

Abstract
An increased number of histaminergic neurons, identified by labeling histidine‐decarboxylase (HDC) its synthesis enzyme, was unexpectedly found in patients with narcolepsy type 1 (NT1). In quest for enlightenment, we evaluate whether an increase in HDC cell number and expression level would be detected in mouse models of the disease, in order to provide proof of concepts reveling possible mechanisms of compensation for the loss of orexin neurons, and/or of induced expression as a consequence of local neuroinflammation, a state that likely accompanies NT1. To further explore the compensatory hypothesis, we also study the noradrenergic wake‐promoting system. Immunohistochemistry for HDC, orexin, and melanin‐concentrating hormone (MCH) was used to count neurons. Quantitative‐PCR of HDC, orexin, MCH, and tyrosine‐hydroxylase was performed to evaluate levels of mRNA expression in the hypothalamus or the dorsal pons. Both quantifications were achieved in genetic and neuroinflammatory models of narcolepsy with major orexin impairment, namely the orexin‐deficient (Orex‐KO) and orexin‐hemagglutinin (Orex‐HA) mice respectively. The number of HDC neurons and mRNA expression level were unchanged in Orex‐KO mice compared to controls. Similarly, we found no change in tyrosine‐hydroxylase mRNA expression in the dorsal pons between groups. Further, despite the presence of protracted local neuroinflammation as witnessed by the presence of reactive microglia, we found no change in the number of neurons nor the expression of HDC in Orex‐HA mice compared to controls. Importantly, no correlation was found in all conditions between HDC and orexin. Our findings indicate that, in mice, the expression of histamine and noradrenalin, two wake‐promoting systems, are not modulated by orexin level whether the lack of orexin is constitutive or induced at adult age, showing thus no compensation. They also show no recruitment of histamine by local neuroinflammation. Further studies will be needed to further define the role of histamine in the pathophysiology of NT1.
Keywords: hypocretin, hypothalamus, orexin, sleep
Histamine do not compensate for the loss of orexin in mice models of narcolepsy type 1, whether the absence of orexin is congenital or acquired at adult age, due to genetic defect or to an induced autoimmune insult.

1. INTRODUCTION
Narcolepsy type 1 (NT1) is a rare disorder of central hypersomnolence, whose main symptoms are excessive daytime sleepiness and cataplexy, caused by the loss of hypocretin/orexin (ORX) neurons, a powerful wake‐promoting neuronal population of the tuberal hypothalamus. The neuronal loss is highly selective and mirrored by low cerebrospinal fluid (CSF) hypocretin‐1 (orexin A) levels in NT1 patients [1, 2]. Convergent studies point toward an autoimmune etiology [3, 4], with recent findings reporting the increased frequency of autoreactive T cells in NT1 patients [5, 6, 7], likely accompanied by wider neuroinflammation.
Interestingly, post‐mortem analyses of NT1 brain tissue revealed that the number of histamine (HIS) neurons identified by labeling of histidine‐decarboxylase (HDC), the HIS synthesis enzyme, is significantly increased in NT1 patients compared to controls [8, 9]. It was unexpected because of HIS is a strong wake‐promoting player and NT1 patients suffer from excessive sleepiness. Also, HIS neurons are excited and disinhibited by ORX [10] suggesting, although not demonstrated, that HIS neuronal firing rate would be rather decreased or unchanged as a consequence of the absence of ORX input. In this regard, CSF histamine content was found to be either decreased [11] or unchanged [12] in adults with NT1 compared to controls. To get a better understanding of these human data, previous authors have also evaluated the number of HIS neurons in animal models of NT1. While one study found no increase in the number of HIS neurons in several animal models of NT1 [8], others reported a mild increase [9, 13]. This observation led the latter ones [9] to advocate for a mechanism compensating for the loss of ORX in order to oppose excessive daytime sleepiness. In addition to HIS, the noradrenergic neurons of the locus coeruleus form another wake‐promoting system that is activated by ORX and relay its wake promoting effect [14]. Cell counting has not been evaluated so far. However a recent study measured monoamine levels in the CSF of 39 NT1 patients (with ORX deficiency) compared to 55 patients with normal ORX level with or without objective sleepiness but found no difference in catecholamine levels and its metabolites between groups [15].
All animal models studied so far were based on genetic impairment of ORX gene or neurons, while human NT1 is thought to be of autoimmune origin. The alternative hypothesis would thus be that HIS level could be modulated by a local neuroinflammatory process accompanying the loss of ORX signaling [16]. Animal models can be of help to test these hypotheses, compensation and/or neuroinflammation, and more broadly to evaluate whether HIS signaling could be modulated by the impairment of ORX signaling.
Thus, to address these questions, we used a genetic model of NT1, the ORX‐knock‐out (Orex‐KO) mice, and the neuroinflammatory model of NT1 we recently developed, the Orex‐HA mice [17].
We first assessed the number of HIS neurons using immunohistochemistry against HDC to compare our results to previous studies. Then, to obtain a more accurate quantification, we evaluated the expression level of HDC mRNA using quantitative PCR. Cell counts and levels of mRNA expression were also assessed for ORX and melanin‐concentrating hormone (MCH) as controls. We verified for the presence of neuroinflammation at the vicinity of HDC neurons in Orex‐HA mice by immunolabeling microglia, and finally, to further explore the compensatory hypothesis, we studied the noradrenergic wake‐promoting system, through quantification of tyrosine‐hydroxylase (TH, synthesis enzyme of catecholamine) expression level in the dorsal pons.
2. MATERIALS AND METHODS
2.1. Mice
Fifty‐seven adult mice were kept in standard conditions until sacrifice (23 ± 1°C, 12:12 light:dark cycle, food and water ad libitum, with littermates and paper cuttings for nesting). All animal experiments were performed in accordance with the European Union guidelines following approval of the local ethics committee (APAFIS#7770‐2017010511127939). We used C57BL/6J Orex‐KO mice backcrossed to C57BL/6J mice for over 14 generations as a genetic model of NT1, and (BALB/cJ × C57BL/6J) Orex‐HA mice as an immune‐mediated model of NT1. The C57BL/6J wild‐type (WT) mice were born from the same litters as the Orex‐KO mice. T cell transfer to Orex‐HA and control mice was performed as previously reported [17]. For immunohistochemistry, we examined the hypothalami of 5 WT C57BL/6J, 5 Orex‐KO, 4 Orex‐HA no‐T (with no T cells transfer), and 6 Orex‐HA‐2xCD8 (transferred twice with autoreactive CD8 T cells) mice. We also used two HDC‐KO mice to test for the specificity of the primary anti‐HDC antibody. They were all young adults of 13 to 26 weeks old. Please note that 13 of the Orex‐HA recipient mice used in cell counting were studied in [17].
For qPCR, all the experimental groups were age and sex matched (10 WT, 26.1 [25.6–27.3] weeks; 11 Orex‐KO, 25.4 [20.6–29.6] weeks; 9 Orex‐HA‐2xCD8, 24.4 [16.9–27.6] weeks; 5 WT‐2xCD8, 27.3 [17.7–27.3] weeks; age expressed as median [Q1–Q3]) (p = 0.89).
2.2. Preparation of mouse brain tissue
Mice were perfused under deep anesthesia with saline followed by 4% paraformaldehyde (PFA). The brains were post‐fixed in 4% PFA for 48 h and then transferred in 30% sucrose until freezing in 2‐methylbutane. Brain tissue used in qPCR experiments were immediately fresh‐frozen in 2‐methylbutane after sacrifice by decapitation under deep anesthesia. All mice were sacrificed between 2 pm and 4 pm, when HDC mRNA expression has been shown to be of intermediate level [18].
2.3. Immunohistochemistry
Brains were sliced in 30 µm‐thick coronal free‐floating sections using a cryostat. The immunohistochemical detection was performed as previously described with minor modifications [19]. For double staining of HDC neurons and microglia, the same procedure was repeated after 24 hours but without nickel enrichment, for Iba1 detection (brown staining). The primary antibodies used were rabbit anti‐MCH 1:100,000 (Phoenix Pharmaceuticals), goat anti‐orexin A (C‐19) 1:10,000 (Santa Cruz Biotechnology), rabbit anti‐HDC 1:1500 (a generous gift from G. Bruneau, France) [20] and rabbit anti‐Iba1 1:5000 (Wako Chemicals USA).
The anti‐orexin A, anti‐MCH, and anti‐Iba1 antibodies have been used in a large number of studies and their specificity extensively evaluated (among others, [21, 22, 23]). Bruneau and colleagues [20] have extensively evaluated the specificity of the anti‐HDC antiserum, and we further tested it on HDC‐KO mice, yielding no staining in HDC‐KO mice and a high‐quality staining of HDC neurons in WT mice (Figure 1G).
FIGURE 1.
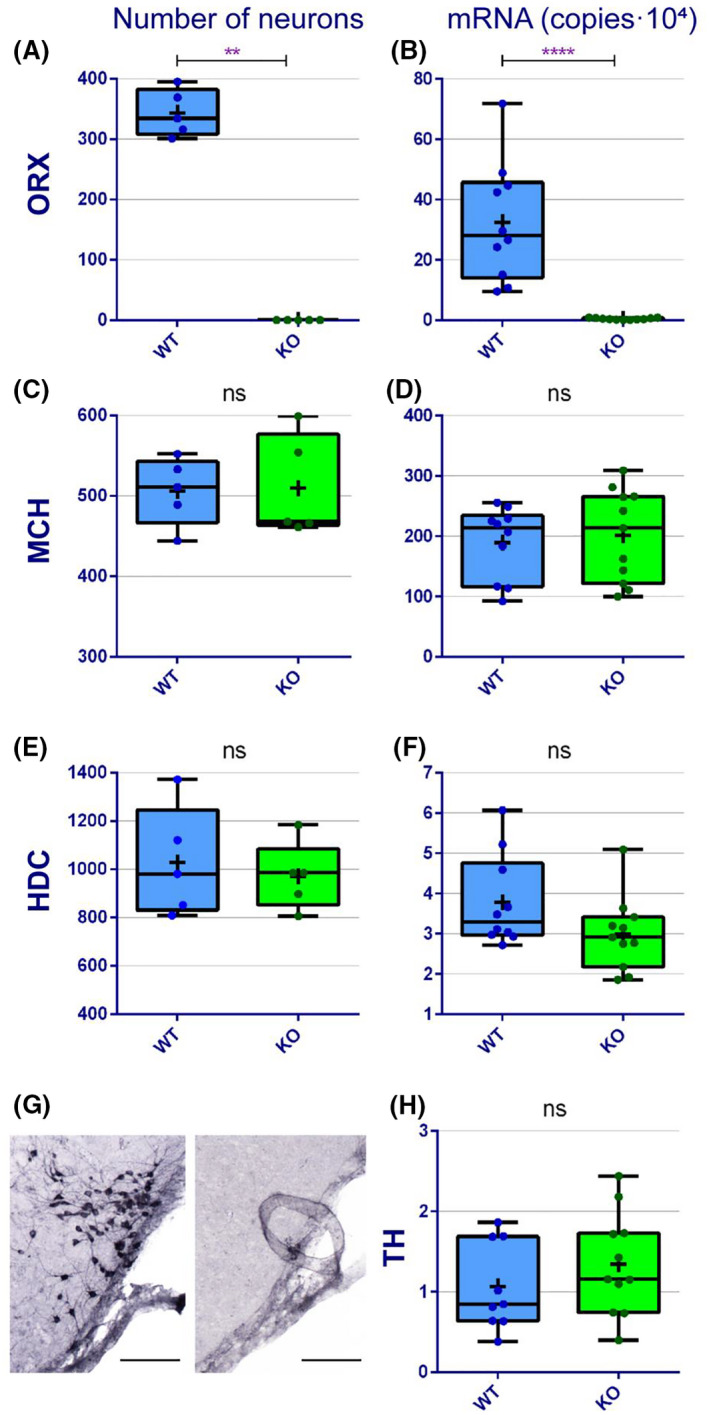
Evaluation of ORX, MCH, HDC, and TH content in a genetic model of NT1, the Orex‐KO mice. Number of ORX (A), MCH (C), and HDC (E) neurons in C57BL/6J WT and Orex‐KO mice. mRNA expression level for ORX (B), MCH (D), HDC (F), and TH (H) in C57BL/6J WT and Orex‐KO mice. Data are shown as median (Q1–Q3), individual data values are represented as dots and the mean is represented as a cross. Mann–Whitney U test, *p < 0.05; **p < 0.01. ns, not significant. (G) Microphotograph illustrating HDC immunostaining in the tuberomammillary nucleus of a WT (left) and a HDC‐KO mouse (right). Scale bars: 110 µm
Neuron counting was performed on 1/4th (for HDC) or 1/12th (for MCH and ORX) of the entire hypothalamus by using Mercator software for stereology (Explora Nova, France). All counts were performed by the same investigator (SM) blinded to the experimental groups.
2.4. Quantitative PCR
The hypothalamus was dissected from four consecutive 500 µm‐thick coronal sections per brain using a surgical blade, and the pons from a single 500 µm‐thick coronal section per brain. Total RNA was extracted using the RNeasy Plus Mini or Micro Kits (QIAGEN) (for hypothalamus and pons respectively) followed by reverse transcription (SuperScript IV Reverse Transcriptase, Invitrogen) starting from 200 ng of RNA for each sample. Only samples with a good quality RNA (A260/A280>1.8) were included in the analyses (NanoDrop 2000, Thermo Scientific). qPCR amplifications were performed in 20 µl reactions containing 5 µl of cDNA, 10 µl of Rotor‐Gene SYBR Green PCR Kit (QIAGEN), and 1 µl of primers mix (10 µM); runs were performed on a Rotor‐Gene Q cycler (QIAGEN). Oligonucleotides used as PCR primers are reported in Table 1.
TABLE 1.
Primers sequences used for qPCR
| Name | Primer sequence |
|---|---|
| HDC‐F | 5′‐GTTTGTGATTCGGTCCTTCG‐3′ |
| HDC‐R | 5′‐AAGGAAGGGTCGCTTCTGAC‐3′ |
| MCH‐F | 5′‐TCTTCCTACATGTTAATGCTGGCT‐3′ |
| MCH‐R | 5′‐TCCTTATGGACTTGGAAGCTGA‐3′ |
| ORX‐F | 5′‐CTTCAGGCCAACGGTAACCAC‐3′ |
| ORX‐R | 5′‐CCGGGGTGCTAAAGCGGTG‐3′ |
| TH‐F | 5′‐ATGCCTCCTCACCTATGCAC‐3′ |
| TH‐R | 5′‐CAGCCAACATGGGTACGTGT‐3′ |
Absolute quantification of cDNA was based on the threshold cycle (Ct) values obtained from the associated Rotor‐Gene Q Series Software (QIAGEN). The concentration in copy number of target gene was obtained relating the Ct value to a standard curve, established using 100‐fold serial dilutions of a reference DNA. Briefly, the reference DNA was prepared pooling the retro‐transcribed total RNA extracted from the hypothalamus (for ORX, MCH, and HDC) or the pons (for TH) of three C57BL/6J WT mice, followed by a step of amplification with the above‐mentioned primers and purification with the QIAquick PCR Purification Kit (QIAGEN).
2.5. Statistical analyses
We run a priori power analysis based on one‐tailed tests using the GPower software to determine the group sizes for Orex‐KO and WT groups, as already done in previous work [13]. Given a desired statistical power >80%, a significance level (α) of 0.05, the effect size based on previous data reporting more HDC neurons in Orex‐KO than in WT mice [9] and a one‐tailed Mann–Whitney test, five animals per group are needed for cell counting. As no data is available considering quantification with qPCR, such test cannot be performed. However, qPCR reactions were performed on larger group size, at least in duplicate for each sample and the mean of the duplicates was used as value for each animal. In light of the groups' size, the different experimental groups were compared using a non‐parametric Mann–Whitney U test when comparing two independent groups, or the non‐parametric Kruskal–Wallis test when comparing more than two independent groups. When comparisons were statistically significant, two‐by‐two comparisons were carried out, using the Dunn's test with correction for multiple comparisons. Note that a one‐tailed Mann–Whitney U test was used when comparing cell counting for Orex‐KO vs WT groups, in accordance with the a priori power analysis performed. However, we performed a two‐tailed test when considering qPCR quantification and cell counts on the Orex‐HA model, given the absence of prior data and hence of directional hypothesis. Correlations between age and HDC or ORX and HDC were performed using the Spearman non‐parametric test of correlation. Statistical significance was accepted at p < 0.05. GraphPad Prism software (Version 6.0) was used to perform analyses and box plots. The data that support the findings of this study are available from the corresponding author upon reasonable request.
3. RESULTS
We found no significant difference in the number of HIS neurons between Orex‐KO and WT mice (p = 0.48) using immunohistochemistry to HDC (Figure 1E,G) and in the mRNA expression of HDC (p = 0.11) using qPCR analysis (Figure 1F). It has been reported that HDC expression may vary with age [24]. Here, the selected mice are all adult mice with limited variation in age. Accordingly, we found no correlation between mRNA HDC expression levels and age within each group of mice (p = 0.8 for WT; p = 0.7 for Orex‐KO).
We also evaluated another wake‐promoting system, the noradrenergic system, and found no significant difference in TH mRNA expression level in the dorsal pons between the Orex‐KO and WT mice (p = 0.26) (Figure 1H).
Both, cell counting and the mRNA quantification confirmed the impairment of the ORX system and the integrity of the MCH system in Orex‐KO mice (Figure 1). Orex‐KO mice showed no presence of ORX‐immunoreactive neurons (p = 0.004) and their ORX mRNA expression was at background level (p < 0.0001) (Figure 1A,B). The number of MCH neurons and MCH expression levels were not different between the Orex‐KO and the WT mice (p = 0.47 and p = 0.55 respectively) (Figure 1C,D).
To explore the hypothesis of a neuroinflammatory process, we next used the immune‐mediated mouse model of NT1 we created, the Orex‐HA [17]. In this model, a specific targeting of the ORX neurons mediated by autoreactive (HA‐specific) T cells was accompanied by a neuroinflammation of the tuberal hypothalamus where ORX neurons are located as evaluated using Iba1 immunostaining of microglia [17]. Here, we further show that microglial cells have prominent hypertrophy of the soma with decreased ramification of distal branches and thickening of proximal processes, all morphological features typical of the activated state, in the posterior hypothalamus that contains HDC cells, in Orex‐HA mice transferred twice with cytotoxic autoreactive CD8 T cells (Figure 2E,F). However, microglial cells showed a resting phenotype with small soma, thin cellular processes, and dense distal arborization in the TMN of both WT (Figure 2A,B) and Orex‐KO (Figure 2C,D) mice. Note that the microglial state was evaluated 60 days after T cell transfer indicating that the inflammatory process was sustained for weeks.
FIGURE 2.
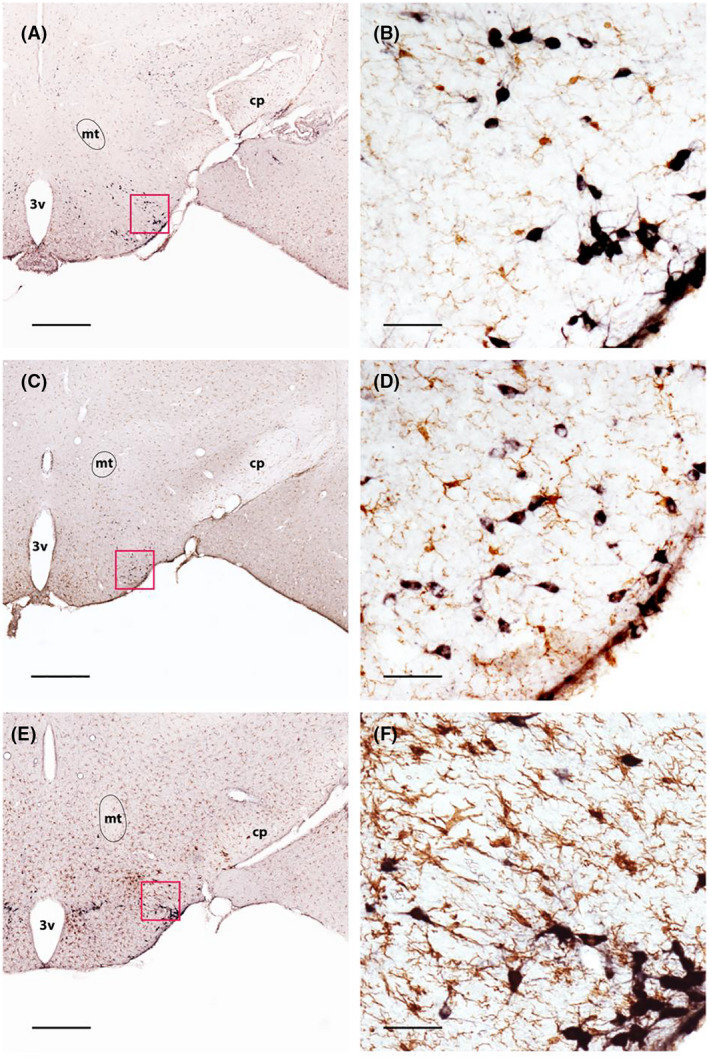
Evaluation of microglial activation in Orex‐KO and Orex‐HA mice. Double immunostaining of HDC (black) and Iba1 (brown) in the hypothalamus of WT (A and B), Orex‐KO (C and D), and Orex‐HA mice transferred twice with autoreactive CD8+ T cells at 60 days post‐injection (E and F). Red squares in A, C, E illustrate the enlarged areas shown in B, D, and F, respectively. Scale bars: 560µm (A, C, and E) or 50µm (B, D, and F). 3v, third ventricle; cp, cerebral peduncle; mt, mammillothalamic tract
Having established the presence of a neuroinflammatory process in the TMN of our immune‐mediated model of NT1, we next evaluated the number of HDC cells and the HDC mRNA expression level in Orex‐HA mice.
Orex‐HA mice being of (BALB/c × C57BL/6) F1 genetic background, we used the untreated Orex‐HA group (Orex‐HA no‐T) as control for subsequent analyses. In order to control for the effect of autoreactive T cell transfer, we also included in the qPCR analyses a group consisting of the WT littermates of the Orex‐HA mice, which were transferred twice with autoreactive cytotoxic T cells (WT‐2xCD8).
Orex‐HA mice transferred twice with cytotoxic autoreactive T cells showed a very significant decrease in the number of ORX neurons (p = 0.009) (Figure 3A). We also found a slight decrease in the number of MCH neurons in this group of mice (p = 0.04) (Figure 3C).
FIGURE 3.
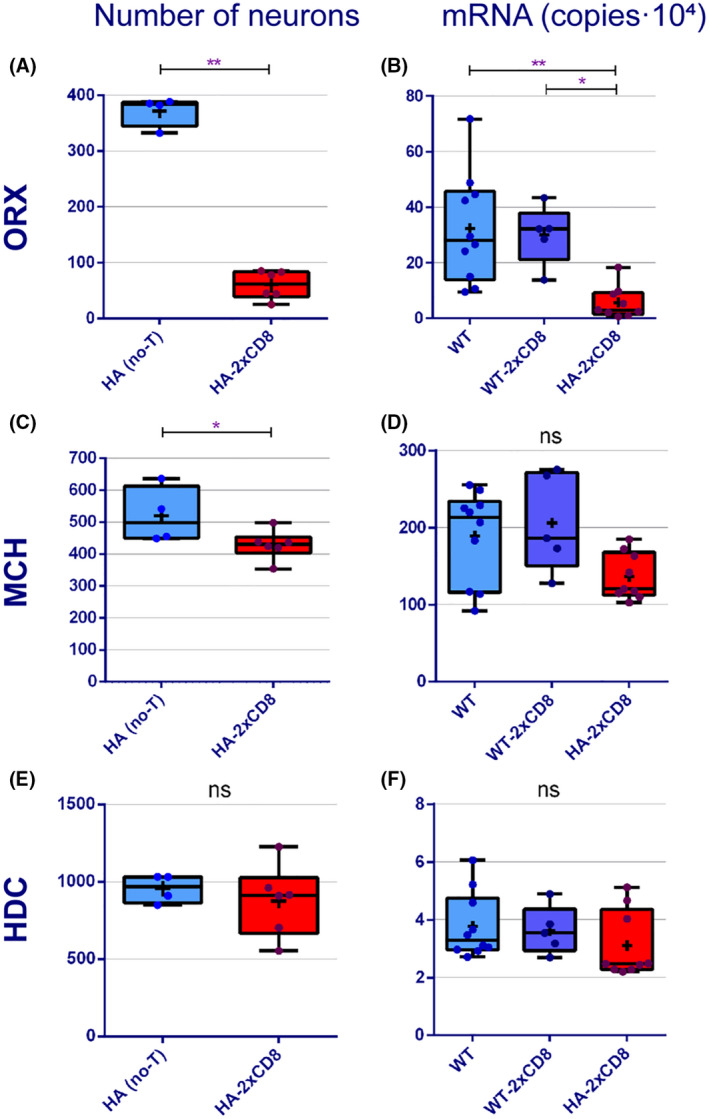
Evaluation of ORX, MCH and HDC content in the immune‐mediated model of narcolepsy. Number of ORX (A), MCH (C), and HDC (E) neurons in WT and in Orex‐HA mice 60 days after T cell transfer. mRNA expression level for ORX (B), MCH (D), and HDC (F) in WT and in Orex‐HA mice 60 days after T cell transfer. Data are shown as median (Q1–Q3), individual data values are represented as dots and the mean is represented as a cross. Mann–Whitney (two groups) or Kruskal–Wallis (three groups) tests, *p < 0.05; **p < 0.01. ns, not significant
Despite the prolonged local inflammation, we found no difference in the number of HDC neurons between groups (p = 0.6) (Figure 3E).
Finally, we found no correlation between the number of ORX and HDC neurons when considering all mice together or considering values within each experimental group (p = 0.9 for Orex‐HA no‐T; p = 0.2 for Orex‐HA‐2xCD8).
In accordance with the immunohistochemistry data, we found that the expression level of ORX was significantly decreased in the Orex‐HA‐2xCD8 group compared to controls (p = 0.001) (Figure 3B) but we found no difference between groups for the expression of HDC (p = 0.2) (Figure 3F). Comparing all groups together, MCH levels of expression were only just significantly different (p = 0.05); but when compared 2 by 2 no significant difference was found between groups (Figure 3D). Similarly to cell counts, we found no correlation between ORX and HDC expression levels (p = 0.2 for Orex‐HA‐2xCD8; p > 0.99 for WT; p = 0.2 for WT‐2xCD8) or HDC expression and age of studied mice in each group (p = 0.4 for Orex‐HA‐2xCD8; p = 0.8 for WT; p = 0.8 for WT‐2xCD8).
4. DISCUSSION
To test whether HDC expression level and/or cell number are modulated by ORX impairment, we used two different mouse models of ORX loss. We found that neither HDC cell numbers nor HDC and TH mRNA expression levels were changed in Orex‐KO mice with constitutive lack of ORX compared to controls. Similarly, despite the drastic acute loss of ORX neurons in Orex‐HA neuroinflammatory mouse model of NT1, we found no change in HDC cell number and mRNA expression level compared to controls. We further found no correlation between HDC and ORX, either looking at the number of neurons or the expression levels.
4.1. Compensation hypothesis
Several wake‐promoting systems have been identified and among them ORX, HIS, and the noradrenergic neurons of the locus coeruleus (NA‐LC) are major players [25]. These neurons are mostly active during wakefulness, slow down their firing rates drastically during non‐REM sleep and are silent during REM sleep [26, 27, 28, 29]. HIS and NA‐LC receive direct excitatory projections from the ORX neurons and application of ORX in the TMN or LC induces activation of the HIS and NA neurons [10]. It was proposed that the increase of HDC neurons seen in human NT1 brains could reflect a homeostatic mechanism compensating for the loss of ORX neurons in order to fight against sleepiness [9] although quite unsuccessfully since excessive daytime sleepiness is the major symptom of NT1. Supporting this hypothesis, ORX, HIS, and NA‐LC axonal pathways project to common brain regions and thus compensate for each other. However, they also show discrepancies—for instance, HIS and NA strongly project to all layers of the cerebral cortex while ORX axons are mainly found in layer 6b of the cortex [19, 30]—and an increasing volume of literature provides evidence that they are involved in different types of wake behaviors (for review, see [25]).
As NT1 patients reported in [8, 9] died more than 40 years after disease onset, we would be looking at long‐term compensatory mechanisms. Therefore, Orex‐KO mice, permanently lacking ORX neuropeptides from the earliest stage of development, are probably the best model to uncover long‐term compensatory mechanisms. However, we and others [8] found no increase in the number of HDC cells in these mice. Conversely, others [9, 13] found a mild increase (39%–53%) in the number of HDC neurons in Orex‐KO mice compared to WT mice. This discrepancy could rise from methodological differences in the counting procedure and/or from inter‐individual variance and small sample size (4 to 6 mice per group) in all of the studies. However, a strength in our study is the confirmation we provide using a different method, the quantitative PCR. We show that the expression level of HDC is not increased in Orex‐KO mice compared to controls.
Other models of induced ORX cell death such as Orexin‐ataxin3, Orexin‐DTA, or Orex‐HA mice transferred twice with CD8 T cells, all displaying a drastic loss of ORX neurons [17, 31, 32], allowed us and others [8, 9] to evaluate the HIS system at different time windows from ORX degeneration but none showed an increase in HIS nor an inverse correlation between the number of HIS and ORX neurons and transcripts. Furthermore, if HIS neurons were compensating for the lack of ORX, one might expect that a similar upregulation could occur in other wake‐promoting systems directly targeted by ORX neurons. We thus quantified the NA‐LC system by measuring the expression level of TH in the dorsal pons in addition to the measure of HDC expression level and found no increase in both. This latter observation is in agreement with recent findings showing no difference in catecholamine CSF levels and their metabolites in NT1 patients compare to patients with or without objective hypersomnolence [15]. Altogether these and our data indicate that histamine expression is not modulated by the ORX amount and therefore do not support the hypothesis of a compensatory mechanism in NT1.
4.2. Neuroinflammation hypothesis
In the great majority of patients, NT1 is a sporadic disorder and accumulating evidence including the HLA genetic predisposition, the detection of anti‐streptolysin O antibodies close to disease onset [33, 34], and the association with H1N1 viral infection or vaccination [35, 36] support the autoimmune hypothesis [3]; environmental factors such as bacterial infection, viral infection or vaccination being triggers of the disease. It may involve a specific immune response to H1N1 with potential molecular mimicry as proposed recently [5, 6, 7]. In addition, the environmental conditions associated with NT1 onset (i.e. vaccination or infections) may cause a state of neuroinflammation, notably in the hypothalamus which is bordered by several areas containing fenestrated capillaries and a leaky blood‐brain barrier and is highly connected with the olfactory bulb [19, 37]. In mice, peripheral challenge with the bacterial endotoxin lipopolysaccharide (LPS) which mimics a bacterial infection, cause neuroinflammation and microglia activation in different brain areas including the hypothalamus [24, 38, 39]. Similarly, intranasal inoculation of H1N1 virus in mice induced microglia activation in the lateral hypothalamus and brainstem, and an increase in the expression of pro‐inflammatory molecules [37, 40]. However, some controversies exist about the presence of hypothalamic neuroinflammation in human NT1. While some found no signs of inflammation or microglial activation at the level of the hypothalamus [1, 41], others reported an increase in GFAP staining reflecting a state of gliosis [42, 43]. This controversy probably rises from the facts that human brains were analyzed tens of years after disease onset and the small sample size. Cytokines and chemokines expressions have also been evaluated in sera and CSF of NT1 patients associated or not with H1N1 vaccination but no consensus between studies was found [44, 45, 46], possibly because of differences in the methodologies used and the difficulty to collect sample at early stages of the disease. Nevertheless, all authors report a tendency of increase in Th1 associated cytokines/chemokines or in the ratio of pro‐ versus anti‐inflammatory in NT1, supporting the hypothesis of a generalized neuroinflammation accompanying a focused assault onto OREX neurons. Interestingly, additional evidences showed that ORX‐A decrease neuroinflammation at the central level, notably reducing the production of cytokines [47, 48, 49], may also suggest that a tendency to neuroinflammation could be exacerbated by the loss of ORX neurons.
Thus, in the attempt to explain the increase of HDC neurons in NT1 patients, an alternative hypothesis was proposed, speculating that HIS could possibly be activated by a non‐specific local neuroinflammation [16]. In our model of immune‐mediated NT1, the Orex‐HA mice transferred twice with cytotoxic autoreactive T cell [17], we showed the presence of reactive microglia, thus a strong and sustained neuroinflammation, a drastic decrease in ORX, but no change in HDC cell number or expression level was observed. It is likely that our immune‐mediated mouse model of NT1 only partially reproduces the human disease, possibly shunting or masking HIS‐dependent mechanisms. Other models of neuroinflammation are thus needed to further evaluate whether HIS expression can be triggered by other neuroinflammation processes. Interestingly, however and supporting our data, it has been reported that the abundant Fos expression in both ORX and HIS neurons during the active period was strongly suppressed following a peripheral pro‐inflammatory challenge with LPS [50] suggesting that HIS system may possibly be dampened by neuroinflammation, at least shortly after bacterial infection, rather than activated. Thus, alternative mechanisms to the compensatory and neuroinflammatory hypotheses should now be explored.
4.3. Technical considerations
Variations in the HDC expression level have been reported based on age [51], circadian time [18], and gender [52]. To take into consideration these confounding factors, we matched groups for age and sex and collected all brains at the same time of day between 2 pm and 4 pm, a time window when the HDC mRNA expression level is intermediate in order to readily detect any possible increase or decrease [18].
Interestingly, a recent study reported a higher HIS level and a lower tele‐methylhistamine level, a direct metabolite of HIS, in the CSF of NT1 children compared to controls [53]. It suggests a decrease in HIS turnover and an impairment of HIS neurotransmission in narcoleptic children. However, such observation was not found in adults using the same techniques for measurements [12]. Here, we only used mice of adult age, paralleling age where the number of HDC neurons was increased [8, 9] while no increase in HIS CSF was found [12]. It is also important to note that CSF measures reflect the amount of HIS released in the extracellular space and not its synthesis rate as evaluated by the quantification of HDC.
We have observed a slight decrease in the number of MCH neurons in Orex‐HA mice, which was not observed in post‐mortem samples from human patients with NT1 [1, 9, 54] nor in our previous study [17] and not confirmed by qPCR. In a previous study, a 25% decrease in the number of MCH neurons was reported in mice after inducing a strong chronic neuroinflammation with LPS [55]. It is thus possible that the slight decrease in the number of MCH neurons we found here is because of the heavy T cells treatment received. We do not know however if the decrease would be transient or maintained over time.
As we performed the first HDC cell counting on the Orex‐HA mouse model, we could not run an appropriate a priori power analysis to define how many mice were needed per group. As a result of the heavy duty of producing such mice, we were limited in the number of animal available. Nevertheless, quantification of HDC expression by qPCR corroborated the absence of difference found by counting the number of HDC cells.
It can also be argued that the time point of 60 days post‐treatment that we chose for our analyses of Orex‐HA mice is not optimum. We previously showed that the T cell infiltration in the hypothalamus of Orex‐HA mice peaked at day 8 post‐injection and declined progressively thereafter [17]. However, local inflammation persisted for at least 60 days in close vicinity to the ORX and HDC neurons as visualized by the presence of reactive microglia, and again human brain samples were analyzed more than 40 years after disease onset, indicating that HIS increase reflects a persistent rather than a transient phenomenon. Nevertheless, although unlikely, we cannot exclude that a change in the HDC brain content would require a longer period of time to become detectable.
To conclude, we report that HDC expression level is not OREX‐dependent in mice using both a genetic and an immune‐mediated model of NT1. In mice, neither HIS nor NA‐LC seem to compensate for the loss or lack of ORX. Further studies, including quantification of HDC mRNA expression in NT1 human post‐mortem brains, will be needed to understand the physiopathology behind the increase in HDC neurons in human NT1.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
AUTHOR CONTRIBUTIONS
SM did the experiments, analyzed the data. ALM helped with immunohistochemistry, CSB helped with qPCR, RL provided the orex‐HA mice, CP designed the experiments, supervised analyses. SM, YD, CP wrote the paper.
ACKNOWLEDGMENTS
The authors are grateful to Gilles Bruneau for generously providing a highly specific antibody against HDC and David Frieser for taking care of the Orex‐HA mice. They thank the Alecs‐SPF and Neurocampus animal facilities for taking care of the orexin‐KO mice and Dr. Luc Gentet for English language revision. The project was funded by the National Agency for Research (ANR‐ and ANR‐18‐CE17‐0014‐02). SM received a fellowship from the French Ministry of Higher Education, Research and Innovation (MESRI).
Melzi S, Morel A‐L, Scoté‐Blachon C, Liblau R, Dauvilliers Y, Peyron C. Histamine in murine narcolepsy: What do genetic and immune models tell us? Brain Pathol. 2022;32:e13027. 10.1111/bpa.13027
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–7. [DOI] [PubMed] [Google Scholar]
- 2. Mignot E, Lammers GJ, Ripley B, Okun M, Nevsimalova S, Overeem S, et al. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Arch Neurol. 2002;59:1553–62. [DOI] [PubMed] [Google Scholar]
- 3. Bassetti CLA, Adamantidis A, Burdakov D, Han F, Gay S, Kallweit U, et al. Narcolepsy—clinical spectrum, aetiopathophysiology, diagnosis and treatment. Nat Rev Neurol. 2019;15:519–39. [DOI] [PubMed] [Google Scholar]
- 4. Barateau L, Liblau R, Peyron C, Dauvilliers Y. Narcolepsy type 1 as an autoimmune disorder: evidence, and implications for pharmacological treatment. CNS Drugs. 2017;31:821–34. [DOI] [PubMed] [Google Scholar]
- 5. Latorre D, Kallweit U, Armentani E, Foglierini M, Mele F, Cassotta A, et al. T cells in patients with narcolepsy target self‐antigens of hypocretin neurons. Nature. 2018;562:63‐68. 10.1038/s41586-018-0540-1 [DOI] [PubMed] [Google Scholar]
- 6. Luo G, Ambati A, Lin L, Bonvalet M, Partinen M, Ji X, et al. Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. Proc Natl Acad Sci U S A. 2018;115:E12323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pedersen NW, Holm A, Kristensen NP, Bjerregaard AM, Bentzen AK, Marquard AM, et al. CD8(+) T cells from patients with narcolepsy and healthy controls recognize hypocretin neuron‐specific antigens. Nat Commun. 2019;10:837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. John J, Thannickal TC, McGregor R, Ramanathan L, Ohtsu H, Nishino S, et al. Greatly increased numbers of histamine cells in human narcolepsy with cataplexy. Ann Neurol. 2013;74:786–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Valko PO, Gavrilov YV, Yamamoto M, Reddy H, Haybaeck J, Mignot E, et al. Increase of histaminergic tuberomammillary neurons in narcolepsy. Ann Neurol. 2013;74:794–804. [DOI] [PubMed] [Google Scholar]
- 10. Schone C, Burdakov D. Orexin/hypocretin and organizing principles for a diversity of wake‐promoting neurons in the brain. Curr Top Behav Neurosci. 2017;33:51–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishino S, Sakurai E, Nevsimalova S, Yoshida Y, Watanabe T, Yanai K, et al. Decreased CSF histamine in narcolepsy with and without low CSF hypocretin‐1 in comparison to healthy controls. Sleep. 2009;32:175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dauvilliers Y, Delallée N, Jaussent I, Scholz S, Bayard S, Croyal M, et al. Normal cerebrospinal fluid histamine and tele‐methylhistamine levels in hypersomnia conditions. Sleep. 2012;35:1359–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berteotti C, Lo Martire V, Alvente S, Bastianini S, Bombardi C, Matteoli G, et al. Orexin/hypocretin and histamine cross‐talk on hypothalamic neuron counts in mice. Front Neurosci. 2021;15:660518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carter ME, Brill J, Bonnavion P, Huguenard JR, Huerta R, de Lecea L. Mechanism for Hypocretin‐mediated sleep‐to‐wake transitions. Proc Natl Acad Sci U S A. 2012;109:E2635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barateau L, Jaussent I, Roeser J, Ciardiello C, Kilduff TS, Dauvilliers Y. Cerebrospinal fluid monoamine levels in central hypersomnolence disorders. Sleep. 2021;44(7):zsab012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shan L, Dauvilliers Y, Siegel JM. Interactions of the histamine and hypocretin systems in CNS disorders. Nat Rev Neurol. 2015;11:401–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bernard‐Valnet R, Yshii L, Queriault C, Nguyen XH, Arthaud S, Rodrigues M, et al. CD8 T cell‐mediated killing of orexinergic neurons induces a narcolepsy‐like phenotype in mice. Proc Natl Acad Sci U S A. 2016;113:10956–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu X, Zecharia A, Zhang Z, Yang Q, Yustos R, Jager P, et al. Circadian factor BMAL1 in histaminergic neurons regulates sleep architecture. Curr Biol. 2014;24:2838–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bruneau G, Batailler M, Belghazi M, Tillet Y, Blanc MR. Evidence that histaminergic neurons are devoid of estrogen receptor alpha in the ewe diencephalon during the breeding season. Gen Comp Endocrinol. 2014;199:86–93. [DOI] [PubMed] [Google Scholar]
- 21. Hanriot L, Camargo N, Courau AC, Leger L, Luppi PH, Peyron C. Characterization of the melanin‐concentrating hormone neurons activated during paradoxical sleep hypersomnia in rats. J Comp Neurol. 2007;505:147–57. [DOI] [PubMed] [Google Scholar]
- 22. Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia‐specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. [DOI] [PubMed] [Google Scholar]
- 23. Deurveilher S, Lo H, Murphy JA, Burns J, Semba K. Differential c‐Fos immunoreactivity in arousal‐promoting cell groups following systemic administration of caffeine in rats. J Comp Neurol. 2006;498:667–89. [DOI] [PubMed] [Google Scholar]
- 24. Skelly DT, Hennessy E, Dansereau MA, Cunningham C. A systematic analysis of the peripheral and CNS effects of systemic LPS, IL‐1Β, TNF‐α and IL‐6 challenges in C57BL/6 mice. PLoS ONE. 2013;8:e69123. 10.1371/journal.pone.0069123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scammell TE, Arrigoni E, Lipton JO. Neural circuitry of wakefulness and sleep. Neuron. 2017;93:747–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steininger TL, Alam MN, Gong H, Szymusiak R, McGinty D. Sleep‐waking discharge of neurons in the posterior lateral hypothalamus of the albino rat. Brain Res. 1999;840:138–47. [DOI] [PubMed] [Google Scholar]
- 27. Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep‐waking cycle. J Neurosci. 2005;25:6716–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aston‐Jones G, Bloom FE. Activity of norepinephrine‐containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep‐waking cycle. J Neurosci. 1981;1:876–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bayer L, Serafin M, Eggermann E, Saint‐Mleux B, Machard D, Jones BE, et al. Exclusive postsynaptic action of hypocretin‐orexin on sublayer 6b cortical neurons. Neuroscience. 2004;24:6760‐6764. 10.1523/jneurosci.1783-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–54. [DOI] [PubMed] [Google Scholar]
- 32. Tabuchi S, Tsunematsu T, Black SW, Tominaga M, Maruyama M, Takagi K, et al. Conditional ablation of orexin/hypocretin neurons: a new mouse model for the study of narcolepsy and orexin system function. J Neurosci. 2014;34:6495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aran A, Lin L, Nevsimalova S, Plazzi G, Hong SC, Weiner K, et al. Elevated anti‐streptococcal antibodies in patients with recent narcolepsy onset. Sleep. 2009;32:979–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cvetkovic‐Lopes V, Bayer L, Dorsaz S, Maret S, Pradervand S, Dauvilliers Y, et al. Elevated tribbles homolog 2‐specific antibody levels in narcolepsy patients. J Clin Invest. 2010;120:713–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Partinen M, Saarenpaa‐Heikkila O, Ilveskoski I, Hublin C, Linna M, Olsén P, et al. Increased incidence and clinical picture of childhood narcolepsy following the 2009 H1N1 pandemic vaccination campaign in Finland. PLoS One. 2012;7:e33723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dauvilliers Y, Montplaisir J, Cochen V, Desautels A, Einen M, Lin L, et al. Post‐H1N1 narcolepsy‐cataplexy. Sleep. 2010;33:1428–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tesoriero C, Codita A, Zhang MD, Cherninsky A, Karlsson H, Grassi‐Zucconi G, et al. H1N1 influenza virus induces narcolepsy‐like sleep disruption and targets sleep‐wake regulatory neurons in mice. Proc Natl Acad Sci U S A. 2016;113:E368–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Palomba M, Seke Etet PF, Veronesi C. Effect of inflammatory challenge on hypothalamic neurons expressing orexinergic and melanin‐concentrating hormone. Neurosci Lett. 2014;570:47–52. [DOI] [PubMed] [Google Scholar]
- 39. Hoogland ICM, Westhoff D, Engelen‐Lee JY, Melief J, Valls Seron M, Houben‐Weerts JHMP, et al. Microglial activation after systemic stimulation with lipopolysaccharide and Escherichia coli . Front Cell Neurosci. 2018;12:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sadasivan S, Zanin M, O'Brien K, Schultz‐Cherry S, Smeyne RJ. Induction of microglia activation after infection with the non‐neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS One. 2015;10:e0124047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Honda M, Arai T, Fukazawa M, Honda Y, Tsuchiya K, Salehi A, et al. Absence of ubiquitinated inclusions in hypocretin neurons of patients with narcolepsy. Neurology. 2009;73:511–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thannickal TC, Siegel JM, Nienhuis R, Moore RY. Pattern of hypocretin (orexin) soma and axon loss, and gliosis, in human narcolepsy. Brain Pathol. 2003;13:340–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dauvilliers Y, Jaussent I, Lecendreux M, Scholz S, Bayard S, Cristol JP, et al. Cerebrospinal fluid and serum cytokine profiles in narcolepsy with cataplexy: a case‐control study. Brain Behav Immun. 2014;37:260–6. [DOI] [PubMed] [Google Scholar]
- 45. Kornum BR, Pizza F, Knudsen S, Plazzi G, Jennum P, Mignot E. Cerebrospinal fluid cytokine levels in type 1 narcolepsy patients very close to onset. Brain Behav Immun. 2015;49:54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lecendreux M, Libri V, Jaussent I, Mottez E, Lopez R, Lavault S, et al. Impact of cytokine in type 1 narcolepsy: role of pandemic H1N1 vaccination? J Autoimmun. 2015;60:20–31. [DOI] [PubMed] [Google Scholar]
- 47. Ogawa Y, Irukayama‐Tomobe Y, Murakoshi N, Kiyama M, Ishikawa Y, Hosokawa N, et al. Peripherally administered orexin improves survival of mice with endotoxin shock. Elife. 2016;5:e21055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Becquet L, Abad C, Leclercq M, Miel C, Jean L, Riou G, et al. Systemic administration of orexin A ameliorates established experimental autoimmune encephalomyelitis by diminishing neuroinflammation. J Neuroinflammation. 2019;16:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li T, Xu W, Ouyang J, Lu X, Sherchan P, Lenahan C, et al. Orexin A alleviates neuroinflammation via OXR2/CaMKKbeta/AMPK signaling pathway after ICH in mice. J Neuroinflammation. 2020;17:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gaykema RP, Goehler LE. Lipopolysaccharide challenge‐induced suppression of Fos in hypothalamic orexin neurons: their potential role in sickness behavior. Brain Behav Immun. 2009;23:926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mazurkiewicz‐Kwilecki IM, Prell GD. Age‐related changes in brain histamine. Agents Actions. 1984;14:554–7. [DOI] [PubMed] [Google Scholar]
- 52. Cacabelos R, Torrellas C, Fernandez‐Novoa L, Aliev G. Neuroimmune crosstalk in CNS disorders: the histamine connection. Curr Pharm Des. 2016;22:819–48. [DOI] [PubMed] [Google Scholar]
- 53. Franco P, Dauvilliers Y, Inocente CO, Guyon A, Villanueva C, Raverot V, et al. Impaired histaminergic neurotransmission in children with narcolepsy type 1. CNS Neurosci Ther. 2019;25:386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thannickal TC, Nienhuis R, Siegel JM. Localized loss of hypocretin (orexin) cells in narcolepsy without cataplexy. Sleep. 2009;32:993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gerashchenko D, Shiromani PJ. Effects of inflammation produced by chronic lipopolysaccharide administration on the survival of hypocretin neurons and sleep. Brain Res. 2004;1019:162–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.