusion, these results suggest that the V5 tag (16-mer amino acid) insertion was tolerated and accessible for immunoprecipitation. Given that V5 tag insertion had less impact on V5Gn229 functionality, i.e. neutralization and immunoprecipitation, the V5Gn229 virus was selected for all downstream experiments.
Figure 4. UBR4 associates with RVFV Gn protein.
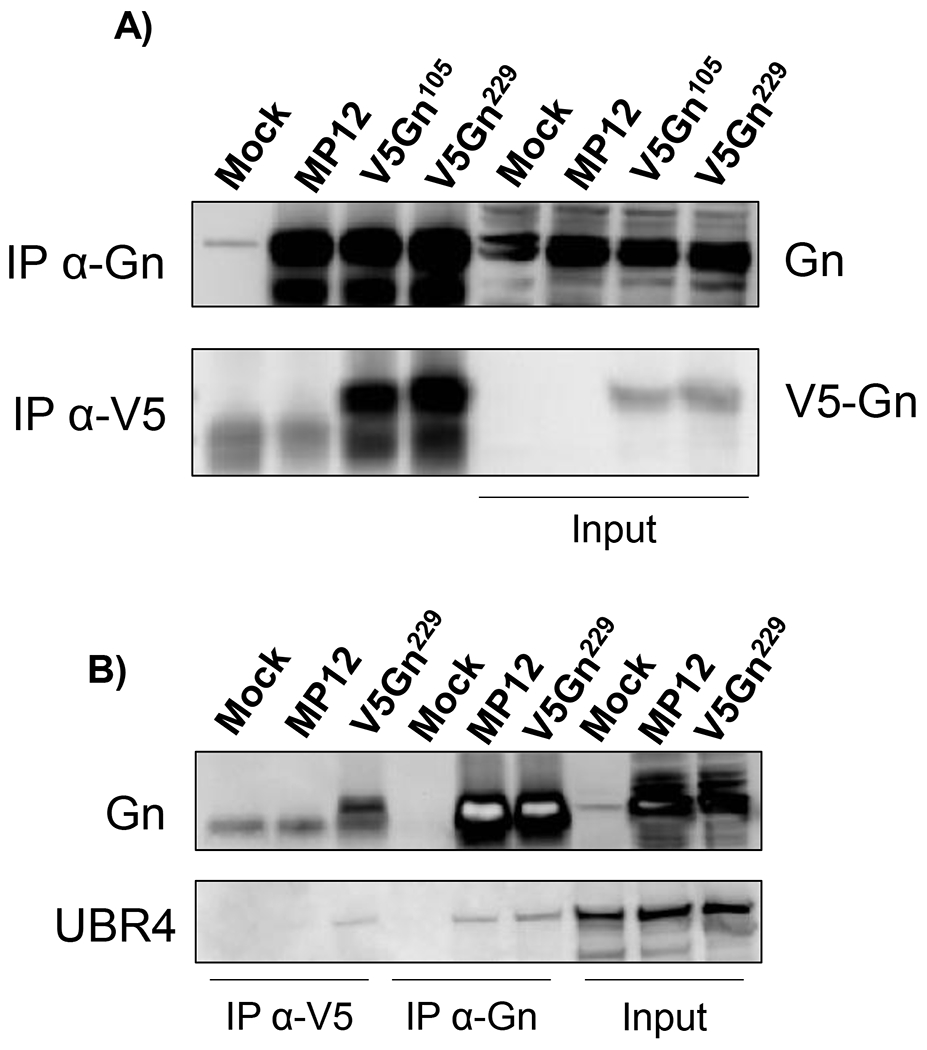
A-B) Huh-7 cells were infected with either the parental MP12, V5Gn105 (panel A only), or V5Gn229 virus at a MOI 1. Mock infection control refers to media alone. At 24 hpi, cells were harvested and whole cell lysates were generated. Immunoprecipitation using V5 epitope or Gn antibodies was performed and probed by western blot for UBR4 (panel B only) and Gn proteins using either an antibody raised against Gn or V5. Fifty micrograms of whole cell lysates were used as input controls.
To determine potential Gn binding partners, immunoprecipitation followed by mass spectrometry (MS) analysis of RVFV Gn-associated complexes was performed using the V5Gn229 and untagged MP12 parental virus. Huh-7 whole cell lysates collected at 24 hpi (MOI 1) were utilized for these experiments. Employing the untagged MP12 parental virus as a control for pull-downs helped to ensure that similar proteomic backgrounds were being used for comparison. After subtracting out cellular factors that were identified in both the untagged and V5-tagged backgrounds, 13 host factors were reproducibly detected (Table 1). A large number of cytoskeleton proteins including actin gamma, myosin, and tropomyosin factors were identified. Prior reports have indicated that bunyaviruses can rearrange cytoskeletal proteins for particle assembly factories. Similar to other viruses, bunyaviruses are heavily dependent on these cytoskeletal proteins for egress (Carnec et al., 2014; Fontana et al., 2008; Nuss et al., 2014; Ward, 2011), thus the high enrichment of these factors is not unexpected. We also observed cellular proteins SEC13 and heat shock 70kDa protein 5 (alias GRP78/BiP). Both proteins have been shown to localize to the ER, suggesting that we were isolating factors necessary for Gn folding and trafficking from the ER. Finally, peptides that mapped to RVFV Gn were also enriched.
The mass spectrometry analysis also identified UBR4 as a potential host-binding partner of Gn. There is mounting evidence that UBR4 can be exploited by other viruses for replication, like DENV (Morrison et al., 2013), IAV (Tripathi et al., 2015) and HPV (Huh et al., 2005), therefore it was chosen as a suitable candidate for confirmation. To substantiate these findings, the same immunoprecipitation utilizing V5 and Gn specific antibodies was performed to isolate Gn associated protein partners followed by western blot analysis (Figure 4B). UBR4 was detected in both the V5-tagged and untagged Gn immunoprecipitated complexes, verifying the mass spectrometry data.
Knockdown and knockout of UBR4 causes decreases in RVFV titers
To determine whether UBR4 is necessary for RVFV replication, an siRNA knockdown of UBR4 was performed in Huh-7 cells and infectious virus titers were measured at 8 and 24 hpi (Figure 5A and 5B). Greater than 80% knockdown of UBR4 was achieved and resulted in a 4- to 5-fold decrease in viral titers. To further examine this phenotype, UBR4 −/− HEK 293T cells were utilized (Tripathi et al., 2015). A significant decrease in the amount of infectious virus produced was observed at 8 and 16 hpi, where titers were decreased by 1 and 0.5 logs, respectively (Figure 5C). However, by 24 hpi the amount of infectious virus recovered in the UBR4 KO cells to a level comparable to the WT cells. These results suggest that UBR4 influences RVFV replication, but is not essential for infectious virus production.
Figure 5. Absence of UBR4 decreases RVFV viral titers.
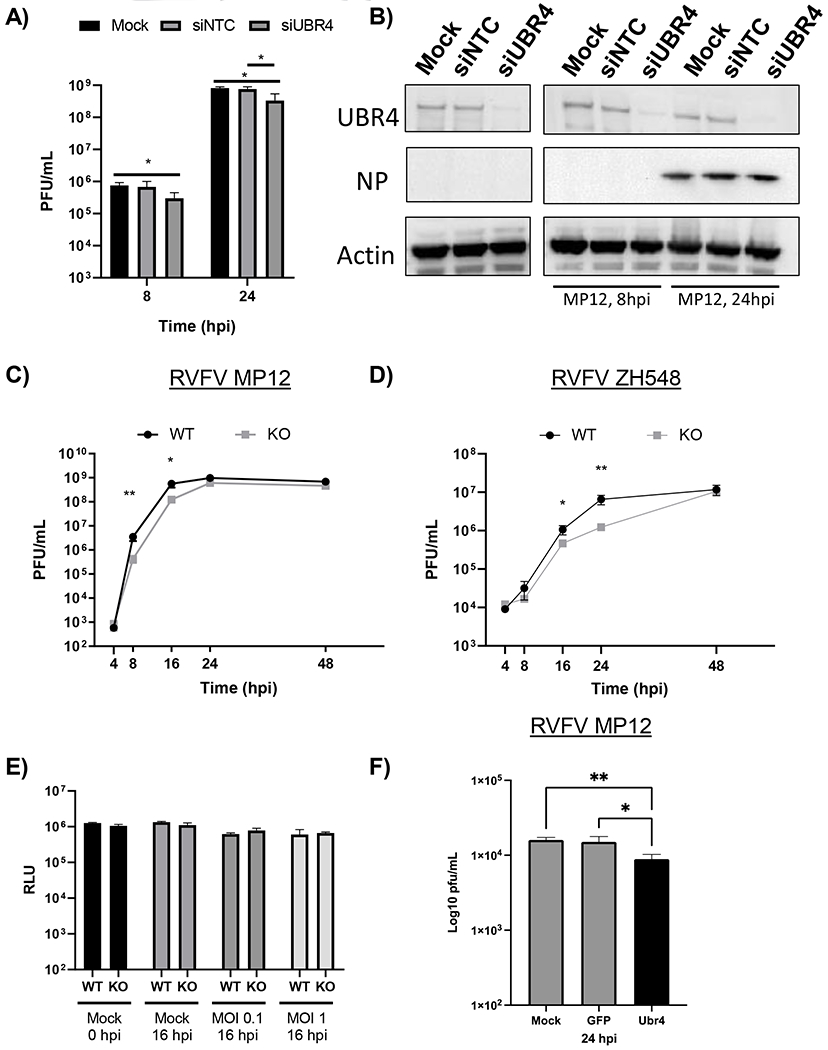
A-B) UBR4 expression in Huh-7 cells was knocked down by siRNA treatment (50 nM). After 72h, cells were infected with MP12 (MOI 0.1). At 8 and 24 hpi, infectious viral titers (A) or protein expression of UBR4, Actin, and RVFV Gn and NP were examined (B). C-D) HEK 293T WT and UBR4 KO cells were infected with untagged parental RVFV attenuated strain, MP12, (C) or the fully virulent strain, ZH548, (D) at a MOI of 0.1. Viral supernatants were collected at various time points post infection and titers were determined by plaque assay. E) HEK 293T WT and UBR4 KO cells were either mock-infected or infected with the parental MP12 virus (MOI 0.1 or 1). Cellular viability was measured either prior to infection or at 16 hpi using Cell Titer-Glo. F) Loss of Ubr4 decreases RVFV replication in mosquito cells. CxQx cells were plated in a 24 well plate and transfected with 100ng of dsRNA per well. The transfected cells were infected with RVFV MP12 (MOI 0.01). The supernatants were collected and viral titer determined by plaque assay. Mock-transfected control cells, GFP- dsRNA transfected cells; and Ubr4- dsRNA transfected cells. Figures 5 A and C–F represent the mean and standard deviation of three biological replicates. *p-value<0.05, **p-value<0.01.
To determine if UBR4 is exploited by additional strains of RVFV, a time course was performed with the virulent strain ZH548 in the UBR4 WT and KO cells (Figure 5D). A significant decrease in the amount of infectious virus produced was once again observed. However, this decrease was shifted towards later time points with a 0.5 log10 decrease at 16 hpi and a 1 log10 decrease at 24 hpi. In both attenuated and virulent RVFV strains, infectious viral titers recovered by 48 hpi. In order to confirm that this effect is not due to a change in growth or viability of the UBR4 KO cells, the amount of ATP present was measured as a proxy for cell viability. No significant variation in the viability of the two cell lines was observed with or without infection (Figure 5E). Finally, a UBR4 siRNA knockdown experiment was performed in Culex quinquefasciatus cells with the RVFV vaccine strain MP-12. A significant decrease of virus replication was observed 24 hpi when UBR4 was knocked down (Figure 5F). Collectively, these data indicate that loss of UBR4 decreases RVFV viral titers.
RVFV egress is not affected in the absence of UBR4
RVFV buds from the Golgi and infectious virus is trafficked to the plasma membrane where it is released via exocytosis. The impact of UBR4 on viral egress was examined by intracellular and extracellular infectious viral titer determination in WT and UBR4 KO cells. If UBR4 impacts viral egress, then an increase in the amount of virus inside the cells would be observed. However, the absence of UBR4 resulted in a decrease of both intracellular and extracellular viral titers (Figure 6A). The ratio of infectious virus inside to outside the cells remained the same and were not significantly different between WT and UBR4 KO cells (Intracellular/Extracellular: WT=0.050 +/− 0.024, KO=0.065 +/−0.027). The total amount of infectious virus produced was calculated by adding the PFU values for both extracellular and intracellular for each respective cell type. These data show that cells lacking UBR4 displayed an overall decrease in viral titers of nearly a 1 log10 (Figure 6B), indicating that UBR4 does not impact viral egress, but rather impacts a stage of viral replication prior to egress.
Figure 6. UBR4 impacts RVFV RNA production and Gn protein levels.
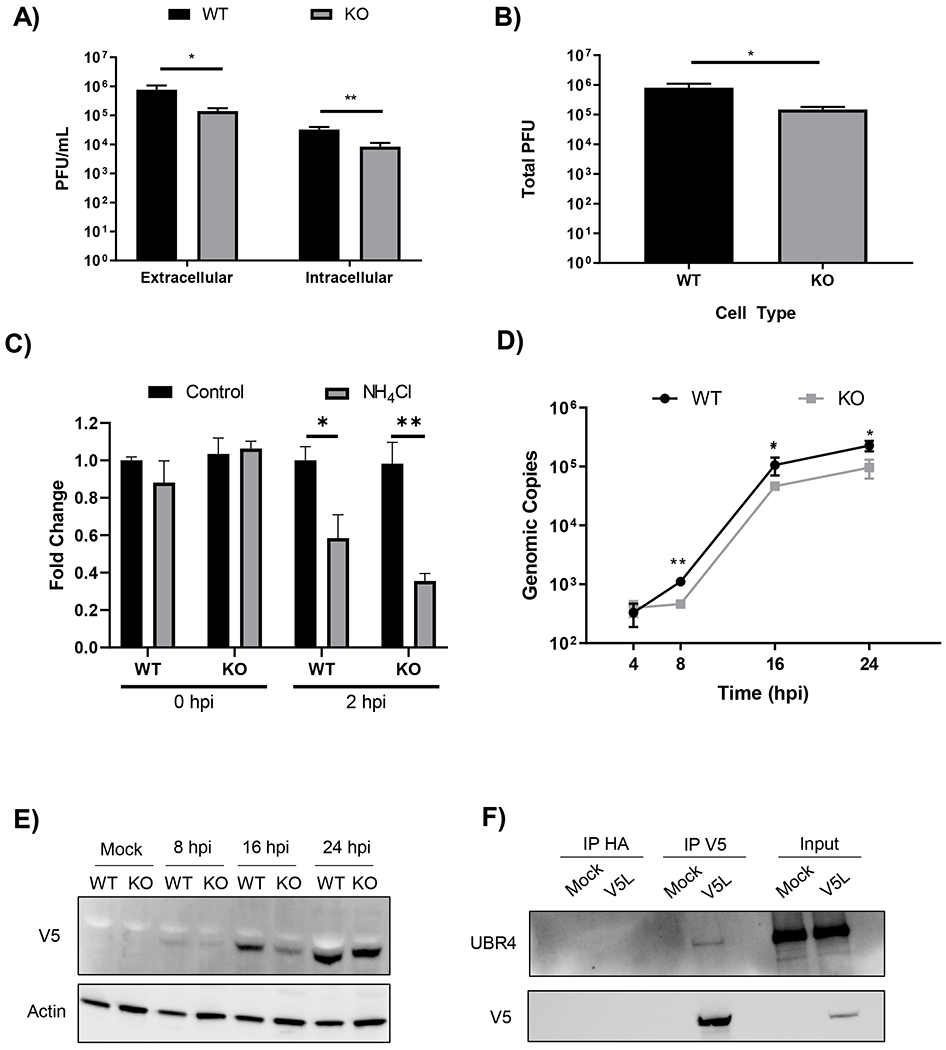
A) HEK 293T WT and UBR4 KO cells were infected with untagged parental RVFV MP12 (MOI 0.1). At 8 hpi, intra- and extracellular infectivity was determined by plaque assay. B) Total infectivity (i.e., the proportional sum of intra- and extracellular infectivity) per infection was also determined. The data shown represents the mean and standard deviation of three biological replicates. C) HEK 293T WT and KO cells were pretreated with ammonium chloride (12mM) for 1 h followed by an infection with RVFV MP12 at an MOI of 10. For the 2 hpi samples cells were post-treated until collection. At zero and 2 hpi cells were lysed using Trizol and RNA was extracted. RT-qPCR was performed using primers and probed against the M segment. Ct values were compared to a standard curve followed by normalization to their respective WT Control. The data shown represents the mean and standard deviation of three biological replicates. D) HEK 293T WT and UBR4 KO cells were infected with RVFV MP12 (MOI 0.01). At 4, 8. 16, and 24 hpi cells were lysed with Trizol and RNA was extracted. RT-qPCR was performed using primers and probe against the M segment. Ct values were compared to a standard curve to determine the viral copy number. The data shown represents the mean and standard deviation of three biological replicates. E) HEK 293T WT and UBR4 KO cells were infected with V5Gn229 (MOI 1). At 8, 16, and 24 hpi whole cell lysates were collected. Lysates were separated by SDS-PAGE and transferred to a PVDF membrane. The membrane was subsequently probed for both V5 and Actin. F) Huh-7 cells were infected with MP12 V5L (MOI 1). Mock infection control refers to media alone. At 24 hpi whole cell lysates were collected. Immunoprecipitation using V5 or HA antibodies was performed followed by a western blot for UBR4 and V5. Fifty micrograms of whole cell lysates were used as input controls. *p-value<0.05, **p-value<0.01.
The absence of UBR4 does not affect viral entry
To further understand the mechanistic impact UBR4 has on the viral life cycle an entry assay was performed using the UBR4 WT and KO cells infected with RVFV MP-12. Previous research has shown that ammonium chloride (NH44Cl) is an inhibitor of the earliest stages of the viral life cycle. Most notably by preventing the acidification of endosomes required for fusion (Brindley and Maury, 2005; Ellenbecker et al., 2014; Farías et al., 1988; Helenius, 2013). Therefore, NH4Cl was used as a control for viral entry inhibition. Treatment was performed 1h prior to, during, and post-infection. Cells were infected for 1 h at 37°C, washed, and post-treated. Zero-hour samples were collected immediately after infection. Intercellular RNA was extracted and RT-qPCR was performed against the viral M segment. A standard curve was used to determine genomic copy numbers and samples were compared to their respective WT control. No change in the amount of viral RNA was observed between the untreated WT and KO cells at both 0 and 2 hpi (Figure 6C). Upon treatment with NH4Cl a significant reduction was seen in both the WT and KO cells at 2 hpi. These data indicate that the virus enters the WT and KO UBR4 cells at a similar rate. Therefore, it is a step after viral entry that UBR4 impacts.
Lack of UBR4 decreases viral RNA production and Gn protein expression
Given the overall viral titers, both intracellular and extracellular, were decreased in the absence of UBR4 but viral egress and entry are not affected, viral RNA production was quantified via RT-qPCR. It was found that the amount of intracellular viral RNA was significantly decreased in UBR4 KO cells compared to the WT cells at 8, 16, and 24 hpi by 0.5 log10 (Figure 6D). Additionally, the protein levels of Gn were also markedly reduced in UBR4 KO cells at 16 hpi (Figure 6E). Notably, viral RNA levels were consistently lower in UBR4 KO cells, while RVFV titers and Gn protein levels recovered by 24 hpi. These data suggest that UBR4 has the most significant impact on viral RNA levels. To understand UBR4’s mechanism of action further its interaction with the L protein of RVFV, the RdRp, was examined. Immunoprecipitation followed by western blot analysis showed that UBR4 interacts with the L protein in Huh-7 cells infected with MP-12 V5L (Figure 6F). These data further substantiate that the effect observed is due to UBR4’s impact on viral RNA production.
Loss of UBR4 effects replication of RNA viruses from distinctive families
Previous studies have established that UBR4 can impact a variety of viruses. To confirm this further, UBR4’s impact on Venezuelan equine encephalitis virus (VEEV), an alphavirus, and ZIVK, a flavivirus, was examined using the UBR4 WT and KO cells. In both VEEV and ZIKV infected cells a significant reduction in infectious titers was observed at various time points post infection (Figure 7A–B). A half log10 reduction was observed with VEEV at 8 hpi, and then over a log10 at 24 hpi. While with ZIKV a log10 reduction was observed at 16 hpi and a half log10 reduction in infectious virus was observed at 24 hpi. These results corroborate and expand on previous findings that multiple viruses utilize UBR4 to facilitate their replication.
Figure 7. The absence of UBR4 effects multiple viruses.
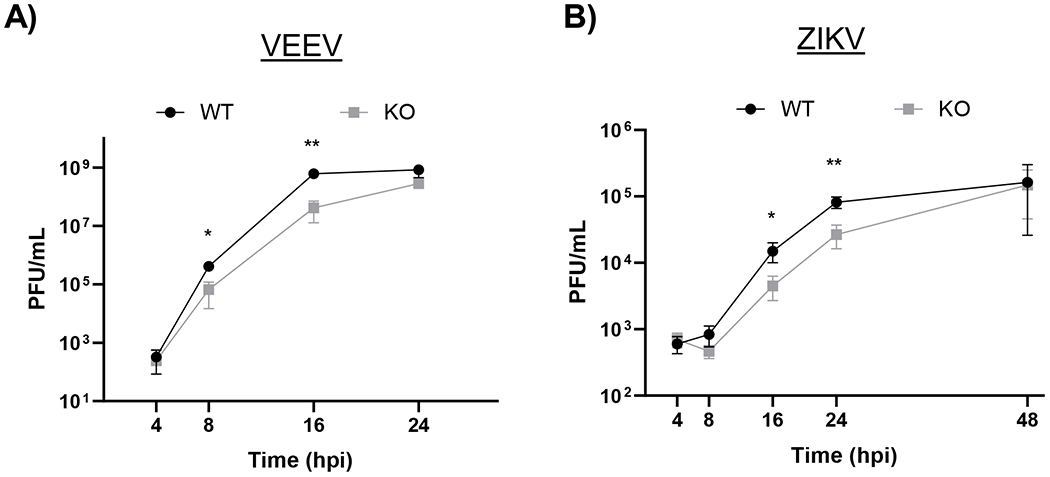
HEK 293T WT and UBR4 KO cells were infected with VEEV TC-83 (A) or ZIKV (B) at a MOI 0.1. At 4, 8, 16, and 24 hpi cellular supernatants were collected and viral titers were determined by plaque assay. The data shown represents the mean and standard deviation of three biological replicates. *p-value<0.05, **p-value<0.001.
Discussion
The incorporation of epitope tags into viral genomes, especially for mass proteomic studies, has yielded several advantages over the reliance of protein specific antibodies. The dependence on foreign epitopes for capture alleviates the potential exclusion of protein partners that bind within the epitope region. Immunoprecipitation conditions such as the cellular proteome at the time of harvest and the antibody and its respective reactivity and background characteristics are then consistent between the experimental tagged virus and the control untagged virus comparison. Also, advantageous is the availability of tag antibodies in a variety of pre-conjugated formats, e.g. fluorescent dyes, magnetic or sepharose beads, for various applications. An obvious caveat to the incorporation of foreign epitope tags is that insertion may disrupt protein structure leading to impaired protein binding and/or functionality. In addition, there has been some interest in the potential of differentiating between naturally infected and vaccinated animals (termed DIVA (Capua et al., 2004; Mansfield et al., 2015)). The incorporation of unique sequences, such as a tag to distinguish between the vaccine and naturally circulating RVFV strains would be useful for tracking.
Our studies indicate that the insertion of a 16-amino acid V5 tag at two different sites within Gn glycoprotein minimally impacted Gn functionality. With C6/36 cells, we observed a minor delay in the percentage of infected cells but not in infectious titers at 18 and 24 hpi for the V5-tagged viruses as compared to the parental MP12 virus (Figure 3B). This suggests that viral spread within the mosquito cell population is slightly delayed during the exponential phase of viral replication. Additionally, tag insertion at both sites slightly increased sensitivity to Gn antibody clone 4D4 neutralization (Figure 3C). However, viral titers, Gn protein levels, and Gn/Gc localization were comparable to the parental MP12 virus suggesting overall that the sites were tolerant of the tag insertion and any impact was minimal.
Our proteomic analysis of Gn interacting partners identified proteins involved with the cytoskeleton, trafficking, and protein folding/stability, all of which could be expected to interact with a viral glycoprotein that is processed through the secretory pathway. One of the identified Gn binding partners, HSPA5 (alias GRP78/BiP), is known to be incorporated into RVFV virions along with other HSP family members, HSP70 and HSP90 (Nuss et al., 2014). siRNA mediated knockdown of HSPA5 resulted in decreased RVFV replication (Nuss et al., 2014). The interaction of HSPA5 with Gn provides a mechanism by which it may be selectively incorporated into RVFV virions. While our method was successful for isolating Gn and some associated proteins, it is still missing the full complement of Gn interactions especially for those localized at the Golgi, the site of RVFV assembly. Future studies will focus on immunoprecipitation of Gn from isolated organelles (e.g. Golgi complex and ER) to facilitate the identification of additional Gn binding partners that may play a role in viral assembly and/or protein trafficking.
Our data confirmed that Gn interacts with UBR4, a 576-kDa E3 ubiquitin ligase that recognizes and binds to specific destabilizing amino acids within a protein’s N-terminal residues (termed N-degrons), resulting in their ubiquitination and subsequent degradation (Sriram et al., 2011). The UBR family is made up of four proteins (UBR1,-2,-4, and -5) that are all E3 ubiquitin ligases that follow the N-end rule (Tasaki et al., 2005). The N-end rule states that some N-terminal destabilized residues are be more likely to be targeted for ubiquitination, which are then recognized by E3 ubiquitin ligases, or N-recognins (Hwang et al., 2011). There are two categories of N-terminal degrons that are recognized within the N-end rule pathway: type 1 substrates which contain acidic residues, and type 2 substrates which contain bulky hydrophobic residues (Kim et al., 2013; Tasaki et al., 2009). Studies have indicated that UBR4 specifically recognizes destabilized N-terminal amino acids, such as arginine, through its evolutionarily conserved UBR domain (Hwang et al., 2011; Kim et al., 2018).
UBR4 interacts with a few viral proteins including DENV NS5 (Morrison et al., 2013), IAV M2 (Tripathi et al., 2015), and HPV E7 (Huh et al., 2005; White et al., 2012). DENV NS5 binds to UBR4 facilitating degradation of STAT2, which enables DENV to efficiently replicate in cells that can mount a type I interferon response. Loss of UBR4 results in decreased replication of both DENV and IAV, but not HSV-1 (Tripathi et al., 2015). Moreover, peptide-conjugated phosphorodiamidate morpholino oligomers (PPMOs) targeting UBR4 protected mice from IAV-induced disease (Tripathi et al., 2015). In IAV infected cells UBR4 localizes to the ER where it interacts with the M2 ion channel and facilitates M2 trafficking to the plasma membrane (Tripathi et al., 2015). The mechanism by which UBR4 enables M2 protein trafficking has yet to be determined, but the authors hypothesize that M2 may be using UBR4 to induce the degradation of a host restriction factor located in the ER, which would prevent viral protein trafficking from the ER to the Golgi complex.
In this study, it was found that loss of UBR4 through siRNA and CRISPR-Cas9 mediated knockout decreases the amount of infectious virus produced. However, it should be noted that by 24 hpi infectious viral titers of RVFV recovered in mammalian cells. This was also observed with two other RNA viruses, VEEV and ZIKV. This means that although UBR4 associates with RVFV Gn, its interaction is either transient or another host-protein can function in its place.
Since UBR4 co-immunoprecipitated with RVFV Gn, it was originally hypothesized that UBR4 might impact viral assembly or egress. However, it was found that a build-up of virus does not occur in the absence of UBR4, instead, less virus is being produced. To understand the mechanistic impact UBR4 has on the RVFV life cycle, viral entry, viral RNA production, and Gn protein expression levels were examined. It was found that UBR4 has no effect on virus entry but a significant reduction was observed for both Gn protein and viral RNA levels in the absence of UBR4. UBR4 had the most significant impact on RVFV RNA levels, which was unexpected given that it was identified as a RVFV Gn interacting partner. One possible explanation for these results is that UBR4 is indirectly interacting with RVFV Gn, and its direct binding partner is another viral protein, such as the L polymerase or NP. Our data lend support to this possibility, as UBR4 coimmunoprecipitated with RVFV L polymerase; however additional experiments are needed to determine if this is a direct interaction. It was previously determined that Gn interacts with NP through its C-terminal tail and also co-localizes with the L polymerase at the Golgi (Hornak et al., 2016). Both the L polymerase and NP play essential roles in RVFV RNA production, with the L polymerase performing cap-snatching and directing RNA transcription and replication (Gaudreault et al., 2019; Olschewski et al., 2020). Future studies will explore UBR4’s role in RVFV RNA production, including the determination of what allows for the recovery of RVFV infectious titers late in infection in cells lacking UBR4.
Supplementary Material
Research Highlights.
Rift Valley fever viruses containing an epitope tag on the Gn protein were generated
The V5-tag minimally impacted Gn functionality
UBR4, an E3 ubiquitin-protein ligase, is a RVFV Gn interacting protein
Depletion of UBR4 significantly decreased RVFV titers and viral RNA production
Acknowledgements.
The authors would like to thank Dr. Shinji Makino (UTMB, USA) for providing the RVFV MP12 reverse genetics system and Dr. Charles M. Rice (Rockefeller University, USA) for the hepatoma cell line, Huh-7 cells. The authors would also like to thank both current and past Kehn-Hall members for thoughtful discussions and suggestions.
Funding Information.
Funding for this study was provided through grants from NBAF Transition Funds from the State of Kansas (JAR), the Department of Homeland Security Center of Excellence for Emerging and Zoonotic Animal Diseases (CEEZAD), under grant number HSHQDC 16-A-B0006 (JAR), and the MCB Core of the Center on Emerging and Zoonotic Infectious Diseases (CEZID), the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health under award number P20GM130448 (JAR). WCW is supported by the USDA, Agricultural Research Service.
Conflicts of Interest
The J.A.R. laboratory received support from Tonix Pharmaceuticals, Xing Technologies, and Zoetis, outside of the reported work. J.A.R. is an inventor on patents and patent applications on the use of antivirals and vaccines for the treatment and prevention of virus infections, owned by Kansas State University, Manhattan, KS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Credit Author Statement
Nicole Bracci: Methodology, Investigation, Writing- Original draft preparation; Cynthia de la Fuente: Conceptualization, Methodology, Investigation, Writing- Original draft preparation; Sahar Saleem: Methodology, Investigation; Chelsea Pinkham: Methodology, Investigation, Writing- Reviewing and Editing; Aarthi Narayanan: Methodology, Investigation; Adolfo García-Sastre: Resources, Methodology; Velmurugan Balaraman: Methodology, Investigation; Juergen A. Richt: Conceptualization, Writing- Reviewing and Editing, Supervision, Project Administration, Funding acquisition, William Wilson: Conceptualization, Writing- Reviewing and Editing, Supervision, Project Administration, Funding acquisition; and Kylene Kehn-Hall: Conceptualization, Writing- Reviewing and Editing, Supervision, Project Administration, Funding acquisition
Publisher's Disclaimer: Disclaimer. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. The conclusions in this report are those of the authors and do not necessarily represent the views of the USDA. USDA is an equal opportunity provider and employer.
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
The J.A.R. laboratory received support from Tonix Pharmaceuticals, Xing Technologies, and Zoetis, outside of the reported work. J.A.R. is an inventor on patents and patent applications on the use of antivirals and vaccines for the treatment and prevention of virus infections, owned by Kansas State University, Manhattan, KS.
References
- Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR, Nibert M, Sabanadzovic S, Sanfacon H, Siddell SG, Simmonds P, Varsani A, Zerbini FM, Gorbalenya AE, Davison AJ, 2017. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2017). Archives of virology 162, 2505–2538. [DOI] [PubMed] [Google Scholar]
- Baer A, Kehn-Hall K, 2014. Viral concentration determination through plaque assays: using traditional and novel overlay systems. J Vis Exp, e52065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer A, Shafagati N, Benedict A, Ammosova T, Ivanov A, Hakami RM, Terasaki K, Makino S, Nekhai S, Kehn-Hall K, 2016. Protein Phosphatase-1 regulates Rift Valley fever virus replication. Antiviral Res 127, 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict A, Bansal N, Senina S, Hooper I, Lundberg L, de la Fuente C, Narayanan A, Gutting B, Kehn-Hall K, 2015. Repurposing FDA-approved drugs as therapeutics to treat Rift Valley fever virus infection. Frontiers in Microbiology 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermúdez-Méndez E, Katrukha EA, Spruit CM, Kortekaas J, Wichgers Schreur PJ, 2021. Visualizing the ribonucleoprotein content of single bunyavirus virions reveals more efficient genome packaging in the arthropod host. Commun Biol 4, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besselaar TG, Blackburn NK, 1991. Topological mapping of antigenic sites on the Rift Valley fever virus envelope glycoproteins using monoclonal antibodies. Archives of virology 121, 111–124. [DOI] [PubMed] [Google Scholar]
- Bird BH, Nichol ST, 2012. Breaking the chain: Rift Valley fever virus control via livestock vaccination. Current opinion in virology 2, 315–323. [DOI] [PubMed] [Google Scholar]
- Bouloy M, Weber F, 2010. Molecular biology of rift valley Fever virus. The open virology journal 4, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindley MA, Maury W, 2005. Endocytosis and a low-pH step are required for productive entry of equine infectious anemia virus. Journal of virology 79, 14482–14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capua I, Cattoli G, Marangon S, 2004. DIVA--a vaccination strategy enabling the detection of field exposure to avian influenza. Dev Biol (Basel) 119, 229–233. [PubMed] [Google Scholar]
- Carnec X, Ermonval M, Kreher F, Flamand M, Bouloy M, 2014. Role of the cytosolic tails of Rift Valley fever virus envelope glycoproteins in viral morphogenesis. Virology 448, 1–14. [DOI] [PubMed] [Google Scholar]
- Collett MS, Purchio AF, Keegan K, Frazier S, Hays W, Anderson DK, Parker MD, Schmaljohn C, Schmidt J, Dalrymple JM, 1985. Complete nucleotide sequence of the M RNA segment of Rift Valley fever virus. Virology 144, 228–245. [DOI] [PubMed] [Google Scholar]
- Crabtree MB, Kent Crockett RJ, Bird BH, Nichol ST, Erickson BR, Biggerstaff BJ, Horiuchi K, Miller BR, 2012. Infection and transmission of Rift Valley fever viruses lacking the NSs and/or NSm genes in mosquitoes: potential role for NSm in mosquito infection. PLoS neglected tropical diseases 6, e1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr N, de la Fuente C, Lecoq L, Guendel I, Chabot PR, Kehn-Hall K, Omichinski JG, 2015. A OmegaXaV motif in the Rift Valley fever virus NSs protein is essential for degrading p62, forming nuclear filaments and virulence. Proceedings of the National Academy of Sciences of the United States of America 112, 6021–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenbecker M, Lanchy J-M, Lodmell JS, 2014. Inhibition of Rift Valley fever virus replication and perturbation of nucleocapsid-RNA interactions by suramin. Antimicrob Agents Chemother 58, 7405–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott RM, 2014. Orthobunyaviruses: recent genetic and structural insights. Nature reviews. Microbiology 12, 673–685. [DOI] [PubMed] [Google Scholar]
- Elliott RM, Brennan B, 2014. Emerging phleboviruses. Current opinion in virology 5, 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faburay B, Gaudreault NN, Liu Q, Davis AS, Shivanna V, Sunwoo SY, Lang Y, Morozov I, Ruder M, Drolet B, Scott McVey D, Ma W, Wilson W, Richt JA, 2016. Development of a sheep challenge model for Rift Valley fever. Virology 489, 128–140. [DOI] [PubMed] [Google Scholar]
- Farías G, Navarrete E, Kiss J, Kuznar J, 1988. Effect of ammonium chloride on the multiplication of infectious pancreatic necrosis virus. Archives of Virology 98, 155–162. [DOI] [PubMed] [Google Scholar]
- Fontana J, Lopez-Montero N, Elliott RM, Fernandez JJ, Risco C, 2008. The unique architecture of Bunyamwera virus factories around the Golgi complex. Cellular microbiology 10, 2012–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiberg AN, Sherman MB, Morais MC, Holbrook MR, Watowich SJ, 2008. Three-dimensional organization of Rift Valley fever virus revealed by cryoelectron tomography. J Virol 82, 10341–10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garry CE, Garry RF, 2004. Proteomics computational analyses suggest that the carboxyl terminal glycoproteins of Bunyaviruses are class II viral fusion protein (beta-penetrenes). Theor Biol Med Model 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreault NN, Indran SV, Balaraman V, Wilson WC, Richt JA, 2019. Molecular aspects of Rift Valley fever virus and the emergence of reassortants. Virus Genes 55, 1–11. [DOI] [PubMed] [Google Scholar]
- Gaudreault NN, Indran SV, Bryant PK, Richt JA, Wilson WC, 2015. Comparison of Rift Valley fever virus replication in North American livestock and wildlife cell lines. Frontiers in microbiology 6, 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A, 2013. Virus entry: What has pH got to do with it? Nature Cell Biology 15, 125–125. [DOI] [PubMed] [Google Scholar]
- Hernandez R, Brown DT, 2010. Growth and maintenance of mosquito cell lines. Curr Protoc Microbiol Appendix 4, 4J. [DOI] [PubMed] [Google Scholar]
- Hornak KE, Lanchy JM, Lodmell JS, 2016. RNA Encapsidation and Packaging in the Phleboviruses. Viruses 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh K-W, DeMasi J, Ogawa H, Nakatani Y, Howley PM, Münger K, 2005. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proceedings of the National Academy of Sciences of the United States of America 102, 11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huiskonen JT, Overby AK, Weber F, Grunewald K, 2009. Electron cryomicroscopy and single-particle averaging of Rift Valley fever virus: evidence for GN-GC glycoprotein heterodimers. Journal of virology 83, 3762–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CS, Sukalo M, Batygin O, Addor MC, Brunner H, Aytes AP, Mayerle J, Song HK, Varshavsky A, Zenker M, 2011. Ubiquitin ligases of the N-end rule pathway: assessment of mutations in UBR1 that cause the Johanson-Blizzard syndrome. PLoS One 6, e24925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, 2012. Molecular biology and genetic diversity of Rift Valley fever virus. Antiviral research 95, 293–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakach LT, Suzich JA, Collett MS, 1989. Rift Valley fever virus M segment: phlebovirus expression strategy and protein glycosylation. Virology 170, 505–510. [DOI] [PubMed] [Google Scholar]
- Kakach LT, Wasmoen TL, Collett MS, 1988. Rift Valley fever virus M segment: use of recombinant vaccinia viruses to study Phlebovirus gene expression. Journal of virology 62, 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan K, Collett MS, 1986. Use of bacterial expression cloning to define the amino acid sequences of antigenic determinants on the G2 glycoprotein of Rift Valley fever virus. Journal of virology 58, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ST, Lee YJ, Tasaki T, Mun SR, Hwang J, Kang MJ, Ganipisetti S, Yi EC, Kim BY, Kwon YT, 2018. The N-recognin UBR4 of the N-end rule pathway is targeted to and required for the biogenesis of the early endosome. Journal of Cell Science 131, jcs217646. [DOI] [PubMed] [Google Scholar]
- Kim ST, Tasaki T, Zakrzewska A, Yoo YD, Sa Sung K, Kim SH, Cha-Molstad H, Hwang J, Kim KA, Kim BY, Kwon YT, 2013. The N-end rule proteolytic system in autophagy. Autophagy 9, 1100–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreher F, Tamietti C, Gommet C, Guillemot L, Ermonval M, Failloux AB, Panthier JJ, Bouloy M, Flamand M, 2014. The Rift Valley fever accessory proteins NSm and P78/NSm-GN are distinct determinants of virus propagation in vertebrate and invertebrate hosts. Emerging microbes & infections 3, e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeker AL, Babiuk S, Pickering BS, Richt JA, Wilson WC, 2020. Livestock Challenge Models of Rift Valley Fever for Agricultural Vaccine Testing. Front Vet Sci 7, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linthicum KJ, Britch SC, Anyamba A, 2016. Rift Valley Fever: An Emerging Mosquito-Borne Disease. Annual review of entomology 61, 395–415. [DOI] [PubMed] [Google Scholar]
- Lozach P-Y, Kühbacher A, Meier R, Mancini R, Bitto D, Bouloy M, Helenius A, 2011. DC-SIGN as a Receptor for Phleboviruses. Cell Host & Microbe 10, 75–88. [DOI] [PubMed] [Google Scholar]
- Lundberg L, Pinkham C, de la Fuente C, Brahms A, Shafagati N, Wagstaff KM, Jans DA, Tamir S, Kehn-Hall K, 2016. Selective Inhibitor of Nuclear Export (SINE) Compounds Alter New World Alphavirus Capsid Localization and Reduce Viral Replication in Mammalian Cells. PLOS Neglected Tropical Diseases 10, e0005122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield KL, Banyard AC, McElhinney L, Johnson N, Horton DL, Hernandez-Triana LM, Fooks AR, 2015. Rift Valley fever virus: A review of diagnosis and vaccination, and implications for emergence in Europe. Vaccine 33, 5520–5531. [DOI] [PubMed] [Google Scholar]
- Morrison J, Laurent-Rolle M, Maestre AM, Rajsbaum R, Pisanelli G, Simon V, Mulder LCF, Fernandez-Sesma A, García-Sastre A, 2013. Dengue Virus Co-opts UBR4 to Degrade STAT2 and Antagonize Type I Interferon Signaling. PLOS Pathogens 9, e1003265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss JE, Kehn-Hall K, Benedict A, Costantino J, Ward M, Peyser BD, Retterer CJ, Tressler LE, Wanner LM, McGovern HF, Zaidi A, Anthony SM, Kota KP, Bavari S, Hakami RM, 2014. Multi-faceted proteomic characterization of host protein complement of Rift Valley fever virus virions and identification of specific heat shock proteins, including HSP90, as important viral host factors. PLoS One 9, e93483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olschewski S, Cusack S, Rosenthal M, 2020. The Cap-Snatching Mechanism of Bunyaviruses. Trends Microbiol 28, 293–303. [DOI] [PubMed] [Google Scholar]
- Paweska JT, 2015. Rift Valley fever. Revue scientifique et technique 34, 375–389. [DOI] [PubMed] [Google Scholar]
- Phoenix I, Nishiyama S, Lokugamage N, Hill TE, Huante MB, Slack OA, Carpio VH, Freiberg AN, Ikegami T, 2016. N-Glycans on the Rift Valley Fever Virus Envelope Glycoproteins Gn and Gc Redundantly Support Viral Infection via DC-SIGN. Viruses 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkham C, An S, Lundberg L, Bansal N, Benedict A, Narayanan A, Kehn-Hall K, 2016. The role of signal transducer and activator of transcription 3 in Rift Valley fever virus infection. Virology 496, 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova TG, Turell MJ, Espina V, Kehn-Hall K, Kidd J, Narayanan A, Liotta L, Petricoin EF 3rd, Kashanchi F, Bailey C, Popov SG, 2010. Reverse-phase phosphoproteome analysis of signaling pathways induced by Rift valley fever virus in human small airway epithelial cells. PLoS One 5, e13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinschen MM, Bharill P, Wu X, Kohli P, Reinert MJ, Kretz O, Saez I, Schermer B, Höhne M, Bartram MP, Aravamudhan S, Brooks BR, Vilchez D, Huber TB, Müller RU, Krüger M, Benzing T, 2016. The ubiquitin ligase Ubr4 controls stability of podocin/MEC-2 supercomplexes. Hum Mol Genet 25, 1328–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolin AI, Berrang-Ford L, Kulkarni MA, 2013. The risk of Rift Valley fever virus introduction and establishment in the United States and European Union. Emerging microbes & infections 2, e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusu M, Bonneau R, Holbrook MR, Watowich SJ, Birmanns S, Wriggers W, Freiberg AN, 2012. An assembly model of rift valley Fever virus. Frontiers in microbiology 3, 254–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman MB, Freiberg AN, Holbrook MR, Watowich SJ, 2009. Single-particle cryo-electron microscopy of Rift Valley fever virus. Virology 387, 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Steele KE, Shamblin J, Honko A, Johnson J, Reed C, Kennedy M, Chapman JL, Hensley LE, 2010. The pathogenesis of Rift Valley fever virus in the mouse model. Virology 407, 256–267. [DOI] [PubMed] [Google Scholar]
- Spiegel M, Plegge T, Pöhlmann S, 2016. The Role of Phlebovirus Glycoproteins in Viral Entry, Assembly and Release. Viruses 8, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram SM, Kim BY, Kwon YT, 2011. The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat Rev Mol Cell Bio 12, 735–747. [DOI] [PubMed] [Google Scholar]
- Tasaki T, Mulder LC, Iwamatsu A, Lee MJ, Davydov IV, Varshavsky A, Muesing M, Kwon YT, 2005. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol Cell Biol 25, 7120–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Zakrzewska A, Dudgeon DD, Jiang Y, Lazo JS, Kwon YT, 2009. The substrate recognition domains of the N-end rule pathway. J Biol Chem 284, 1884–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, Yanguez E, Andenmatten D, Pache L, Manicassamy B, Albrecht RA, Gonzalez MG, Nguyen Q, Brass A, Elledge S, White M, Shapira S, Hacohen N, Karlas A, Meyer TF, Shales M, Gatorano A, Johnson JR, Jang G, Johnson T, Verschueren E, Sanders D, Krogan N, Shaw M, Konig R, Stertz S, Garcia-Sastre A, Chanda SK, 2015. Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaney MC, Rey FA, 2011. Class II enveloped viruses. Cell Microbiol 13, 1451–1459. [DOI] [PubMed] [Google Scholar]
- Ward BM, 2011. The taking of the cytoskeleton one two three: how viruses utilize the cytoskeleton during egress. Virology 411, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Reisen WK, 2010. Present and future arboviral threats. Antiviral research 85, 328–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingartl HM, Zhang S, Marszal P, McGreevy A, Burton L, Wilson WC, 2014. Rift Valley fever virus incorporates the 78 kDa glycoprotein into virions matured in mosquito C6/36 cells. PLoS One 9, e87385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EA, Sowa ME, Tan MJA, Jeudy S, Hayes SD, Santha S, Munger K, Harper JW, Howley PM, 2012. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proceedings of the National Academy of Sciences of the United States of America 109, E260–E267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichgers Schreur PJ, Kormelink R, Kortekaas J, 2018. Genome packaging of the Bunyavirales. Current Opinion in Virology 33, 151–155. [DOI] [PubMed] [Google Scholar]
- Wichgers Schreur PJ, Kortekaas J, 2016. Single-Molecule FISH Reveals Non-selective Packaging of Rift Valley Fever Virus Genome Segments. PLoS pathogens 12, e1005800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WC, Davis AS, Gaudreault NN, Faburay B, Trujillo JD, Shivanna V, Sunwoo SY, Balogh A, Endalew A, Ma W, Drolet BS, Ruder MG, Morozov I, McVey DS, Richt JA, 2016. Experimental Infection of Calves by Two Genetically-Distinct Strains of Rift Valley Fever Virus. Viruses 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WC, Kim IJ, Trujillo JD, Sunwoo SY, Noronha LE, Urbaniak K, McVey DS, Drolet BS, Morozov I, Faburay B, Schirtzinger EE, Koopman T, Indran SV, Balaraman V, Richt JA, 2018. Susceptibility of White-Tailed Deer to Rift Valley Fever Virus. Emerg Infect Dis 24, 1717–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.