

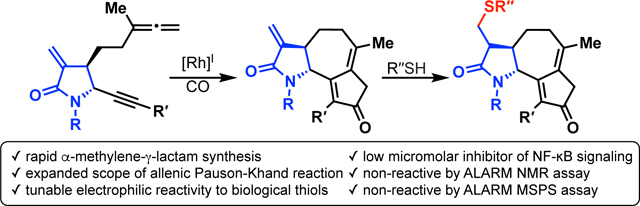
Abstract
α-Methylene-γ-lactones are present in ~3% of known natural products, and compounds comprising this motif display a range of biological activities. However, this reactive lactone limits informed structure-activity relationships for these bioactive molecules. Herein, we describe chemically tuning the electrophilicity of the α-methylene-γ-lactone by replacement with an α-methylene-γ-lactam. Guaianolide analogs having α-methylene-γ-lactams are synthesized using the allenic Pauson–Khand reaction. Substitution of the lactam nitrogen with electronically different groups affords diverse thiol reactivity. Cellular NF-κB inhibition assays for these lactams were benchmarked against parthenolide and a synthetic α-methylene-γ-lactone showing a positive correlation between thiol reactivity and bioactivity. Cytotoxicity assays show good correlation at the outer limits of thiol reactivity, but less so for compounds with intermediate reactivity. ALARM NMR and mass spectrometry peptide sequencing assays with the La antigen protein demonstrate that lactam analogs with muted non-specific thiol reactivities constitute a better electrophile for rational chemical probe and therapeutic molecule design.
Graphical Abstract
Introduction
α-Methylene-γ-lactones are present in ~3% of all known natural products.1 Given this abundance, and their validated biological function, it is curious that not a single compound with this motif has received FDA approval.2–4 This absence is due in part to the electrophilicity of the α,β-unsaturated carbonyl of the lactone that reacts rapidly and non-selectively with biothiols via a hetero-Michael addition, which limits the value of structure–activity relationship (SAR) studies and can result in off-target effects.1, 2, 5–8 We hypothesized that substituting the α-methylene-γ-lactone (X = O) of a compound predisposed to non-covalent binding to a particular protein target (Ki = koff/kon), with an α-methylene-γ-lactam (X = NR), would afford analogs with muted thiol reactivity (kinact) leading to useful SAR data (Figure 1).9 Our approach to tuning the electrophilic reactivity of the exocyclic methylene group utilizes the lactam nitrogen that is substituted with electronically different R groups.10–15 Using this R group on the nitrogen, which is distal from the reacting methylenyl group, serves as a way to parse out electronic effects from steric effects on kinact.
Figure 1.

Initial non-covalent binding specificity (Ki = koff/kon) of a covalent inhibitor to a target protein and second order rate constant (kinact) for covalent bond formation.
To test this hypothesis we turned to the guaianolides, a subclass of sesquiterpene lactone natural products, with rich bioactivity largely attributed to the α-methylene-γ-lactone.1–4, 16–18 Moreover, guaianolides have been shown to target the NF-κB p65 protein at Cys38, making the NF-κB signaling pathway an excellent model for testing the bioactivity of these novel α-methylene-γ-lactam-containing compounds.19–22 This assertion is further supported by results from our labs where several guaianolide analogs were prepared and tested for inhibition of NF-κB signaling.23, 24 Compound 1 represents one member from a series of guaianolide analogs differing in substitution at C14 and stereochemistry at C6, C7, and C8 (Figure 2A). All analogs from this series demonstrated high levels of inhibition of the NF-κB signaling pathway at low micromolar concentrations but with little evidence of SAR.24 Furthermore, the criticality of the α-methylene-γ-lactone for inhibition was demonstrated by loss of all inhibitory activity when the α-methylene group of one of these analogs was reduced to a methyl substituent.24
Figure 2.

Guaianolide analogs: potent inhibitors of NF-κB signaling. (A) Example structure of a guaianolide analog, where the series varies by the substitution at the C14 position, and the stereochemistry at the C6, C7, and C8 positions. (B) Retrosynthetic analysis suggests that lactam 3 can be prepared by an allenic Pauson–Khand reaction (APKR) of allene-yne 5 based upon precedent established for the preparation of 2 from 4.
Testing our hypothesis, where an electrophilic α-methylene-γ-lactone is replaced with an electronically tunable α-methylene-γ-lactam to control thiol reactivity, required synthetic access to lactam-containing guaianolide analogs. Previously, we demonstrated that the allenic Pauson–Khand reaction (APKR) could be used to prepare the carbocyclic skeleton of lactone 2 from allene-yne 4 tethered with an α-methylene-γ-lactone (Figure 2B).23, 25 Expanding the scope of the APKR to include allene-yne 5 equipped with an α-methylene-γ-lactam would provide rapid access to the molecularly complex and electronically tunable 5,7,5-fused ring system 3. Additionally, this APKR approach offers access to lactam analogs structurally similar to previously studied lactones, enabling direct comparisons of electrophilic reactivity towards thiols and inhibitory activity of the NF-κB signaling pathway.23, 24
Results and Discussion
Synthesis of α-Methylene-γ-lactam Guaianolide Analogs
Our approach to the 5,7,5-fused structure 6 was designed around the intramolecular APKR of lactam-containing allene-yne 7 (Scheme 1).23, 25 Functionalization of the nitrogen of α-methylene-γ-lactam 7 late in the synthetic sequence was deemed necessary as it minimizes potential reactivity issues imparted by an electrophilic group. Naturally occurring guaianolides typically possess a methyl group at C15; however, here we chose to incorporate aryl and silyl groups (R1=Ar or SiR3) at this position due to their electronic and steric versatility. The allenyl group of 7 should be available from the methyl ketone 8 using a 3-step reaction sequence involving a tertiary propargyl carboxylate.26, 27 In turn, access to the α-methylene-γ-lactam 8 should be possible by reaction of the imine 10, formed in situ from 3-phenyl- or 3-silyl-2-propynal and ammonium hydroxide, and allylboronate 9. While we have utilized a similar reaction for the preparation of the corresponding lactones, formation of lactam 8 by this approach will represent the first example of a consecutive allylboration and lactamization process involving the imine of a 2-alkynal.23, 28
Scheme 1.

APKR Approach to α-Methylene-γ-lactam Guaianolide Analogs
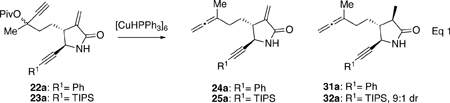
To test the feasibility of this APKR strategy for the preparation of α-methylene-γ-lactam guaianolide analogs, synthesis of allene-ynes 24a and 25a begins with alkynoate 11 prepared from 2,4-pentanedione (S1) in 3 steps on multigram scale (Scheme 2A) (See Supporting Information, SI). Conversion of alkynoate 11 to allylboronate 9 was accomplished by addition of 11 to a solution of methyl lithium, copper iodide, hexamethylphosphoramide (HMPA), and diisobutylaluminum hydride (DIBALH) followed by addition of chloromethyl pinacolboronate.29,30 This reaction was performed several times affording allylboronate 9 with an average yield of 81% yield and a 2:1 Z:E isomeric mixture that also included an alkenoate byproduct S4 (5–10%, see SI) resulting from protonation of the intermediate alkenyl metal species.23, 25, 30 Allylboronate isomers 9 and the alkenoate byproduct S4 were taken on as a mixture to the next step as chromatographic separation of the mixture resulted in a substantially reduced yield of the product. Addition of this mixture to 3-phenyl-2-propynal (12) and ammonium hydroxide afforded lactams 14a,b in 58% yield as a 5–2:1 mixture of the trans:cis isomers. Addition of mixture 9 to 3-triisopropysilyl-2-propynal (13) and ammonium hydroxide afforded lactams 15a,b in an average yield of 71% with a ratio of 3.5–2:1 for the trans:cis isomers. 3-Trimethylsilyl-2-propynal (R1=SiMe3) proved unstable to the lactamization reaction conditions, presumably due to loss of the trimethylsilyl group under the basic reaction conditions (not shown). These two allylboration/lactamization reactions represent the first examples of a 3-component process using 2-alkynals to prepare alkynyl functionalized α-methylene-γ-lactams.28, 31 We hypothesize that the (Z)-allylboronate reacts via Zimmerman–Traxler transition state 16 affording trans lactams 14a and 15a, while the minor (E)-allylboronate isomer reacts similarly (not shown) to afford cis lactams 14b and 15b (Scheme 2A).28 However, because the trans:cis lactam ratio (14a:14b, 4:1 and 15a:15b, 3.5:1) is higher than the Z:E ratio for the allyboronate precursor, it is likely that for the minor (E)-allylboronate isomer, the relatively small alkynyl group can adopt and react through an axial orientation as depicted by transition state 17. Alternatively, boat-like transition states have been invoked for secondary aldimines to explain mixture ratios.30, 32 These stereochemical assignments of the lactams were initially assigned by analogy to similar compounds and later confirmed by X-ray crystallography of the APKR product (vide infra).23, 25, 28 The alkenoate byproduct S4 could be readily separated from lactams 14a,b and 15a,b by column chromatography. Lactams 14a, 14b, 15a, and 15b were separated for characterization purposes, but in most cases the lactams were advanced through the synthetic sequence as mixtures of cis and trans isomers, then separated near the end of the sequence and only the trans lactam isomers were tested in biological assays.
Scheme 2. Synthesis of α-Methylene-γ-lactam Tethered Allene-ynes.

Reagents and conditions: (a) CuI, MeLi in Et2O, THF, −30 °C, 30 min; then toluene, HMPA, DIBALH in hexanes, −30 °C, 2 h; then 11, −20 °C, 5 h; then 2-(chloromethyl)-4,4,5,5-tetramethyl-1,3-dioxaborolane (PinBCH2Cl), −20 °C to rt, 16 h, 76% as a 3:1; Z:E mixture along with methyl (Z,E)-5-(2-methyl-1,3-dioxolan-2-yl)pent-2-enoate (S4), 10%; (b) 12 or 13, ammonium hydroxide, ethanol, rt, 16 h; (c) PPTS, acetone:water (15:1), reflux, 16 h; (d) ethynyl magnesium bromide, THF, 0 °C, 3 h; (e) scandium triflate, pivalic anhydride, CH3CN, rt, 16 h; (f) triphenylphosphine copper hydride hexamer (Stryker’s reagent), toluene, −10 °C, 2 h; (g) tetra-n-butyl ammonium fluoride, THF, 0 °C, 45 min; (h) sodium hydride, iodomethane, DMF, 0 °C to rt, 15 min; (i) acetic anhydride, Et3N, DMAP, CH2Cl2, 0 °C to rt, 3 h.
Next, removal of the ketal protecting group of 14a,b and 15a,b by acid-catalyzed hydrolysis using pyridinium para-toluenesulfonate (PPTS) in refluxing acetone and water (15/1, v/v) afforded keto lactams 18a,b in 83% yield and 19a,b in 73% yield. Both were afforded as a 4:1 mixture of trans:cis lactam isomers. Addition of ethynyl magnesium bromide to ketones 18a,b and 19a,b in THF at 0 °C gave tertiary propargyl alcohol 20a,b in 75% yield and 21a,b in 82% yield. Each was afforded as a ~1:1 diastereomeric mixture at the newly created stereocenter. Conversion of the tertiary hydroxyl group to pivalate 22a,b and 23a,b was carried out using pivalic anhydride and a substoichiometric quantity of scandium triflate (Sc(OTf)3, 0.4 equiv) at rt for 16 h. These conditions afforded chemoselective pivaloylation of the hydroxyl group over the secondary lactam nitrogen, whereas acetylation conditions (Ac2O, NEt3, DMAP) resulted in the N-acetylated product. At this point the cis and trans lactam isomers 22a and 22b were separated via column chromatography to afford a 61% yield of the trans isomer 22a and 16% yield of the cis isomer 22b, each as a 1:1 mixture of diastereomers at C10. Similarly, pivalates 23a (39%) and 23b (16%) were separated by column chromatography. Only the trans isomers were taken on for the remainder of the synthetic sequence.
Conversion of the propargyl pivalate group to a 3,3-disubstituted allene was investigated. Propargyl pivalate 22a was reacted with triphenylphosphine copper hydride hexamer (Stryker’s reagent) at −10 °C to afford a 33% yield of allene 24a.33 Propargylic pivalate 23a gave a 46% yield of desired allene 25a when subjected to Stryker’s reagent. Removal of the TIPS substituent from the alkyne terminus by treatment of lactam 25a with tetra-n-butyl ammonium fluoride (TBAF) provided terminal alkyne 26 in 79% yield. A competing reduction of the α-methylene group of the lactam during allene formation is the reason for the low yields. Progress towards solving this chemoselectivity challenge is discussed in more detail in the following section.
To examine the potential impact of the free NH of the secondary lactam on this reaction sequence, 14a (containing a small amount of cis lactam 14b) was converted to the corresponding tertiary lactam by the addition of sodium hydride and iodomethane (Scheme 2B). Removal of the ketal protecting group as described above afforded 27a in 73% yield. Addition of ethynyl magnesium bromide gave a 62% yield of 28a as a 7:1 trans:cis ratio and a 1:1 diastereomeric mixture at the newly formed stereogenic carbon. Conversion of 28a to the propargyl acetate 29a was accomplished in 54% yield. Reaction of propargyl acetate 29a with Stryker’s reagent gave a 53% yield of desired allene 30a with no evidence of reduced lactam methylene. Thus, formation of the allene was the only step in the reaction sequence impacted by the free NH of the secondary lactam. We presume that complexation of the less sterically hindered secondary lactam of 22a/23a with copper increases the electrophilic reactivity of the methylene group over that of the tertiary lactam of 29a.
Optimizing the Formation of 3,3-Disubstituted Allenes from Propargyl Pivalates
Efforts to increase chemoselectivity and yield for the transformation of propargyl pivalates 22a and 23a to allenes 24a and 25a involved changing the reaction temperature, time, scale, and equivalents and source of Stryker’s reagent (Eq 1, Table 1). Performing the reduction with 23a using a previously-opened, aged container of Stryker’s reagent at 0 °C in degassed toluene (0.04 M) with 2 equiv of water gave a 64% yield of 25a and only trace amounts of reduced methyl lactam 32a (Table 1, entry 1). However, when a newly opened container of Stryker’s reagent was used under identical conditions, a 60:40 ratio of allene 25a to methyl lactam 32a was obtained in 73% yield (entry 2). Lowering the equiv of Stryker’s reagent (0.8 equiv, new bottle) and temperature (−10 °C) provided a slightly improved ratio of 25a to 32a (63:25), but with unreacted starting material (entry 3). Decreasing the temperature further (−20 °C) with 0.9 equiv of Stryker’s reagent afforded 25a to 32a (80:20) in 58% yield with no starting material (entry 4). Performing the reaction on larger scale (220 mg) with 0.9 equiv Stryker’s reagent and −10 °C (conditions used in remaining entries) gave a 77:23 ratio of 25a to 32a in 78% yield (entry 5). Increasing the scale of the reaction to 440 mg provided a ratio of 72:19:9 of 25a:32a:23a in a 49% yield (entry 6). The phenyl substituted alkyne 22a afforded a 70:16 ratio of 24a to 31a in a 70% yield, but reduction of the alkyne to a cis alkene was observed and accounted for ~14% of the product mixture (entry 7).33 Decreasing the reaction time (1.5 h) led to a 56% yield of 24a and 31a in a 90:10 ratio with minimal reduction of the phenyl alkyne (entry 8).
Table 1.
Optimization of Allene Formation with Stryker’s Reagent ([CuHPPh3]6).
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | SM | Equiv Stryker’s | Temp (°C) | Time (h) | Total yield (%) | (24a/25a):(31a/32a):SM | Scale (mg) |
|
| |||||||
| 1 | 23a | 1.8 | 0 | 1.5 | 64 | 100:trace:0 | 110a |
|
| |||||||
| 2 | 23a | 1 | 0 | 1 | 73 | 60:40:0 | 110 |
|
| |||||||
| 3 | 23a | 0.8 | −10 | 2 | 66 | 63:25:12 | 110 |
|
| |||||||
| 4 | 23a | 0.9 | −20 | 2 | 58 | 80:20:0 | 110 |
|
| |||||||
| 5 | 23a | 0.9 | −10 | 2 | 78 | 77:23:0 | 220 |
|
| |||||||
| 6 | 23a | 0.9 | −10 | 2 | 49 | 72:19:9 | 440 |
|
| |||||||
| 7 | 22a | 0.9 | −10 | 2 | 70 | 70:16:0b | 110 |
|
| |||||||
| 8 | 22a | 0.9 | −10 | 1.5 | 56 | 90:10:0 | 122 |
SM = starting material
Reaction performed with a previously-opened aged container of Stryker’s reagent; all other reactions were performed with the newly opened container
14% of the total yield was a product resulting from reduction of the alkyne.
Thus, optimal conditions minimizing byproduct formation involved 0.9 equiv of Stryker’s reagent, 0.04 M solution of propargyl carboxylate in toluene, 2 equiv of water at −10 °C, and a reaction time of 1.5 to 2 h. Reaction of cis-lactams 22b or 23b with Stryker’s reagent gave lower yields (30–35%) of the corresponding allenes 24b or 25b (see SI) where the over-reduction product was afforded predominantly. A selective reduction of the exocyclic methylene of the unsaturated lactams of 22b and 23b may be due to one face of the cis-lactam isomers being more accessible to the copper hydride complex. Alternative conditions for the formation of 3,3-disubstituted allenes were examined but were not productive.34–36
Functionalization of the Lactam Nitrogen
Functionalization of the lactam nitrogen of 24a with electronically different groups was examined. Para-substituted aryl groups were selected so that any changes in electrophilicity of the α-methylene-γ-lactam would result from electronic contributions, to the degree that it is possible to separate electronic from steric contributions. A Buchwald–Hartwig coupling reaction was performed on lactam 24a with aryl iodides having an electronically neutral group (33, R3=H), electron withdrawing groups (34, R3=CN; 35, R3=CF3), and an electron donating group (36, R3=OMe) (Scheme 3).37, 38 N-Arylation of allene-yne 24a with iodobenzene (33) gave N-phenyl allene-yne 37 in 44% yield using cesium carbonate as the base; a lower yield (27%) was observed with potassium carbonate. N-Arylation of 24a with 4-iodobenzonitrile (34), 4-iodobenzotrifluoride (35), or 4-iodoanisole (36) provided 38, 39, or 40 in 42%, 47%, or 63% yield, respectively (Eq 2). These same conditions were used to N-arylate 24a with 2-iodothiophene (41) to afford the N-heteroaryl lactam 42 with an average yield of 59% (Eq 3). The moderate yields for the N-arylation and -heteroarylation products are attributed to the sterically hindered nature of the amide nitrogen.38 Two additional electron withdrawing groups were introduced by treatment of 24a with sodium hydride and TsCl to give N-Ts lactam 43 in 29% yield (Eq 4), and lactam 24a was converted to a carbamate using di-tert-butyl dicarbonate to give the boc-functionalized lactam 44 with an average yield of 47% (Eq 5). These N-functionalized lactams provide a spectrum of electronically disparate groups informing on the impact of electrophilic reactivity of the methylene lactam.
Scheme 3. Functionalization of the Lactam Nitrogen.

(a) copper iodide (CuI, 20 mol%), N,N’-dimethylethylenediamine (40 mol%), cesium carbonate (Cs2CO3), ArI, toluene, 80 °C, 20 h; (b) sodium hydride (NaH, 60% dispersion), para-toluenesulfonyl chloride (p-TsCl), DMF, 0 °C 2 h; (c) di-tert-butyl dicarbonate (boc2O), N,N-dimethylaminopyridine (DMAP), Et3N, CH2Cl2, 0 °C to rt, 2 h.
Examining the Feasibility and Scope of the Allenic Pauson–Khand Reaction
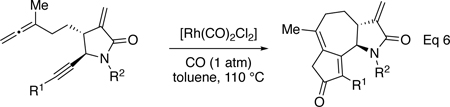
With a number of α-methylene-γ-lactam tethered allene-ynes in hand, the feasibility of the APKR was tested. Because these substrates have a methyl-substituted allene, we used conditions previously developed in our group that minimize dimer formation by slowly adding the allene-yne to the Rh(I) catalyst.39 First, allene-yne 24a was diluted in toluene and added dropwise over 1 h to rhodium biscarbonyl chloride dimer ([Rh(CO)2Cl]2, 10 mol%) in toluene under carbon monoxide (1 atm) at 110 °C (Eq 6). Heating the reaction for an additional 30 min after the addition of the allene-yne afforded lactam 45 in 72% yield (Table 2, entry 1). The structure of this compound was confirmed by X-ray crystallography (Figure 3). Reaction of TIPS-substituted alkyne 25a using similar conditions afforded 46 in 46% yield (entry 2). The reduced yield for 46 compared to lactam 45 is attributed to a developing A1,3 interaction between the large TIPS group on the terminus of the alkyne and the lactam during cyclization. This steric argument is further supported by a longer reaction time for 25a compared to 24a (3 h vs 1.5 h). In addition, a structurally related α-methylene-γ-lactone-tethered allene-yne with a less bulky trimethylsilyl group on the alkyne underwent an APKR in high yield with a reaction time similar to a phenyl substituted alkyne.25 A terminal alkyne was tolerated in the APKR, as evidenced by the reaction of allene-yne 26 to provide lactam 47 in 53% yield (entry 3). Next, we examined the impact of the substitution on the lactam nitrogen on the APKR yield and reaction time. Allene-yne 30a with an N-methyl group afforded the APKR product 48 in 75% yield (entry 4), showing that the free NH has minimal impact on the efficiency of the APKR (compare entries 1 and 4). Allene-ynes 37-40, where the lactam nitrogen was substituted with an N-phenyl, N-4-cyanophenyl, N-4-trifluoromethyl phenyl, or N-4-methoxyphenyl gave yields of 67, 72, 70, and 77% of tricyclic structures 49-52, respectively (entries 5–8). Under these conditions, neither the reaction time nor the yield was significantly impacted by the electronic nature of the aryl group on the lactam nitrogen. The N-2-thiophenyl substituted allene-yne 42 provided lactam 53 in 67% yield (entry 9). The APKR of N-Ts and N-boc allene-ynes 43 and 44 gave lactams 54 and 55 in yields of 71% and 40%, respectively (entries 11–12). The lower yield of lactam 55 is attributed to the thermal instability of the boc group at 110 °C.40
Table 2.
APKR with α-Methylene-γ-lactam Tether.
| ||||
|---|---|---|---|---|
| Entry | Allene-yne | R1 | R2 | APKR yield %a |
|
| ||||
| 1 | 24a | Ph | H | 45 (72)b,c |
|
| ||||
| 2 | 25a | TIPS | H | 46 (46)b,d |
|
| ||||
| 3 | 26 | H | H | 47 (53)b |
|
| ||||
| 4 | 30a | Ph | Me | 48 (75)e |
|
| ||||
| 5 | 37 | Ph | Ph | 49 (67)b,c |
|
| ||||
| 6 | 38 | Ph | 4-CN-C6H4 | 50 (72)e |
|
| ||||
| 7 | 39 | Ph | 4-CF3-C6H4 | 51 (70)b |
|
| ||||
| 8 | 40 | Ph | 4-OMe-C6H4 | 52 (77)b |
|
| ||||
| 9 | 42 | Ph | 2-thiophenyl | 53 (67)b |
|
| ||||
| 10 | 43 | Ph | Ts | 54 (71)b,c |
|
| ||||
| 11 | 44 | Ph | boc | 55 (40)b,c |
Dropwise addition of allene-yne to rhodium catalyst over 1 h, then heated for an additional 30 min
Average yield for two runs
Similar yields were obtained when allene-yne was added in a single portion.
Reaction was heated for 3 h upon completion of allene-yne addition
Yield based upon one run.
Figure 3.

Crystal structure of APKR product 45.
Next, we tested the impact of the α-methylene group on the efficiency of the APKR using allene-yne α-methyl-γ-lactams 56 and 57 (Scheme 4). Trans, trans-lactam 56 (8:1 dr at C11) underwent the APKR in 1.5 h to afford 58 in 78% yield as 8:1 mixture of diastereomers (Eq 7). The APKR of α-methyl-γ-lactam 56 afforded a moderately higher yield than that of the α-methylene-γ-lactam 26, each possessing a terminal alkyne (78% vs 62%, respectively). Cis, trans-lactam 57 afforded the APKR product 59 in 77% yield as a single diastereomer (Eq 8). This latter reaction was performed by the addition of the allene-yne to the rhodium catalyst in a single portion. The stereochemistry at C11 for both 58 and 59 was assigned by comparing calculated coupling constants to experimental values.
Scheme 4.

APKR with methyl lactams.
In summary, the reaction of allene-ynes shown in Table 2 represent the first examples of an APKR with an α-methylene-γ-lactam tether. Substituents on the alkyne terminus had a moderate influence on the yield and reaction time (Table 2, entries 1–3). The groups on the nitrogen of the lactam had little impact on the yield or reaction time (entries 4–11) demonstrating that the APKR is tolerant of N-alkyl, N-aryl (neutral, electron donating or withdrawing), N-thiophenyl, N-sulfonyl, N-carbamate, and N-H α-methylene-γ-lactam tethers. Comparison of APKR reaction times of 56 and 57 and yields for lactams 58 and 59 suggest that the α-methylenyl group has some influence on the yield when compared with α-methyl lactams.
Electrophilic Reactivity of α-Methylene-γ-Lactams Towards Thiols
A subset of these newly synthesized α-methylene-γ-lactams was reacted with excess cysteamine to obtain pseudo-first order reaction kinetics as a way to quantify the electrophilic reactivity of these analogs towards a biologically-relevant thiol.15 Given that 13C NMR chemical shifts for the Cβ resonance of the methylene group can be a good predictor of electrophilicity in some systems,12 we selected lactams 45 (R1 = H, 13Cβ δ = 115.3); 49 (R1 = Ph, 13Cβ δ = 117.1); 51 (R1 = 4-CF3-C6H4, 13Cβ δ = 118.2); 52 (R1 = 4-MeO-C6H4, 13Cβ δ = 116.7); 54 (R1 = Ts, 13Cβ δ = 121.4); 60 (R1 = Ac, 13Cβ δ = 120.6) that show a range of 13Cβ chemical shifts. Cysteamine was selected as the biologically-relevant thiol nucleophile given the strong preference for the sulfhydryl group reacting over the amino group at pH 7.4 and its compatibility with organic solvents.41, 42 Each reaction was performed by adding a solution of the lactam and cysteamine (15 equiv) in CDCl3 to an NMR tube at rt (22 °C). The reaction progress was monitored by 1H NMR with spectra acquired at regular intervals, and the reaction tube held in the autosampler between measurements.42 Reaction of α-methylene-γ-lactams 45 and 49 with cysteamine each shows a half-life of 2.1 d (entries 1–2, Table 3). The trifluoromethyl aryl lactam 51 reacted with cysteamine with a half-life of 9.8 h, and the methoxy aryl lactam 52 reacted with a half-life of 8.5 d (entries 3 and 4). N-Ts-substituted lactam 54 reacted completely in less than 10 min; attempts were not made to determine half-lives for this lactam (entry 5). Lactam 60 reacted with cysteamine with a half-life of 5 min (entry 6). A chemoselective addition to the lactam α-methylenyl group is supported by 1H NMR showing the complete disappearance of alkenyl proton resonances (see SI). Finally, a structurally related lactone S17 (see SI for structure) reacted with cysteamine with a half-life of 7 min (entry 7). The α-methylene-γ-butyrolactone parthenolide (PTL) was previously shown to react completely with cysteamine in less than 5 min (entry 8).42
Table 3.
Reaction of Cysteamine with α-Methylene-γ-lactams.
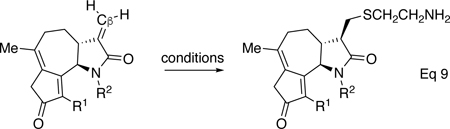
Structural confirmation of thiol adducts was thwarted by their instability to column chromatography. Thus, N-methyl lactam 48 was reacted with tert-butyl thiol to form the thiol adduct S11 after 30 min in 23% yield (dr 1.6:1). The major diastereomer was assigned by analogy to methyl lactam 58, where calculations predict this isomer to be thermodynamically more stable by ~2 kcal/mol.43 Thiol adduct S11 showed a single equivalent of thiol was added to the exocyclic alkene of 48.
In summary, groups on the lactam nitrogen greatly impact the reactivity of the α-methylene-γ-lactams towards cysteamine where reaction half-lives ranged from days, for electron-neutral and -donating groups, to minutes for electron-withdrawing groups. The chemical shift for the Cβ in the 13C NMR is an excellent predictor of reactivity; a slow rate of reaction is observed for compounds with a Cβ chemical shift in the range of 115.3–117.1 and requires days to complete one reaction half-life. For compounds with Cβ chemical shifts in the range of 120.6–121.4, a reaction half-life is achieved in minutes. Notably, lactam 51, N-substituted with a trifluoroaryl group and a Cβ chemical shift of 118.2, showed moderate reactivity with a half-life measured in hours.
NF-κB Bioactivity and Cytotoxicity of α-Methylene-γ-Lactams
Several natural products and synthetic compounds containing α-methylene-γ-lactones are known to inhibit the NF-κB pathway; however, these highly reactive lactones limit the availability of informed SAR as they react readily with accessible thiols.5, 19, 45–50 We have shown with our 1H NMR studies that the electrophilic reactivity of α-methylene-γ-lactams towards thiols can be modulated depending upon the electronic character of N-substituents; this tunable electrophilicity should contribute to a better understanding of the role of covalent adduct formation to inhibition of the NF-κB signaling pathway. To characterize the biological utility of the α-methylene-γ-lactams, we performed cellular NF-κB inhibition assays with a representative subset of the synthesized compounds and benchmarked activities to the known α-methylene-γ-lactone NF-κB inhibitor PTL.50, 51 We used two NF-κB reporter cell lines for our studies: A549 cells bearing a stably transfected NF-κB-driven luciferase reporter gene and HEK293 cells containing a stably transfected NF-κB-driven secreted alkaline phosphatase (SEAP) reporter gene. These orthogonal assays with different readouts were used to corroborate data to ensure inhibition was not occurring at the enzymatic readout level (i.e. direct inhibition of luciferase or SEAP). Direct inhibition of luciferase by sesquiterpene lactones at high micromolar concentrations has been reported.52 Our assays also inform how different substituents containing electron-withdrawing, electron-donating, or electronically neutral functional groups attached to the lactam nitrogen affect inhibition of the NF-κB pathway. Select compounds were incubated with cells for 30 min at 5–20 μM for the luciferase reporter assay and 1–7.5 μM for the SEAP reporter assay, before induction with 15 ng/mL or 22.5 ng/mL TNF-α, respectively, for 8 h and subsequent read-out of the reporter gene (Figure 4).53
Figure 4.

Modulation of canonical NF-κB signaling by α-methylene-γ-lactams. (A) Compounds were tested at 20, 10, and 5 μM and NF-κB signaling was induced with TNF-a (15 ng/mL) in A549 cells containing a NF-κB driven luciferase gene. (B) Compounds were tested at 7.5, 5.0, 2.5, and 1.0 μM and NF-κB signaling was induced with TNF-a (22.5 ng/mL) in HEK293 cells containing a NF-κB driven secreted alkaline phosphatase (SEAP) gene. All wells in both assays were induced with TNF-α except for non-induced (N) control wells. Relative NF-κB activities (referenced to the induced, I, control set to 100%) are shown in dark colors. Accompanying cellular cytotoxicity measurements were made using Alamar Blue viability dye and are shown behind NF-κB inhibition in light colors. Cytotoxicity was normalized to the induced control, which was set at 100%. Columns marked with NT (non-toxic) indicate instances in which NF-κB activity (dark bars) obstructs the cellular cytotoxicity (light bars). Occluded values range from 88–101% (Table S1). Values shown are mean ± S.D. for n ≥ 3 biological replicates. PTL = parthenolide. See SI Tables S1 and S2 for numerical values.
Compounds with no substituents on the lactam nitrogen (45 and 47) did not inhibit NF-κB signaling at 20 μM, the highest concentration tested; a result supported by the cysteamine study showing low thiol reactivity. Lactams 48, 49, and 52 containing a N-methyl, N-phenyl, and N-4-methoxy phenyl substituents, respectively, also showed no inhibitory activities. However, compounds with electron-withdrawing substituents did inhibit NF-κB activation, with lactam 54 containing a N-Ts substituent displaying potent activity with complete inhibition of induced NF-κB signaling at 10 μM in the luciferase reporter assay and at 7.5 μM in the SEAP reporter assay. Cytotoxicity was observed with 54 at 10 μM treatment in A549/NF-κB-luc cells (64% cell viability at 8 h), although less toxicity for 54 was observed in HEK293/NF-κB-SEAP cells at the concentrations tested (82% cell viability at 7.5 μM). Derivatives 51 and 60 with N-(4-trifluoromethyl)phenyl and N-acetyl groups, respectively, showed similar inhibitory activities in the luciferase reporter assay (21% [51] and 28% [60] residual NF-κB activity at 20 μM, as well as 62% [51] and 59% [60] residual NF-κB activity at 10 μM). Both compounds maintained inhibitory activity in the SEAP reporter assay, but derivative 60 was more potent (44% [51] and 19% [60] residual NF-κB activity at 7.5 μM). In A549/NF-κB-luc cells, significant toxicities were not observed for lactams 51 and 60 (88% [51] and 82% [60] cellular viability at 20 μM). In HEK293/NF-κB-SEAP cells, lactam 51 was non-toxic at all concentrations; conversely, lactam 60 demonstrated toxicities at 7.5 μM (69% cell viability) and 5.0 μM (76% cell viability) treatments. In comparison, a lactone analogue of 45 was previously reported to reduce NF-κB activity completely in A549/NF-κB-luc cells when tested at 20 μM, with limited cytotoxic effects.23 The compounds were also benchmarked against the α-methylene-γ-butyrolactone parthenolide (PTL), a known NF-κB inhibitor. Previously published results show PTL at 10 μM concentration reduces NF-κB to 53% residual activity in the A549/NF-κB-luc assay with no cytotoxicity.23 These data are consistent with HEK293/NF-κB-SEAP assay results, which show a 20% residual NF-κB activity with a moderate level of cytotoxicity at 7.5 μM. From these results we conclude that α-methylene-γ-lactams containing electron-withdrawing substituents increase inhibitory activity toward the NF-κB pathway in both assays compared to electronically neutral and electron-donating substituents, which display minimal or no effect on NF-κB inhibition.
To further investigate the bioactivity of our α-methylene-γ-lactam derivatives, their cytotoxicity was assessed in a model non-cancerous cell line for a longer treatment duration (Table 4). Vero is a kidney epithelial cell line derived from an African green monkey that is commonly used as a standard for cytotoxicity in healthy cells.54, 55 Lactam derivatives were incubated with cells for 48 h at concentrations ranging from 0.1–200 μM and cellular viability was measured by Alamar Blue assays.53 Derivative 47 showed little or no inhibition of the NF-κB pathway in either of the two reporter assays, and displayed low cytotoxicity. Compounds 49 and 52, which had little NF-κB inhibitory activity, displayed cytotoxicity to Vero cells (IC50 = 21.9 and 44.0 μM, respectively). Lactams 51 and 60, two of the more potent derivatives in the NF-κB inhibitory assays that were also non-toxic to the host cell lines, were cytotoxic to Vero cells (51: IC50 = 15.7 μM, 60: IC50 = 7.8 μM); however, 51 was approximately 2-fold less toxic than 60 and constitutes the most compelling lactam for further mechanistic and structural optimization studies. A more comprehensive evaluation of the toxicity of 51 will be pursued in future in vivo pharmacokinetics studies in mice.
Table 4. Cellular Toxicity of Lactams to Vero Cells.
Cellular viabilities were measured by Alamar Blue viability dye. IC50 values shown are mean ± S.D. for n ≥ 3 biological replicates.
| Compound | IC50 (μM) |
|---|---|
| 47 | > 200 |
| 49 | 21.9 ± 1.6 |
| 51 | 15.7 ± 1.2 |
| 52 | 44.0 ± 5.7 |
| 60 | 7.8 ± 0.9 |
| S17 | 6.2 ± 1.0 |
A comparison of the half-life for the reaction of an α-methylene-γ-lactam with cysteamine and NF-κB inhibition showed good correlation between thiol reactivity of the lactam and its inhibition (Table 3). For example, lactams 45, 49, and 52 displayed low thiol reactivity towards cysteamine with t1/2 = 2.1 d, 2.1 d, and 8.5 d, respectively, and each demonstrated no NF-κB inhibition. Lactam 51 displayed intermediate thiol reactivity (t1/2 = 9.8 h) showed 62% residual NF-κB activity for the luciferase assay (10 μM) and 44% for the SEAP reporter assay (7.5 μM). These inhibitory values were balanced by less overall cytotoxicity in the Vero model (IC50 = 15.7 μM) in comparison to lactam 60 (t1/2 = 5 min for cysteamine reactivity), which was 2-fold more cytotoxic to Vero cells (IC50 = 7.8 μM), yet similarly potent towards inhibiting NF-κB signaling (59% residual NF-κB activity in the luciferase assay [at 10 μM] and 19% residual NF-κB activity for the SEAP reporter assay [at 7.5 μM]). As expected, lactam 52 displaying low thiol reactivity also showed lower relative cytotoxicity (IC50 = 44.0 μM).
Measuring Thiol-Reactivity of Lactam Inhibitors with ALARM NMR
To determine the thiol-reactivity of this series of lactam compounds in a proteinaceous environment, we turned to ALARM NMR (A La Assay to detect Reactive Molecules by Nuclear Magnetic Resonance), an assay using the La antigen, which contains a cysteinyl group (C245) that is highly reactive towards electrophilic compounds.56–58 This bioassay involves monitoring the changes in the chemical shifts of 13C-labeled methyl groups (L249, L294, and L296) by 2D 1H-13C HMQC that occur when C245 is modified. The α-methylene-γ-lactam 51 was selected for this study as it shows moderate reactivity with cysteamine when compared to the other lactams (see Table 3) and is the most balanced lactam with respect to NF-κB inhibitory activity and cellular cytotoxicity. Reaction of excess 51 with 13C-labeled La antigen at 37 °C for 90 min in the presence and absence of dithiothreitol (DTT, 20 mM) resulted in no changes in the HMQC spectra, similar to that seen for the negative control compound, fluconazole (Figure 5). Thus, lactam 51 is characterized as ALARM NMR negative and non-reactive, as this experiment provides compelling evidence that the La antigen protein does not react with the α-methylene-γ-lactam. Reaction of the analogous α-methylene-γ-lactone S17 shows high reactivity with the La antigen thiol, similar to that seen for the positive control compound, CPM (N-[4-(7-diethylamino-4-methylcoumarin-3-yl)phenyl]maleimide). This latter result demonstrates that the La protein conformation is perturbed as evidenced by the disappearance of the characteristic chemical shifts of 13C-labeled methyl groups of L249, L294, and L296 in the absence of DTT (Figure 5). Therefore, S17 reacts with the La antigen protein and is characterized as ALARM NMR positive and reactive. These experiments support the premise that α-methylene-γ-lactam-containing compounds may react discriminately with proteins containing cysteine residues, whereas the α-methylene-γ-lactones have the propensity to react more indiscriminately.
Figure 5.

ALARM NMR thiol-reactivity assays. Shown are the 1H-13C HMQC spectra of key 13C-labeled methyl groups of the La antigen protein incubated with DMSO; CPM (N-[4-(7-diethylamino-4-methylcoumarin-3-yl)phenyl]maleimide), positive thiol-reactive compound control; Fluconazole, negative thiol-reactive and aggregation compound control; α-methylene-γ-lactone S17; and α-methylene-γ-lactam 51. Compounds tested at 400 μM final concentrations, with a La antigen protein concentration of 50 μM; spectra for S17 and 51 are from the t = 9 h timepoint, spectra for the DMSO, CPM, and fluconazole spectra are representative.
Measuring Thiol-Reactivity and Reversibility of α-Methylene-γ-Lactam 51 and α-Methylene-γ-Lactone S17 Using Mass Spectrometry Peptide Sequencing
To corroborate the thiol reactivity properties of lactam 51 and lactone S17, a novel ALARM MSPS (A La Assay to detect Reactive Molecules by Mass Spectrometry Peptide Sequencing) assay of the La antigen was developed, offering a complementary assay to ALARM NMR. There are two possible thiol adduction sites on the La antigen: C232 and C245. Consistent with previously reported literature and with our own experiments, C245 is the more reactive cysteine (further discussed in the SI).56 Although the La antigen has been previously analyzed by mass spectrometry (MS), whole-protein analysis was performed, which does not distinguish covalent adduction between C232 and C245.56 In fact, whole-protein MS studies with La antigen and electrophilic compounds revealed both single and double adducted products in the same sample, supporting the hypothesis that there is a difference in thiol reactivity between C232 and C245. Therefore, we turned to MS analysis of tryptic peptides of the La antigen to characterize only those compounds that adduct C245, which is more reactive. Moreover, our new approach, ALARM MSPS, provides a method for evaluation of compounds that may function as reversible covalent inhibitors. Recently, a similar assay with isotopic iodoacetamide was reported to study protein dynamics, which supports our approach.59
The experimental workflow of the ALARM MSPS assay involves the following steps (Figure 6, panel A): 1) Incubation of the La antigen, containing the reactive cysteine C245, with test compounds 51 or S17; 2) Addition of iodoacetamide (IAD) to carbamidomethylate cysteine residues that were not adducted; 3) Protein denaturation; 4) Incubation with “heavy” d4-IAD to distinguish between two possible reactions, one where C245 did not react with an electrophile (labeled with IAD), versus the other where the adducted C245 of the La antigen then underwent a retro-hetero-Michael addition under the denaturation conditions (where the liberated C245 is subsequently adducted with heavy d4-IAD); 5) Digestion of adducted antigen with trypsin; and 6) MS/MS analysis of the adducted peptide FSGDLDDQTCR containing the C245 residue. (For information regarding the peptide containing C232, see SI.)
Figure 6.

ALARM MSPS assay analysis of non-specific compound adduction to La antigen C245. (A) Experimental workflow to identify thiol reactivity and reversible/irreversible covalent binding of compounds to C245 of the La antigen. Compounds added at 50 mM final concentration, La antigen final concentration of 30 μM, incubated for 1 h at room temperature in the dark. (B) Heat map indicating the number of samples that afforded IAD-adducted peptide FSGDLDDQTC245R. Three biological replicates for each compound were run in technical triplicate. Fluconazole was used as a non-reactive negative control and CPM was used as a thiol-reactive positive control, as in the ALARM NMR assay. Data on the additional peptides analyzed containing C232 can be found in the SI. aThe C245 peptide was not identified in one technical replicate of one of the biological replicates, but did appear as IAD-adducted in the other two technical replicates of that sample. Therefore, the biological replicate was considered positive for IAD adduction. bThe FSGDLDDQTC245R peptide was not observed in one biological replicate, likely due to low peptide abundance since expected d2-IAD adductions were clearly observed in other biological replicates, as well as for C232-containing peptides (see Table S12).
Subjecting lactone S17 to the ALARM MSPS assay afforded only the heavy (d2-IAD) C245 adduct and none of the IAD adduct (Figure 6B, green color), providing support that α-methylene-γ-lactone S17 reacted with C245 but undergoes a retro-Michael addition upon protein denaturation. The same results were obtained for the positive control experiment using CPM, where only the d2-IAD-C245 adduct was observed. In contrast, α-methylene-γ-lactam 51 showed only IAD-labeled C245 (Figure 6B, red color) indicating that 51 either does not react with C245 or reacts with very slow kinetics. Similar results were observed for the negative control compound, fluconazole. This newly developed ALARM MSPS assay provides compelling data to support that α-methylene-γ-lactam-containing compounds may react discriminately with cysteinyl-containing proteins, whereas the α-methylene-γ-lactones react more indiscriminately. Our data is consistent with previous ALARM NMR profiling of the covalent inhibitor of Bruton’s tyrosine kinase, Ibrutinib, which also tests as ALARM NMR negative.58 Therefore, although compounds may engage protein targets covalently, molecules with less reactive electrophiles, such as 51, can be identified with ALARM-based assays.
Conclusions
In this report, the scope of the APKR was expanded to allene-ynes tethered by an α-methylene-γ-lactam, and the 3-component allyboration/lactamization reaction sequence extended to 2-propynals. The electrophilic reactivity of α-methylene-γ-lactams towards thiols in the hetero-Michael addition reaction is impacted by the electronic nature of the group on the nitrogen of the lactam. For example, substituents on the lactam nitrogen of the 5,7,5-fused ring system of the APKR products can be used to control thiol reactivity (kinact) of the α-methylene-γ-lactam where electron-neutral and -donating N-substituents are slow to react with cysteamine, whereas lactams with electron-withdrawing N-substituents are faster to react. This ability to tune the electrophilic reactivity towards thiols contrasts with α-methylene-γ-lactone-containing compounds, where the electrophilic reactivity is high and cannot be readily tuned. We have shown that α-methylene-γ-lactam guaianolide analogs function as small molecule regulators of the NF-κB signaling pathway with reasonable cellular toxicity. The NF-κB inhibitory activity for the lactam analogs was found to positively correlate with thiol reactivity. Cytotoxicity shows a positive correlation at the outer most limits of thiol reactivity. Further studies evaluating structure activity relationships of the α-methylene-γ-lactams described herein having intermediate thiol reactivity are warranted. Finally, because guaianolides are well-known NF-κB inhibitors, these proof-of-concept studies show that modulating the thiol reactivity profile is beneficial from an inhibitor optimization standpoint. Further, these findings support our hypothesis that NF-κB signaling is an ideal system for evaluating an approach where an electronically tunable methylene lactam combined with a small molecule scaffold predisposed to protein-target binding can afford discriminant covalent inhibitors. Finally, these results inform our next step to design and synthesize electronically tuned lactam analogs for SAR studies, which are expected to have enhanced potential as lead compounds for drug design.
Experimental Section
Chemistry.
Commercially available compounds were used as received unless otherwise noted. Dichloromethane (CH2Cl2), tetrahydrofuran (THF), and diethyl ether (Et2O) were purified by passing through alumina using the Sol-Tek ST-002 solvent purification system. Triethylamine was distilled from calcium hydride (CaH2) and stored over 4 Å molecular sieves. Acetic anhydride (Ac2O) was shaken with phosphorus pentoxide (P2O5), decanted, fractionally distilled from anhydrous potassium carbonate (K2CO3) and stored over 4 Å molecular sieves. Hexamethylphosphoramide (HMPA) was vacuum distilled from CaH2 and stored over 4 Å molecular sieves. Deuterated chloroform (CDCl3) was stored over anhydrous K2CO3. All designated temperatures are bath temperatures unless specified otherwise. All reactions are performed under an atmosphere of nitrogen unless indicated otherwise. Silica gel (40–63 μm particle size, 60 Å pore size) purchased from Sorbent Technologies is used for the purification of compounds by flash chromatography. TLC analyses were performed on Silicycle SiliaPlate G silica gel glass plates (250 μm thickness). 1H NMR and 13C NMR spectra were recorded on Bruker AVANCE 300 MHz, 400 MHz, or 500 MHz spectrometers. Spectra were referenced to residual chloroform (7.26 ppm, 1H, 77.16 ppm, 13C). Chemical shifts are reported in ppm, multiplicities are indicated by s (singlet), br (broad signal), d (doublet), t (triplet), q (quartet), quint (quintet), and m (multiplet), dt (doublet of triplets). Coupling constants, J, are reported in Hertz (Hz). All NMR spectra were obtained at rt unless otherwise specified. IR spectra were obtained using a Nicolet Avatar E.S.P. 360 FT-IR. ESI mass spectroscopy was performed on a Waters Q-TOF Ultima API, Micromass UK Limited high-resolution mass spectrometer. The purity of representative final compounds was checked by HPLC and ranged from 91–99% (see Table S11).
Hex-5-yn-2-one (S2):
Compound S2 was synthesized according to a literature procedure.60 A flame-dried, 250-mL, single-necked, round-bottomed flask, equipped with a magnetic stir bar, septum, and nitrogen inlet needle was charged with 2,4-pentanedione (S1, 30 mL, 290 mmol) and ethanol (120 mL, 2.2 M). Propargyl chloride (37 mL, 260 mmol) was added via syringe followed by potassium carbonate (48 g, 350 mmol) in a single portion. The septum was removed, and a reflux condenser equipped with a septum and nitrogen inlet needle was attached. The reaction was heated to reflux (oil bath temperature 85 °C) for 24 h. Upon completion of the reaction as observed by TLC, the mixture was cooled to rt and filtered via vacuum filtration to remove the solids. The solid was washed with ethyl acetate. Ethyl acetate and ethanol were removed by simple distillation at atmospheric pressure. The residue was diluted with diethyl ether (100 mL), transferred to a separatory funnel, washed with deionized water (50 mL), then brine (50 mL), dried over magnesium sulfate, gravity filtered, and concentrated by simple distillation at atmospheric pressure. The residue was further purified by simple, vacuum distillation (45 mmHg, 85–90 °C) to give 12.5 g of product in a 49% yield. The spectral data matched literature values.60
1H NMR (300 MHz, CDCl3): δ 2.67, (t, J = 7.2 Hz, 2H, CH2), 2.42 (dt, J = 2.7, 7.1 Hz, 2H, °CCH2), 2.16 (s, 3H, CH3), 1.93 (t, J = 2.7 Hz, 1H, °CH); 13C NMR (100 MHz, CDCl3) δ 206.3 (C=O), 82.7, 68.6, 42.0, 29.8, 12.8; TLC Rf = 0.46 (25% EtOAc/hexanes) [silica gel, UV, KMnO4].
2-(But-3-ynyl)-2-methyl-1,3-dioxolane (S3).
Following a procedure analogous to that previously reported,61 a flame-dried, 200-mL, single-necked, round-bottomed flask equipped with a magnetic stir bar was charged with hex-5-yn-2-one (S2, 12.5 g, 131 mmol), benzene (130 mL), ethylene glycol (8.8 mL, 160 mmol), and p-toluenesulfonic acid (0.497 g, 2.61 mmol). The flask was equipped with a Dean-Stark trap that was attached to a condenser. The solution was heated at reflux (100 °C). After 15 h, TLC showed complete consumption of the starting material. The solution was allowed to cool to rt, diluted with diethyl ether (80 mL), transferred to a separatory funnel, washed successively with saturated sodium bicarbonate (100 mL) and brine (100 mL). The organic layer was dried over magnesium sulfate, gravity filtered, and diethyl ether and benzene were removed by simple distillation at atmospheric pressure. The residue was purified by vacuum distillation at 30 mmHg. Two fractions were collected: the first contained benzene with trace amounts of product S3 (bp = 40–50 °C, 30 mmHg); the second contained product with less than 10% benzene (bp = 113–120 °C, 30 mmHg, 13.0 g, 71% yield), as determined by integration of the benzene resonance at 7.19 ppm and terminal alkyne resonance at 1.87 ppm. 1H NMR (300 MHz, CDCl3): δ 3.96–3.86 (m, 4H, OCH2CH2O), 2.25 (dt, J = 2.7, 7.5 Hz, 2H, °CCH2), 1.92–1.88 (m, 2H, CH2), 1.87 (s, 1H, °CH), 1.30 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 109.2, 84.5, 67.9, 65.0 (2C), 38.1, 23.9, 13.3; TLC Rf = 0.55 (25% EtOAc/hexanes) [silica gel, UV, KMnO4].
Methyl 5-(2-methyl-1,3-dioxolan-2-yl)pent-2-ynoate (11):
Following a procedure analogous to that previously reported,62 a flame-dried, 200-mL, single-necked, round-bottomed flask equipped with a magnetic stir bar, septum, and nitrogen inlet needle, was charged with alkyne S3 (2.10 g, 15.0 mmol) and tetrahydrofuran (75 mL) then cooled to −78 °C. n-Butyl lithium (1.6 M in hexanes, 11.3 mL, 18.0 mmol) was added dropwise via syringe. Upon completion of addition the reaction was maintained at −78 °C for an additional 30 min. Methyl chloroformate (1.5 mL, 19.5 mmol) was added dropwise via syringe and maintained at −78 °C for an additional 30 min then warmed to rt. After 3 h, TLC showed complete disappearance of the starting material. Saturated aqueous ammonium chloride (30 mL) was added, and the solution was transferred to a separatory funnel. The layers were separated, and the aqueous layer was extracted with diethyl ether (3 × 50 mL). The organic layers were combined, washed with brine (50 mL), dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation (30 °C), and purified by silica gel flash column chromatography eluting with 20% diethyl ether in hexanes to yield alkynoate 11 (2.14 g, 72% yield) as a clear liquid. 1H NMR (300 MHz, CDCl3): δ 3.98–3.89 (m, 4H, OCH2CH2O), 3.74 (s, 3H, OCH3), 2.42 (t, J = 7.9 Hz, 2H, CH2), 1.95 (t, J = 7.9 Hz, 2H, CH2), 1.31 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 154.4 (C=O), 108.8, 89.7, 72.7, 65.0 (2C), 52.7,36.9, 24.0, 13.6; TLC: Rf = 0.23 (20% diethyl ether/hexanes) [silica gel, UV, KMnO4].
2-(Chloromethyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (S5):
Following a procedure analogous to that previously reported,63 a flame-dried, single-necked, 250-mL, round-bottomed flask equiped with a stir bar, septum, and nitrogen inlet needle was charged with trimethyl borate (8.6 mL, 77 mmol) and bromochloromethane (5.5 mL, 85 mmol) and cooled to −78 °C. n-Butyl lithium (1.6 M in hexanes, 53 mL, 85 mmol) was added dropwise via syringe pump over 35 min. Upon completion of addition, the solution was maintained at −78 °C for an additional 30 min. Chlorotrimethylsilane (12 mL, 92 mmol) was added dropwise at −78 °C and the reaction was warmed to rt by removal of the dry ice/acetone bath. The reaction was allowed to stand for 16 h (without stirring), then pinacol (10.0 g, 85 mmol) was added with stirring at rt. The reaction was maintained for 1 h then poured into a separatory funnel containing water (100 mL) and diethyl ether (100 mL). The organic layer was separated, washed with brine (50 mL), dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation (37 °C) and the residue transferred to a 25-mL, single-necked, round-bottomed flask. This residue was fractionally distilled under reduced pressure using a short, jacketed vigreux column connected to a short-path distillation head to give chloromethylpinacol boronate S5 as a colorless liquid (108–115 °C, 14 mmHg, 8.89 g, 65% yield). 1H NMR (300 MHz, CDCl3): δ 2.94 (s, 2H, CH2Cl), 1.28 (s, 12H, (CH3)2CC(CH3)2); 13C NMR (100 MHz, CDCl3): δ 84.7, 24.8 (4C); 11B NMR (128 MHz, CDCl3): δ 31.42; IR: 3459, 2980, 2935, 1352, 1273, 1143 cm−1; TLC : Rf = 0.45 (10% diethyl ether/hexanes) [silica gel, KMnO4].
Methyl 5-(2-methyl-1,3-dioxolan-2-yl)-2-((4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)methyl)pent-2-enoate (9).
Prepared using a procedure analogous to that previously reported.30, 64 Run 1: A flame-dried, 100-mL, single-necked, round-bottomed flask equipped with a magnetic stir bar, septum, and nitrogen inlet needle was charged with copper iodide (48 mg, 0.25 mmol) and tetrahydrofuran (8.4 mL), then cooled to −30 °C. Methyl lithium (1.6 M solution in diethyl ether, 0.16 mL, 0.25 mmol) was added dropwise over 1 min. Upon completion of addition the solution was maintained at −30 °C for an additional 20 min at which time it turned from dark brown to black. Toluene (18 mL) was added slowly to the reaction mixture over 10 min via syringe, followed by hexamethylphosphoramide (0.88 mL, 5.1 mmol). Diisobutylaluminum hydride (1 M solution in hexanes, 3.8 mL, 3.8 mmol) was added dropwise via syringe over 10 min at −30 °C, a temperature that was maintained for an additional 2 h. Alkynoate 11 (500 mg, 2.52 mmol) in toluene (12 mL) was added dropwise to the reaction mixture over 10 min via syringe, and the solution was allowed to warm to −20 °C, a temperature that was maintained for 5 h. The reaction progress was monitored by 1H NMR as the alkene byproduct S4 and alkynoate 11 have the same Rf by TLC. Freshly distilled 2-(chloromethyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (S5) (623 mg, 3.53 mmol) in toluene (6 mL) was added dropwise via syringe over 5 min at −20 °C. The reaction was allowed to warm to rt and maintained for 16 h. The reaction was diluted with diethyl ether (10 mL), and 1N hydrochloric acid (2 mL) was added dropwise over 5 min. The layers were separated, and the organic layer was washed sequentially with 1N hydrochloric acid (3 × 3 mL), saturated aqueous sodium bicarbonate (1 × 5 mL), water (2 × 5 mL), and brine (1 × 10 mL), dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation, and passed through a plug of silica gel eluting with 20% diethyl ether in hexanes to yield allylboronate 9 (755 mg, 88% yield) as a pale yellow oil as a 2:1 ratio of Z:E isomers. The Z:E ratio was determined by integration of the alkene resonances at 5.92 ppm for the Z-isomer and 6.71 for the E-isomer. The allylboronate 9 was contaminated with about 5% of alkene byproducts S4 (2:1 Z:E), as determined by integration of the alkene resonances at 6.25 (dt) and 5.75 (dt) ppm for 9Z:S4 and 6.96 (dt) and 5.90 (dt) for 9E:S4. Attempts to separate allylboronate 9 from S4 by column chromatography resulted in a greatly reduced yield of allylboronate. Run 2: alkynoate 11 (3.02 g, 15.1 mmol), chloromethylpinacolboronate S5 (3.74 g, 21.2 mmol), copper iodide (288 mg, 1.51 mmol), methyl lithium (1.5 M in diethyl ether, 1.1 mL, 1.5 mmol), diisobutylaluminum hydride (1 M in hexanes, 23 mL, 23 mmol), hexamethylphosphoramide (5.3 mL, 30 mmol), toluene (216 mL), and THF (50 mL) provided allylboronate 9 (3.73 g, 73% yield, 2:1 Z:E containing ~10% of the alkene byproduct S4) as a pale yellow oil. 1H NMR (400 MHz, CDCl3): δ 5.92 (t, J = 7.6 Hz, 1H, =CH), 3.92–3.88 (m, 4H, OCH2CH2O), 3.67 (s, 3H, OCH3), 2.56 (app q, J = 7.6 Hz, 2H, CH2), 1.82 (br s, 2H, CH2B), 1.77–1.71 (m, 2H, CH2), 1.30 (s, 3H, CH3), 1.20 (s, 12H, (CH3)2CC(CH3)2); E-isomer, where distinguishable: δ 6.71 (t, J = 7.6 Hz, 1H, =CH), 2.25–2.19 (m, 2H, CH2), 1.85 (br s, 2H, CH2B); 13C NMR (100 MHz, CDCl3): δ 168.2 (C=O), 142.9, 140.6, 129.3, 128.0, 109.9, 83.4, 64.8 (2C), 51.2, 38.6, 24.8, 24.0 (4C) ppm; E-isomer, where distinguishable: δ 168.7 (C=O), 142.9, 141.0, 129.2, 128.0, 109.6, 83.3, 64.8 (2C), 51.7, 37.8, 24.7, 23.7 (4C) ppm; IR (thin film) 2981, 2884, 2240, 1716, 1645, 1437, 1351, 1257, 1145, 1055 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C17H30BO6 341.2130; Found 341.2118; TLC Rf = 0.31 (25% EtOAc/hexanes), visualized with UV and p-anisaldehyde stain.
3-Phenyl-2-propynal (12).
Aldehyde 12 was prepared as previously described, and the spectral data matched that reported.65 Active γ-manganese dioxide was prepared from manganese sulfate and potassium permanganate according to the previously reported method.66 Alternatively, aldehyde 12 was also prepared according to the procedure used for the preparation of 3-triisopropylsilyl-2-propynal (13).67 1H NMR (400 MHz, CDCl3): δ 9.43 (s, 1H, CHO), 7.62–7.60 (m, 2H, Ph-H), 7.51–7.47 (m, 1H, Ph-H), 7.43–7.39 (m, 2H, Ph-H); 13C NMR (100 MHz, CDCl3): δ 177.0 (C=O), 133.4 (2C), 131.4, 128.9 (2C), 119.6, 95.3, 88.6.
3-Triisopropylsilyl-2-propynal.
Aldehyde 13 was prepared as previously described, and the spectral data matched that reported.67 1H NMR (400 MHz, CDCl3): δ 9.2 (s, 1H, CHO), 1.12–1.07 (m, 21H, Si(CH(CH3)2)3); 13C NMR (100 MHz, CDCl3): δ 176.8 (C=O), 104.6, 101.0, 18.6, 11.1.
General Procedure A:
α-Methylene-γ-Lactam Formation from Allylboration/Lactamization Sequence Using 2-Propynals.
This procedure was modified from the procedure reported by Hall and Elford.28 Run 1: A 5-mL, single-necked, round-bottomed flask equipped with a magnetic stir bar, septum, and nitrogen inlet needle was charged with 3-phenyl-2-propynal (12) (72 mg, 0.55 mmol) and ethanol (1 mL). Ammonium hydroxide (28–30% ammonia in water, 0.74 mL, 5.5 mmol) was added via syringe in a single portion at rt. The nitrogen inlet needle was removed and the reaction maintained for 20 min at rt. Allylboronate 9 (171 mg, 0.50 mmol, 2:1 Z:E) in ethanol (1 mL) was added dropwise over 1 min and the reaction was stirred in the sealed flask for 5 h at rt. At this time, complete consumption of allylboronate was indicated by TLC, then 1N HCl (~5 mL) was slowly added to afford a solution with a final pH of 1.5–2 (pH paper). The resulting solution was transferred to a separatory funnel and extracted with diethyl ether (4 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation, and purified by silica gel flash column chromatography eluting with 30–50% ethyl acetate in hexanes to give lactams 14a (trans) and 14b (cis) (96 mg, 59%) in a 4:1 ratio. The trans lactam was taken on to the next step as a single isomer or as a mixture. This reaction was performed using an isomeric mixture of allylboronate 9 and alkene S4 without significant effects on the yield; however, purification was difficult when the alkene comprises >20% of the molar ratio. This reaction was repeated eleven times with the yields ranging from 45 to 86% with an average yield of 58%. The trans:cis lactam ratio ranged from 2:1 to 5:1 even though the allylboronate Z:E ratio was ~ 2:1. Run 2: allylboronate 9 (1.66 g, 4.86 mmol, 2:1 Z:E), 3-phenyl-2-propynal (12) (696 mg, 5.35 mmol), ammonium hydroxide (28–30% ammonia in water, 7.2 mL, 54.0 mmol), and ethanol (20 mL) provided lactam 14a,b (1.38 g, 86% yield, 5:1 trans:cis) as a brown oil after column chromatography. Run 3: allylboronate 9 (1.48 g, 4.35 mmol, 2:1 Z:E), 3-phenyl-2-propynal (12) (623 mg, 4.79 mmol), ammonium hydroxide (28–30% ammonia in water, 6.7 mL, 48.0 mmol), and ethanol (18 mL) provided trans lactam 14a (349 mg, 24% yield), a mixture of lactams 14a,b (380 mg, 27% yield, 3:1 trans:cis), and a mixture of lactams 14b,a (75 mg, 5% yield, 2:1 cis:trans) after column chromatography.
(4S*,5S*)-4-(2-(2-Methyl-1,3-dioxolan-2-yl)ethyl)-3-methylene-5-(phenylethynyl)pyrrolidin-2-one (14a).
1H NMR (300 MHz, CDCl3): δ 7.40–7.31 (m, 2H, Ph-H), 7.30–7.27 (m, 3H, Ph-H), 7.04 (br s, 1H, NH), 6.10 (d, J = 2.8 Hz, 1H, =CH), 5.40, (d, J = 1.6 Hz, 1H, =CH), 4.24, (d, J = 4.0 Hz, 1H, CH), 3.98–3.89 (m, 4H, OCH2CH2O), 3.11–3.09 (m, 1H, CH), 1.88–1.71 (m, 4H, CH2CH2), 1.33 (s, 3H, CH3) and some baseline impurities in spectra; 13C NMR (100 MHz, CDCl3): δ 169.7 (C=O), 142.2, 131.8 (2C), 128.7, 128.4 (2C), 122.3, 117.3, 109.7, 87.8, 84.5, 64.8 (2C), 48.8, 46.8, 35.8, 28.2, 24.1; IR (thin film) 2983, 1703, 1659, 1491, 1324, 1063 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C19H22NO3 312.1594; Found 312.1585; TLC Rf = 0.17 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde stain.
(4R*,5S*)-4-(2-(2-Methyl-1,3-dioxolan-2-yl)ethyl)-3-methylene-5-(phenylethynyl)pyrrolidin-2-one (14b).
1H NMR (400 MHz, CDCl3): δ 7.41–7.38 (m, 3H, Ph-H), 7.31–7.28 (m, 2H, Ph-H), 6.31 (br s, 1H, NH), 6.11 (d, J = 2.4 Hz, 1H, =CH), 5.39 (d, J = 2.4 Hz, 1H, =CH), 4.67 (d, J = 8.0 Hz, 1H, CH), 3.93–3.87 (m, 4H, OCH2CH2O), 3.12–3.08 (m, 1H, CH), 1.94–1.84 (m, 2H, CH2), 1.76–1.66 (m, 2H, CH2), 1.33 (s, 3H, CH3), spectra were obtained as a mixture of isomers and baseline impurities were present in the 1H NMR. 13C NMR (100 MHz, CDCl3): δ 170.3 (C=O), 141.8, 131.8 (2C), 129.0, 128.5 (2C), 122.3, 117.1, 109.9, 86.7, 85.0, 64.8, 64.7, 47.7, 42.7, 36.4, 24.7, 24.1; IR (thin film) 2983, 2245, 1704, 1659, 1490, 1321, 1269, 1062 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C19H22NO3 312.1594; Found 312.1586; TLC Rf = 0.32 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
Prepared according to General Procedure A. Run 1: 3-Triisopropylsilyl-2-propynal (13) (340 mg, 1.62 mmol) ethanol (6 mL), ammonium hydroxide (28–30% ammonia in water, 2.2 mL, 16.0 mmol), allylboronate 9 (500 mg, 1.47 mmol, 2:1 Z:E, contained 15% S4) were stirred for 5 h. 2N HCl (10 mL) was added to the solution then transferred to a separatory funnel and extracted with diethyl ether (3 × 20 mL). Column chromatography (gradient elution with 10–40% ethyl acetate in hexanes) provided trans lactam 15a (126 mg, 18% yield), cis lactam 15b (23 mg, 3% yield, contaminated with 25% trans isomer), and a 2.3:1 trans (15a):cis (15b) mixture (217 mg, 31% yield) as yellow oils in a 52% overall yield and a ratio of 3.5:1 trans:cis lactams. The trans and cis isomers were separated for characterization purposes but were taken on as a mixture to the next step. Run 2: allylboronate 9 (1.67 g, 4.86 mmol, 2:1 Z:E), 3-triisopropylsilyl-2-propynal (13) (1.13 g, 5.35 mmol), ammonium hydroxide (28–30% ammonia in water, 7.2 mL), ethanol (20 mL), provided lactams 15a,b (1.72 g, 90% yield, 2:1 trans:cis) as a brown oil after column chromatography.
(4S*,5S*)-4-(2-(2-Methyl-1,3-dioxolan-2-yl)ethyl)-3-methylene-5-((triisopropylsilyl)ethynyl)pyrrolidin-2-one (15a).
1H NMR (400 MHz, CDCl3): δ 6.06 (d, J = 2.6 Hz, 1H, =CH), 6.02 (br s, 1H, NH), 5.37 (d, J = 2.6 Hz, 1H, =CH), 4.02 (d, J = 5.2 Hz, 1H, CH), 3.98–3.87 (m, 4H, OCH2CH2O), 3.01–2.97 (m, 1H, CH), 1.91–1.63 (m, 4H, CH2CH2), 1.31 (s, 3H, CH3), 1.05 (br s, 21H, Si(CH(CH3)2)3); 13C NMR (100 MHz, CDCl3): δ 169.3 (C=O), 142.0, 117.0, 109.6, 106.0, 86.2, 64.9 (2C), 48.9, 47.4, 36.2, 27.7, 25.0, 18.7 (6C), 11.2 (3C); IR (thin film) 2943, 2175, 1708, 1660, 1462, 1376, 1325, 1150, 1063; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C22H38NO3Si 392.2616; Found 392.2616; TLC Rf = 0.30 (50% EtOAc/hexanes), visualized with UV and KMnO4.
(4R*,5S*)-4-(2-(2-Methyl-1,3-dioxolan-2-yl)ethyl)-3-methylene-5-((triisopropylsilyl)ethynyl)pyrrolidin-2-one (15b).
1H NMR (300 MHz, CDCl3): δ 6.23 (br s, 1H, NH), 6.05 (d, J = 2.0 Hz, 1H, =CH), 5.34 (d, J = 2.0 Hz, 1H, =CH), 4.45 (d, J = 7.6 Hz, 1H, CH), 3.95–3.79 (m, 4H, OCH2CH2O), 3.00–2.97 (m, 1H, CH), 1.90–1.70 (m, 4H, CH2CH2), 1.31 (s, 3H, CH3), 1.02 (br s, 21H, Si(CH(CH3)2)3); 13C NMR (100 MHz, CDCl3): δ 170.3 (C=O), 142.1, 116.8, 103.2, 109.9, 88.4, 64.8, 64.7, 47.9, 42.8, 36.4, 24.3, 23.9, 18.7 (6C), 11.2 (3C); IR (thin film) 3213, 2866, 2175, 1660, 1377, 1221, 1143, 1112 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C22H38NO3Si 392.2616; Found 392.2616; TLC Rf = 0.21 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(4S*,5S*)-1-Methyl-4-(2-(2-methyl-1,3-dioxolan-2-yl)ethyl)-3-methylene-5 - (phenylethynyl)pyrrolidin-2-one (S8).
Run 1: A flame-dried, 10-mL, single-necked, round-bottomed flask equipped with a magnetic stir bar, septum, and nitrogen inlet needle was charged with 14a,b (337 mg, 1.03 mmol, ~10:1 trans:cis) dissolved in dimethylformamide (5.2 mL) and cooled to 0 °C in an ice bath. Sodium hydride (60% in mineral oil, 62 mg, 1.5 mmol) was added in a single portion and the reaction was stirred 15 min at 0 °C. Iodomethane (0.13 mL, 2.1 mmol) was added dropwise over 1 min and the reaction was stirred at 0 °C for 15 min before removing the ice bath and allowing the reaction to warm to rt for 15 min. Upon disappearance of starting material, the reaction mixture was poured into a separatory funnel containing saturated aqueous ammonium chloride solution (20 mL). The mixture was extracted with diethyl ether (3 × 25 mL). The organic layers were combined, washed with brine (10 mL), dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation, and purified by filtration through a plug of silica gel (elution with 50% ethyl acetate and hexanes) to provide the N-methyl lactam S8 (225 mg, 67% yield, contains 6% of the cis isomer and some other baseline impurities) as a yellow oil. This reaction was repeated three times with the yields ranging from 67 to 74%. Run 2: Ketal 14a,b (430 mg, 1.3 mmol, 4:1 trans:cis), sodium hydride (60% in mineral oil, 105 mg, 2.6 mmol), iodomethane (0.21 mL, 3.3 mmol), and DMF (7 mL) provided ketal S8 (321 mg, 71% yield, 4:1 trans:cis) as a yellow oil after column chromatography. 1H NMR (400 MHz, CDCl3): δ 7.42–7.40 (m, 2H, Ph-H), 7.34–7.31 (m, 3H, Ph-H), 6.08 (d, J = 2.4 Hz, 1H, =CH), 5.36 (d, J = 2.4 Hz, 1H, =CH), 4.12 (d, J = 4.0 Hz, 1H, CH), 3.99–3.90 (m, 4H, OCH2CH2O), 3.05–3.04 (m, 1H, CH), 3.03 (s, 3H, NCH3), 1.87–1.69 (m, 4H, CH2CH2), 1.33 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 167.2, 142.3, 131.8 (2C), 128.9, 128. 5 (2C), 122.2, 116.4, 109.7, 86.2, 85.7, 64.9 (2C), 55.4, 44.6, 35.9, 29.8, 28.5, 24.1; IR (thin film) 2925, 1697, 1660, 1427, 1396, 1145, 1082 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C20H24NO3 326.1751; Found 326.1752; TLC Rf = 0.39 (75% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
General Procedure B:
Methyl Ketone Formation via Hydrolysis of Ketal Protecting Group.
Run 1: A 10-mL, single-necked, round-bottomed flask equipped with a magnetic stir bar was charged with ketal 14a,b (100 mg, 0.304 mmol, 5:1 trans:cis), acetone (4 mL), and water (0.3 mL). Pyridinium para-toluene sulfonate (38 mg, 0.152 mmol) was added in a single portion, a reflux condenser capped with a septum and nitrogen inlet needle was attached, and the reaction was refluxed for 16 h (oil bath temperature 70 °C). Upon completion of the reaction as observed by TLC, the reaction was allowed to cool to rt, diluted with ethyl acetate (20 mL), transferred to a separatory funnel, washed consecutively with water (2 × 5 mL) and brine (1 × 5 mL), dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation, then eluted through a plug of silica gel with 75% ethyl acetate in hexanes to yield ketone 18a,b (63 mg, 73% yield, 5:1 trans:cis) as a light yellow oil. For larger scale reactions, the reaction was concentrated by rotary evaporation prior to dilution with ethyl acetate. The ratios of 18a:18b was based upon integrated values of trans-lactam methine (18a, CHNH at 4.21 ppm) and cis-lactam methine (18b, CHNH at 4.68 ppm). This reaction was repeated nine times with yields ranging from 71 to 91% with an average yield of 83%. The trans and cis isomers were generally taken on as a mixture but separated for characterization purposes. This reaction was performed successfully on gram scale. Run 2:, ketal 14a,b (1.66 g, 5.33 mmol, 3:1 trans:cis), pyridinium para-toluenesulfonate (670 mg, 2.7 mmol), acetone (72 mL), and water (4 mL) provided ketone 18a,b (1.3 g, 91% yield, 3:1 trans:cis) as a pale yellow oil after column chromatography.
(4S*,5S*)-3-Methylene-4-(3-oxobutyl)-5-(phenylethynyl)pyrrolidin-2-one (18a).
1H NMR (300 MHz, CDCl3): δ 7.38–7.37 (m 2H, Ph-H), 7.32–7.29 (m, 3H, Ph-H), 6.77 (br s, 1H, NH). 6.11 (d, J = 2.4 Hz, 1H, =CH), 5.40 (d, J = 2.4 Hz, 1H, =CH), 4.21 (d, J = 4.8 Hz, 1H, CH), 3.16–3.08 (m, 1H, CH), 2.64 (t, J = 7.8 Hz, 2H, CH2), 2.14 (s, 3H, CH3), 2.14–2.13 (m, 1H), 1.92–1.80 (m, 1H), 16% cis-isomer 18b, where distinguishable: δ 4.68 (d, J = 7.5 Hz, 1H, CH); 13C NMR (100 MHz, CDCl3): δ 207.5, 169.5, 141.8, 131.8 (2C), 128.9, 128.5 (2C), 122.1, 117.4, 87.4, 84.9, 48.8, 46.1, 39.9, 30.3, 27.0; IR (thin film) 3434, 2088, 1643 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C17H18NO2 268.1344; Found 268.1332; TLC Rf = 0.32 (75% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(4R*,5S*)-3-Methylene-4-(3-oxobutyl)-5-(phenylethynyl)pyrrolidin-2-one (18b).
Prepared according to General Procedure B. Ketal 14a,b (426 mg, 1.30 mmol, 3:1 trans:cis), pyridinium para-toluene sulfonate (163 mg, 0.648 mmol), acetone (18 mL), and deionized water (1 mL) provided ketone 18a,b (307 mg, 88% yield, 3:1 trans:cis) as a clear oil after filtration through a silica gel plug (elution with 50% ethyl acetate in hexanes). Re-purification of the mixture by column chromatography afforded fractions with predominantly the cis isomer which were used for characterization purposes. 1H NMR (300 MHz, CDCl3): δ 7.39–7.37 (m, 2H, Ph-H), 7.34–7.30 (m, 3H, Ph-H), 6.39 (br s, 1H, NH), 6.13 (d, J = 2.3 Hz, 1H, =CH), 5.42 (d, J = 2.3 Hz, 1H, =CH), 4.70 (d, J = 7.5 Hz, 1H, CH), 3.17–3.13 (m, 1H, CH), 2.72–2.58 (m, 2H, CH2), 2.16 (s, 3H, CH3), 2.15–2.09 (m, 2H, CH2), 10% trans-isomer 18a, where distinguishable: δ 4.21 (d, J = 4.8 Hz, 1H, CH); 13C NMR (100 MHz, CDCl3): δ 207.9, 169.9, 141.5, 131.9 (2C), 129.0, 128.6 (2C), 122.0, 117.6, 87.2, 84.6, 47.5, 41.7, 40.5, 30.2, 24.5; IR (thin film) 3245, 2927, 1708, 1657, 1418, 1360, 1273, 1166 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C17H18NO2 268.1332; Found 268.1327; TLC Rf = 0.28 (75% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(4S*,5S*)-1-Methyl-3-methylene-4-(3-oxobutyl)-5-(phenylethynyl)pyrrolidin-2-one (27a).
Prepared according to General Procedure B. Run 1: trans-Ketal S8 (290 mg, 0.882 mmol, contained 6% cis isomer), pyridinium para-toluene sulfonate (PPTS) (111 mg, 0.441 mmol), acetone (12 mL), and water (0.6 mL) provided the ketone 27a (164 mg, 66%) as a yellow oil after column chromatography. Run 2: ketal S8 (490 mg, 1.4 mmol, 5:1 trans:cis), PPTS (180 mg, 0.72 mmol, acetone (19 mL), and water (1 mL) to provide trans ketone 27a (310 mg, 73% yield, 19:1 trans:cis) and cis ketone (18 mg, 4% yield, 7:1 cis:trans) each as clear oils after column chromatography. 1H NMR (500 MHz, CDCl3): δ 7.42–7.40 (m, 2H, Ph-H), 7.35–7.32 (m, 3H, Ph-H), 6.11 (d, J = 2.5 Hz, 1H, =CH), 5.36 (d, J = 2.5 Hz, 1H, =CH), 4.08 (d, J = 4.5 Hz, 1H, CH), 3.08–3.04 (m, 1H, CH), 3.03 (s, 3H, NCH3), 2.66–2.63 (app t, J = 7.5 Hz, 2H, CH2), 2.17 (s, 3H, CH3), 2.12–2.04 (m, 1H), 1.90–1.84 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 207.5, 167.1, 142.0, 131.9 (2C), 129.0, 128.6 (2C), 122.1, 116.5, 86.1, 85.9, 55.4, 44.1, 40.1, 30.3, 28.5, 27.3; IR (thin film) 2924, 1696, 1424, 1396, 1265 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C18H20NO2 282.1489; Found 282.1489; TLC Rf = 0.48 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(4S*,5S*)-3-Methylene-4-(3-oxobutyl)-5-((triisopropylsilyl)ethynyl)pyrrolidin-2-one (19a).
Prepared according to General Procedure B. Run 1: Ketal 15a (120 mg, 0.306 mmol, pure trans), pyridinium para-toluene sulfonate (PPTS, 39 mg, 0.15 mmol), acetone (4.1 mL), and water (0.3 mL) provided ketone 19a (74 mg, 70% yield) as a pale yellow oil after filtration through a plug of silica (elution with 50% ethyl acetate in hexanes). This reaction was repeated six times with the yields ranging from 58 to 85% with an average yield of 73%. The cis and trans isomers were taken on to the next step as a mixture but were separated for characterization purposes. Run 2: ketal 15a,b (1.74 g, 4.4 mmol, 2:1 trans:cis), PPTS (552 mg, 2.2 mmol), acetone (60 mL), and water (3 mL) provided ketone 19a,b (1.05 g, 68%, 2:1 trans:cis) as a pale yellow oil after column chromatography. Run 3: ketal 15a,b (350 mg, 0.89 mmol, 2:1 trans:cis), PPTS (112 mg, 0.45 mmol), acetone (13 mL), and water (1 mL) to provide ketone 19a,b (265 mg, 85% yield, 1.5:1 trans:cis) as a clear oil after column chromatography. 1H NMR (400 MHz, CDCl3): δ 6.26 (br s, 1H, NH), 6.08 (d, J = 2.6 Hz, 1H, =CH), 5.38 (dd, J = 2.6 Hz, 0.8 Hz, 1H, =CH), 4.00 (d, J = 4.8 Hz, 1H, CH), 3.03–2.97 (m, 1H, CH), 2.68–2.58 (m, 2H, CH2), 2.16 (s, 3H, CH3), 2.15–2.06 (m, 1H), 1.85–1.75 (m, 1H), 1.05 (br s, 21H, Si(CH(CH3)2)3); 13C NMR (100 MHz, CDCl3): δ 207.3, 169.1, 141.6, 117.2, 105.9, 86.5, 48.9, 46.6, 40.3, 30.1, 26.9, 18.7 (6C), 11.2 (3C); IR (thin film): 2943, 2865, 2175, 1709, 1659, 1463, 1366, 1323 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C20H34O2NSi 348.2359; Found 348.2373; TLC Rf = 0.48 (75% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(4R*,5S*)-3-Methylene-4-(3-oxobutyl)-5-((triisopropylsilyl)ethynyl)pyrrolidin-2-one (19b).
Prepared according to General Procedure B. Ketal 15a,b (1.72 g, 4.39 mmol, 4:1 trans:cis), PPTS (552 mg, 2.20 mmol), acetone (60 mL), and deionized water (3 mL) provided a first fraction of ketone 19a,b (1.22 g, 80% yield, 4:1 trans:cis) and a second fraction of ketone 19b,a (30 mg, 2% yield, 5:1 cis:trans) after column chromatography (gradient elution with 25–75% ethyl acetate in hexanes) as pale yellow oils. 1H NMR (400 MHz, CDCl3): δ 6.48 (br s, 1H, NH), 6.07 (d, J = 2.4 Hz, 1H, =CH), 5.37 (d, J = 2.4 Hz, 1H, =CH), 4.46 (d, J = 8.0 Hz, 1H, CH), 3.04–2.99 (m, 1H, CH), 2.69–2.51 (m, 2H, CH2), 2.14 (s, 3H, CH3), 2.13–1.97 (m, 2H, CH2), 1.0 (br s, 21H, Si(CH(CH3)2)3), NMR contains ~20% of trans isomer; 13C NMR (100 MHz, CDCl3): δ 207.5, 170.0, 141.5, 117.3, 102.8, 88.8, 47.5, 41.3, 40.5, 30.0, 24.3, 18.6 (6C), 11.2 (3C); IR (thin film): 3175, 2909, 2832, 2145, 1690, 1639, 1446, 1348, 1308, 1257, 1151 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C20H34O2NSi 348.2353; Found 348.2363; TLC Rf = 0.41 (75% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
General Procedure C:
Propargyl Alcohol Formation via Addition of Ethynyl Magnesium Bromide to Methyl Ketone.
Run 1: A flame-dried, 100-mL, single-necked, round-bottomed flask equipped with a magnetic stir bar, a septum, and a nitrogen inlet needle was charged with ketone 18a,b (558 mg, 2.08 mmol, ~3:1 trans:cis) dissolved in tetrahydrofuran (25 mL). The solution was cooled to 0 °C in an ice bath, then ethynyl magnesium bromide (0.5 M solution in tetrahydrofuran, 17 mL, 8.5 mmol) was added dropwise via syringe over 15 min. The reaction was maintained at 0 °C for 3 h. Next, 1N HCl (25 mL) was added dropwise at 0 °C. The resulting solution was transferred to a separatory funnel and extracted with diethyl ether (4 × 40 mL). The organic layers were combined, washed with brine (20 mL), dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation, and purified by silica gel flash column chromatography eluting with 40–60% ethyl acetate in hexanes to provide trans propargyl alcohol 20a (41 mg, 7% yield), cis propargyl alcohol 20b (1 mg, 1% yield, cis), and a 4:1 trans:cis mixture of propargyl alcohols 20a and 20b (385 mg, 63% yield) each as clear oils and as a 1:1 ratio of diastereomers as determined by 13C NMR. The cis and trans isomers were taken on as a mixture but were separated for characterization purposes. This reaction was repeated six times with the yields ranging from 65 to 81%. The trans:cis ratio of propargyl alcohol products reflected the ratio of the ketone starting material. Run 2: Ketone 18a,b (1.30 g, 4.9 mmol, 3:1 trans:cis), ethynyl magnesium bromide (0.5 M in THF, 38 mL, 19 mmol), and THF (60 mL) provided propargyl alcohols 20a and 20b (1.13 g, 78% yield, 3:1 trans:cis) as a clear oil after column chromatography. Run 3: Ketone 18a, b (640 mg, 2.4 mmol, 4:1 trans:cis), ethynyl magnesium bromide (0.5 M in THF, 19 mL, 9.5 mmol), and THF (29 mL) provided propargyl alcohols 20a and 20b in two fractions (528 mg, 75% yield, 6:1 trans:cis, and 39 mg, 6% yield, 2:1 cis:trans) each as clear oils after column chromatography.
(4S*,5S*)-4-(3-Hydroxy-3-methylpent-4-ynyl)-3-methylene-5-(phenylethynyl) pyrrolidin-2-one (20a).
1H NMR (500 MHz, CDCl3): δ 7.40–7.38 (m, 2H, Ph-H), 7.31–7.27 (m, 3H, Ph-H), 6.98 (br s, 1H, NH), 6.12 (d, J = 2.0 Hz, 1H, =CH), 5.43 (d, J = 2.0 Hz, 1H, =CH), 4.26 (d, J = 4.5 Hz, 1H, CH), 3.14–3.11 (m, 1H, CH), 2.62 (br s, 1H, OH), 2.46 (s, 1H, °CH), 2.04–1.96 (m, 1H), 1.90–1.79 (m, 3H), 1.52 (s, 3H, CH3); diethyl ether at δ 3.48 and 1.21; 13C NMR (125 MHz, CDCl3): δ 169.8, 142.0, 131.8 (2C), 128.8, 128.5 (2C), 122.2, 117.6, 117.5*, 87.7, 87.4, 84.7, 72.01, 71.97*, 67.67, 67.65*, 48.9, 48.8*, 46.80, 46.78*, 39.9, 39.8*, 30.3, 30.2*, 28.9, 28.8*, *Discernible signals for one of two diastereomers at δ 65.9; IR (thin film): 3448, 1652, 1156 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C19H20NO2 294.1489; Found 294.1477; TLC Rf = 0.36 (75% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(4R*,5S*)-4-(3-Hydroxy-3-methylpent-4-yn-1-yl)-3-methylene-5-(phenylethynyl)pyrrolidin-2-one (20b).
1H NMR (500 MHz, CDCl3): δ 7.41–7.39 (m, 2H, Ph-H), 7.33–7.27 (m, 3H, Ph-H), 6.19 (br s, 1H, NH), 6.13 (d, J = 2.3 Hz, 1H, =CH), 5.44 (d, J = 2.3 Hz, 1H, =CH), 5.42 (d, J = 2.0 Hz, 1H, =CH)*, 4.70 (d, J = 7.5 Hz, 1H, CH), 4.69 (d, J = 8.0 Hz, 1H, CH)*, 3.16–3.13 (m, 1H, CH), 2.36 (s, 1H, °CH), 2.16 (s, 1H, °CH)*, 2.14–2.06 (m, 2H), 1.95–1.90 (m, 1H), 1.84–1.78 (m, 1H), 1.53 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 170.2, 141.7, 131.9 (2C), 131.85*(2C), 129.0, 128.9*, 128.6 (2C), 128.5*, 122.2, 122.0*, 117.3, 117.2*, 87.3, 87.2*, 85.0, 84.9*, 72.1, 71.9*, 68.0, 67.8*, 47.74, 47.71*, 47.4, 46.2*, 42.74, 42.71*, 40.55, 40.51*, 30.4, 30.3*, 30.2, 30.1*, *Discernible signals for one of two diastereomers; IR: (thin film): 2929, 2231, 2145, 1686, 1660, 1490, 1435, 1400, 1276, 1082 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C19H20NO2 294.1489; Found 294.1477; TLC Rf = 0.29 (75% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(4S*,5S*)-4-(3-Hydroxy-3-methylpent-4-ynyl)-1-methyl-3-methylene-5-(phenylethynyl)pyrrolidin-2-one (28a).
Prepared according to General Procedure C. Ketone 27a (310 mg, 1.04 mmol, trans:cis 7:1), THF (14 mL) and ethynyl magnesium bromide (0.5 M solution in THF, 6.3 mL, 3.1 mmol) provided propargyl alcohol 28a (198 mg, 62% yield, trans:cis 7:1, 1:1 ratio of diastereomers) as a white, sticky solid after column chromatography (gradient elution with 20–60% ethyl acetate in hexanes). The diastereomeric ratio was based on the 13C NMR, as the diastereomers were indistinguishable by 1H NMR. 1H NMR (400 MHz, CDCl3): δ 7.42–7.39 (m, 2H, Ph-H), 7.34–7.31 (m, 3H, Ph-H), 6.09 (d, J = 2.6 Hz, 1H, =CH), 5.38 (d, J = 2.6 Hz, 1H, =CH), 4.13 (d, J = 4.0 Hz, 1H, CH), 3.07–3.05 (m, 1H, CH), 3.03 (s, 3H, NCH3), 2.47 (s, 1H, °CH), 2.28 (br s, 1H, OH), 2.06–1.95 (m, 1H), 1.87–1.78 (m, 3H), 1.53 (s, 3H, CH3); NMR contains ~7% of the ketone starting material; 13C NMR (100 MHz, CDCl3): δ 167.2, 142.14, 142.12*, 131.9 (2C), 128.9, 128.5 (2C), 122.1, 116.64, 116.66*, 87.2, 86.1, 85.9, 72.13, 72.09*, 67.8, 67.7*, 55.4, 55.3*, 44.6, 44.5*, 39.9, 39.8*, 30.4, 30.3*, 29.2, 29.0*, 28.6, *Discernible signals for one of two diastereomers; IR (thin film): 3298, 2979, 2929, 2231, 2108, 1680, 1659, 1490, 1432, 1292, 1159, 1084 cm−1; HRMS (TOF MS ES+) m/e: [M+H]+ Calcd for C20H22NO2 308.1651; Found 308.1658; TLC Rf = 0.60 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(4S*,5S*)-4-(3-Hydroxy-3-methylpent-4-ynyl)-3-methylene-5-((triisopropylsilyl) ethynyl)pyrrolidin-2-one (21a).
Prepared according to General Procedure C. Run 1: Ketone 19a (270 mg, 0.78 mmol, pure trans), ethynyl magnesium bromide (0.5 M solution in tetrahydrofuran, 6.2 mL, 3.1 mmol), and tetrahydrofuran (9.3 mL) yielded the propargyl alcohol 21a (265 mg, 91% yield) as a pale yellow oil and a 1:1 mixture of diastereomers based on the 13C NMR. This reaction was repeated five times with the yields ranging from 64 to 91% with an average yield of 82%. The trans:cis ratio remained the same as the starting material. 1H NMR (500 MHz, CDCl3): δ 6.09 (d, J = 3.0 Hz, 1H, =CH), 5.90 (br s, 1H, NH), 5.42–5.40 (m, 1H, =CH), 4.05 (d, J = 5.0 Hz, 1H, CH), 3.05–3.04 (m, 1H), 2.47 (s, 1H, °CH)*, 2.46 (s, 1H, °CH), 2.16–2.03 (m, 1H), 1.93–1.77 (m, 4H), 1.24 (s, 3H, CH3), 1.05 (s, 21H, Si(CH(CH3)2)3); 13C NMR (125 MHz, CDCl3): δ 169.1, 141.8, 117.13, 117.09*, 105.9, 87.2, 87.1*, 86.4, 72.1, 67.80, 67.77*, 49.0, 48.9*, 47.4, 47.3*, 30.5, 30.3*, 28.45, 28.40*, 25.0, 18.7 (6C), 11.2 (3C), *Discernible signals for one of two diastereomers; IR (thin film): 2943, 2865, 1703, 1660, 1462, 1323, 1260, 1093, 1019 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C22H36NO2Si 374.2515; Found 374.2508; TLC Rf = 0.33 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(4R*,5S*)-4-(3-Hydroxy-3-methylpent-4-yn-1-yl)-3-methylene-5-((triisopropyl silyl)ethynyl)pyrrolidin-2-one (21b).
Prepared according to General Procedure C. Ketone 19a,b (1.22 g, 3.51 mmol, 4:1 trans:cis), ethynyl magnesium bromide (0.5 M solution in THF, 28 mL, 14 mmol), and THF (42 mL) provided one fraction of predominantly trans propargyl alcohol 21a (1.07 g, 82% yield, 4:1 trans:cis) and one fraction of predominantly cis propargyl alcohol 21b (52 mg, 4% yield, 2:1 cis:trans) each as a white sticky solid after column chromatography (gradient elution with 50–75% ethyl acetate in hexanes). The ratio of diastereomers was estimated to be about 1.5:1 based on the ratio of 13C NMR peaks, but the peaks could not be integrated separately in the 1H NMR. 1H NMR (400 MHz, CDCl3): δ 6.52 (br s, 1H, NH), 6.06 (d, J = 1.8 Hz, 1H, =CH), 5.38 (d, J = 1.8 Hz, 1H, =CH), 4.50–4.45 (m, 1H, CH), 3.04–3.00 (m, 1H, CH), 2.42 (s, 1H, °CH), 2.41*(s, 1H, °CH), 2.04–1.95 (m, 2H), 1.89–1.75 (m, 2H), 1.50 (s, 3H, CH3), 1.49* (s, 3H, CH3), 1.02 (s, 21H, Si(CH(CH3)2)3), NMR contains ~56% of trans isomer; 13C NMR (100 MHz, CDCl3): δ 170.3, 170.2*, 141.8, 117.2, 116.9*, 103.2, 103.0*, 88.7, 88.5*, 87.7, 87.3*, 72.0, 71.7*, 67.9, 67.7*, 48.0, 47.9*, 42.8, 42.7*, 40.4, 40.2*, 30.2, 30.0*, 25.2, 25.0*, 18.70 (6C), 18.67*, 11.2 (3C), *Discernible signals for one of two diastereomers; IR (thin film) 2943, 2246, 2172, 1703, 1658, 1544, 1462, 1321, 1172, 1072 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C22H36NO2Si 308.1651; Found 308.1658; TLC Rf = 0.22 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
3-Methyl-5-((2S*,3S*)-1-methyl-4-methylene-5-oxo-2-(phenylethynyl)pyrrolidin-3-yl)pent-1-yn-3-yl acetate (29a).
A flame-dried, 5-mL, single-necked, round-bottomed flask equipped with a stir bar, septum, and nitrogen inlet needle was charged with 4-dimethylaminopyridine (DMAP) (4 mg, 0.03 mmol) and propargyl alcohol 28a (86 mg, 0.27 mmol) dissolved in CH2Cl2 (1.3 mL) and cooled to 0 °C. Triethylamine (0.37 mL, 2.7 mmol) was added dropwise via syringe over 2 min, followed by dropwise addition of acetic anhydride (0.12 mL, 1.3 mmol) over 1 min via syringe. The ice bath was removed, and the mixture was allowed to warm to rt for 3 h, at which time the TLC showed consumption of the starting material. The mixture was diluted with CH2Cl2 (25 mL) and transferred to a separatory funnel. The organic layer was washed with a saturated solution of aqueous ammonium chloride (5 mL), then brine (5 mL), dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation, and purified by flash column chromatography on silica gel (gradient elution with 40–50% ethyl acetate in hexanes) to provide lactam 29a (50 mg, 54% yield, dr 1:1) as a clear oil. Following General Procedure D afforded a 30% yield of 29a after column chromatography.
1H NMR (400 MHz, CDCl3): δ 7.42–7.40 (m, 2H, Ph-H), 7.34–7.29 (m, 3H, Ph-H), 6.13 (d, J = 2.4 Hz, 1H, =CH), 5.37 (d, J = 2.4 Hz, 1H, =CH), 4.11 (d, J = 4.0 Hz, 1H, CH), 3.08–3.05 (m, 1H, CH), 3.03 (s, 3H, NCH3), 2.58 (s, 1H, °CH), 2.16–2.07 (m, 1H), 2.03 (s, 3H, CH3), 1.98–1.81 (m, 3H), 1.70 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 169.42, 169.39*, 167.1, 142.0, 131.9 (2C), 129.0, 128.5 (2C), 122.1, 116.6, 116.5*, 86.0, 83.42, 83.39*, 74.51, 74.48*, 74.02, 74.00*, 55.3, 55.2*, 44.5, 44.4*, 38.23, 38.19*, 28.63, 28.57, 28.5, 26.71, 26.66*, 22.01, *Discernible signals for one of two diastereomers; IR (thin film) 3292, 2937, 2243, 2120, 1742, 1697, 1661, 1427, 1243, 1173, 1082 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C22H24NO3 350.1751; Found 350.1751; TLC Rf = 0.26 (50% EtOAc/hexanes), visualized with UV and KMnO4.
General Procedure D:
Propargyl Pivalate Formation from Propargyl Alcohol using Scandium(III) Trifluoromethanesulfonate and Trimethyl Acetic Anhydride.
Run 1: This reaction was performed on a mixture of cis and trans propargyl alcohols 20a,b or 21a,b. A flame-dried, 10-mL test tube equipped with a magnetic stir bar, septum, and nitrogen inlet needle was charged with propargyl alcohol 20a (56 mg, 0.18 mmol, pure trans) dissolved in acetonitrile (0.72 mL). Trimethyl acetic anhydride (0.05 mL, 0.2 mmol) was added via syringe followed by scandium(III) trifluoromethanesulfonate (36 mg, 0.073 mmol). The reaction was stirred at rt for 16 h at which time the TLC showed consumption of the starting material. The reaction was diluted with diethyl ether (20 mL), transferred to a separatory funnel, then washed with saturated sodium bicarbonate solution (10 mL). The aqueous layer was extracted with diethyl ether (2 × 10 mL), the organic layers were combined, washed with brine, dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation, and purified by silica gel flash column chromatography (gradient elution with 15–30% ethyl acetate in hexanes) to provide propargyl pivalate 22a (54 mg, 76%, dr 1:1) as a clear oil. Run 2: Propargyl alcohol 20a,b (368 mg, 1.25 mmol, 3.5:1 trans:cis), pivalic anhydride (0.36 mL, 1.8 mmol), scandium(III) trifluoromethanesulfonate (246 mg, 0.50 mmol), and acetonitrile (5 mL) provided trans pivalate 22a (299 mg, 61% yield) and cis pivalate 22b (79 mg, 16% yield) after column chromatography. Run 3: propargyl alcohol 20a,b (1.13 g, 3.85 mmol, 3:1 trans:cis), pivalic anhydride (0.94 mL, 4.62 mmol), scandium(III) trifluoromethanesulfonate (758 mg, 1.54 mmol), and acetonitrile (15 mL) provided trans pivalate 22a (490 mg, 34% yield), cis pivalate 22b (163 mg, 11% yield), and a 4:1 trans:cis mixture of 22a,b (176 mg, 12% yield) each as a clear oil after column chromatography.
3-Methyl-5-((2S*,3S*)-4-methylene-5-oxo-2-(phenylethynyl)pyrrolidin-3-yl)pent-1-yn-3-yl pivalate (22a).
1H NMR (500 MHz, CDCl3): δ 7.35–7.34 (m, 2H, Ph-H), 7.27–7.21 (m, 3H, Ph-H), 6.98 (s, 1H, NH), 6.08 (d, J = 2 Hz, 1H, =CH), 5.36 (s, 1H, =CH), 4.21 (d, J = 4.5 Hz, 1H, CH), 3.10–3.09 (m, 1H, CH), 2.50 (s, 1H, °CH), 2.12–1.77 (m, 4H, CH2CH2), 1.64 (s, 3H, CH3), 1.13 (s, 9H, C(CH3)3); 13C NMR (125 MHz, CDCl3): δ 176.6, 169.6, 142.0, 131.8 (2C), 128.8, 128.4 (2C), 122.2, 117.3, 117.2*, 87.5, 84.8, 83.52, 83.47*, 74.0, 73.9*, 73.7, 73.6*, 48.8, 48.7*, 46.7, 39.3, 38.34, 38.27*, 28.2, 27.1 (3C), 26.6, *Discernible signals for one of two diastereomers; IR (thin film): 2926, 1702, 1711, 1456, 1153 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C24H28NO3 378.2064; Found 378.2049; TLC Rf = 0.47 (50% EtOAc/hexanes), visualized with UV and KMnO4.
3-Methyl-5-((2S*,3R*)-4-methylene-5-oxo-2-(phenylethynyl)pyrrolidin-3-yl)pent-1-yn-3-yl pivalate (22b).
Propargyl pivalate 22b was prepared according to General Procedure D. A mixture of propargyl alcohols 20a and 20b (140 mg, 0.48 mmol, 5:1 trans:cis), trimethyl acetic anhydride (0.12 mL, 0.57 mmol), scandium(III) trifluoromethanesulfonate (94 mg, 0.19 mmol), and acetonitrile (1.9 mL) provided trans isomer 22a (99 mg, 55% yield) as a clear oil, cis isomer 22b (20 mg, 11% yield) as a clear oil, and a 2:1 trans:cis mixture of 22a,b (7 mg, 4% yield) each as a clear oil after column chromatography (gradient elution with 15–30% ethyl acetate in hexanes). A 1.5:1 dr was determined by integration of terminal alkyne resonances in the 1H NMR at 2.48 (major) and 2.43 ppm (minor). 1H NMR (500 MHz, CDCl3): δ 7.39–7.38 (m, 2H, Ph-H), 7.31–7.28 (m, 3H, Ph-H), 6.58 (br s, 1H, NH), 6.57* (br s, 1H, NH), 6.13–6.12 (m, 1H, =CH), 5.41–5.40 (m, 1H, =CH), 4.71 (d, J = 7.5 Hz, 1H, CH), 4.70* (d, J = 7.5 Hz, 1H), 3.12–3.08 (m, 1H, CH), 2.48 (s, 1H, °CH), 2.43* (s, 1H, °CH), 2.17–2.01 (m, 4H, CH2CH2), 1.68 (s, 3H, CH3), 1.45*(s, 9H, C(CH3)3), 1.11 (s, 9H, C(CH3)3); 13C NMR (125 MHz, CDCl3) δ 176.7, 170.20, 170.15*, 141.8, 141.7*, 131.9 (2C), 128.9, 128.4 (2C), 122.2, 122.18*, 117.1, 87.03, 86.99*, 84.9, 84.8*, 83.8, 83.5*, 74.2, 73.9*, 73.7, 73.5*, 47.8, 42.70, 42.68*, 30.32, 39.30*, 39.1, 39.0*, 27.1 (3C), 26.7, 26.6*, 25.0, 24.8*, *Discernible signals for one of two diastereomers; IR (thin film): 2974, 2934, 1705, 1660, 1491, 1479, 1444, 1322, 1285, 1150, 1103 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C24H28NO3 378.2064; Found 378.2068; TLC Rf = 0.37 (50% EtOAc/hexanes), visualized with UV and KMnO4.
3-Methyl-5-((2S*,3S*)-4-methylene-5-oxo-2-((triisopropylsilyl)ethynyl)pyrrolidin-3-yl)pent-1-yn-3-yl pivalate (23a).
Prepared according to General Procedure D. Run 1: Propargyl alcohol 21a (100 mg, 0.27 mmol, pure trans), scandium(III) trifluoromethanesulfonate (53 mg, 0.11 mmol), pivalic anhydride (0.07 mL, 0.35 mmol), and acetonitrile (1.1 mL) provided pivalate 23a (118 mg, 96% yield) as a clear oil after column chromatography (gradient elution with 20–50% ethyl acetate in hexanes). 13C NMR showed a 1:1 mixture of diastereomers. Run 2: Propargyl alcohol 21a,b (230 mg, 0.62 mmol, 2.5:1 trans:cis), scandium(III) trifluoromethanesulfonate (121 mg, 0.25 mmol), pivalic anhydride (0.16 mL, 0.80 mmol), and acetonitrile (2.5 mL) provided trans pivalate 23a (110 mg, 39% yield) and cis pivalate 23b (44 mg, 16% yield) each as clear oils after column chromatography in a total yield of 55%. 1H NMR (500 MHz, CDCl3): δ 6.49 (s, 1H, NH), 6.08 (d, J = 2.6 Hz, 1H, =CH), 5.38 (d, J = 2.6 Hz, 1H, =CH), 5.37* (d, J = 2.4 Hz, 1H, =CH), 4.08–4.00 (m, 1H, CH), 3.05–3.01 (m, 1H, CH), 2.53 (s, 1H, °CH), 2.53* (s, 1H, °CH), 2.15–1.76 (m, 4H, CH2CH2), 1.67 (s, 3H, CH3), 1.18 (s, 9H, C(CH3)3), 1.02 (s, 21H, Si(CH(CH3)2)3); 13C NMR (125 MHz, CDCl3): δ 176.6, 169.4, 141.83, 141.81*, 117.0, 116.9*, 105.75, 105.74*, 86.4, 83.43, 83.40*, 73.9, 73.8*, 73.65, 73.60*, 49.0, 48.9*, 47.0, 39.3, 38.73*, 38.69, 38.57*, 27.9, 27.8*, 27.3 (3C), 27.2* (3C), 26.51, 26.49*, 18.7 (6C), 11.2 (3C), *Discernible signals for one of two diastereomers; IR (thin film) 2943, 2866, 1709, 1660, 1462, 1367, 1325, 1285, 1101 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C27H44NO3Si 458.3090; Found 458.3059; TLC Rf = 0.37 (25% EtOAc/hexanes), visualized with UV and KMnO4.
3-Methyl-5-((2S*,3R*)-4-methylene-5-oxo-2-((triisopropylsilyl)ethynyl)pyrrolidin-3-yl)pent-1-yn-3-yl pivalate (23b).
Propargyl pivalate 23b was prepared according to General Procedure D. Run 3: Propargyl alcohol 21a,b (1.07 g, 2.87 mmol, 4:1 trans:cis), pivalic anhydride (0.70 mL, 3.5 mmol), scandium (III) trifluoromethanesulfonate (565 mg, 1.15 mmol), and acetonitrile (12 mL) provided trans lactam 23a (565 mg, 43% yield), cis lactam 23b (139 mg, 11% yield), and a 2:1 trans:cis mixture (72 mg, 5% yield) each as a clear oil after column chromatography (gradient elution with 5–25% ethyl acetate in hexanes). The ratios of 23a:23b was based upon integrated values of trans-lactam methylene (23a, =CH at 5.38 ppm) and cis-lactam methylene (23b, =CH at 5.40 ppm). 1H NMR (500 MHz, CDCl3): δ 6.11–6.07 (m, 1H, =CH), 5.84 (br s, 1H, NH), 5.40 (d, J = 2.0 Hz, 1H, =CH), 5.38* (d, J = 1.5 Hz, 1H, =CH), 4.53–4.49 (m, 1H, CH), 3.05–3.02 (m, 1H, CH), 2.52 (s, 1H, °CH), 2.51*(s, 1H, °CH), 2.08–1.88 (m, 4H, CH2CH2), 1.68 (s, 3H, CH3), 1.66*(s, 3H, CH3), 1.183 (s, 9H, C(CH3)3), 1.177*(s, 9H, C(CH3)3), 1.04 (s, 21H, Si(CH(CH3)2)3), 1.03*(s, 21H, Si(CH(CH3)2)3), 13C NMR (125 MHz, CDCl3:) δ 176.7, 169.85, 169.77*, 141.65, 141.61*, 117.3, 117.2*, 103.04, 102.99*, 89.0, 88.9*, 83.9, 83.4*, 74.3, 73.9*, 73.8, 73.4*, 47.9, 42.8, 42.6*, 39.4, 39.3*, 39.0, 38.8*, 27.20 (3C), 27.19*, 26.6, 26.3*, 25.0, 24.8*, 18.7 (6C), 11.2 (3C), *Discernible signals for one of two diastereomers; IR (thin film) 3270, 2909, 2832, 2219, 2150, 1684, 1640, 1445, 1308 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C27H44NO3Si 458.3085; Found 458.3095. TLC Rf = 0.29 (25% EtOAc/hexanes), visualized with UV and KMnO4.
General Procedure E:
3,3-Disubstituted Allene Formation From Propargyl Pivalate Using (Triphenylphosphine)copper Hydride Hexamer.
A flame-dried, 15-mL, round-bottomed flask equipped with a stir bar, septum, and nitrogen inlet needle was charged with propargyl pivalate 22a (107 mg, 0.272 mmol), toluene (7 mL), and deionized water (0.01 mL). This solution was degassed by bubbling nitrogen through the solution for 10 min, then the solution was cooled to −10 °C with a slurry of ice and sodium chloride. (Triphenylphosphine)copper hydride hexamer (533 mg, 0.272 mmol) was weighed in a glove box under a N2 atmosphere into a weighing boat, removed from the glovebox and exposed to air for ~1 min. The septum was removed from the flask, and the (triphenylphosphine)copper hydride hexamer was added in a single portion. The flask was evacuated and filled with nitrogen (3x) and stirred under nitrogen for 2 h. The reaction progress was monitored by removing aliquots from the reaction and measuring the disappearance of the terminal alkyne proton at 2.5 ppm by 1H NMR. Upon completion, the mixture was poured into a cooled (0 °C) solution of saturated ammonium chloride. The mixture was diluted with diethyl ether (10 mL) and stirred open to air for 30 min, then poured into a separatory funnel and extracted with diethyl ether (3 × 10 mL). The organic layers were dried over magnesium sulfate, filtered through a plug of silica gel eluting with diethyl ether, concentrated by rotary evaporation, then purified by silica gel flash column chromatography eluting with 15–30% ethyl acetate in hexanes to give allene 24a (24 mg, 33%) as a clear oil.
When performing this reaction with a previously opened container of Stryker’s reagent purchased from Sigma-Aldrich (90%) that was stored in the glovebox, reduction of the α-methylene group was not observed. When a newly opened bottle of Stryker’s reagent purchased from Acros (97%) was used, significant amounts of the over-reduction product (24a:31a;1:1) were obtained when performing the reaction at rt; however, lowering the temperature to −10 °C gave better ratios (24a:31a;10:1). Temperature was a major contributing factor to this product ratio as all other commercial sources and batches of Stryker’s reagent (Sigma-Aldrich 90%, Acros 97%), or reagent that was freshly synthesized from copper(II) acetate, triphenylphosphine, and diphenylsilane68 afforded little over-reduction product when the reaction was conducted at −10 °C. The sources of Stryker’s reagent varied in appearance: Acros (97%) was bright to dark orange depending on batch, Sigma-Aldrich (90%) was brick red to light brown depending on batch, and freshly prepared Stryker’s reagent68 was a reddish orange color similar to that purchased from Acros.
(4S*,5S*)-3-Methylene-4-(3-methylpenta-3,4-dienyl)-5-(phenylethynyl)pyrrolidin-2-one (24a).
1H NMR (400 MHz, CDCl3): δ 7.41–7.38 (m, 2H, Ph-H), 7.33–7.30 (m, 3H, Ph-H), 6.77 (br s, 1H, NH), 6.11 (d, J = 2.2 Hz, 1H, =CH), 5.41 (d, J = 2.2 Hz, 1H, =CH), 4.66–4.64 (m, 2H, =CH2), 4.26 (d, J = 4.0 Hz, 1H, CH), 3.17–3.16 (m, 1H, CH), 2.15–2.08 (m, 2H), 1.92–1.86 (m, 1H), 1.81–1.74 (m, 1H), 1.71 (t, J = 3.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 206.1, 169.7, 142.2, 131.8 (2C), 128.8, 128.5 (2C), 122.3, 117.3, 97.7, 87.8, 84.5, 75.3, 49.0, 46.4, 31.9, 29.9, 19.1; IR (thin film) 2922, 1741, 1736, 1445, 1372, 1270, 1216, 1110, 1042 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C19H20NO 278.1539; Found 278.1533; TLC Rf = 0.44 (50% EtOAc/hexanes), visualized with UV and vanillin.
(4S*,5S*)-1-Methyl-3-methylene-4-(3-methylpenta-3,4-dien-1-yl)-5-(phenylethynyl) pyrrolidin-2-one (30a):
Prepared according to General Procedure E. Propargyl acetate 29a (69 mg, 0.20 mmol), (triphenylphosphine)copper hydride hexamer (385 mg, 0.196 mmol), toluene (5.0 mL), and deionized water (0.01 ml) provided allene 30a (30 mg, 53% yield) as a clear oil after column chromatography (gradient elution with 10–20% ethyl acetate in hexanes). Some baseline impurities from triphenylphosphine byproducts of Stryker’s reagent were observed in the aromatic regions of the 1H and 13C NMR spectra. 1H NMR (500 MHz, CDCl3): δ 7.41–7.40 (m, 2H, Ph-H), 7.34–7.30 (m, 3H, Ph-H), 6.07 (d, J = 1.5 Hz, 1H, =CH), 5.35 (s, 1H, =CH), 4.66– 4.64 (m, 2H, =CH2), 4.11 (d, J = 4.0 Hz, 1H, CH), 3.10 – 3.01 (m, 1H, CH), 3.03 (s, 3H, CH3), 2.14–2.08 (m, 2H), 1.90–1.84 (m, 1H), 1.77–1.73 (m, 1H), 1.71 (t, J = 3.0 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 201.2, 167.3, 142.4, 131.9 (2C), 128.9, 128.5 (2C), 122.2, 116.3, 97.7, 86.2, 85.7, 75.2, 55.5, 44.2, 32.2, 30.0, 28.5, 19.1; IR (thin film) 2078, 1633, 1414, 1381, 1260, 1058 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C20H22NO 292.1701; Found 292.1712; TLC Rf = 0.66 (50% EtOAc/hexanes), visualized with UV and KMnO4.
(4R*,5R*)-3-Methyl-4-(3-methylpenta-3,4-dien-1-yl)-5-(phenylethynyl)pyrrolidin-2-one (31a)
The structure of the reduced product 31a was not confirmed due to our inability to separate this compound from 24a during the column chromatography; however, two resonances in the 1H NMR at 1.03 ppm (d, J = 7.0 Hz, 3H, CH3) and 4.23 ppm (d, J = 4.5 Hz, 1H, CHNH) closely match that of lactams 32a and 56.
(4R*,5S*)-3-Methyl-4-(3-methylpenta-3,4-dien-1-yl)-5-(phenylethynyl)pyrrolidin-2-one (57).
Prepared according to General Procedure E. Cis lactam 22b (75 mg, 0.19 mmol), (triphenylphosphine)copper hydride hexamer (373 mg, 0.19 mmol), toluene (5 mL), and deionized water (0.01 mL) provided allene 57 (19 mg, 36% yield) after column chromatography (gradient elution with 15–25% ethyl acetate in hexanes) as a clear oil and as the only product. 1H NMR (500 MHz, CDCl3): δ 7.40–7.38 (m, 2H, Ph-H), 7.33–7.31 (m, 3H, Ph-H), 5.77 (br s, 1H, NH), 4.64–4.61 (m, 2H, =CH2), 4.51 (d, J = 7.0 Hz, 1H, CH), 2.27–2.14 (m, 3H), 2.03–1.98 (m, 1H), 1.92–1.84 (m, 2H), 1.71 (t, J = 3.0 Hz, 3H, CH3), 1.20 (d, J = 7.0 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 206.3, 179.8, 131.8 (2C), 128.8, 128.5 (2C), 122.5, 97.8, 86.4, 85.3, 74.7, 47.6, 46.8, 40.3, 31.2, 28.0, 18.8, 13.8; IR (thin film) 3245, 2895, 1937, 1681, 1426, 1361 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C19H22NO 280.1696; Found 280.1698; TLC Rf = 0.33 (50% EtOAc/hexanes), visualized using UV and vanillin.
(4S*,5S*)-3-Methylene-4-(3-methylpenta-3,4-dienyl)-5-((triisopropylsilyl)ethynyl)pyrrolidin-2-one (25a).
Prepared according to General Procedure E using a reaction temperature of −20 °C. Propargyl pivalate 23a (110 mg, 0.24 mmol), (triphenylphosphine)copper hydride hexamer (424 mg, 0.22 mmol), and toluene (6 mL) yielded allenes 25a (39 mg, 46% yield) and 32a (9 mg, 11% yield) in a 4:1 ratio each as a clear oil after column chromatography (gradient elution with 5–25% ethyl acetate in hexanes). Other runs gave allene 25a in 31–53% yield along with 32a 0–31% yield. Baseline impurities in the alkenyl and aryl region were present in 1H and 13C NMR. 1H NMR (400 MHz, CDCl3): δ 6.24 (br s, 1H, NH), 6.05 (d, J = 2.4 Hz, 1H, =CH), 5.36 (d, J = 2.4 Hz, 1H, =CH), 4.66–4.63 (m, 2H, =CH2), 4.03, (d, J = 4.8 Hz, 1H, CH), 3.08–3.03 (m, 1H, CH), 2.16–2.09 (m, 1H), 2.06–1.99 (m, 1H), 1.93–1.84 (m, 1H), 1.72–1.61 (m, 1H) 1.69 (t, J = 3.0 Hz, 3H, CH3), 1.04 (s, 21H, Si(CH(CH3)2)3); 13C NMR (100 MHz, CDCl3): δ 206.0, 169.5, 142.1, 117.0, 106.2, 97.9, 86.1, 75.4, 49.1, 47.0, 31.7, 30.1, 19.2, 18.7 (6C), 11.2 (3C); IR (thin film) 2926, 2865, 1704, 1658, 1462, 1324 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C22H36NOSi 358.2561; Found 358.2566; TLC Rf = 0.32 (25% EtOAc/hexanes), visualized with UV and KMnO4.
(4S*,5S*)-3-Methyl-4-(3-methylpenta-3,4-dien-1-yl)-5-((triisopropylsilyl)ethynyl) pyrrolidin-2-one (32a).
The diastereomeric ratio was based upon integrated values of trans-lactam methine (32a, CHNH at 4.02 ppm) and cis-lactam methine (CHNH at 3.95 ppm). Peaks reported for major diastereomer only. 1H NMR (500 MHz, CDCl3): δ 5.77 (br s, 1H, NH), 4.64–4.62 (m, 2H, =CH2), 3.95 (d, J = 8.0 Hz, 1H, CH), 2.18–2.01 (m, 4H), 1.80–1.74 (m, 1H), 1.69 (t, J = 2.7 Hz, 3H, CH3), 1.69–1.60 (m, 1H), 1.24 (d, J = 10.5 Hz, 3H, CH3), 1.05 (s, 21H, Si(CH(CH3)2)3); 13C NMR (125 MHz, CDCl3): δ 206.0, 178.5, 106.3, 98.2, 85.9, 75.3, 51.5, 49.7, 42.4, 31.1, 31.0, 19.1, 18.7 (6C), 15.1, 11.3 (3C); IR (thin film) 2908, 2831, 2156, 1961, 1685, 1443 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C22H38NOSi 360.2717; Found 360.2708; TLC Rf = 0.38 (35% EtOAc/hexanes), visualized with UV and vanillin.
General Procedure F:
Terminal Alkyne Formation from Triisopropylsilyl Substituted Alkyne using Tetra-n-butylammonium Fluoride.
Run 1: A flame-dried, 5-mL, round-bottomed flask equipped with a stir bar, septum, and nitrogen inlet needle was charged with TIPS allene 25a (38 mg, 0.11 mmol, pure trans isomer) dissolved in THF (1.1 mL) and cooled to 0 °C in an ice water bath. Tetra-n-butylammonium fluoride (1 M solution in THF, 0.15 mL, 0.15 mmol) was added dropwise via syringe and the reaction was stirred for 45 min when TLC showed consumption of the starting material. Saturated aqueous ammonium chloride (2 mL) was added to the cooled reaction mixture and the biphasic mixture was transferred to a separatory funnel. The aqueous layer was extracted with ethyl acetate (4 × 5 mL). The combined organic layers were washed with brine (1 × 5 mL), dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation, and purified by silica gel flash column chromatography (gradient elution with 25–75% ethyl acetate in hexanes) to provide allene 26 (12 mg, 57% yield) as a clear oil.
This reaction was repeated four times with yields ranging from 57 to 93% with an average yield of 79%. Run 2: TIPS allene 25a (40 mg, 0.11 mmol, trans isomer), tetra-n-butylammonium fluoride (1 M solution in THF, 0.17 mL, 0.17 mmol), THF (1.1 mL) provided allene 26 (18 mg, 82% yield) as a clear oil after column chromatography
(4S*,5S*)-5-Ethynyl-3-methylene-4-(3-methylpenta-3,4-dien-1-yl)pyrrolidin-2-one (26).
1H NMR (400 MHz, CDCl3): δ 6.79 (br s, 1H, NH), 6.07 (d, J = 2.2 Hz, 1H, =CH), 5.38 (d, J = 2.2 Hz, 1H, =CH), 4.65–4.63 (m, 2H, =CH2), 4.02 (dd, J = 2.0, 4.0 Hz, 1H, CH), 3.09–3.05 (m, 1H, CH), 2.39 (d, J = 2.0 Hz, 1H, °CH), 2.10–2.02 (m, 2H), 1.88–1.80 (m, 1H), 1.73–1.71 (m, 1H), 1.70 (t, J = 3.0 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 206.1, 169.7, 141.9, 117.4, 97.6, 82.7, 75.3, 72.9, 48.1, 46.1, 31.9, 29.8, 19.0; IR (thin film) 2094, 1936, 1633, 1411, 1308, 1061 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C13H16NO 202.1226; Found 202.1227; TLC Rf = 0.36 (50% EtOAc/hexanes), visualized using UV and p-anisaldehyde.
(4S*,5S*)-5-Ethynyl-3-methyl-4-(3-methylpenta-3,4-dien-1-yl)pyrrolidin-2-one (56).
Prepared according to General Procedure F. Lactam 32a (26 mg, 0.072 mmol, 9:1 dr), tetra-n-butylammonium fluoride (1 M in THF, 0.11 mL, 0.10 mmol), and THF (0.8 mL) yielded terminal alkyne 56 (14 mg, 86% yield, 9:1 dr) as a clear oil after column chromatography (gradient elution with 25–50% ethyl acetate in hexanes). The diastereomeric ratio was based upon integrated values of trans-lactam methine (56, CHNH at 3.92 ppm) and cis-lactam methine (CHNH at 3.93 ppm). 1H NMR (500 MHz, CDCl3): δ 6.04 (br s, 1H, NH), 4.65–4.63 (m, 2H, =CH2), 3.92 (dd, J = 7.5, 1.7 Hz, 1H, CH), 2.39 (d, J = 1.7 Hz, 1H, °CH), 2.17–2.01 (m, 4H), 1.79–1.68 (m, 2H), 1.70 (t, J = 3.2 Hz, 3H, CH3), 1.24 (d, J = 7.0 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ 206.1, 178.7, 98.0, 82.8, 75.3, 72.8, 50.6, 48.8, 42.2, 31.0, 30.7, 19.0, 15.3; IR (thin film) 3190, 2889, 2060, 1936, 1679, 1440, 1364 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C13H18NO 204.1383; Found 204.1378; TLC Rf = 0.24 (50% EtOAc/hexanes), visualized using UV and vanillin.
General Procedure G:
N-Arylated Lactam Formation using Buchwald-Hartwig Cross Coupling Conditions.
This procedure is modified from the method reported for the amidation of aryl halides.38 A flame-dried, 10-mL, test tube equipped with a magnetic stir bar was charged with copper iodide (4 mg, 0.02 mmol) and cesium carbonate (50 mg, 0.15 mmol). The test tube was capped with a septum, then evacuated and filled with nitrogen (3x). Iodobenzene (13 μL, 0.11 mmol) and trans-N,N’-dimethylcyclohexane-1,2-diamine (8 μL, 0.05 mmol), were added via syringe. Lactam 24a (21 mg, 0.076 mmol) was dissolved in toluene (0.8 mL) and added to this test tube via syringe. The test tube was evacuated and filled with nitrogen (3x), and the nitrogen inlet needle was removed. The test tube was then placed in an oil bath (preheated 80 °C) for 20 h at which point TLC showed a new spot. The reaction was allowed to cool to rt, filtered through a plug of silica gel eluting with 50% ethyl acetate in hexanes, concentrated, then purified by silica gel flash column chromatography eluting with 5–15% ethyl acetate in hexanes to give lactam 37 as a clear oil (12 mg, 44% yield).
Changing the ligand to N,N’-dimethylethylenediamine and the base to potassium carbonate afforded a lower yield with recovery of starting material. Lactam 24a (10 mg, 0.033 mmol), iodobenzene (5 μL, 0.04 mmol), copper iodide (5 mg, 0.03 mmol), trans-N,N’-dimethylcyclohexane-1,2-diamine (4 μL, 0.03 mmol) potassium carbonate (10 mg, 0.065 mmol), and toluene (0.4 mL) provided lactam 37 (3 mg, 27% yield, PAJ 8–6) as a clear oil after column chromatography.
(4S*,5S*)-3-Methylene-4-(3-methylpenta-3,4-dien-1-yl)-1-phenyl-5-(phenylethynyl)pyrrolidin-2-one (37).
1H NMR (400 MHz, CDCl3): δ 7.77–7.75 (m, 2H, Ph-H), 7.44–7.40 (m, 2H, Ph-H), 7.30–7.20 (m, 6H, Ph-H), 6.25 (d, J = 2.2 Hz, 1H, =CH), 5.51 (d, J = 2.2 Hz, 1H, =CH), 4.69–4.66 (m, 3H), 3.26–3.23 (m, 1H, CH), 2.18–2.13 (m, 2H), 1.91–1.83 (m, 2H), 1.73 (t, J = 3.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 206.2, 166.3, 143.0, 138.3, 131.8 (2C), 129.0 (2C), 128.8 (2C), 128.4, 125.7, 122.2 (2C), 118.4, 97.6, 87.1, 85.8, 76.8, 75.4, 55.1, 44.6, 32.5, 30.0, 19.1; IR (thin film) 2924, 2227, 1959, 1701, 1660, 1498, 1372 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C25H24NO 354.1852; Found 354.1837; TLC Rf = 0.86 (50% EtOAc/hexanes), visualized with UV and vanillin.
4-((4S*,5S*)-3-Methylene-4-(3-methylpenta-3,4-dien-1-yl)-2-oxo-5-(phenylethynyl)pyrrolidin-1-yl)benzonitrile (38).
Prepared according to General Procedure G. Lactam 24a (33 mg, 0.12 mmol), 4-iodobenzonitrile (38 mg, 0.17 mmol), copper iodide (5 mg, 0.02 mmol), N,N’-dimethylethylenediamine (5 μL, 0.05 mmol), cesium carbonate (78 mg, 0.24 mmol), and toluene (1.2 mL) provided lactam 38 (19 mg, 42% yield) as a pale yellow oil after column chromatography (gradient elution with 5–15% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3): δ 7.99 (app dd, J = 2.0, 6.8 Hz, 2H, Ph-H), 7.70 (app dd, J = 2.0, 6.8 Hz, 2H, Ph-H), 7.33–7.28 (m, 5H, Ph-H), 6.30 (d, J = 2.2 Hz, 1H, =CH), 5.59 (d, J = 2.2 Hz, 1H, =CH), 4.71 (d, J = 3.2 Hz, 1H, CH), 4.68–4.66 (m, 2H, =CH2), 3.28–3.27 (m, 1H, CH), 2.17–2.13 (m, 2H), 1.89–1.83 (m, 2H), 1.73 (t, J = 3.2 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 206.2, 166.5, 142.2, 133.0 (2C), 131.8 (2C), 129.2, 128.5 (2C), 121.6, 121.0 (2C), 120.0, 118.9, 108.11, 97.4, 86.5, 85.9, 75.5, 54.4, 44.3, 32.4, 29.9, 19.1; IR (thin film) 3432, 2101, 1642, 1511 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C26H23N2O 379.1732; Found 379.1735; TLC Rf = 0.43 (25% EtOAc/hexanes), visualized with UV and vanillin.
(4S*,5S*)-3-Methylene-4-(3-methylpenta-3,4-dien-1-yl)-5-(phenylethynyl)-1-(4-(trifluoromethyl)phenyl)pyrrolidin-2-one (39).
Prepared according to General Procedure G. Lactam 24a (24 mg, 0.087 mmol), 4-benzotrifluoromethyl iodide (0.02 mL, 0.1 mmol), copper iodide (2 mg, 0.02 mmol), N,N’-dimethylethylenediamine (4 μL, 0.04 mmol), cesium carbonate (57 mg, 0.17 mmol), and toluene (0.9 mL) provided lactam 39 (17 mg, 47%) as a clear oil after column chromatography (gradient elution with 5–10% ethyl acetate in hexanes). When potassium carbonate was used only 14–16% of lactam 39 was isolated and 20–25% of the starting material was recovered (PAJ 8–100, PAJ 8–192). 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 8.6 Hz, 2H, Ph-H), 7.67 (d, J = 8.6 Hz, 2H, Ph-H), 7.32–7.28 (m, 5H, Ph-H), 6.29 (d, J = 2.0 Hz, 1H, =CH), 5.56 (d, J = 2.0 Hz, 1H, =CH), 4.72 (d, J = 3.2 Hz, 1H, CH), 4.68–4.66 (m, 2H, =CH2), 3.29–3.26 (m, 1H, CH), 2.19–2.13 (m, 2H), 1.90–1.83 (m, 2H), 1.73 (t, J = 3.0 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 206.2, 166.5, 142.5, 141.3, 131.8 (2C), 129.1 (2C), 128.5 (2C), 127.2, 126.08 (q, J = 4.0 Hz, 1C), 125.6, 121.9, 121.2 (2C), 119.5, 97.5, 86.3, 75.5, 54.7, 44.5, 32.5, 29.9, 19.1; IR (thin film) 2942, 2381, 1715, 1325, 1213, 1122, 1068 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C26H23F3NO 422.1726; Found 422.1710; TLC Rf = 0.86 (50% EtOAc/hexanes), visualized with UV and vanillin.
(4S*,5S*)-1-(4-Methoxyphenyl)-3-methylene-4-(3-methylpenta-3,4-dien-1-yl)-5-(phenylethynyl)pyrrolidin-2-one (40).
Prepared according to General Procedure G. Lactam 24a (29 mg, 0.11 mmol), 4-iodoanisole (37 mg, 0.16 mmol), copper iodide (4 mg, 0.02 mmol), N,N’-dimethylethylenediamine (5 μL, 0.04 mmol), cesium carbonate (68 mg, 0.21 mmol), and toluene (1 mL) provided lactam 40 (25 mg, 63% yield) as a pale yellow oil after column chromatography (gradient elution with 5–20% ethyl acetate in hexanes).
Performing this reaction with potassium carbonate as a base: Lactam 24a (20 mg, 0.072 mmol), 4-iodoanisole (20 mg, 0.087 mmol), copper iodide (3 mg, 0.01 mmol), N,N’-dimethylethylenediamine (4 μL, 0.03 mmol), potassium carbonate (20 mg, 0.14 mmol), and toluene (0.72 mL) provided lactam 40 (5 mg, 19% yield) as a pale yellow oil and starting material 24a (5 mg, 25%) after column chromatography. 1H NMR (500 MHz, CDCl3): δ 7.60 (d, J = 8.7 Hz, 2H, Ph-H), 7.32–7.27 (m, 5H, Ph-H), 6.94 (d, J = 8.7 Hz, 2H, Ph-H), 6.22 (d, J = 1.5 Hz, 1H, =CH), 5.48 (d, J = 1.5 Hz, 1H, =CH), 4.68–4.66 (m, 2H, =CH2), 4.62 (d, J = 3.0 Hz, 1H, CH), 3.82 (s, 3H, OCH3), 3.27–3.21 (m, 1H, CH), 2.18–2.13 (m, 2H), 1.93–1.82 (m, 2H), 1.73 (t, J = 2.7 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 206.3, 166.3, 157.7, 143.1, 131.8 (2C), 131.3, 128.8, 128.4 (2C), 124.5 (2C), 122.3, 117.8, 114.3 (2C), 97.7, 87.3, 85.8, 75.3, 55.7, 55.6, 44.7, 32.5, 30.0, 19.1; IR (thin film) 2924, 1995, 1697, 1512, 1377, 1300, 1249 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C26H26NO2 384.1958; Found 384.1968; TLC Rf = 0.59 (50% EtOAc/hexanes), visualized with UV and Vanillin.
(4S*,5S*)-3-Methylene-4-(3-methylpenta-3,4-dien-1-yl)-5-(phenylethynyl)-1-(thiophen-2-yl)pyrrolidin-2-one (42).
Prepared according to General Procedure G. Run 1: Lactam 24a (21 mg, 0.076 mmol), copper iodide (3 mg, 0.02 mmol), trans-N,N’-dimethylcyclohexane-1,2-diamine (5 μL, 0.03 mmol), cesium carbonate (50 mg, 0.15 mmol), 2-iodothiophene (12 μL, 0.10 mmol), and toluene (0.8 mL) provided lactam 42 (15 mg, 56% yield) as a pale yellow oil after column chromatography (5–10% ethyl acetate in hexanes gradient elution). Run 2: Lactam 24a (23 mg, 0.083 mmol), copper iodide (3 mg, 0.02 mmol), trans-N,N’-dimethylcyclohexane-1,2-diamine (4 μL, 0.03 mmol), cesium carbonate (52 mg, 0.16 mmol), iodothiophene (10 μL, 0.11 mmol), and toluene (0.8 mL) provided lactam 42 (19 mg, 63% yield) as a pale yellow oil after column chromatography. 1H NMR (400 MHz, CDCl3): δ 7.38–7.36 (m, 2H, Ph-H), 7.32–7.28 (m, 3H, Ph-H), 7.03–7.02 (m, 1H, HetAr-H), 6.97–6.94 (m, 2H, HetAr-H), 6.27 (d, J = 2.0 Hz, 1H, =CH), 5.54 (d, J = 2.0 Hz, 1H, =CH), 4.70 (d, J = 2.8 Hz, 1H, CH), 4.68–4.66 (m, 2H, =CH2), 3.34–3.29 (m, 1H, CH), 2.15–2.11 (m, 2H), 1.84 (m, 2H), 1.73 (t, J = 3.2 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 206.2, 164.3, 141.5, 139.4, 131.9 (2C), 129.0, 128.5 (2C), 124.2, 122.0, 119.3, 119.2, 113.0, 97.4, 86.1, 86.0, 75.4, 55.4, 44.9, 33.2, 29.8, 19.1; IR (thin film) 2924, 2227, 1959, 1695, 1534, 1451, 1399 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C23H22NOS 360.1417; Found 360.1401; TLC Rf = 0.58 (25% EtOAc/hexanes), visualized with UV and p-anisaldehyd
(4S*,5S*)-3-Methylene-4-(3-methylpenta-3,4-dien-1-yl)-5-(phenylethynyl)-1-tosylpyrrolidin-2-one (43):
Run 1: A flame-dried test tube equipped with a stir bar, septum, and nitrogen inlet needle was charged with lactam 24a (20 mg, 0.07 mmol) and DMF (0.75 mL) and cooled to 0 °C under a nitrogen atmosphere. Sodium hydride (60% dispersion in mineral oil, 6 mg, 0.14 mmol) was added in a single portion and the reaction mixture was stirred for 15 min at 0 °C. para-Toluene sulfonyl chloride (28 mg, 0.14 mmol) was added in a single portion, the ice bath was removed, and the solution was allowed to warm to rt for 2.5 h. TLC analysis revealed consumption of the starting material. The reaction mixture was poured into a separatory funnel containing saturated aqueous ammonium chloride (5 mL). The aqueous layer was extracted with ethyl acetate (3 × 10 mL). The organic layers were combined, dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation, and purified by silica gel flash column chromatography eluting with 10–25% ethyl acetate in hexanes. Lactam 43 was obtained as a pale yellow oil (9 mg, 29% yield). Run 2: This reaction was repeated, but the temperature was maintained at 0 °C for 1 h after addition of para-toluene sulfonyl chloride, then allowed to warm to rt for only 1 h before workup. Lactam 24a (18 mg, 0.069 mmol), sodium hydride (60% dispersion in mineral oil, 5 mg, 0.1 mmol), para-toluene sulfonyl chloride (25 mg, 0.13 mmol), and DMF (0.7 mL) provided lactam 43 (14 mg, 48% yield) as a yellow oil after column chromatography; unidentified contaminants are observed in the 1H NMR. Additional purification by column chromatography provided allene-yne 43 (4 mg, 18% yield) as a clear oil. 1H NMR (500 MHz, CDCl3): δ 8.08 (d, J = 8.0 Hz, 2H, Ph-H), 7.25–7.28 (m, 5H, Ph-H), 7.22 (d, J = 8.0 Hz, 2H, Ph-H), 6.23 (d, J= 1.7 Hz, 1H, =CH), 5.54 (d, J = 1.7 Hz, 1H, =CH), 4.98 (d, J = 1.5 Hz, 1H, CH), 4.68–4.67 (m, 2H, =CH2), 3.11–3.08 (m, 1H, CH), 2.38 (s, 3H, CH3), 2.08–2.05 (m, 2H), 1.88–1.84 (m, 1H), 1.74–1.69 (m, 1H), 1.71 (t, J = 3.3 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 206.3, 164.9, 145.3, 141.1, 135.6, 131.9 (2C), 131.7, 129.8 (2C), 129.5 (2C), 128.5 (2C), 122.6, 121.9, 97.2, 86.3, 85.9, 75.4, 53.6, 45.5, 32.8, 29.8, 21.8, 19.0; IR (thin film) 2891, 2218, 1937, 1697, 1579, 1429, 1356, 1160, 1078, 804, 750 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C26H26NO3S 432.1628; Found 432.1641; TLC Rf = 0.66 (25% EtOAc/hexanes), visualized with UV and vanillin.
tert-Butyl (4S*,5S*)-3-methylene-4-(3-methylpenta-3,4-dien-1-yl)-2-oxo-5-(phenylethynyl) pyrrolidine-1-carboxylate (44):
Run 1: A 5-mL, flame-dried, test tube equipped with a stir bar, septum, and nitrogen inlet needle was charged with allene-yne 24a (15 mg, 0.054 mmol) and CH2Cl2 (0.3 mL). 4-Dimethylaminopyridine (1 mg, 0.008 mmol) was added and the solution was cooled to 0 °C. Triethylamine (0.08 mL, 0.5 mmol) was added dropwise followed by addition of di-tert-butyl dicarbonate (59 mg, 0.27 mmol) in a single portion. The ice bath was removed, and the solution was allowed to warm to rt and stirred for 2 h. After consumption of the starting material was observed by TLC, the solution was diluted with CH2Cl2 (15 mL), transferred to a separatory funnel, and washed sequentially with saturated aqueous ammonium chloride (5 mL) and brine (5 mL). The organic layer was dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation, and purified by silica gel flash column chromatography (gradient elution with 5–25% ethyl acetate in hexanes) to provide allene-yne 44 (8 mg, 40% yield) as a clear oil. Run 2: Lactam 24a (23 mg, 0.083 mmol), dimethylaminopyridine (1 mg, 0.008 mmol), triethylamine (0.12 mL, 0.83 mmol), di-tert-butyl dicarbonate (90 mg, 0.41 mmol), and CH2Cl2 (0.4 mL) provided lactam 44 (17 mg, 54% yield) as a pale yellow oil after column chromatography. 1H NMR (500 MHz, CDCl3): δ 7.38–7.36 (m, 2H, Ph-H), 7.31–7.29 (m, 3H, Ph-H), 6.30 (d, J = 1.7 Hz, 1H, =CH), 5.54 (d, J = 1.7 Hz, 1H, =CH), 4.67–4.65 (m, 3H), 3.08–3.05 (m, 1H, CH), 2.07–2.05 (m, 2H), 1.80–1.71 (m, 2H), 1.71 (t, J = 3.2 Hz, 3H, CH3), 1.57 (s, 9H, C(CH3)3); 13C NMR (125 MHz, CDCl3): δ 206.3, 165.6, 149.9, 142.1, 131.8 (2C), 128.7, 128.5 (2C), 122.4, 121.6, 97.4, 87.5, 84.0, 83.7, 75.3, 52.8, 43.6, 32.8, 29.8, 28.2 (3C), 19.0; IR (thin film) 2969, 2915, 1793, 1759, 1717, 1401, 1302, 1153 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C24H28NO3 378.2069; Found 378.2082; TLC Rf = 0.69 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
General Procedure H:
Fused 5,7,5-ring Structure Formation Using Rh(I)-Catalyzed Allenic Pauson–Khand Reaction Conditions (Slow Addition of Allene-yne to Rhodium Catalyst).
Run 1: A flame-dried, 10-mL, 2-necked, round-bottomed flask equipped with a magnetic stir bar, a condenser capped with septum, and a septum was charged with rhodium biscarbonyl chloride dimer ([Rh(CO)2Cl]2, 2 mg, 0.004 mmol) and toluene (3.4 mL). The apparatus was placed under vacuum for 2–3 seconds by piercing the septum with a needle attached to a Schlenk line then filled with carbon monoxide via a needle attached to a balloon filled with carbon monoxide. This process was repeated three times. The reaction flask was lowered into an oil bath (preheated to 110 °C). Allene-yne 24a (13 mg, 0.044 mmol) was dissolved in toluene (1.1 mL) and added dropwise to the stirring solution of rhodium biscarbonyl chloride dimer over 1 h using a syringe pump. After the addition was complete, the reaction was stirred an additional 30 min at 110 °C at which time TLC showed consumption of the starting material. The flask was removed from the oil bath and allowed to cool to rt, then polymer bound triphenylphosphine (3 mmol/g, 60 mg) was added, and the reaction stirred (4 h). The polymer was removed by vacuum filtration, the solution was concentrated, and the residue purified by silica gel flash column chromatography with gradient elution using 25–75% ethyl acetate in hexanes to yield lactam 45 as a white solid (9 mg, 64% yield). Run 2: Allene-yne 24a (21 mg, 0.076 mmol), rhodium biscarbonyl chloride dimer (3 mg, 0.008 mmol), and toluene (7.6 mL) provided lactam 45 (19 mg, 79% yield) as a white solid after column chromatography. Compound 45 was crystallized by vapor diffusion using ethyl acetate and pentanes, and X-ray crystallography confirmed compound structure 45.
General Procedure I:
Fused 5,7,5-ring Structure Formation Using Rh(I)-Catalyzed Allenic Pauson–Khand Reaction Conditions.
Run 3: To a flame-dried, 2-necked, 25-mL, round-bottomed flask equipped with a septum, condenser capped with a septum, and a magnetic stir bar was added rhodium biscarbonyl chloride dimer (4 mg, 0.01 mmol) and toluene (7 mL). The apparatus was placed under vacuum for 2–3 seconds by piercing the septum with a needle attached to a Schlenk line then filled with carbon monoxide via a needle attached to a balloon filled with carbon monoxide. This process was repeated three times. The reaction flask was lowered into an oil bath (preheated to 110 °C). Allene 24a (30 mg, 0.11 mmol) was dissolved in toluene (4 mL) and added to the stirring solution of rhodium catalyst via syringe in a single portion. Consumption of the starting material was evident by TLC after 1 h, and the reaction mixture was concentrated by rotary evaporation. The crude residue was purified by silica gel flash column chromatography eluting with a gradient of 30–75% ethyl acetate in hexanes to give lactam 45 as a white solid (25 mg, 75% yield).
(3aS*,9bS*)-6-Methyl-3-methylene-9-phenyl-3,3a,4,5-tetrahydro-1H-azuleno[4,5-b]pyrrole-2,8(7H,9bH)-dione (45).
1H NMR (400 MHz, CDCl3): δ 7.46–7.40 (m, 3H, Ph-H), 7.25–7.24 (m, 2H, Ph-H), 5.98 (d, J = 3.2 Hz, 1H, =CH), 5.58 (br s, 1H, NH), 5.27 (d, J = 3.2 Hz, 1H, =CH), 4.82 (d, J = 8.4 Hz, 1H, CH), 3.18, 3.11 (AB q, J = 20.0 Hz, 2H, CH2), 3.05–3.02 (m, 1H, CH), 2.77–2.72 (m, 1H), 2.42–2.35 (m, 2H), 1.99 (s, 3H, CH3), 1.91–1.83 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 202.0, 168.6, 165.5, 143.1, 140.8, 135.7, 131.8, 131.3, 129.3 (4C), 128.4, 115.3, 56.2, 44.3, 40.9, 31.8, 26.6, 24.8; IR (thin film) 3419, 2950, 2865, 1697, 1675, 1460, 1380, 1305, 1201 cm−1; HRMS (TOF MS ESI+) m/z: [M + H]+ Calcd for C20H20NO2 306.1489; Found 306.1479; TLC Rf = 0.22 (75% EtOAc/hexanes) visualized with UV and p-anisaldehyde.
(3aS*,9bS*)-6-Methyl-3-methylene-9-(triisopropylsilyl)-3,3a,4,5-tetrahydro-1H-azuleno[4,5-b]pyrrole-2,8(7H,9bH)-dione (46).
Prepared according to General Procedure H. Run 1: Allene-yne 25a (20 mg, 0.056 mmol), rhodium dicarbonyl chloride dimer (2 mg, 0.006 mmol), and toluene (5.7 mL) provided lactam 46 (11 mg, 50% yield) as a clear oil after column chromatography (gradient elution with 15–30% ethyl acetate in hexanes). Consumption of the starting material was evident by TLC after 3 h. Run 2: Allene-yne 25a (25 mg, 0.078 mmol), rhodium biscarbonyl chloride dimer (3 mg, 0.008 mmol), and toluene (8 mL) provided lactam 46 (11 mg, 41% yield) as a white solid after column chromatography. 1H NMR (300 MHz, CDCl3): δ 6.19 (br s, 1H, NH), 6.07 (d, J = 3.1 Hz, 1H, =CH), 5.31 (d, J = 3.1 Hz, 1H, =CH), 4.65 (d, J = 8.1 Hz, 1H, CH), 3.05–3.00 (m, 1H, CH), 3.00 (s, 2H, CH2), 2.99–2.65 (m, 1H), 2.45–2.27 (m, 2H), 1.95 (s, 3H, CH3), 1.90–1.80 (m, 1H), 1.63–1.53 (m, 3H), 1.13 (d, J = 2.0 Hz, 9H), 1.09 (d, J = 2.0 Hz, 9H); 13C NMR (100 MHz, CDCl3): δ 208.2, 179.9, 169.3, 143.1, 137.9, 134.1, 133.9, 115.7, 56.8, 44.2, 41.4, 31.9, 27.1, 25.4, 19.5 (3C), 19.4 (3C), 13.0 (3C); IR (thin film) 2943, 2881, 1719, 1688, 1518, 1504 cm−1; HRMS (TOF MS ESI+) m/z: [M + H]+ Calcd for C23H36NO2Si 386.2510 Found 386.2515; TLC Rf = 0.37 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(3aS*,9bS*)-6-Methyl-3-methylene-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (47):
Prepared according to General Procedure H. Run 1: Allene-yne 26 (12 mg, 0.060 mmol), rhodium biscarbonyl chloride dimer (3 mg, 0.006 mmol), and toluene (6 mL) provided 47 (9 mg, 64% yield) as a white solid after column chromatography (gradient elution with 50–100% ethyl acetate in hexanes). Run 2: Allene-yne 26 (17 mg, 0.085 mmol), rhodium dicarbonyl chloride dimer (4 mg, 0.008 mmol), and toluene (8.6 mL) provided lactam 47 (8 mg, 42% yield) as a white solid after column chromatography. 1H NMR (500 MHz, CDCl3): δ 7.86 (br s, 1H, NH), 6.14 (s, 1H, =CH-CO), 6.06 (d, J = 3.0 Hz, 1H, =CH), 5.32 (d, J = 3.0 Hz, 1H, =CH), 4.58 (d, J = 8.5 Hz, 1H, CH), 3.00 (s, 2H, CH), 2.86–2.82 (m, 1H), 2.76–2.71 (m, 1H), 2.42–2.31 (m, 2H), 1.94 (s, 3H, CH3), 1.93–1.87 (m, 1H); 13C NMR (126 MHz, CDCl3): δ 204.0, 173.0, 170.7, 143.9, 137.1, 131.6, 126.6, 115.8, 56.1, 43.4, 41.3, 32.0, 26.8, 24.9; IR (thin film) 2895, 1685, 1556, 1420, 1327, 1231, 1149 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C14H16NO2 230.1179; Found 230.1176; TLC Rf = 0.15 (100% EtOAc), visualized with UV and p-anisaldehyde.
(3aS*,9bS*)-1,6-Dimethyl-3-methylene-9-phenyl-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (48).
Prepared according to General Procedure H. Allene 30a (30 mg, 0.1 mmol), rhodium biscarbonyl chloride dimer (4 mg, 0.009 mmol), and toluene (8.6 mL) provided lactam 48 (24 mg, 75% yield) as a white solid after column chromatography (gradient elution with 25–75% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3): δ 7.33–7.31 (m, 3H, Ph-H), 7.11–7.10 (m, 2H, Ph-H), 6.10 (d, J = 3.2 Hz, 1H, =CH), 5.34 (d, J = 3.2 Hz, 1H, =CH), 4.52 (d, J = 8.0 Hz, 1H, CH), 3.17, 3.10 (AB q, J = 21.2 Hz, 2H, CH2), 3.20–3.07 (m, 1H), 2.93–2.84 (m, 1H), 2.57–2.46 (m, 1H), 2.43–2.38 (m, 1H), 2.01 (s, 3H, CH3), 1.99 (s, 3H, CH3), 1.96–1.88 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 202.8, 168.8, 165.5, 143.2, 137.6, 136.6, 130.6, 130.3 (2C), 128.4, 127.7 (2C), 115.6, 62.1, 41.7, 40.5, 32.3, 30.4, 29.8, 28.5, 25.8; IR (thin film) 2885, 1670, 1638, 1422, 1378, 1296, 1257, 1141, 1080 cm−1; HRMS (TOF MS ESI+) m/z: [M + H]+ Calcd for C21H22NO2 320.1645; Found 320.1651; TLC Rf = 0.36 (75% EtOAc/hexanes), visualized with silica gel, UV, p-anisaldehyde.
(3aS*,9bS*)-6-Methyl-3-methylene-1,9-diphenyl-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (49):
Prepared according to General Procedure I. Run 1: Allene-yne 37 (11 mg, 0.031 mmol), rhodium biscarbonyl chloride dimer (1.2 mL of a solution of Rh(I)-catalyst in toluene, 1 mg Rh/1 mL toluene, 0.003 mmol), and toluene (3.1 mL) provided lactam 49 (8 mg, 67% yield ) as a white solid after column chromatography (gradient elution with 25–50% ethyl acetate in hexanes). Run 2: Allene-yne 37 (3 mg, 0.008 mmol), rhodium biscarbonyl chloride dimer (0.5 mL of a 1 mg/mL solution in toluene), and toluene (0.5 mL) provided lactam 49 (2 mg, 66% yield) as a white solid after column chromatography. 1H NMR (500 MHz, CDCl3): δ 7.10–6.86 (m, 8H, Ph-H), 6.53–6.52 (m, 2H, Ph-H), 6.24 (d, J = 3.3 Hz, 1H, =CH), 5.46 (d, J = 3.3 Hz, 1H, =CH), 5.34 (d, J = 8.0 Hz, 1H, CH), 3.31–3.29 (m, 1H, CH), 3.17, 3.08 (AB q, J = 21.0 Hz, 2H, CH2), 3.08–3.02 (m, 1H), 2.63–2.60 (m, 1H), 2.52–2.47 (m, 1H), 2.05 (s, 3H, CH3), 2.08–2.03 (m, 1H); 13C NMR (126 MHz, CDCl3): δ 202.5, 166.4, 163.3, 143.6, 139.4, 137.6, 136.4, 130.6, 130.5, 129.6 (2C), 128.1 (2C), 127.5, 127.4 (2C), 124.1, 119.3 (2C), 117.1, 59.1, 40.9, 40.6, 32.4, 28.7, 25.9; IR (thin film) 2891, 1675, 1631, 1477, 1356 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C26H24NO2 382.1802; Found 382.1807; TLC Rf = 0.35 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
4-((3aS*,9bS*)-6-Methyl-3-methylene-2,8-dioxo-9-phenyl-2,3,3a,4,5,7,8,9b-octahydro-1H-azuleno[4,5-b]pyrrol-1-yl)benzonitrile (50).
Prepared according to General Procedure H. Allene-yne 38 (17 mg, 0.045 mmol), rhodium biscarbonyl chloride dimer (2 mg, 0.0045 mmol), and toluene (4.5 mL) provided lactam 50 (13 mg, 72% yield) as a light yellow solid after column chromatography (gradient elution with 15–35% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3): δ 7.27–7.25 (m, 2H, Ph-H), 7.13–7.03 (m, 5H, Ph-H), 6.61–6.45 (m, 2H, Ph-H), 6.30 (d, J = 3.4 Hz, 1H, =CH), 5.55 (d, J = 3.4 Hz, 1H, =CH), 5.31 (d, J = 8.4 Hz, 1H, CH), 3.35–3.31 (m, 1H, CH), 3.19, 3.10 (AB q, J = 21.0, 36.6 Hz, 2H, CH2), 3.06–3.01 (m, 1H,), 2.68–2.59 (m, 1H), 2.55–2.49 (m, 1H), 2.10–2.03 (m, 1H), 2.08 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 202.1, 166.6, 162.2, 142.6, 141.4, 139.2, 137.0, 132.1 (2C), 130.3, 130.2, 129.7 (2C), 127.8, 127.5 (2C), 119.6 (2C), 119.0, 118.8, 107.0, 58.9, 40.8, 40.5, 32.2, 28.6, 25.9; IR (thin film) 2925, 2854, 2224, 1704, 1659, 1604, 1509, 1345, 1179 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C27H23N2O2 407.1754; Found 407.1753; TLC Rf = 0.27 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(3aS*,9bS*)-6-Methyl-3-methylene-9-phenyl-1-(4-(trifluoromethyl)phenyl)-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (51).
Prepared according to General Procedure H. Run 1: Allene-yne 39 (17 mg, 0.04 mmol), rhodium biscarbonyl chloride dimer (2 mg, 0.004 mmol), and toluene (4 mL) provided lactam 51 (13 mg, 72% yield) as a white solid after column chromatography (gradient elution with 15–50% ethyl acetate in hexanes). Run 2: Allene-yne 39 (6 mg, 0.01 mmol), rhodium biscarbonyl chloride dimer (0.5 mL of a 1 mg/mL solution in toluene), and toluene (0.9 mL) provided lactam 51 (4 mg, 67% yield) as a white solid after column chromatography. 1H NMR (500 MHz, CDCl3): δ 7.20 (d, J = 8.5 Hz, 2H, Ph-H) 7.10 (app t, J = 7.5 Hz, 1H, Ph-H), 7.00 (d, J = 9.0 Hz, 4H, Ph-H), 6.55–6.43 (m, 2H, Ph-H), 6.28 (d, J = 3.5 Hz, 1H, =CH), 5.52 (d, J = 3.5 Hz, 1H, =CH), 5.33 (d, J = 8.5 Hz, 1H, CH), 3.34–3.31 (m, 1H, CH), 3.17, 3.10 (ABq, J = 21.0 Hz, 2H, CH2), 3.09–3.02 (m, 1H), 2.66–2.59 (m, 1H), 2.53–2.49 (m, 1H), 2.10–1.99 (m, 1H), 2.06 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 202.3, 166.5, 162.5, 143.0, 140.4, 139.3, 136.9, 130.38, 130.36, 129.6 (2C), 127.6 (2C), 127.5 (2C), 125.8, 125.6 (q, J = 32.5 Hz, 1C), 125.2 (q, J = 3.7 Hz, 1C), 122.8, 119.3, 118.2, 59.1, 40.9, 40.5, 32.3, 28.6, 25.9; IR (thin film) 2930, 1698, 1615, 1325, 1168, 1117 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C27H23NO2F3 450.1675; Found 450.1655; TLC Rf = 0.36 (50% EtOAc/hexanes), visualized with UV and vanillin.
(3aS*,9bS*)-1-(4-Methoxyphenyl)-6-methyl-3-methylene-9-phenyl-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (52):
Prepared according to General Procedure I. Run 1: Allene-yne 40 (6 mg, 0.2 mmol), rhodium dicarbonyl chloride dimer (0.5 mL of a solution of Rh(I)-catalyst in toluene, 1 mg Rh/1 mL toluene, 0.001 mmol), and toluene (1.3 mL) provided lactam 52 (5 mg, 76%) as a white solid after column chromatography (gradient elution with 15–35% ethyl acetate in hexanes). Run 2: Allene-yne 40 (25 mg, 0.065 mmol), rhodium biscarbonyl chloride dimer (3 mg, 0.006 mmol), and toluene (6.6 mL) provided lactam 52 (20 mg, 77%) as a white solid after column chromatography. 1H NMR (500 MHz, CDCl3) δ 7.12–7.10 (m, 1H, Ph-H), 7.06–7.03 (m, 2H, Ph-H), 6.79–6.77 (m, 2H, Ph-H), 6.58–6.57 (m, 2H, Ph-H), 6.51–6.49 (m, 2H), 6.22 (d, J = 3.2 Hz, 1H, =CH), 5.45 (d, J = 3.2 Hz, 1H, =CH), 5.31 (d, J = 8.5 Hz, 1H, CH), 3.72 (s, 3H, OCH3), 3.31–3.28 (m, 1H), 3.17, 3.08 (AB q, J = 21.0 Hz, 2H, CH2), 3.09–3.01 (m, 1H), 2.62–2.58 (m, 1H), 2.51–2.46 (m, 1H), 2.10–2.00 (m, 1H), 2.05 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 202.4, 166.2, 163.4, 156.3, 143.7, 139.3, 136.4, 131.2, 130.7, 129.6 (2C), 127.6, 127.4 (2C), 120.6 (2C), 116.7, 113.5 (2C), 59.2, 55.6, 41.0, 40.6, 32.4, 29.9, 28.7, 25.8; IR(thin film) 2924, 1700, 1511, 1250 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C27H26NO3 412.1907; Found 412.1901; TLC Rf = 0.23 (50% EtOAc/hexanes) [silica gel, UV, vanillin].
(3aS*,9bS*)-6-Methyl-3-methylene-9-phenyl-1-(thiophen-2-yl)-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (53).
Prepared according to General Procedure I. Run 1: Allene-yne 42 (12 mg, 0.033 mmol), rhodium biscarbonyl chloride dimer (1.3 mL of a solution of Rh(I)-catalyst in toluene, 1 mg Rh/1 mL toluene, 0.003 mmol), and toluene (3.3 mL) provided lactam 53 (7 mg, 54% yield) as a pale yellow solid after column chromatography (gradient elution with 25–50% ethyl acetate in hexanes). Run 2: Allene-yne 42 (18 mg, 0.050 mmol), rhodium biscarbonyl chloride dimer (2 mg, 0.005 mmol), and toluene (5 mL) provided lactam 53 (15 mg, 79%) as a pale yellow solid after column chromatography. 1H NMR (400 MHz, CDCl3) δ 7.10–7.04 (m, 3H, Ph-H), 6.70 (dd, J = 1.4, 5.4 Hz, 1H, HetAr-H), 6.65–6.59 (m, 3H), 6.25 (d, J = 3.6 Hz, 1H, HetAr-H), 6.24 (d, J = 3.2 Hz, 1H, =CH), 5.49 (d, J = 3.2 Hz, 1H, =CH), 5.16 (d, J = 8.0 Hz, 1H, CH), 3.40–3.34 (m, 1H, CH), 3.21, 3.12 (ABq, J = 21.0 Hz, 2H, CH2), 3.08–2.98 (m, 1H), 2.69–2.58 (m, 1H), 2.55–2.48 (m, 1H), 2.06 (s, 3H, CH3), 2.03–1.99 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 202.4, 164.6, 163.1, 141.8, 139.5, 139.0, 136.2, 130.4, 130.1, 129.3 (2C), 127.8, 127.2 (2C), 123.0, 119.2, 117.8, 111.8, 60.8, 41.9, 40.5, 32.2, 28.5, 25.9; IR (thin film) 3434, 1693, 1575, 1360, 1305 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C24H22NO2S 388.1366; Found 388. 1357; TLC Rf = 0.31 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(3aS*,9bS*)-6-Methyl-3-methylene-9-phenyl-1-tosyl-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (54).
Prepared according to General Procedure I. Run 1: Allene-yne 43 (4 mg, 0.009 mmol), rhodium biscarbonyl chloride dimer (0.5 mL of a solution of Rh(I)-catalyst in toluene, 1 mg Rh/1 mL toluene, 0.001 mmol), and toluene (1 mL) provided lactam 54 (3 mg, 75% yield) as a white solid after column chromatography (gradient elution with 25–50% ethyl acetate in hexanes). Run 2: Allene-yne 43 (9 mg, 0.02 mmol), rhodium biscarbonyl chloride (1 mg, 0.002), and toluene (2.1 mL) provided lactam 54 (6 mg, 67% yield) as a white solid after column chromatography. 1H NMR (500 MHz, CDCl3): δ 7.64 (d, J = 8.5 Hz, 2H, Ph-H), 7.40–7.38 (m, 3H, Ph-H), 7.21 (d, J = 8.5 Hz, 2H, Ph-H), 7.18–7.14 (m, 2H, Ph-H), 6.16 (d, J = 3.5 Hz, 1H, =CH), 5.43 (d, J = 3.5 Hz, 1H, =CH), 5.07 (d, J = 8.5 Hz, 1H, CH), 3.23–3.22 (m, 1H, CH), 3.23, 3.10 (ABq, J = 20.5 Hz, 2H, CH2), 2.96–2.86 (m, 1H), 2.56–2.47 (m, 1H), 2.43–2.38 (m, 1H), 2.37 (s, 3H, CH3), 2.01 (s, 3H, CH3), 1.80–1.74 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 202.6, 166.5, 164.6, 145.3, 140.7, 139.3, 134.8, 134.7, 132.0, 131.4 (2C), 130.9, 130.4, 129.7 (2C), 128.5 (2C), 127.8 (2C), 121.4, 60.2, 41.5, 40.7, 31.9, 29.3, 25.7, 21.8; IR (thin film) 2888, 1714, 1674, 1358, 1160 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C27H26NO4S 460.1577; Found 460.1577; TLC Rf = 0.23 (50% EtOAc/hexanes), visualized with UV and vanillin.
tert-Butyl (3aS*,9bS*)-6-methyl-3-methylene-2,8-dioxo-9-phenyl-2,3,3a,4,5,7,8,9b-octahydro-1H-azuleno[4,5-b]pyrrole-1-carboxylate (55).
Prepared according to General Procedure H. Run 1: Allene-yne 44 (7 mg, 0.018 mmol), rhodium biscarbonyl chloride dimer (1 mg, 0.002 mmol), and toluene (1.9 mL) provided lactam 55 (3 mg, 40% yield) as a pale yellow solid after column chromatography (gradient elution with 15–50% ethyl acetate in hexanes). Lactam 55 was also prepared according to General Procedure I. Run 2: Allene-yne 44 (16 mg, 0.042 mmol), rhodium biscarbonyl chloride dimer (2 mg, 0.004 mmol), and toluene (4.2 mL) provided lactam 55 (7 mg, 41% yield) as a pale yellow solid after column chromatography. 1H NMR (500 MHz, CDCl3): δ 7.35–7.27 (m, 3H, Ph-H), 7.05–7.04 (m, 2H, Ph-H), 6.29 (d, J = 3.5 Hz, 1H, =CH), 5.50 (d, J = 3.5 Hz, 1H, =CH), 5.12 (d, J = 9.0 Hz, 1H, CH), 3.17, 3.06 (AB q, J = 21.0 Hz, CH2), 3.14–3.11 (m, 1H, CH), 3.01–2.92 (m, 1H), 2.56–2.50 (m, 1H), 2.43–2.39 (m, 1H), 2.00 (s, 3H, CH3), 1.95–1.92 (m, 1H), 1.18 (s, 9H, C(CH3)3); 13C NMR (125 MHz, CDCl3): δ 202.7, 165.0, 164.8, 150.4, 142.8, 137.9, 135.7, 131.0 (2C), 130.6, 130.2, 127.9 (2C), 119.9, 83.5, 60.5, 58.5, 40.6, 39.8, 31.9, 28.9, 27.9 (3C), 25.7; IR (thin film) 2969, 2915, 1793, 1757, 1717, 1401, 1302, 1153 cm−1; HRMS (TOF MS ESI+) m/z: [M + H]+ Calcd for C25H28NO4 406.2013 Found 406.2013; TLC Rf = 0.65 (50% EtOAc/hexanes), visualized with UV and p-anisaldehyde.
(3aS*,9bS*)-3,6-Dimethyl-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (58):
Prepared according to General Procedure H. Allene-yne 56 (12 mg, 0.059 mmol, 9:1 dr), rhodium biscarbonyl chloride dimer (2 mg, 0.005 mmol), and toluene (5 mL) provided lactam 58 (11 mg, 78% yield, 7.3:1 dr) as a white solid after column chromatography (gradient elution with 25–100% ethyl acetate in hexanes). The diastereomeric ratio was determined based on integration of the resonances corresponding to the lactam methine (4.56, d, CHNH) and lactam methine (58, 4.51, d, CHNH). The stereochemistry of the carbon adjacent to the lactam carbonyl was not assigned. 1H NMR (500 MHz, CDCl3): δ 7.47 (br s, 1H, NH)*, 7.36 (br s, 1H, NH), 6.10 (s, 1H, CHCO)*, 6.07 (s, 1H, CHCO), 4.56 (d, J = 10.5 Hz, 1H, CH)*, 4.51 (d, J = 10.0 Hz, 1H, CH), 3.00 (s, 2H, CH2), 2.76–2.70 (m, 1H), 2.31–2.18 (m, 3H), 1.91 (s, 3H, CH3), 1.95–1.87 (m, 1H), 1.77–1.70 (m, 1H), 1.20 (d, J = 7.0 Hz, 3H, CH3) *Minor diastereomer where distinguishable; 13C NMR (126 MHz, CDCl3): δ 204.0, 179.5, 173.3, 137.5, 131.2, 126.6, 57.1*, 56.6, 47.3, 45.0, 42.0*, 41.5*, 41.4, 33.3*, 32.7, 28.1, 24.8, 23.7*, 13.8, 12.0*; IR (thin film) 3196, 2893, 1680, 1553, 1438, 1231, 1156 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C14H18NO2 232.1259; Found 232.1339; TLC Rf = 0.11 (100% EtOAc), visualized with UV and p-anisaldehyde.
(3aR*,9bS*)-3,6-Dimethyl-9-phenyl-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (59).
Prepared according to General Procedure I. Allene-yne 57 (17 mg, 0.061 mmol), rhodium dicarbonyl chloride dimer (3 mg, 0.0061 mmol), and toluene (6 mL) yielded lactam 59 (14 mg, 77% yield) as a white solid after purification by column chromatography (gradient elution with 50–100% ethyl acetate in hexanes). The stereochemistry of the carbon adjacent to the lactam carbonyl was not assigned. 1H NMR (400 MHz, CDCl3): δ 7.44–7.38 (m, 3H, Ph-H), 7.17–7.15 (m, 2H, Ph-H), 5.21 (br s, 1H, NH), 4.98 (d, J = 7.6 Hz, 1H, CH), 3.19, 3.12 (AB q, J = 20.8 Hz, 2H, CH2), 2.59–2.46 (m, 2H), 2.35–2.24 (m, 2H), 2.09–2.02 (m, 1H), 1.90 (s, 3H, CH3), 1.86–1.81 (m, 1H), 1.22 (d, J = 7.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ 202.5, 179.8, 163.0, 144.7, 140.9, 131.2, 129.5 (2C), 129.0, 128.8 (2C), 128.7, 55.3, 45.6, 42.4, 42.1, 32.7, 31.9, 24.2, 15.6; IR (thin film) 3374, 2902, 1673, 1431 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C20H22NO2 308.1645; Found 308.1649; TLC Rf = 0.15 (75% EtOAc/hexanes), visualized with UV and vanillin.
General Procedure J:
N-Acetyl Lactam Formation From 2° Lactam. (3aS*,9bS*)-1-Acetyl-6-methyl-3-methylene-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (60).
A flame-dried test tube equipped with a stir bar, septum, and nitrogen inlet needle was charged with lactam 47 (8 mg, 0.03 mmol) dissolved in DCM (0.4 mL). 4-Dimethylaminopyridine (1 mg, 0.008 mmol) was added, and the solution was cooled to 0 °C. Triethylamine (0.05 mL, 0.3 mmol) was added dropwise via syringe followed by dropwise addition of acetic anhydride (0.02 mL, 0.2 mmol) via syringe. The reaction was allowed to warm to rt with stirring for 2 h, at which point TLC showed consumption of starting material. The reaction was diluted with DCM (10 mL) and transferred to a separatory funnel. The organic layer was washed with saturated ammonium chloride (5 mL), brine (5 mL), dried over magnesium sulfate, gravity filtered, concentrated by rotary evaporation, and purified by silica gel flash column chromatography (gradient elution with 50–75% ethyl acetate in hexanes) to provide N-acetyl lactam 60 (7 mg, 78% yield) as a white, sticky solid. 1H NMR (400 MHz, CDCl3): δ 6.27 (d, J = 3.6 Hz, 1H, =CH), 5.63 (s, 1H, CHCO), 5.49 (d, J = 3.6 Hz, 1H, =CH), 5.03 (d, J = 9.2 Hz, 1H, CH), 3.02, 2.94 (AB q, J = 20.8 Hz, 2H, CH2), 2.89–2.75 (m, 2H), 2.65 (s, 3H, CH3), 2.56–2.41 (m, 1H), 2.37–2.31 (m, 1H), 1.94 (s, 3H, CH3), 1.87–1.79 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 203.6, 172.4, 171.9, 167.6, 142.4, 136.1, 131.8, 126.9, 120.3, 57.8, 41.0, 39.6, 31.2, 27.7, 25.8, 24.9; IR (thin film) 2890, 1710, 1653, 1555, 1351, 1299, 1145 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C16H18NO3 272.1281; Found 272.1277; TLC Rf = 0.57 (100% EtOAc), visualized with UV and p-anisaldehyde.
(3aS*,9bS*)-1-Acetyl-6-methyl-3-methylene-9-phenyl-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (S9).
Prepared according to General Procedure J. Lactam 45 (9 mg, 0.03 mmol), dimethylaminopyridine (1 mg, 0.008 mmol), triethylamine (0.04 mL, 0.3 mmol), acetic anhydride (0.02 mL, 0.2 mmol), DCM (0.2 mL), and 20 h at rt provided N-acetyl lactam S9 (6 mg, 60% yield) as a pale yellow solid after column chromatography (gradient elution with 15–50% ethyl acetate in hexanes). 1H NMR (500 MHz, CDCl3): δ 7.35–7.29 (m, 3H, Ph-H), 7.04–6.49 (m, 2H, Ph-H), 6.29 (d, J = 3.2 Hz, 1H, =CH), 5.54 (d, J = 3.2 Hz, 1H, =CH), 5.16 (d, J = 8.5 Hz, 1H, CH), 3.17, 3.06 (AB q, J = 21.0 Hz, 2H, CH2), 3.12–3.10 (m, 1H, CH), 2.99–2.91 (m, 1H), 2.59–2.51 (m, 1H), 2.44–2.39 (m, 1H), 2.00 (s, 3H, CH3), 1.91–1.88 (m, 1H), 1.59 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 202.7, 171.8, 166.4, 164.5, 142.8, 138.3, 135.8, 131.2, 131.0 (2C), 130.6, 127.8 (2C), 120.6, 57.4, 40.5, 39.7, 31.8, 29.7, 28.6, 25.7, 23.9; IR (thin film) 3390, 2892, 1689, 1355, 1273, 1164 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C22H22NO3 348.1521; Found 348.1518; TLC Rf = 0.31 (50% EtOAc/hexanes), visualized with UV and KMnO4.
(3aS*,9bS*)-1-Acetyl-3,6-dimethyl-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (S10).
Prepared according to General Procedure J. Lactam 58 (11 mg, 0.05 mmol, 9:1 ratio of diastereomers), dimethylaminopyridine (1 mg, 0.008 mmol), triethylamine (0.07 mL, 0.5 mmol), acetic anhydride (0.02 mL), DCM (0.5 mL), and 6.5 h at rt provided N-acetyl lactam S10 (6 mg, 46% yield) as a white solid after column chromatography (gradient elution with 25–75% ethyl acetate in hexanes). The diastereomeric ratio was determined based on integration of the resonances corresponding to the lactam methine (5.08, d, CHNAc) and lactam methine (S10, 5.02, d, CHNAc). 1H NMR (500 MHz, CDCl3): δ 5.59 (s, 1H, CHCO), 5.08 (d, J = 5.5 Hz, 1H, CH)*, 5.02 (d, J = 10.5 Hz, 1H, CH), 3.01, 2.94 (AB q, J = 21.0 Hz, 2H, CH2), 2.87–2.81 (m, 1H), 2.57 (s, 3H, CH3), 2.41–2.26 (m, 3H), 1.93 (s, 3H, CH3), 1.87–1.79 (m, 1H), 1.71–1.65 (m, 1H), 1.24 (d, J = 7.0 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 203.7, 177.1, 172.5, 171.7, 136.5, 131.7, 127.1, 66.0, 58.3, 45.7, 42.3*, 41.0, 31.6, 29.0, 25.4, 24.7, 15.4*, 13.7, * Discernible signals for 1 of 2 diastereomers; IR (thin film) 2893, 1721, 1681, 1661, 1544, 1353, 1254 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C16H20NO3 274.1438; Found 274.1438; TLC Rf = 0.46 (75% EtOAc/hexanes), visualized with UV and vanillin.
Synthesis of α-Methylene-γ-Lactone S17
4-(3-hydroxybutyl)-3-methylene-5-(phenylethynyl)dihydrofuran-2(3H)-one (S13).
A flame-dried, round-bottomed flask equipped with a Teflon-coated stir-bar was charged with S12 (1.2 g, 2.9 mmol, E:Z = 2.5:7.5), 3-phenyl-2-propynal (12) (757 mg, 5.8 mmol) and toluene (7.8 mL) and cooled to 0 °C. The resulting solution was treated with trifluoromethanesulfonic acid (43.6 mg, 0.29 mmol) and maintained at 0 °C under a nitrogen atmosphere for 12 h. The mixture was then diluted with NH4Cl (aq):NH4OH (9:1, v/v, 58 mL) and extracted with diethyl ether (3 × 60 mL). The combined extracts were washed with brine (2 × 30 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The crude residue was loaded onto a 100 g SNAP column and purified using a Biotage normal phase automated purification system with a gradient of 15–90% diethyl ether/pentane to afford the title compound S13 (0.375 g, 48%) as a yellow oil. The product was obtained as a mixture of isomers in a trans:cis 8.5:2.5 ratio. A pure fraction of the trans-lactone was collected for characterization purposes. 1H NMR (CDCl3, 500 MHz): δ 7.43–7.42 (m, 2H, Ph-H), 7.32–7.30 (m, 3H, Ph-H), 6.32 (d, J = 2.5 Hz, 1H, =CH), 5.68 (d, J = 2.5 Hz, 1H, =CH), 4.99–4.98 (m, 1H, CH), 3.87–3.85 (m, 1H), 3.20–3.18 (m, 1H), 2.23 (br s, 1H, OH), 1.83–1.79 (m, 1H), 1.63–1.58 (m, 3H), 1.23–1.21 (m, 3H); 13C NMR (CDCl3, 100 MHz): δ 169.3, 137.4, 131.6 (2C), 129.0, 128.2 (2C), 123.0, 121.3, 87.7, 85.0, 72.3, 67.3, 46.8, 35.3, 29.3, 23.5; IR (thin film) 3465, 2966, 2921, 2855, 2230, 1772, 1670, 1491, 1446, 1405, 1446, 1368, 1269, 1135, 968 cm−1; HRMS (TOF MS ESI+) m/z: [M+K]+ Calcd for C17H18O3K1 309.0893; Found 309.0901; TLC Rf = 0.3 (80% diethyl ether/pentane), visualized with UV and KMnO4.
3-methylene-4-(3-oxobutyl)-5-(phenylethynyl)dihydrofuran-2(3H)-one (S14).
A flame-dried, round-bottomed flask equipped with a Teflon-coated stir-bar was charged with S13 (0.11 g, 0.41 mmol, pure trans) and CH2Cl2 (5.8 mL). The resulting solution was treated with Dess-Martin periodinane (0.21 g, 0.49 mmol). The progress of the reaction was monitored by TLC and upon completion (3 h), the mixture was concentrated under reduced pressure. The crude residue was loaded onto a 25 g SNAP column and purified using a Biotage normal phase automated purification system with a gradient of 50–100% diethyl ether/pentane to afford the title compound S14 (96 mg, 88%) as a slightly yellow oil. 1H NMR (CDCl3, 500 MHz): d 7.43–7.31 (m, 5H, Ph-H), 6.32 (d, J = 2.5 Hz, 1H, =CH), 5.67 (d, J = 2.5 Hz, 1H, =CH), 4.92 (d, J = 6 Hz, 1H, CH), 3.22–3.20 (m, 1H), 2.68–2.63 (m, 2H), 2.15 (s, 3H, CH3), 2.10–2.05 (m, 1H), 1.91–1.84 (m, 1H); 13C NMR (CDCl3, 125 MHz): d 206.9, 169.8, 137.2, 131.7 (2C), 129.2, 129.4 (2C), 123.0, 121.2, 88.0, 84.8, 72.1, 46.0, 39.4, 30.0, 26.0; IR (thin film) 3056, 2925, 2235, 1773, 1715, 1659, 1487, 1437, 1413, 1368, 1270, 1140, 980 cm−1; HRMS (TOF MS ES+) m/z: [M+K]+ Calcd for C17H16O3K1 307.0737; Found 307.0745; TLC Rf = 0.7 (80% diethyl ether/pentane), visualized with UV and KMnO4.
4-methylene-5-oxo-2-(phenylethynyl)tetrahydrofuran-3-yl)butan-2-yl acetate (S15).
A flame-dried, 50-mL Schlenk tube equipped with a Teflon-coated stir-bar was charged with Cerium(III) trichloride (anhydrous beads, 1.13 g, 4.56 mmol) in a nitrogen-filled glove box. The Schlenk tube was removed from the glove box, THF (8.7 mL) was added, and the suspension was sonicated for 4 h. The resulting solution was stirred at rt for 12 h, cooled to 0 °C, treated with ethynyl magnesium bromide (0.5 M in THF, 9.62 mL, 4.80 mmol) and stirred at 0 °C for 45 min. The solution was then cooled to − 78 °C and cannulated into a round-bottom flask containing a solution of ketone S14 (0.129 g, 0.48 mmol) in THF (11.7 mL) cooled to − 78 °C. After stirring for 30 min at − 78 °C, the solution was then diluted with saturated ammonium chloride (5 mL) and diethyl ether (10 mL). After stirring for 1 h at rt, the mixture was filtered through a Celite plug and the filter cake was rinsed with diethyl ether (60 mL). The organic phase was separated and the aqueous layer was extracted with diethyl ether (2 × 20 mL). The combined organic layers were washed with brine (30 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to afford S15 (0.178 g, 0.60 mmol). The propargyl alcohol was not stable to column chromatography, so it was taken on immediately to the next step. Crude S15 was dissolved in CH2Cl2 (2.6 mL) and 4-dimethylaminopyridine (7.42 mg, 0.061 mmol), triethylamine (0.85 mL, 6.07 mmol) and acetic anhydride (0.29 mL, 3.03 mmol) were added. The resulting solution was stirred at rt for 12 h, diluted with ammonium chloride (5 mL), filtered through a Celite plug and rinsed with CH2Cl2. The phases were separated and the aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over Na2SO4, filtered, concentrated under reduced pressure and purified by flash chromatography (25% diethyl ether/pentane) to afford the title compound S15 (0.132 g, 82%) as a slightly orange oil. 1H NMR (CDCl3, 400 MHz): d 7.45–7.31 (m, 5H, Ph-H), 6.34 (d, J = 2.8 Hz, 1H, =CH), 5.67 (d, J = 2.8 Hz, 1H, =CH), 4.98 (d, J = 5.2 Hz, 1H, CH), 3.22–3.20 (m, 1H, CH), 2.55 (s, 1H, ≡CH), 2.07–2.02 (m, 1H), 2.00 (d, J = 1.2 Hz, 3H, COCH3), 1.88–1.70 (m, 3H), 1.68 (d, J = 0.8 Hz, 3H, CH3); 13C NMR (CDCl3, 125 MHz): d 169.2, 169.0, 137.3, 131.8 (2C), 129.2, 128.4 (2C), 123.3, 121.4, 88.1, 84.9, 83.0, 74.2, 74.1, 72.2, 46.8, 38.1, 27.7, 26.6, 21.9; IR (thin film) 3289, 2929, 2855, 2231, 2116, 1769, 1736, 1667, 1491, 1438, 1368, 1242, 1131, 1103, 1013 cm−1; HRMS (TOF MS ES+) m/z: [M+Na]+ Calcd for C21H20O4Na1 359.1259; Found 359.1255; TLC Rf = 0.7 (80% diethyl ether/pentane), visualized with silica gel, UV, KMnO4 stain.
3-methylene-4-(3-methylpenta-3,4-dienyl)-5-(phenylethynyl)dihydrofuran-2(3H)-one (S16):
A flame-dried, round-bottomed flask equipped with a Teflon-coated stir-bar was charged with S15 (115 mg, 0.33 mmol) and THF (3.3 mL). The resulting solution was treated with dimethylamine (0.45 mL, 0.83 mmol, 2M in THF), stirred at rt under a nitrogen atmosphere for 11 h and concentrated under reduced pressure to afford the crude Michael adduct (139 mg, 0.37 mmol). After dilution with toluene (5.7 mL, degassed), (triphenylphosphine)copper hydride hexamer (Stryker’s reagent) (716 mg, 0.37 mmol) was added and the resulting solution was stirred at rt under a nitrogen atmosphere for 3 h, quenched with saturated ammonium chloride (5 mL) and stirred opened to air for 12 h. The crude mixture was filtered through a Celite plug and rinsed with diethyl ether. The phases were separated, and the aqueous layer was extracted with diethyl ether (2 × 20 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, filtered, concentrated under reduced pressure to afford crude allene-yne (153 mg, 0.47 mmol). The latter was diluted with CH2Cl2 (18 mL), treated with silica gel (4 g, 66 mmol), capped with a glass stopper and stirred at rt for 11 h. The solution was then filtered, rinsed with CH2Cl2, concentrated under reduced pressure and purified by flash chromatography (10% diethyl ether/pentane) to afford the title compound S16 (36.1 mg, 39%) as a colorless oil. 1H NMR (CDCl3, 500 MHz): d 7.44–7.32 (m, 5H, Ph-H), 6.35 (d, J = 2.5 Hz, 1H, =CH), 5.69 (d, J = 2.5 Hz, 1H, =CH), 4.98 (d, J = 5.5 Hz, 1H, CH), 4.62–4.60 (m, 2H, =CH2), 3.20–3.18 (m, 1H, CH), 2.02–2.00 (m, 1H), 1.81–1.76 (m, 2H), 1.69–1.67 (t, J = 3.0 Hz, 3H, CH3), 1.66–1.64 (m, 2H); 13C NMR (CDCl3, 175 MHz): d 206.0, 169.2, 137.7, 131.7 (2C), 129.0, 128.3 (2C), 122.6, 121.5, 97.6, 87.7, 85.1, 74.6, 72.4, 46.8, 32.8, 32.5, 18.7; IR (thin film) 3289, 2925, 2844, 2231, 1957, 1769, 1491, 1442, 1262, 1131 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C19H19O2 279.1385; Found 279.1375; TLC Rf = 0.27 (20% diethyl ether/pentane), visualized with UV and KMnO4.
3-methylene-9-phenyl-3,3a,4,5-tetrahydroazuleno[4,5-b]furan-2,8(7H,9bH)-dione (S17):
A flame-dried vial (15 × 45 mm) equipped with a Teflon-coated stir-bar and a septum cap was charged with allene-yne S16 (15 mg, 0.054 mmol, 1 equiv) and toluene (1.6 mL, toluene degassed by bubbling with nitrogen for ~ 5 min). The tube was placed under vacuum for 3–5 sec. and refilled with carbon monoxide (3 ×). To the allene-yne solution was added [Rh(CO)2Cl]2 (1 mg, 5.6 × 10−3 mmol, 0.10 equiv) in one portion by temporary removal of the septum, and the vial was placed under vacuum and refilled with carbon monoxide (3 ×). The vial was placed in an oil bath (preheated to 90 °C) and stirred under carbon monoxide. After 25 min, TLC indicated completion, and the mixture was cooled to rt, passed through a short plug of Celite using diethyl ether, and concentrated in vacuo. Purification of the residue by flash chromatography (75% diethyl ether/pentane) afforded the title compound S17 (8.8 mg, 54%) as a slightly yellow oil. 1H NMR (CDCl3, 300 MHz): d 7.37–7.33 (m, 3H, Ph-H), 7.26–7.23 (m, 2H, Ph-H), 6.22 (d, J = 3.3 Hz, 1H, =CH), 5.54 (d, J = 3.3 Hz, 1H, =CH), 5.45 (d, J = 9.9 Hz, 1H, CH), 3.16–3.13 (m, 2H, COCH2), 3.13–3.06 (m, 1H, CH), 2.77–2.68 (m, 1H), 2.46–2.35 (m, 2H), 1.99 (s, 3H, CH3), 1.91–1.85 (m, 1H); 13C NMR (175 MHz, CDCl3): d 202.1, 168.1, 162.1, 140.9, 138.9, 135.2, 131.2, 130.2, 129.7 (2C), 127.9, 127.3 (2C), 120.7, 78.3, 43.9, 40.6, 31.3, 25.9, 24.6; IR (thin film) 2929, 2860, 2247, 1777, 1691, 1446, 1311, 1258, 1131, 1037, 1004 cm−1; HRMS (TOF MS ES+) m/z: [M+H]+ Calcd for C20H19O3 307.1334; Found 307.1339; TLC Rf = 0.3 (70% diethyl ether/pentane), visualized with UV and KMnO4.
General Procedure K: Reaction of α-Methylene-γ-Lactams and Lactone S17 with Cysteamine and Monitoring Reaction Progress by 1H NMR.
Sample preparation for pseudo-first order reaction kinetic studies.
Pseudo-first order rate constants were determined using a procedure modified from the original report.42 A 1-dram scintillation vial was charged with 1–2 mg of the lactam (1 equiv) and dissolved in CDCl3 to afford a concentration of 11.5 mM. A second 1-dram scintillation vial was charged with cysteamine (15 equiv) and dissolved in CDCl3. (The volume of CDCl3 used for this dilution is equal to that used to dissolve the lactam). These two solutions were transferred via glass pipet to a third 1-dram scintillation vial making the final lactam concentration 5.7 mM. This vial is capped, mixed for 10 sec using a vortex mixer, and transferred to a 5 mm NMR tube.
Monitoring reaction progress by 1H NMR without internal standard.
A 1H NMR spectrum was obtained immediately after transferring the prepared sample to the NMR tube, and at regular intervals of time thereafter. The solution was maintained at ambient temperature (22 °C) over the course of the reaction. The progress of the reaction was monitored by the disappearance of methylene resonances Ha, Hb and methine Hc and the simultaneous appearance of Hd of the thio-adduct (See eq S1 for identity of Ha, Hb, Hc, Hd). For each timepoint the integration value corresponding to Ha was set to 1.0, and the fraction of remaining lactam was calculated following eq S2 using the integration values of Ha, Hb, Hc, and Hd. The natural log-transformed values of Fraction Remaining were plotted against time to afford a linear plot. Using the linear form of the first-order rate equation, the slope of the line-of-best-fit is equal to the pseudo-first order rate constant (kpseudo1st) (eq S3). The reaction half-life (t1/2) is calculated using eq S4.
Monitoring reaction progress by 1H NMR with internal standard.
Two lactams, 49 and 52, were re-analyzed following the procedure above with an internal standard, hexamethylbenzene (1 equiv). The half-lives for these reactions with and without internal standard were nearly identical indicating that monitoring the reactions in the absence of internal standard is reliable (vide infra).
Reaction of α-Methylene-γ-Lactam 45 with Cysteamine.
This reaction was performed according to General Procedure K. Tricyclic lactam 45 (8 mg, 0.026 mmol), cysteamine (30 mg, 0.39 mmol), and chloroform-d (1 mL) were used (See Table S3 and Figures S1–S3).
Reaction of α-Methylene-γ-Lactam 49 with Cysteamine.
This reaction was performed according to General Procedure K. Lactam 49 (1.65 mg, 0.0043 mmol), cysteamine (5.0 mg, 0.0649 mmol), and chloroform-d (0.76 mL) were used (see Table S4 and Figures S4–S6).
Reaction of α-Methylene-γ-Lactam 49 with Cysteamine Using an Internal Standard.
This reaction was performed and monitored using an internal standard according to General Procedure K. Lactam 49 (1.7 mg, 0.0043 mmol), cysteamine (5 mg, 0.065 mmol), hexamethylbenzene (0.7 μL mmol), and chloroform-d (0.76 mL) were used. Fraction Remaining was calculated using eq S5 as the initial NMR showed 2:1 integrative ratio of hexamethylbenzene:lactam (see Table S5 and Figures S7–S9).
Reaction of α-Methylene-γ-Lactam 51 with Cysteamine.
This reaction was monitored according to General Procedure K. Lactam 51 (0.98 mg, 0.0022 mmol), cysteamine (2.8 mg, 0.0334 mmol), and chloroform-d (0.4 mL) were used (see Table S6 and Figures S10–12).
Reaction of α-Methylene-γ-Lactam 52 with Cysteamine.
This reaction was performed according to General Procedure K. Lactam 52 (1.2 mg, 0.0029 mmol), cysteamine (3.4 mg, 0.044 mmol), and chloroform-d (0.5 mL) were used (see Table S7 and Figures S13–15).
Reaction of α-Methylene-γ-Lactam 52 with Cysteamine using an internal standard.
This reaction was performed and monitored using an internal standard according to General Procedure K. Lactam 52 (1.2 mg, 0.0029 mmol), cysteamine (3.4 mg, 0.044 mmol), hexamethylbenzene (0.4 μL, 0.0029 mmol), and chloroform-d (0.05 mL) were used. Fraction Remaining was calculated using eq S6 as the initial NMR showed a 1:1 integrative ratio of hexamethylbenzene:lactam (see Table S8 and Figures S16–18).
Reaction of α-Methylene-γ-Lactam 54 with Cysteamine.
Lactam 54 (2 mg, 0.004 mmol) was dissolved in chloroform-d (0.7 mL) and transferred to an NMR tube. Cysteamine (5 mg, 0.07 mmol) was dissolved in chloroform-d separately, transferred to the tube containing lactam 54 via pipet and the tube was capped and shaken vigorously by hand for 2 min. The progress of the reaction was monitored 1H NMR, specifically the disappearance of the methylene resonances (Ha and Hb) that are observed as doublets at δ 6.17 and 5.44. Complete disappearance of these resonances was observed in less than 10 min.
Reaction of α-Methylene-γ-Lactam 60 with Cysteamine.
This reaction was monitored according to General Procedure K. Lactam 60 (1.1 mg, 0.0041 mmol), cysteamine (5.0 mg, 0.065 mmol), and chloroform-d (0.80 mL) were used (see Table S9 and Figures 19–21).
Reaction of α-Methylene-γ-Lactone S17 with Cysteamine.
This reaction was performed according to General Procedure K. Lactone S17 (1.0 mg, 0.0033 mmol), cysteamine (3.8 mg, 0.049 mmol), and chloroform-d (0.66 mL) were used (see Table S10 and Figures 23–24).
Structural Confirmation of tert-Butyl Thiol Adduct
(3aS*,9bS*)-3-((tert-Butylthio)methyl)-1,6-dimethyl-9-phenyl-1,3a,4,5,7,9b-hexahydro-2H-azuleno[4,5-b]pyrrole-2,8(3H)-dione (S11):
A 2-mL vial equipped with a magnetic stir bar was charged with tricyclic lactam 48 (10 mg, 0.03 mmol), tert-butyl thiol (0.02 mL, 0.2 mmol), acetone (0.5 mL), and phosphate buffered saline (pH = 8.0, 0.5 mL). The vial was capped with a septum and allowed to stir at rt for 24 h. Only starting material was detected by TLC analysis, so the pH was raised to 12 (pH paper) by addition of potassium hydroxide (2 mg, 0.04 mmol) and the starting material was consumed within 30 min as observed by TLC. The solution was poured into a separatory funnel containing deionized water (3 mL) and DCM (10 mL). The layers were separated. The aqueous layer was extracted with DCM (2 × 5 mL). The organic layers were combined, washed with brine (5 mL), dried over magnesium sulfate, filtered, and concentrated by rotary evaporation and purified by flash column chromatography on silica gel (gradient elution with 25–50% ethyl acetate in hexanes) to provide thiol adduct S11 (3 mg, 23%, 1.6:1 diastereomeric ratio) as a clear oil. The diastereomeric ratio was determined by integration of resonances corresponding to Ha at δ 4.66 (J = 10.0 Hz) for the minor diastereomer and δ 4.50 (J = 8.5 Hz) for the major diastereomer. 1H NMR (500 MHz, CDCl3) δ 7.33–7.27 (m, 3H), 7.23–7.21 (m, 2H), 4.66 (d, J = 10.0 Hz, 1H)*, 4.50 (d, J = 8.5 Hz, 1H), 3.17–3.07 (m, 3H), 2.94–2.79 (m, 2H), 2.75–2.69 (m, 1H), 2.51–2.41 (m, 2H), 2.37–2.30 (m, 2H), 1.98 (s, 3H), 1.90 (s, 3H), 1.37 (s, 9H), *Discernible signals for 1 of 2 diastereomers; 13C NMR (100 MHz, CDCl3) δ 202.92, 202.86*, 175.3, 166.1, 138.5, 137.7, 137.1, 130.8, 130.73*, 130.67 (2C), 128.5, 128.4*, 127.7 (2C), 127.6*, 62.7, 61.7*, 50.3, 44.2, 43.2*, 42.7, 42.6*, 40.6, 34.3, 33.4*, 32.6, 31.1, 31.0*, 30.8, 30.4*, 30.32, 30.30*, 29.4, 25.7, 25.6*, 21.0, *Discernible signals for 1 of 2 diastereomers; IR (thin film) 3407, 2927, 1686, 1445, 1079 cm−1; HRMS TOF MS ES+ [M+H]: C25H32NO2S, Calc: 410.2148 Found: 410.2144; TLC Rf = 0.32 (50% EtOAc/hexanes) visualized with UV and vanillin.
Biological Materials and Methods
Preparation and Storage of Compound Stock Solutions.
Compound stock solutions were prepared in biological grade DMSO (40 mM to 100 mM concentrations) and stored at −20 °C when not in use. Compound purities were assessed prior to conducting biological assays by analytical reverse-phase HPLC. Fresh solutions were prepared as needed.
General Protocol for HPLC Analysis of Synthesized Compounds.
DMSO stock solutions of synthesized compounds were dissolved in a 1:1 mixture of methanol and distilled and deionized water (ddH2O) containing trifluoroacetic acid (TFA, 0.1% v/v) and analyzed on an Agilent 1200 series instrument equipped with a diode array detector and Zorbax SB-C18 column (4.6 × 150 mm, 5 μm, Agilent Technologies). The analysis method (1 mL/min flow rate) starts with an isocratic eluent system of 1:9 MeCN (containing TFA, 0.1% v/v; Solvent A):ddH2O (containing TFA, 0.1% v/v; Solvent B) from 0–2 minutes followed by a linear gradient of 10:90 to 85:15 A:B from 2–24 minutes, followed by 85:15 to 95:5 A:B from 24–26 minutes, and finally an isocratic eluent system of 95:5 A:B from 26–30 minutes. Wavelengths monitored = 215 nm (See Table S11 and Figures S25–S32).
Protocol for Mammalian Cell Culture.
Cell lines were kept in a humidified 19% O2, 5% CO2, and 37 °C environment. A549/NF-κB-luciferase cells were cultured as previously described.53 HEK293/NF-κB-SEAP (Novus Biologicals #NBP2–26260) were cultured in DMEM media (Corning) supplemented with 10% v/v FBS (Gibco), 100 I.U. penicillin (ATCC), 100 μg/mL streptomycin (ATCC), and 0.5 mg/mL G418 (Geneticin).
NF-κB Luciferase Reporter Assay.
Performed as previously described by our laboratory.53
NF-κB Secreted Placental Alkaline Phosphatase (SEAP) Reporter Assay.
HEK293/NF-κB-SEAP cells (Novus Biologicals #NBP2–26260) were seeded in standard 96-well cell culture plates (Costar) at a density of 5,000 cells/well (50 μL/well) 24 hours before dosing with compounds. Compounds serially diluted in pre-warmed media were dosed to cells (50 μL/well, final DMSO concentration: 0.5%) and following a 30 min incubation period, all wells except for the non-induced (N) control were induced with TNF-a (10 μL; final well concentration: 22.5 ng/mL; Invitrogen). Non-induced control wells received 1X PBS (10 μL). After 8 h, 25 μL of media was harvested from each well and plated into a second, 96-well white plate with clear bottom (Costar). Using the NovaBright™ Phospha-Light™ EXP Assay Kit for SEAP Reporter Gene Detection (Invitrogen #N10577) and a BioTek H1 Synergy microplate reader, SEAP was quantified per the manufacturer’s protocol. Each experimental condition had three technical replicates and each experiment was performed in biological triplicate. Activity values were obtained by averaging the mean activity values from each biological replicate. The standard deviation is obtained by propagating the standard deviations of each of the individual biological replicates. Statistical analyses were performed using Microsoft excel and plotted in GraphPad Prism (v. 5.0b).
Vero Cell Viability Assay.
Performed as previously described by our laboratory,53 with one change: compounds were dosed to cells for 30 min, followed by induction with TNF-α for 8 h.
ALARM NMR Assay.
Lactone S17 and lactam 51 were tested by ALARM NMR as previously described with minor modifications.58 Briefly, test compounds (400 μM final concentration) were incubated with 13C-methyl labeled La antigen (50 μM final concentration) at 37 °C for 90 minutes. Each compound was tested in the absence and presence of 20 mM DTT. Samples were loaded into Bruker 3 mM SampleJet tubes with 160 μL total sample volumes and stored at ambient temperature while in queue. Data were recorded at 25 °C on a Bruker 700 MHz NMR spectrometer equipped with a cryoprobe (Bruker). Data were collected for each compound every hour for 10 hours. Nonreactive compounds were identified by the absence of chemical shifts (13C-methyl) independent of the presence of DTT. Reactive compounds were identified by the presence of chemical shifts (13C-methyl) and peak attenuations in certain diagnostic peaks in the absence of DTT.
La Antigen Expression.
The La antigen protein was expressed and purified similar to a previously reported protocol.58 The protocol specifies the expression of 13C-labeled protein, but for mass spectrometry experiments, non-13C-labeled (unlabeled) protein was prepared by skipping the secondary culture spin down and resuspension in defined media steps.
La Antigen Mass Spectrometry Sample Preparation.
The tryptic digestion of purified recombinant protein was adapted from a previously published protocol.69 All solutions utilized in this section were purified by centrifugation with a 3 kDa MWCO (molecular weight cut-off) filter (Amiconâ, MilliporeSigma) and collection of the flow-through. Recombinant La antigen (2.5 μL; 2 μg/μL in aqueous 25 mM Na2HPO4/NaH2PO4 buffer, pH 7.0) was added to a solution of the test compound (15.5 μL of 10 mM DMSO stock). The sample was further diluted to 310 μL with 20 mM Tris buffer (pH 8.0) and incubated for 1 hr at 37°C. Next, unlabeled iodoacetamide (IAD) was added to a final concentration of 50 mM and the solution was incubated in the dark for 1 hr at room temperature. The protein was then isolated by spinning down the samples in a 3 kDa MWCO filter, diluting the sample with distilled and deionized H2O (ddH2O, 500 μL), and isolating the protein again through the MWCO filter (3x total). The protein was then evaporated to dryness overnight (SpeedVac). The dried protein-compound samples were resuspended in 20 μL of a freshly prepared aqueous denaturing solution (8 M urea, 50 mM ammonium bicarbonate, 50 mM DTT) and incubated at 37°C for 1 hr. A solution of 50 mM d4-IAD (Cambridge Isotope Laboratories, item #DLM-7249-PK) in ddH2O was prepared and 20 μL was added to each sample. The samples were incubated for 1 hr in the dark at room temperature. Next, 138 μL of 100 mM ammonium bicarbonate aqueous solution was added to each sample in order to dilute the urea concentration for trypsin digestion. A 1 μg/μL solution of trypsin (Promega, catalog #V5280) in 50 mM acetic acid was diluted to 100 ng/μL with 100 mM aqueous ammonium bicarbonate, and 10 μL of the resulting solution was added to each sample. Acetonitrile was immediately added to each sample to a final concentration of 10% (v/v). Samples were incubated in a rotating (800 rpm) heat block overnight at 37°C. Glacial acetic acid was added until the pH of each sample was less than 4.0 (~5 μL), then the samples were desalted using C18 resin pipette tips according to the manufacturer’s protocol (Pierce™ C18 Tips 10 μL bed, ThermoFisher). Desalted samples were evaporated to dryness (SpeedVac) and then reconstituted in 20 μL of 98:2 LC-MS grade H2O:MeCN containing 0.1% formic acid (v/v).
La Antigen Mass Spectrometry Analysis.
Peptides within each sample were separated with a front-end Dionex UltiMate 3000 ultra-high performance liquid chromatography (UHPLC) instrument. Peptides were separated using a home-packed analytical Luna C18 (100 Å pore, 5 μm particles) reverse phase column (75 μm ID × 200 mm, 10 μm emitter orifice, Phenomenex), where mass spectrometry (MS)-grade water with 0.1% formic acid was eluent A and acetonitrile with 0.1% formic acid was eluent B at room temperature. Initially, an isocratic 2% B elution was run for 5 minutes (1 μL/min flow rate). Then, the gradient elution (0.3 μL/min flow rate for all gradient steps) began by running from 2% B to 10 % B over 5 minutes, followed by 10% B to 25% B over 40 minutes, and then 25% B to 40% B over 10 minutes. The elution gradient further increased from 40% B to 90% B over one minute, then held at 90% B for 4 minutes before decreasing from 90% B to 2% B over 0.5 minutes and re-equilibrating at 2% B for 4.5 minutes (1 μL/min flow rate). Eluted peptides were analyzed with a LTQ Orbitrap Velos™ (ThermoFisher) in the Nth Order Double Play mode. The mass spectrometer utilized an electrospray ionization source with a source voltage of +2.5 kV. MS1 scan range was m/z = 220.0–1800.0. For MS2, the precursor ion mass of peptides containing the target cysteines in the La antigen were predicted using Skyline software.70 The m/z of the double and triple-charged peptides were calculated for three peptides: FSGDLDDQTC245R, IGC232LLK, AND SLEEKIGC232LLK. Both IGCLLK and SLEEKIGCLLK were searched, as our initial experiments suggested that the SLEEKIGCLLK peptide may be more readily identified with the small carbamidomethylation modifications, but the IGCLLK peptide may be more readily identified with the larger compound modifications. Seven possible cysteine modifications were evaluated: carbamidomethylation (+57.0215 Da), d2-carbamidomethylation (+59.0340 Da), d4-carbamidomethylation (+61.0466 Da), 2-chloro-1,4-napthoquinone (+306.1041 Da), CPM (N-[4-(7-diethylamino-4-methylcoumarin-3-yl)phenyl]maleimide) (+402.1580 Da), lactone S17 (+306.1256 Da), and lactam 51 (+449.1602 Da). In total, 42 precursor ions were selected for MS2 analysis (calculated MS1 precursors are listed in the Calculated Precursor Ions and MS2 y- and b-Ions List Excel supplementary file). Only the most intense ion from the specified parent ion list were selected for MS2, repeated for the top 12 peaks. Already examined precursor ions were excluded for 15 seconds. Ions with an unassigned charge state or a +1 charge state were rejected. Selected precursor ions (minimum signal = 5000) were analyzed using higher-energy C-trap dissociation (HCD) with a normalized collision energy of 35 V, an isolation width of 2, and the first mass value of m/z = 100.00.
Raw data was analyzed manually using Xcalibur™ Quan Browser software (ThermoFisher). Raw data files and associated peak list files were uploaded to the MassIVE UCSD public repository, and can be downloaded at ftp://massive.ucsd.edu/MSV000085781/. MS2 spectra of precursor ions were searched for peaks matching the expected y- and b-ion series based on peptide sequences (calculated y- and b-ion masses are identified in the Calculated Precursor Ions and MS2 y- and b-Ions List Excel supplementary file). Peptides were considered positively identified if all or all but one y-ions and at least two b-ions could be identified in a given spectra. Example spectra are included below (Figures S27–S37). CSV files containing the m/z and intensity of peaks of each positively identified MS2 spectrum can be found as supplemental Excel files. Each sample was run in technical triplicate and analyzed independently.
Peptides SLEEKIGC232LLK and IGC232LLK were searched and identified in the raw data, but the results indicate that C232 is less reactive than C245. For example, in the DMSO control samples, C232 peptides are found adducted with both IAD and d2-IAD, indicating that C232 is inefficiently labeled with IAD in the initial alkylation step with the properly folded La antigen protein. Consequently, C232 thiols are then alkylated with d4-IAD following protein denaturation. The same pattern of sluggish reactivity of C232 was observed in the fluconazole negative control. These data are also consistent with published ALARM NMR literature, in which titration experiments indicated C245 reacts more quickly than C232.56 Additional whole protein mass spectrometry studies from the same paper indicate both single- and double-adduct species, likely from the partial adduction at one cysteine. Taking these reactivity features into consideration, we elected to only use the FSGDLDDQTC245R peptide for our studies. However, the data for C232 peptides are shown below in Table S12 for general reference.
Supplementary Material
Acknowledgements
The authors acknowledge Todd Rappe and the Minnesota NMR center for assistance with the ALARM NMR assay. The authors acknowledge Jayme Dahlin for technical assistance and thoughtful discussion. We thank Dr. Steve Geib (Pitt) for X-ray data collection and refinement.
Funding
This work was supported by the NIH (R01-GM110129 to DAH) and the University of Pittsburgh. Funding for NMR instrumentation used for ALARM NMR assay was provided by the UMN Office of the Vice President for Research, UMN Medical School, UMN College of Biological Science, NIH, NSF and the Minnesota Medical Foundation. Protein mass spectrometry was performed at the Analytical Biochemistry Core Facility of the UMN Masonic Cancer Center, which is supported by the NIH (P30-CA77598). KFJ was supported by the National Science Foundation Graduate Research Fellowship Program.
Abbreviations
- μM
micromolar
- ALARM MSPS
a La assay to detect reactive molecules by nuclear magnetic resonance by mass spectrometry peptide sequencing
- APKR
allenic Pauson–Khand reaction
- Bpin
boron pinacolate
- Cs2CO3
cesium carbonate
- CDCl3
deuterochloroform
- CF3
trifluoromethyl
- CN
cyano
- CO
carbon monoxide
- CPM
(N-[4-(7-diethylamino-4-methylcoumarin-3-yl)phenyl]maleimide)
- CuH
copper hydride
- dr
diastereomeric ratio
- Et2O
diethyl ether
- EtOAc
ethyl acetate
- HCl
hydrochloric acid
- HEK
human embryonic kidney
- IAD
iodoacetamide
- K2CO3
potassium carbonate
- kinact
inactivation rate constant
- koff
dissociation rate constant
- kon
association rate constant
- KMnO4
potassium permanganate
- MeLi
methyl lithium
- mg
milligram
- Na2SO3
sodium sulfite
- NaI
sodium iodide
- NEt3
trimethylamine
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- ng
nanogram
- NH4Cl
ammonium chloride
- NH4OH
ammonium hydroxide
- OMe
methoxy
- p65
65 kDa polypeptide
- Ph
phenyl
- PTL
parthenolide
- PPh3
triphenylphosphine
- [Rh(CO)2Cl]2
Rhodium(bis)carbonylchloride dimer
- ScOTf3
scandium(III) triflate
- SEAP
secreted embryonic alkaline phosphatase
- SiMe3
trimethylsilane
- Tips
triisopropylsilyl
- TNF-α
tumor necrosis factor alpha
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is free of charge.
Molecular Formula Strings (CSV) have been uploaded.
1H NMR and 13C NMR spectra are provided for compounds 9, 11–13, 14a, 14b, 15a, 15b, 18a, 18b, 19a, 19b, 20a, 20b, 21a, 21b, 22a, 22b, 23a, 23b, 24a, 25a, 26, 27a, 28a, 29a, 32a, 30a, 37–40, 42, 43, 44–60, 64, 65, S2–S3, S5, S8–S10, S13–S17, PTL.
Accession Codes
Mass spectrometry raw data was deposited at the MassIVE UCSD Public Repository and can be accessed at ftp://massive.ucsd.edu/MSV000085781.
References
- 1.Kitson RR; Millemaggi A; Taylor RJ The Renaissance of α-Methylene-γ-Butyrolactones: New Synthetic Approaches. Angew. Chem. Int. Ed. 2009, 48, 9426–9451. [DOI] [PubMed] [Google Scholar]
- 2.Simonsen H; Weitzel C; Christensen S, Guaianolide Sesquiterpenoids: Pharmacology and Biosynthesis. In Natural Products, Ramawat KG; Mérillon J-M, Eds.; Springer; Berlin Heidelbergm, 2013; pp 3069–3098. [Google Scholar]
- 3.Drew DP; Krichau N; Reichwald K; Simonsen HT Guaianolides in Apiaceae: Perspectives on Pharmacology and Biosynthesis. Phytochem. Rev. 2009, 8, 581–599. [Google Scholar]
- 4.Ghantous A; Gali-Muhtasib H; Vuorela H; Saliba NA; Darwiche N What Made Sesquiterpene Lactones Reach Cancer Clinical Trials? Drug Discov. Today 2010, 15, 668–678. [DOI] [PubMed] [Google Scholar]
- 5.Kempema AM; Widen JC; Hexum JK; Andrews TE; Wang D; Rathe SK; Meece FA; Noble KE; Sachs Z; Largaespada DA; Harki DA Synthesis and Antileukemic Activities of C1–C10-Modified Parthenolide Analogues. Bioorg. Med. Chem. 2015, 23, 4737–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdeldayem A; Raouf YS; Constantinescu SN; Moriggl R; Gunning PT Advances in Covalent Kinase Inhibitors. Chem. Soc. Rev. 2020, 49, 2617–2687. [DOI] [PubMed] [Google Scholar]
- 7.Gehringer M; Laufer SA Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [DOI] [PubMed] [Google Scholar]
- 8.Dalton SE; Campos S Covalent Small Molecules as Enabling Platforms for Drug Discovery. ChemBioChem 2020, 21, 1080–1100. [DOI] [PubMed] [Google Scholar]
- 9.Lonsdale R; Ward RA Structure-Based Design of Targeted Covalent Inhibitors. Chem. Soc. Rev. 2018, 47, 3816–3830. [DOI] [PubMed] [Google Scholar]
- 10.Xu J; Chen A; Joy J; Xavier VJ; Ong EH; Hill J; Chai CL, Rational Design of Resorcylic Acid Lactone Analogues as Covalent MNK1/2 Kinase Inhibitors by Tuning the Reactivity of an Enamide Michael Acceptor. ChemMedChem 2013, 8, 1483–1494. [DOI] [PubMed] [Google Scholar]
- 11.Xu J; Ong EH; Hill J; Chen A; Chai CL Design, Synthesis and Biological Evaluation of FLT3 Covalent Inhibitors with a Resorcylic Acid Core. Bioorg. Med. Chem. 2014, 22, 6625–6637. [DOI] [PubMed] [Google Scholar]
- 12.Cee VJ; Volak LP; Chen Y; Bartberger MD; Tegley C; Arvedson T; McCarter J; Tasker AS; Fotsch C Systematic Study of the Glutathione (GSH) Reactivity of N-Arylacrylamides: 1. Effects of Aryl Substitution. J. Med. Chem. 2015, 58, 9171–9178. [DOI] [PubMed] [Google Scholar]
- 13.Flanagan ME; Abramite JA; Anderson DP; Aulabaugh A; Dahal UP; Gilbert AM; Li C; Montgomery J; Oppenheimer SR; Ryder T; Schuff BP; Uccello DP; Walker GS; Wu Y; Brown MF; Chen JM; Hayward MM; Noe MC; Obach RS; Philippe L; Shanmugasundaram V; Shapiro MJ; Starr J; Stroh J; Che Y Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors. J. Med. Chem. 2014, 57, 10072–10079. [DOI] [PubMed] [Google Scholar]
- 14.Jackson PA; Widen JC; Harki DA; Brummond KM Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem. 2017, 60, 839–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin JS; MacKenzie CJ; Fletcher D; Gilbert IH Characterising Covalent Warhead Reactivity. Bioorg. Med. Chem. 2019, 27, 2066–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monika IK; Balbina JP Lactones: Generic Inhibitors of Enzymes? Mini-Rev. Med. Chem. 2005, 5, 73–95. [DOI] [PubMed] [Google Scholar]
- 17.Irmgard M Perspectives on Sesquiterpene Lactones in Inflammation and Cancer. Curr. Drug Targets 2011, 12, 1560–1573. [DOI] [PubMed] [Google Scholar]
- 18.Ma G-H; Chen K-X; Zhang L-Q; Li Y-M Advance in Biological Activities of Natural Guaiane-Type Sesquiterpenes. Med. Chem. Res. 2019, 28, 1339–1358. [Google Scholar]
- 19.Siedle B; García-Piñeres AJ; Murillo R; Schulte-Mönting J; Castro V; Rüngeler P; Klaas CA; Da Costa FB; Kisiel W; Merfort I Quantitative Structure−Activity Relationship of Sesquiterpene Lactones as Inhibitors of the Transcription Factor NF-κB. J. Med. Chem. 2004, 47, 6042–6054. [DOI] [PubMed] [Google Scholar]
- 20.Buchele B; Zugmaier W; Lunov O; Syrovets T; Merfort I; Simmet T Surface Plasmon Resonance Analysis of Nuclear Factor-κB Protein Interactions with the Sesquiterpene Lactone Helenalin. Anal. Biochem. 2010, 401, 30–37. [DOI] [PubMed] [Google Scholar]
- 21.Hayden MS; Ghosh S Shared Principles in NF-κB signaling. Cell 2008, 132, 344–362. [DOI] [PubMed] [Google Scholar]
- 22.Chaturvedi MM; Sung B; Yadav VR; Kannappan R; Aggarwal BB NF-κB Addiction and its Role in Cancer: ‘One Size Does Not Fit All’. Oncogene 2011, 30, 1615–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wen B; Hexum JK; Widen JC; Harki DA; Brummond KM A Redox Economical Synthesis of Bioactive 6,12-Guaianolides. Org. Lett. 2013, 15, 2644–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wells SM The Synthesis of 6,12-Guaianolide Analogs and Related Chemical Tools for Probing their Mechanism of NF-κB Inhibition, PhD Thesis. University of Pittsburgh, 2016. [Google Scholar]
- 25.Grillet F; Huang C; Brummond KM An Allenic Pauson-Khand Approach to 6,12-Guaianolides. Org. Lett. 2011, 13, 6304–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brummond KM; Lu J A Short Synthesis of the Potent Antitumor Agent (±)-Hydroxymethylacylfulvene Using an Allenic Pauson-Khand Type Cycloaddition. J. Am. Chem. Soc. 1999, 121, 5087–5088. [Google Scholar]
- 27.Tsuji J; Sugiura T; Minami I Preparation of 1,2-Dienes by the Palladium-Catalyzed Hydrogenolysis of 3-Methoxycarbonyloxy-1-alkynes with Ammonium Formate. Synthesis 1987, 7, 603–606. [Google Scholar]
- 28.Elford TG; Hall DG Imine Allylation Using 2-Alkoxycarbonyl Allylboronates as an Expedient Three-Component Reaction to Polysubstituted α-exo-Methylene-γ-Lactams. Tetrahedron. Lett. 2008, 49, 6995–6998. [Google Scholar]
- 29.To obtain high yields consistently for this reaction, HMPA was distilled from CaH2, then stored over 4 Å molecular sieves, and chloromethyl pinacolboronate was freshly distilled.
- 30.Chataigner I; Zammattio F; Lebreton J; Villiéras J Enantioselective Addition of β-Functionalized Allylboronates to Aldehydes and Aldimines. Stereocontrolled Synthesis of α-Methylene-γ-lactones and Lactams. Tetrahedron 2008, 64, 2441–2455. [Google Scholar]
- 31.Elford TG; Ulaczyk-Lesanko A; Pascale GD; Wright GD; Hall DG Diversity-Oriented Synthesis and Preliminary Biological Screening of Highly Substituted Five-Membered Lactones and Lactams Originating From an Allylboration of Aldehydes and Imines. J. Comb. Chem. 2009, 11, 155–168. [DOI] [PubMed] [Google Scholar]
- 32.Chataigner I; Zammattio F; Lebreton J; Villiéras J Enantioselective Synthesis of α-Methylene-γ-Lactams. Nucleophilic Addition of a Chirally Modified β-Funtionalized Allylboronate Reagent to Imines. Synlett 1998, 275–276. [Google Scholar]
- 33.Daeuble JF; McGettigan C; Stryker JM Selective Reduction of Alkynes to Cis-Alkenes by Hydrometallation Using [(Ph3P)CuH]6. Tet. Lett 1990, 31, 2397–2400. [Google Scholar]
- 34.Petasis NA; Hu Y-H Allenation of Carbonyl Compounds with Alkenyltitanocene Derivatives. J. Org. Chem. 1997, 62, 782–783. [Google Scholar]
- 35.Tsuji J; Sugiura T; Yuhara M; Minami I Palladium-Catalysed Preparation of 1,2-Dienes by Selective Hydrogenolysis of Alk-2-ynyl Carbonates with Ammonium Formate. J. Chem. Soc., Chem. Commun. 1986, 12, 922–924. [Google Scholar]
- 36.Liu Z; Liao P; Bi X Lewis and Bronsted Acid Cocatalyzed Reductive Deoxyallenylation of Propargylic Alcohols with 2-Nitrobenzenesulfonylhydrazide. Chem. Eur. J. 2014, 20, 17277–17281. [DOI] [PubMed] [Google Scholar]
- 37.Yin J; Buchwald SL Pd-Catalyzed Intermolecular Amidation of Aryl Halides: The Discovery that Xantphos can be Trans-Chelating in a Palladium Complex. J. Am. Chem. Soc. 2002, 124, 6043–6048. [DOI] [PubMed] [Google Scholar]
- 38.Klapars A; Huang X; Buchwald SL A General and Efficient Copper Catalyst for the Amidation of Aryl Halides. J. Am. Chem. Soc. 2002, 124, 7421–7428. [DOI] [PubMed] [Google Scholar]
- 39.Wells SM; Brummond KM Conditions for a Rh(I)-catalyzed [2+2+1] Cycloaddition Reaction with Methyl Substituted Allenes and Alkynes. Tetrahedron. Lett. 2015, 56, 3546–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kallepalli VA; Shi F; Paul S; Onyeozili EN; Maleczka RE; Smith MR Boc Groups as Protectors and Directors for Ir-Catalyzed C−H Borylation of Heterocycles. J. Org. Chem. 2009, 74, 9199–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petri L; Ábrányi-Balogh P; Varga PR; Imre T; Keserű GM Comparative Reactivity Analysis of Small-Molecule Thiol Surrogates. Bioorg. Med. Chem. 2020, 7, 115357. [DOI] [PubMed] [Google Scholar]
- 42.Avonto C; Taglialatela-Scafati O; Pollastro F; Minassi A; Marzo VD; Petrocellis LD; Appendino G An NMR Spectroscopic Method to Identify and Classify Thiol-Trapping Agents: Revival of Michael Acceptors for Drug Discovery. Angew. Chem. Int. Ed. 2011, 50, 467–471. [DOI] [PubMed] [Google Scholar]
- 43.Spartan ‘16 MMFF was used to determine the lowest energy conformer and equilibrium geometries were established using DFT calculations (B3LYP) and 6–31G* basis set. All calculations were performed in the gas phase.
- 44.Castañeda-Acosta J; Fischer NH; Vargas D Biomimetic Transformations of Parthenolide. J. Nat. Prod. 1993, 56, 90–98. [DOI] [PubMed] [Google Scholar]
- 45.Widen JC; Kempema AM; Villalta PW; Harki DA Targeting NF-κB p65 with a Helenalin Inspired Bis-electrophile. ACS Chem. Biol. 2017, 12, 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rana S; Blowers EC; Tebbe C; Contreras JI; Radhakrishnan P; Kizhake S; Zhou T; Rajule RN; Arnst JL; Munkarah AR; Rattan R; Natarajan A Isatin Derived Spirocyclic Analogues with α-Methylene-γ-Butyrolactone as Anticancer Agents: A Structure–Activity Relationship Study. J. Med. Chem. 2016, 59, 5121–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.García-Piñeres AJ; Lindenmeyer MT; Merfort I Role of Cysteine Residues of p65/NF-κB on the Inhibition by the Sesquiterpene Lactone Parthenolide and N-Ethyl Maleimide, and on its Transactivating Potential. Life Sci. 2004, 75, 841–856. [DOI] [PubMed] [Google Scholar]
- 48.Pande V; Sousa SF; Ramos MJ Direct Covalent Modification as a Strategy to Inhibit Nuclear Factor-κB. Curr. Med. Chem. 2009, 16, 4261–4273. [DOI] [PubMed] [Google Scholar]
- 49.Widen JC; Kempema AM; Baur JW; Skopec HM; Edwards JT; Brown TJ; Brown DA; Meece FA; Harki DA Helenalin Analogues Targeting NF-κB p65: Thiol Reactivity and Cellular Potency Studies of Varied Electrophiles. ChemMedChem 2018, 13, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kwok BH; Koh B; Ndubuisi MI; Elofsson M; Crews CM The Anti-Inflammatory Natural Product Parthenolide from the Medicinal Herb Feverfew Directly Binds to and Inhibits IκB Kinase. Chem. Biol. 2001, 8, 759–766. [DOI] [PubMed] [Google Scholar]
- 51.Freund RRA; Gobrecht P; Fischer D; Arndt H-D Advances in Chemistry and Bioactivity of Parthenolide. Nat. Prod. Rep. 2020, 37, 541–565. [DOI] [PubMed] [Google Scholar]
- 52.Lindenmeyer MT; García-Piñeres AJ; Castro V; Merfort I Sesquiterpene Lactones Inhibit Luciferase but not β-Galactosidase Activity in vitro and ex vivo. Anal. Biochem. 2004, 328, 147–154. [DOI] [PubMed] [Google Scholar]
- 53.Hexum JK; Tello-Aburto R; Struntz NB; Harned AM; Harki D Bicyclic Cyclohexenones as Inhibitors of NF-kB Signaling. ACS Med. Chem. Lett. 2012, 3, 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moraski GC; Markley LD; Hipskind PA; Boshoff H; Cho S; Franzblau SG; Miller MJ Advent of Imidazo[1,2-a]pyridine-3-carboxamides with Potent Multi- and Extended Drug Resistant Antituberculosis Activity. ACS Med. Chem. Lett 2011, 2, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X-C; Babu KS; Jacob MR; Khan SI; Agarwal AK; Clark AM Natural Product-Based 6-Hydroxy-2,3,4,6-tetrahydropyrrolo[1,2-a]pyrimidinium Scaffold as a New Antifungal Template. ACS Med. Chem. Lett. 2011, 2, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huth JR; Mendoza R; Olejniczak ET; Johnson RW; Cothron DA; Liu Y; Lerner CG; Chen J; Hajduk PJ ALARM NMR: A Rapid and Robust Experimental Method to Detect Reactive False Positives in Biochemical Screens. J. Am. Chem. Soc. 2005, 127, 217–224. [DOI] [PubMed] [Google Scholar]
- 57.Huth JR; Song D; Mendoza RR; Black-Schaefer CL; Mack JC; Dorwin SA; Ladror US; Severin JM; Walter KA; Bartley DM; Hajduk PJ Toxicological Evaluation of Thiol-Reactive Compounds Identified Using A La Assay to Detect Reactive Molecules by Nuclear Magnetic Resonance. Chem. Res. Toxicol. 2007, 20, 1752–1759. [DOI] [PubMed] [Google Scholar]
- 58.Dahlin JL; Cuellar M; Singh G; Nelson KM; Strasser J; Rappe T; Xia Y; Veglia G; Walters MA ALARM NMR for HTS Triage and Chemical Probe Validation. Curr. Protoc. Chem. Biol. 2018, 10, 91–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Potter ZE; Lau HT; Chakraborty S; Fang L; Guttman M; Ong SE; Fowler DM; Maly DJ Parallel Chemoselective Profiling for Mapping Protein Structure. Cell Chem. Biol. 2020, 8, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Görl C; Alt HG The Combination of Mononuclear Metallocene and Phenoxyimine Complexes to Give Trinuclear Catalysts for the Polymerization of Ethylene. J. Organomet. Chem. 2007, 692, 5727–5753. [Google Scholar]
- 61.Abidi SL Direct Conversion of Terpenylalkanolamines to Ethylidyne N-Nitroso Compounds. J. Org. Chem. 1986, 51, 2687–2694. [Google Scholar]
- 62.Balog A; Curran DP Ring-Enlarging Annulations. A One-Step Conversion of Cyclic Silyl Acyloins and ω-Alkynyl Acetals to Polycyclic Enediones. J. Org. Chem. 1995, 60, 337–344. [Google Scholar]
- 63.Whiting A A Convenient Preparation of β-boronate Carbonyl Derivatives. Evidence For the Intervention of Boronate “ate”-Complexes in Enolate Alkylations. Tetrahedron. Lett. 1991, 32, 1503–1506. [Google Scholar]
- 64.Grillet F; Huang C; Brummond KM An Allenic Pauson-Khand Approach to 6,12-Guaianolides. Org. Lett. 2011, 13, 6304–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Serra S; Fuganti C Benzannulation of Substituted 3-Alkoxycarbonylhex-3-en-5-ynoic Acids: A New Route to 4-Substituted 3,5-Dihydroxybenzoic Acids Derivatives. Synlett 2002, 2002, 1661–1664. [Google Scholar]
- 66.Fatiadi AJ Active Manganese Dioxide Oxidation in Organic Chemistry - Part I. Synthesis 1976, 2, 65–104. [Google Scholar]
- 67.Kern N; Hoffmann M; Blanc A; Weibel JM; Pale P Gold(I)-Catalyzed Rearrangement of N-Aryl 2-Alkynylazetidines to Pyrrolo[1,2-a]indoles. Org. Lett. 2013, 15, 836–839. [DOI] [PubMed] [Google Scholar]
- 68.Lee D; Yun J Direct Synthesis of Stryker’s Reagent from a Cu(II) Salt. Tetrahedron. Lett. 2005, 46, 2037–2039. [Google Scholar]
- 69.Carlson TL; Moini M; Eckenrode BA; Allred BM; Donfack J Protein Extraction from Human Anagen Head Hairs 1-Millimeter or Less in Total Length. BioTechniques 2018, 64, 170–176. [DOI] [PubMed] [Google Scholar]
- 70.MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. J. Bioinform. 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.