

Abstract
Meningiomas are the most frequent primary intracranial tumors. The considerable variety of histological subtypes has been expanded by the definition of molecular alterations, which can improve both diagnostic accuracy and determination of individual patient's outcome. According to the upcoming WHO classification of brain tumors, the in‐time analysis of frequent molecular events in meningiomas may become mandatory to define meningioma subtypes. We have compiled a custom‐made amplicon‐based next generation sequencing (NGS) meningioma panel covering the most frequent known recurrent mutations in 15 different genes. In an unselected consecutive meningioma cohort (109 patients) analyzed over a period of 12 months, we detected mutations in 11 different genes, with most frequent alterations in NF2 (43%), AKT1 E17K (15%), and TRAF7 (13%). In 39 tumors (36%), two different mutations were detected, with NF2 and SUFU (n = 5) and KLF4 and TRAF7 (n = 5) being the most frequent combinations. No alterations were found in POLR2A, CDKN2A, CDKN2B, and BAP1, and no homozygous CDKN2A/B deletion was detected. NF2 mutations were found in tumors of all WHO grades, whereas mutations in KLF4, TRAF7, and SMO were restricted to WHO grade I meningiomas. In contrast, SMARCE1 and TERT mutations were associated with WHO grade II meningiomas (according to the WHO classification 2016). The distribution of mutations across histological subtypes or tumor localization was in line with the existing literature, with typical combinations like KLF4K409Q/TRAF7 for secretory meningiomas and preferential skull base localization of meningiomas harboring SMO and AKT1 E17K mutations. Thus, we present a custom‐made NGS meningioma panel providing a time and cost‐efficient reliable detection of relevant somatic molecular alterations in meningiomas suitable for daily routine.
Keywords: amplicon‐based targeted sequencing, meningioma, next generation sequencing
Description of a novel custom‐made NGS panel for rapid molecular characterization of meningiomas.

1. INTRODUCTION
Meningiomas are the most frequent primary intracranial tumors [1]. Beside the majority of tumors belonging to the benign WHO [World Health Organization] grade I group of meningiomas, approximately 20% are aggressive WHO grade II tumors, and about 1% are graded as malignant WHO grade III meningiomas, characterized by aggressive biology and reduced progression free and overall survival [2, 3, 4]. To understand the biology of aggressive meningiomas and to identify potential treatment targets, key molecular alterations need to be defined. Moreover, the variety of histological subtypes even among WHO grade I meningiomas has raised the question about diverse underlying molecular alterations.
For several decades, the tumor suppressor NF2 had been identified as the only key tumor driver being present in roughly half of sporadic meningiomas [5]. Only the introduction of highly sensitive next generation sequencing (NGS) techniques has provided novel insight in the molecular basis of meningiomas and has resulted in the refinement of non‐NF2 meningiomas, which are characterized by different recurrent mutations, sometimes in close relation to histological subtypes [4, 6, 7, 8]. The frequency of individual mutations is mostly below 10% of meningiomas, but some alterations like combined KLF4/TRAF7 mutations, which are highly specific for secretory meningiomas [9], have opened the road for a molecularly‐supported diagnosis of meningiomas. Moreover, some alterations are associated with reduced time to tumor recurrence [10, 11], whereas the presence of others like KLF4 might indicate favorable clinical course [12, 13].
Beside the application of time‐ and cost‐intensive genome‐wide DNA methylation analyses to classify meningiomas according their biological features and risk of recurrence [14], comprehensive gene panels covering the most essential genetic alterations in meningiomas are desirable, especially if intended to be incorporated into daily routine practice. Moreover, alterations with potential therapeutic impact can be identified, for instance the AKT1 E17K mutation as a drug target [15]. These aspects are becoming relevant with the new WHO classification of brain tumors, linking the diagnosis of certain meningioma subtypes with characteristic molecular aberrations [16].
2. MATERIALS AND METHODS
2.1. Human tumor material
A total of 109 consecutive meningioma samples were sequenced in the time period between February 2020 and February 2021. All samples were derived from tumor surgery and submitted to the Institute of Neuropathology Magdeburg, Germany, for diagnostic purposes. Tumors were routinely embedded in paraffin by standard methods and characterized using standard staining procedures. Tumors were classified according to the current 2016 WHO classification of brain tumors [17].
2.2. Preparation of DNA
The relevant tumor region was labeled on a routine H&E‐stained slice to allow DNA isolation from a histologically inspected tumor area. After dewaxing with Protax Clear (Quartett, Germany), DNA was extracted applying the Nucleospin Tissue kit (Macherey & Nagel, Düren, Germany), according to the manufacturer's instructions. Immediately prior to library preparation, DNA was then quantified by a Qubit fluorometer (Thermofisher, Germany) using the high sensitivity chemistry (dsHS assay kit). DNA was used immediately for library preparation.
2.3. NGS procedure
2.3.1. Platform and panel construction
Amplicon based NGS sequencing (AmpliSeq) of a custom meningioma gene panel (Illumina) was performed on a MiniSeq platform (Illumina, San Diego, USA), utilizing the Mid Output flow cell and sequencing cartridge. The new custom meningioma panel contained 10 genes as complete cds (see Table 1), which had been reported to be mutated in meningiomas according to COSMIC database and literature with varying frequency. It included the most frequently mutated gene NF2, as well as CDKN2A, the homozygous deletion of which can cause rare, but important losses of the p16 and p14 tumor suppressors [18]. It also contained PIK3CA and PIK3R1, which can carry mutations that alter the PI3K‐AKT signaling pathway in non‐NF2 meningiomas.
TABLE 1.
Summary of meningioma‐related genes covered by the NGS meningioma panel
| Gene | Region | Frequency (%) current study | Frequency (%) literature | References |
|---|---|---|---|---|
| NF2 | cds | 43 | 43 | [6] |
| AKT1 | E17K | 15 | 5–13.6 [30] | [6, 7] ([11]) |
| TRAF7 | cds | 13 | 20–28.9 | [7, 19] |
| PIK3CA | cds | 10 | 6 | [8] |
| KLF4 | K409Q | 6 | 8.8 | [7] |
| SMO | L412F, W535L | 6 | 5 [28] | [6] ([10]) |
| SUFU | cds | 6 | Somatic rare | [7] |
| PIK3R1 | cds | 3 | 0.6 | [7] |
| SMARCE1 | cds | 1 | rare | [20] |
| SMARCB1 | cds | 1 | 6 | [6, 8] |
| TERT | C228T, C250T | 1 | 6.4 | [21] |
| POLR2A | hotspots | 0 | 6 | [8] |
| CDKN2A/B | cds | 0 | 5 | [18] |
| BAP1 | cds | 0 | Somatic rare | [22] |
Abbreviation: cds, coding DNA sequence and splice sites.
The panel was complemented by hot spots of five additional genes, including those in SMO, KLF4, and AKT1, most often mutated in benign WHO‐grade I non‐NF2 meningiomas. The latter protein kinase often carries a constitutively activating missense mutation (E17K), which enhances PI3K‐AKT signaling and reaches mutation frequencies around 30% in the subgroup growing at the skull base [11]. The panel also includes the two TERT promoter hot spots C228T and C250T. Although frequent in gliomas, these promoter activating mutations are rare, but important to be analyzed in meningiomas due to their potentially negative prognostic influence [21].
The complete target region is 32,784 bp in length. 96.1% are covered by the designed amplicons, reaching from 125 to 175 bp with mean and median values of 157 and 163 bp, respectively. Primers for multiplex PCR of these amplicons were distributed in two pools of 190 and 186 amplicons. Due do the necessity to include the highly GC rich and difficult TERT region and in order to avoid more than 2 oligo pools, a panel of low stringency was accepted, which may be expected to generate somewhat lower sequencing depth due to a higher percentage of non‐targeted multiplex PCR products. However, the panel turned out to work properly in practical sequencing, yielding excellent mean sequencing depth (typically 2000–5000), and most often acceptable, but low depth in TERT (typically <150).
2.4. Library synthesis and sequencing
Library synthesis was performed with the AmpliSeq Library Pius Kit (Illumina) according to the manufacturer's instructions and using the corresponding adaptor plates, containing in each well a premix of two index‐adaptors, each with an index length of 8 bp (Illumina). If possible, 10 to 100 ng of genomic DNA were used per oligo pool for the initial multiplex PCR reaction, usually yielding high library concentrations of 10 to 20 ng/µl (Qubit). In cases of low DNA availability, also 3 ng per pool mostly yielded weaker, but acceptable libraries, which could be well sequenced in the majority of cases. Lower template amounts rarely worked.
According to the manufacturer's instructions, libraries were pooled at a concentration of 10 nM, then the pool concentration was measured again (Qubit) and the pool was diluted accordingly to 1 nM, denatured according to the kit protocol and then diluted with hybridization buffer (delivered with MiniSeq cartridges) to a final concentration of 1.6 pM. Usage of this final library concentration for sequencing tended to generate a cluster density, moderately higher than the intended maximum of 230,000 clusters per mm2, but without loss of quality (clusters passing filter, percentage Q30) or sequencing yield. Runs were governed by LRM (local run manager, Illumina) and bi‐directional sequencing with 2x 151 cycles was chosen with 2 × 8 bp index reads.
2.5. Data processing
Data collection, base calling and variant calling were governed by LRM (Illumina). When initializing a run, the allele frequency filter was set to 5% in the mode ‘somatic’. Variant call depth filter was set to 10 and BWA chosen as aligner. Indel alignment was put on and annotation was labeled ‘RefSeq’. After picture acquisition, base calling and de‐complexing, the obtained sequences were automatically compared with the defined 32.8 kb target region of the human reference sequence (GRCh37, hg19). Output variant tables were transferred into Excel format and visually inspected for potentially relevant mutations, that is, known or likely pathogenic mutations by utilizing the automatically delivered hints from ClinVar or COSMIC database. Known pathogenic TERT promoter mutations were identified according to their coordinates in hg19, delivered in the variant tables. For relevance of a detected mutation, we exploited literature data for classical hot spot and other mutations repeatedly observed in meningiomas and besides that pathogenic score delivered by COSMIC in case of doubtful missense mutations. We used information delivered by the database ClinVar, if available.
When no SMO, KLF4, AKT1, and TERT hotspot mutations were detected in a sample, sufficient sequencing depth was controlled specifically at the corresponding hg19 coordinates, using the coverage tables, delivered by LRM. In addition, coverage tables were checked for homozygous deletion of CDKN2A/B, which was not observed in any of the analyzed 109 meningiomas, but could be repeatedly detected in the malignant intraosseous IOMM‐Lee meningioma cell line, which served as a positive control [23, 24].
In general, sequencing and variant calling had been initially checked by NGS re‐sequencing of meningiomas with known AKT1, SMO, and KLF4 mutations (analyzed earlier by Sanger sequencing) and by re‐sequencing of several glioma samples with known TERT mutations, previously sequenced by NGS with a published glioma gene panel [19]. Some known NF2 mutated cases from other sources had been used to check sequencing of this gene.
In the variant tables, variant quality (automatically generated in the variant tables) was checked for all detected pathogenic mutations. It was 100% in all cases listed in the present manuscript. Number of reads for each variant and allele frequency (AF) were checked. All AFs were compatible with somatic mutations and extremely low AF, suggesting the mutation to be present only in a fraction of tumor cells, did not occur in the meningioma mutations described here. Generally, mean sequencing depth and sequencing homogeneity were controlled for each sample in the control dataset of LRM.
2.6. Statistical analysis
Frequencies of subgroups are reported as absolute counts and percentages. Associations between categorical variables have been described by cross‐tabulation and tested in Pearson's chi‐squared tests with the option for the use of the exact test distribution. Age has been compared between subgroups in Kruskal‐Wallis tests. Because of the exploratory analysis strategy, all tests have been carried out at an unadjusted level of 5%. The analyses have been carried out with IBM SPSS Statistics, version 26.
3. RESULTS
3.1. Basic characteristics of the cohort
The cohort consisted of 109 consecutive tumors samples from 48 males (mean age: 64 years, range 39–88 years) and 61 females (mean age: 63 years, range 30–88 years). Tumor localization was categorized as intracranial tumors (convexity or skull base) or spinal tumor (Figure 1A). Ninety (82.6%) tumors were WHO grade I, 16 (14.7%) WHO grade II, and 3 (2.8%) WHO grade III tumors. All WHO grade II tumors were classified as atypical meningiomas, and all WHO grade III tumors were anaplastic meningiomas. For each patient, one sample was analyzed. In six cases (5.5%), samples were derived from tumor recurrences.
FIGURE 1.
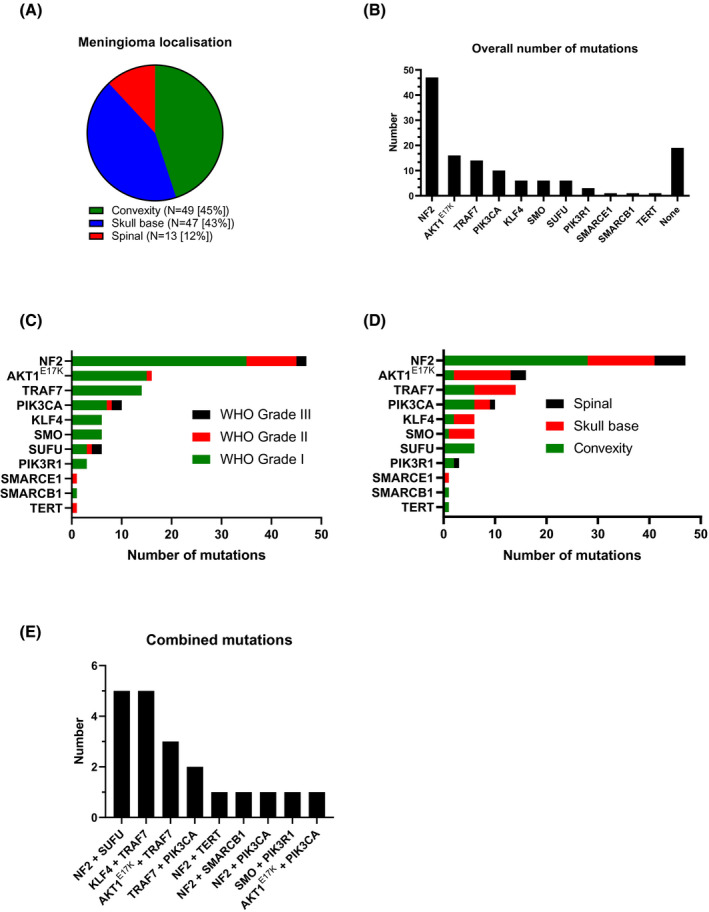
Distribution of meningioma localization (A). Overall number of somatic mutations detected within all 109 samples (B). Distribution of mutations within all three WHO grades (C) and separated by tumor localization (D). Combined mutations within the cohort (E)
3.2. Frequency and distribution of mutations detected
Using the newly created NGS panel, we detected mutations in 89 tumors (82%), while in 20 tumors (18%) no alterations were found. Within the 89 tumors with mutations, 11 different mutated genes were detected (Figure 1B). No alterations were detected in the following four genes: POLR2A, CDKN2A, CDKN2B, and BAP1. Two different mutations were detected in 39/109 samples (35.8%), and three mutations were found in one sample (0.9%). The most frequently affected gene was NF2 (43% among 109 samples), followed by AKT1 E17K (15%) and TRAF7 (13%). Combined mutations of KLF4/TRAF7 were detected in 5 samples (3 secretory meningiomas, 2 meningothelial meningiomas). No differences of the mutation frequency in relation to the age were found.
3.3. Distribution by tumor grade and location
According to the general dominance of tumors of WHO grade I, most mutations occurred in meningiomas of this grade. However, the particularly frequent inactivating NF2 mutations (mostly frameshifts, nonsense or splice site mutations), but also missense mutations in the gene encoding the catalytic subunit of the important signal transducing phosphoinositide‐3 kinase (PIK3CA), as well as the relatively rare SUFU mutations occurred across all WHO grades. The rare SMARCE1 and activating TERT promoter mutations were restricted to atypical meningiomas WHO grade II (Figure 1C). SUFU mutations were overrepresented among malignant meningiomas as compared to WHO grade I or II meningiomas (p = 0.000). To a lesser extent, the same relative overrepresentation in anaplastic WHO grade III meningiomas as compared to grades I and II occurred with respect to mutations in the PIK3CA gene (p = 0.02). In contrast, TRAF7 (missense) and the hotspot KLF4 K409Q (or both in combination) mutations, as well the two known missense hotspot SMO mutations were found selectively in WHO grade I meningiomas (Figure 1C).
The analysis of tumor localization (Figure 1D) largely reflected the knowledge of the current literature. NF2 inactivation occurred in tumors of all localizations, reflecting the general importance of this tumor suppressor in meningiomas. Still, NF2 mutations were significantly more often located at the convexity as compared to skull base or spinal meningiomas (p = 0.014).
The hotspot mutations in AKT1 and SMO showed a clear preference for the skull base (Figure 1D). Because the AKT1 hotspot mutation E17K was the second most frequent alteration observed in our series, a meaningful statistical analysis could be performed with respect to localization. It indeed statistical analysis confirmed the preference for skull base over convexity and spinal localizations (p = 0.02). In addition, the association of SUFU mutations with the convexity was significant (p = 0.02). TRAF7 mutations (most often amino acid exchanges in the C‐terminal WD40 repeat region) occurred at comparable frequencies at skull base and convexity locations, reflecting the similar percentage of both locations among the 109 cases analyzed.
For histological subtypes, significant associations (p < 0.001) were found between KLF4 and secretory subtype, NF2 and fibroblastic/transitional subtype, and TRAF7 and meningothelial subtype. Despite the low number of cases, recurrent tumors showed higher frequencies of PIK3CA (p < 0.001) and SUFU mutations (p = 0.002).
Several combinations of multiple mutations, that is, more than one in a single tumor were detected as shown in Figure 1E. The status of ‘multiple mutations’ was significantly associated with the genes KLF4, TRAF7, SUFU, and PIK3CA (p < 0.05).
3.4. Role of patient's age at diagnosis
Patients with NF2 mutations were significantly older than patients without (60.5 vs 67.2 years, p = 0.007), whereas patients with TRAF7 mutations were significantly younger than those without (54.4 years vs. 64.7 years; p = 0.006). There was no significant association between sex and type of mutation.
3.5. NF2 mutations
As expected from databases, NF2 mutations were distributed along the full coding region of the NF2 gene and were by far most frequently frameshifts, nonsense, or splice site mutations (Figure 2A), together representing 93.5% of all NF2 mutations detected. The primary causes of these events were base exchanges or small deletions or insertions. As shown in Figure 2B, nearly all histological subtypes of meningiomas were affected by these mutations. They occurred frequently in the major histologic subtypes of WHO grade I, that is, meningothelial, fibroblastic, and transitional meningiomas, but also in atypical (grade II), anaplastic (grade III), and some rarer subtypes of meningiomas. They were absent in secretory meningiomas, which usually carried the KLF4K 409Q mutation that is largely absent in NF2 mutated meningiomas, as it is the case for other recently detected non‐NF2 mutations.
FIGURE 2.
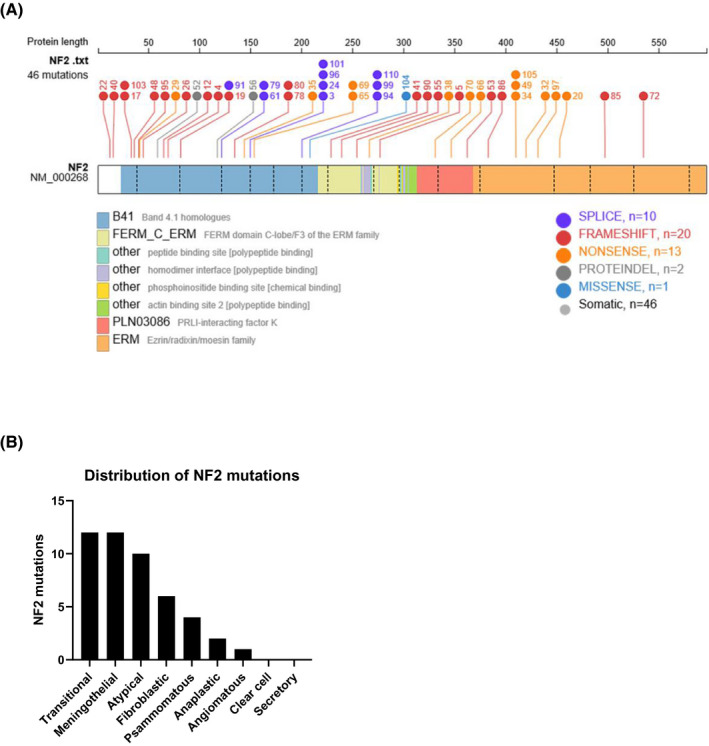
Alterations of NF2 in the cohort. Distribution and type of NF2 mutations across the coding region of NF2 (A). Distribution of NF2 mutations related to the histological subtype (B)
3.6. TRAF7 mutations
TRAF7 mutations were by far most often missense mutations, distributed over large parts of the coding region, but enriched in the carboxy‐terminal WD40 repeat region (Figure 3A). This gene harbored only slightly lower mutation counts as compared to AKT1, the second most frequently mutated gene in our tumor series. Amino acid exchanges in TRAF7 occurred in secretory meningiomas (in combination with KLF4 K409Q), but also as single mutation or in other combinations in meningothelial and transitional meningiomas (Figure 3B).
FIGURE 3.
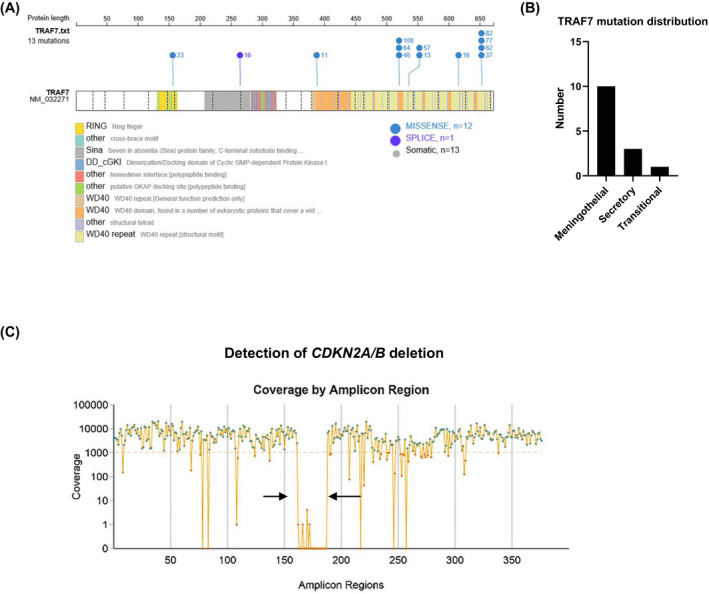
Distribution of TRAF7 mutations across the coding region of the TRAF7 gene (A) and among histological subtypes (B). Detection of CDKN2A/B deletions (arrows) with the NGS panel (C) (example from malignant IOMM‐Lee meningioma cells with known CDKN2A deletion [24])
3.7. Detection of homozygous CDKN2A/B deletions
In the whole series of 109 meningiomas, we did not detect any case harboring a homozygous CDKN2A/B deletion, that is, the functional loss of the gene coding for the p16INK4a cell‐cycle regulating tumor suppressor in one of its reading frames and for the p14arf tumor suppressor in a second one. The constructed NGS panel is expected to detect such homozygous deletion as a dramatic decrease of read frequencies of the amplicons covering this region of chromosome 9 (GRCh37 coordinates chr9:21,967,751–21,995,323). In case of nearly pure tumor tissue, read frequencies are expected to be close to zero. As a positive control, we sequenced IOMM‐Lee meningioma cells, a cell line established from an intraosseous malignant meningioma [23] and known to harbor a homozygous CDKN2A/B deletion [24]. As shown in Figure 3C, the mean coverage of amplicons in that run was several thousand‐fold, as it is the case also for routine FFPE meningioma samples in our hands in a standard run, the precise coverage depending on the number of samples in the run. The homogeneity of coverage is quite good, despite some (expected) and irrelevant drops in single amplicons. However, the CDKN2A/B region (arrows in Figure 3C) is selectively missing with coverages usually of zero and of up to five reads in a few cases. In other runs of IOMM‐Lee cell clones, the maximum read frequency in a few amplicons of this region was 1, but zero in nearly all amplicons. Such a picture did neither occur in any of the 109 tumors of the series, nor since then in routine diagnostics. However, it has been reproduced several times in single clones of IOMM‐Lee cells. Thus, our panel is able to detect homozygous CDKN2A/B deletions in general.
3.8. The meningioma panel in laboratory practice
Since the introduction of this custom‐made meningioma panel, it turned out to be a robust sequencing tool for routine FFPE material, fresh frozen tumor samples, or cell lines. For FFPE material, routine DNA extraction from labeled tissues can be used without further modifications. Library preparations using starting DNA amounts between 10 and 100 ng of genomic DNA (QuBit) per oligo pool delivered nearly always libraries of intermediate to high concentrations (QuBit), yielding high quality sequencing results. Library preparations starting with 3 to <10 ng of genomic DNA usually delivered libraries of lower concentration, which could nevertheless be sequenced with high quality in most cases. Starting concentrations below 3 ng only occasionally worked, although 1 ng per oligo pool might theoretically be acceptable according to the manufacturer's instructions (Illumina). In order to avoid overclustering, it is reasonable to load library pools of 1.5 to 1.6 pM onto the flow cell, instead of 1.8 pM as described in the manufacturer's instructions.
After labeling of histologic slices (H&E) by the neuropathologist and DNA extraction (1 day), the preparation of libraries, which we perform in 8‐well strips (ultrathin), takes 1.5 days. On the same or the next day pooling, denaturation and dilution of libraries can be performed and the sequencing run can be started, which will be finished overnight. Therefore, the complete laboratory procedure can be finished within 4–5 days. With the current version of the LRM (local run manager), the analysis of the runs up to the level of variant tables can be done automatically by the MiniSeq itself within the overnight period for sequencing, thus requiring no additional time for IT work.
4. DISCUSSION
Here we present a meningioma‐specific 15 gene next generation sequencing (NGS) panel, which allows convenient detection of relevant somatic mutations in meningiomas. Timely detection of tumor‐defining mutations or mutations with an impact on tumor recurrence or overall survival will become mandatory with the introduction of the 2021 WHO classification of brain tumors [16].
Meningiomas are the most frequent intracranial tumors [1]. While the vast majority belongs to the WHO grade I group (80%) with low proliferative potential of the tumor and good clinical outcome, a significant fraction belongs to the more aggressive WHO grade II or III meningiomas, characterized by more frequent tumor recurrences, increased morbidity, and impaired overall survival [2, 25]. The quantitative predominance of benign meningiomas was reflected in our series and the frequencies of the various mutations observed was thus expected to correspond roughly to distributions described in recent literature for non‐ selected meningiomas. Taking into account the naturally occurring variability for rare mutations, our results reflect the landscape described by the literature fairly well.
4.1. Comparison of the mutational landscape with the literature
For the most frequently mutated gene NF2, the result of 43% affected tumors (Table 1), covering all grades, fits exactly with the NGS study of Brastianos and colleagues [6]. In our series, the AKT1 E17K hotspot was on the upper edge of observed frequencies in unselected series [6, 7], while it was mutated twice as frequently in a series of pure skull base meningiomas [11]. It seems that AKT1‐driven tumors were moderately over‐represented in our fraction of non‐NF2 tumors, while amino acid exchanges in TRAF7 exhibited a relatively lower frequency, as compared to other studies [7, 26]. For the other genes besides AKT1, which code for components of the PI3K‐AKT‐mTOR pathway and can exhibit activating oncogenic missense mutations, the results were sufficiently compatible with the landscape delivered by the literature, as can be seen for the catalytic and regulatory subunits of PI3K, that is, PIK3CA and PIK3R1 [8].
Altered hedgehog signaling by missense mutations in SMO hotspots exhibited low frequencies in our series, which were in accordance with prior NGS studies [6]. In contrast, the tumor suppressor SUFU, an element of hedgehog signaling downstream of SMO [27], seemed to exhibit somewhat more mutations in our series (5%), though enriched in high‐grade tumors. Germline mutations of this suppressor may in rare cases underlie a familial tumor syndrome with multiple meningiomas, outside the setting of neurofibromatosis type 2 [28], but can also occur occasionally as somatic mutations in sporadic meningiomas [7, 8]. A good correspondence with previous data occurred also for the KLF4 hotspot, which often characterized secretory meningiomas and was often combined with amino acid exchanges in TRAF7, as described by others [7, 9]. The predominance of missense mutations in the WD40 regions (Figure 3A) also fits with the literature [9].
The rare mutations in SMARCB1 [6] and SMARCE1, the latter known to be restricted to clear cell meningiomas and frequently found in younger patients [20, 29], were both observed only at frequencies around 1% in our series. The single TERT promoter mutation occurred in a high‐grade (atypical) meningioma, in accordance with literature, where the activating mutations of the promoter, C228T and C250T, are largely restricted to WHO grades II and III, where they occur at moderate frequencies around 4%–5% (grade II) and 16%–20% (grade III) [21, 30]. They can be regarded as indicators of highly aggressive tumor growth requiring closer surveillance. This is underlined by the dramatically shorter median time to progression in cases with (10.1 months) as compared to cases without TERT promoter mutation (179 months) [21].
Homozygous deletions of the tumor suppressors p16 and ARF14 (alternative reading frames of CDKN2A) and p15 (encoded by the neighboring gene CDKN2B on chromosome 9), which are all involved in negative regulation of the cell cycle, are rare events and restricted to high grade meningiomas [18]. It is thus not surprising that deletions of both neighboring regions were not detected in our series, but could be demonstrated in the cell line IOMM‐Lee, derived from a malignant intraosseous meningioma and known to harbor this deletion [23, 24]. Occasionally, point mutations (e.g. missense) of a less clear impact were found in these genes in addition by some studies. However, point mutations were not detected in the whole cds in our series. On the other hand, the occasionally described heterozygous deletions of these genes cannot be completely excluded in our series, because NGS procedure and mode of data analysis were not sensitive to moderate CNV (copy number variation), the detection of which was not the aim of this panel.
Germline mutations in BAP1 are known to underlie sometimes melanomas, renal cell carcinomas, malignant mesotheliomas, and some other cancers [31]. They may rarely occur also in high‐grade meningiomas. In the latter tumors they may even occasionally occur as somatic events and the cds of BAP1 was thus represented on our meningioma panel [22, 32]. Given the rarity of these events, it was not surprising that no mutation was found.
Another rare event in meningiomas are mutations in the RNA polymerase II gene (POLR2A), responsible for synthesis of mRNAs and many small RNAs, thereby being an important regulator of the tumor cell transcriptome [33]. No mutations in some previously described hotspots were detected in our series, although they occurred in 6% of meningiomas in one study [8].
A recent study clearly demonstrated the significant role of molecular characterization of meningiomas in the future by elaborating the strong impact of molecular subgroups on outcome [13]. For example, PI3K‐activated tumors recurred earlier than other subgroups and subgroups with alterations in NF2, PI3K, and hedgehog signaling were particularly aggressive, with 22 times more frequent recurrences than subgroups defined by KLF4, POLR2A, and SMARCB1 alterations. Such results clearly demonstrate the impact of mutational screening by NGS to define prognostic markers in meningiomas.
Taken together, we propose a custom‐made NGS panel detecting all relevant mutational events currently regarded as relevant for accurate diagnosis of meningioma with known characteristic molecular features, as well as for information about a potentially aggressive clinical course with reduced time to tumor recurrence.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
AUTHOR CONTRIBUTIONS
Christian Mawrin: writing, data analysis, concept; Ralf Koch: data analysis; Natalie Waldt: data analysis; I. Erol Sandalcioglu: patient data; Werner E. K. Braunsdorf: patient data; Jan‐Peter Warnke: patient data; Felix Goehre: patient data; Hans‐Jürgen Meisel: patient data; Christian Ewald: patient data; Sina Neyazii: graphics; Ulrich Schüller: writing, graphics; Elmar Kirches: data analysis, writing.
ACKNOWLEDGEMENTS
We thank Ms. Jessica Zajontz and Ms. Gabriele Schulze for their enthusiastic work while establishing laboratory details and procedures for the meningioma NGS panel. We also thank Dr. Tekaya‐Ammar for helpful discussion. U.S. was supported by the Fördergemeinschaft Kinderkrebszentrum Hamburg and S.N. by a fellowship of the University Cancer Center Hamburg.
Mawrin C, Koch R, Waldt N, Sandalcioglu IE, Braunsdorf WEK, Warnke J‐P, et al. A new amplicon‐based gene panel for next generation sequencing characterization of meningiomas. Brain Pathol. 2022;32:e13046. 10.1111/bpa.13046
DATA AVAILABILITY STATEMENT
Raw data from sequencing are available upon request.
REFERENCES
- 1. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz‐Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro‐Oncology. 2018;20(suppl_4):iv1–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Durand A, Labrousse F, Jouvet A, Bauchet L, Kalamarides M, Menei P, et al. WHO grade II and III meningiomas: a study of prognostic factors. J Neurooncol. 2009;95(3):367–75. [DOI] [PubMed] [Google Scholar]
- 3. Mawrin C, Perry A. Pathological classification and molecular genetics of meningiomas. J Neurooncol. 2010;99(3):379–91. [DOI] [PubMed] [Google Scholar]
- 4. Preusser M, Brastianos PK, Mawrin C. Advances in meningioma genetics: novel therapeutic opportunities. Nat Rev Neurol. 2018;14(2):106–15. [DOI] [PubMed] [Google Scholar]
- 5. Ruttledge MH, Sarrazin J, Rangaratnam S, Phelan CM, Twist E, Merel P, et al. Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat Genet. 1994;6(2):180–4. [DOI] [PubMed] [Google Scholar]
- 6. Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet. 2013;45(3):285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clark VE, Erson‐Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, et al. Genomic analysis of non‐NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339(6123):1077–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clark VE, Harmanci AS, Bai H, Youngblood MW, Lee TI, Baranoski JF, et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat Genet. 2016;48(10):1253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reuss DE, Piro RM, Jones DT, Simon M, Ketter R, Kool M, et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. 2013;125(3):351–8. [DOI] [PubMed] [Google Scholar]
- 10. Boetto J, Bielle F, Sanson M, Peyre M, Kalamarides M. SMO mutation status defines a distinct and frequent molecular subgroup in olfactory groove meningiomas. Neuro‐oncology. 2017;19(3):345–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yesiloz U, Kirches E, Hartmann C, Scholz J, Kropf S, Sahm F, et al. Frequent AKT1E17K mutations in skull base meningiomas are associated with mTOR and ERK1/2 activation and reduced time to tumor recurrence. Neuro‐oncology. 2017;19(8):1088–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. von Spreckelsen N, Waldt N, Poetschke R, Kesseler C, Dohmen H, Jiao HK, et al. KLF4(K409Q)‐mutated meningiomas show enhanced hypoxia signaling and respond to mTORC1 inhibitor treatment. Acta Neuropathol Commun. 2020;8(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Youngblood MW, Miyagishima DF, Jin L, Gupte T, Li C, Duran D, et al. Associations of meningioma molecular subgroup and tumor recurrence. Neuro‐oncology. 2021;23(5):783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sahm F, Schrimpf D, Stichel D, Jones DT, Hielscher T, Schefzyk S, et al. DNA methylation‐based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 2017;18(5):682–94. [DOI] [PubMed] [Google Scholar]
- 15. Weller M, Roth P, Sahm F, Burghardt I, Schuknecht B, Rushing EJ, et al. Durable control of metastatic AKT1‐mutant WHO grade 1 meningothelial meningioma by the AKT inhibitor, AZD5363. J Natl Cancer Inst. 2017;109(3):1–4. [DOI] [PubMed] [Google Scholar]
- 16. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella‐Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro‐oncology. 2021;23(8):1231–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella‐Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;1–18. [DOI] [PubMed] [Google Scholar]
- 18. Sievers P, Hielscher T, Schrimpf D, Stichel D, Reuss DE, Berghoff AS, et al. CDKN2A/B homozygous deletion is associated with early recurrence in meningiomas. Acta Neuropathol. 2020;140(3):409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zacher A, Kaulich K, Stepanow S, Wolter M, Köhrer K, Felsberg J, et al. Molecular diagnostics of gliomas using next generation sequencing of a glioma‐tailored gene panel. Brain Pathol. 2017;27(2):146–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sievers P, Sill M, Blume C, Tauziede‐Espariat A, Schrimpf D, Stichel D, et al. Clear cell meningiomas are defined by a highly distinct DNA methylation profile and mutations in SMARCE1. Acta Neuropathol. 2021;141(2):281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sahm F, Schrimpf D, Olar A, Koelsche C, Reuss D, Bissel J, et al. TERT promoter mutations and risk of recurrence in meningioma. J Natl Cancer Inst. 108(5):djv377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shankar GM, Abedalthagafi M, Vaubel RA, Merrill PH, Nayyar N, Gill CM, et al. Germline and somatic BAP1 mutations in high‐grade rhabdoid meningiomas. Neuro‐oncology. 2017;19(4):535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee WH. Characterization of a newly established malignant meningioma cell line of the human brain: IOMM‐Lee. Neurosurgery. 1990;27(3):389–95; discussion 96. [PubMed] [Google Scholar]
- 24. Mei Y, Bi WL, Greenwald NF, Agar NY, Beroukhim R, Dunn GP, et al. Genomic profile of human meningioma cell lines. PLoS One. 2017;12(5):e0178322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perry A, Scheithauer BW, Stafford SL, Lohse CM, Wollan PC. “Malignancy” in meningiomas: a clinicopathologic study of 116 patients, with grading implications. Cancer. 1999;85(9):2046–56. [DOI] [PubMed] [Google Scholar]
- 26. Yuzawa S, Nishihara H, Tanaka S. Genetic landscape of meningioma. Brain Tumor Pathol. 2016;33(4):237–47. [DOI] [PubMed] [Google Scholar]
- 27. Laurendeau I, Ferrer M, Garrido D, D'Haene N, Ciavarelli P, Basso A, et al. Gene expression profiling of the hedgehog signaling pathway in human meningiomas. Mol Med. 2010;16(7–8):262–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aavikko M, Li SP, Saarinen S, Alhopuro P, Kaasinen E, Morgunova E, et al. Loss of SUFU function in familial multiple meningioma. Am J Hum Genet. 2012;91(3):520–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kirches E, Sahm F, Korshunov A, Bluecher C, Waldt N, Kropf S, et al. Molecular profiling of pediatric meningiomas shows tumor characteristics distinct from adult meningiomas. Acta Neuropathol. 2021;142(5):873–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koelsche C, Sahm F, Capper D, Reuss D, Sturm D, Jones DT, et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013;126(6):907–15. [DOI] [PubMed] [Google Scholar]
- 31. Walpole S, Pritchard AL, Cebulla CM, Pilarski R, Stautberg M, Davidorf FH, et al. Comprehensive study of the clinical phenotype of germline BAP1 variant‐carrying families worldwide. J Natl Cancer Inst. 2018;110(12):1328–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shankar GM, Santagata S. BAP1 mutations in high‐grade meningioma: implications for patient care. Neuro‐oncology. 2017;19(11):1447–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hansen AW, Arora P, Khayat MM, Smith LJ, Lewis AM, Rossetti LZ, et al. Germline mutation in POLR2A: a heterogeneous, multi‐systemic developmental disorder characterized by transcriptional dysregulation. HGG Adv. 2021;2(1):100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw data from sequencing are available upon request.