

Abstract
Although the molecular mechanisms underlying amyotrophic lateral sclerosis (ALS) are not yet fully understood, several studies report alterations in tau phosphorylation in both sporadic and familial ALS. Recently, we have demonstrated that phosphorylated tau at S396 (pTau‐S396) is mislocalized to synapses in ALS motor cortex (mCTX) and contributes to mitochondrial dysfunction. Here, we demonstrate that while there was no overall increase in total tau, pTau‐S396, and pTau‐S404 in ALS post‐mortem mCTX, total tau and pTau‐S396 were increased in C9ORF72‐ALS. Additionally, there was a significant decrease in pTau‐T181 in ALS mCTX compared controls. Furthermore, we leveraged the ALS Knowledge Portal and Project MinE data sets and identified ALS‐specific genetic variants across MAPT, the gene encoding tau. Lastly, assessment of cerebrospinal fluid (CSF) samples revealed a significant increase in total tau levels in bulbar‐onset ALS together with a decrease in CSF pTau‐T181:tau ratio in all ALS samples, as reported previously. While increases in CSF tau levels correlated with a faster disease progression as measured by the revised ALS functional rating scale (ALSFRS‐R), decreases in CSF pTau‐T181:tau ratio correlated with a slower disease progression, suggesting that CSF total tau and pTau‐T181 ratio may serve as biomarkers of disease in ALS. Our findings highlight the potential role of pTau‐T181 in ALS, as decreases in CSF pTau‐T181:tau ratio may reflect the significant decrease in pTau‐T181 in post‐mortem mCTX. Taken together, these results indicate that tau phosphorylation is altered in ALS post‐mortem mCTX as well as in CSF and, importantly, the newly described pathogenic or likely pathogenic variants identified in MAPT in this study are adjacent to T181 and S396 phosphorylation sites further highlighting the potential role of these tau functional domains in ALS.
Keywords: amyotrophic lateral sclerosis, biomarker, tau
Short abstract
Although the molecular mechanisms underlying amyotrophic lateral sclerosis (ALS) are not yet fully understood, recent studies report alterations in tau phosphorylation in ALS. Our study builds on these findings and demonstrates that tau phosphorylation is altered in post‐mortem ALS motor cortex and highlights new and ALS‐specific variants in MAPT, the gene encoding tau. Lastly, we report alterations in phosphorylated tau in ALS cerebrospinal fluid that may function as a predictive biomarker for ALS.
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that primarily affects both cortical and spinal motor neurons [1]. Several genes have been implicated in ALS pathogenesis [2]; however, mutations in these genes account for a minority of cases, and the etiology of the disease remains to be elucidated. Therefore, understanding the exact molecular mechanisms leading to motor neuron loss is crucial for the development of new therapeutic approaches and the discovery of novel and useful biomarkers of disease.
Recent studies have begun to link alteration in tau phosphorylation to ALS pathogenesis with tau pathology reported in both sporadic and familial cases [3, 4]. Tau protein is a member of the microtubule‐associated protein (MAP) family and plays a critical role in stabilizing microtubules, the major component of the eukaryotic cytoskeleton involved in cell processes such as cell division, mobility, and intracellular organization, and trafficking of organelles [5]. Tau hyperphosphorylation, accumulation, and mutations have been linked to a group of progressive neurodegenerative diseases collectively known as tauopathies [6, 7, 8], in which hyperphosphorylation of key epitopes on tau promotes its disassembly from microtubules, aggregation, and subcellular mislocalization, leading to the formation of inclusions and neurofibrillary tangles (NFTs) in both neurons and glia [6, 8].
Although the exact molecular mechanisms underlying tau toxicity are not yet fully understood, the main consequence of the accumulation of toxic tau is the disruption of neuronal transport [9, 10, 11]. This impairment is an early pathogenic event in neurodegeneration, and tau‐mediated alterations in neuronal trafficking have been described in several neurodegenerative diseases [9, 10, 11]. Furthermore, deficits in neuronal transport were shown to disrupt several cellular functions, including but not limited to alterations in both trafficking and function of mitochondria, synapse loss, excitotoxicity, and cell death [9, 11, 12].
In ALS, there is a significant increase in total tau as well as cytoplasmic inclusions of hyperphosphorylated tau (T175, T217, S208/210, S212, S396, and S404) in post‐mortem motor cortex (mCTX) and spinal cord of ALS patients [13, 14, 15]. Moreover, alterations in tau and pTau:tau ratio have been reported in ALS cerebrospinal fluid (CSF) [16, 17, 18, 19]. Importantly, tau‐induced alterations in cellular processes such as excitotoxicity, mitochondrial dysfunction, synapse loss, and impairments of nucleocytoplasmic transport, are also pathogenic features of ALS [1, 20, 21], suggesting that alterations in tau could underlie these molecular events in ALS. Accordingly, recent findings from our group have revealed that hyperphosphorylated tau at S396 (pTau‐S396) mislocalizes to synapses in mCTX across ALS subtypes and contributes to mitochondrial fragmentation through interaction with the pro‐fission GTPase dynamin‐related protein 1 (DRP1) [22], further supporting a pathogenic role for tau in ALS.
Here, we used a large cohort of ALS post‐mortem mCTX samples to further investigate whether there were alterations in tau phosphorylation in ALS. In addition, we sought to determine whether there were novel genetic variants in MAPT, the gene encoding tau, in ALS. Lastly, we measured tau and pTau in CSF derived from people living with ALS and healthy controls given the contradictory results from previous biomarker studies in ALS.
2. MATERIALS AND METHODS
2.1. Human tissue samples
Post‐mortem mCTX from ALS and controls were provided by the Massachusetts Alzheimer's Disease Research Center (ADRC) and from the Veterans Affairs Biorepository Brain Bank (VABBB; Merit review BZ002466) with approval from the Massachusetts General Hospital Institutional Review Board (IRB). In total, we assessed 52 ALS and 25 non‐neurological control mCTX as well as entorhinal cortex (EC) from two Alzheimer's disease (AD) cases as positive controls. Sixteen ALS and seven control mCTX were used for immunohistochemistry (IHC): the mean age was 68.9 years (SD = 14.6) for control and 62.4 years (SD = 11.5) for ALS. Control samples were 57.1% male, while the ALS group was 68.8% male. Twelve ALS cases were diagnosed with limb onset disease, while two were diagnosed with bulbar onset. Region of ALS onset was unknown for two samples. One case was diagnosed with ALS/frontotemporal dementia (ALS/FTD); however, post‐mortem evaluation revealed no positive staining for either β‐amyloid or α‐synuclein as well as no NFTs or Lewy bodies. Two of the ALS cases were positive for C9ORF72 repeat expansion. Genetic status of all other ALS cases was unknown. All 16 ALS mCTX demonstrated TDP43 proteinopathy. Post‐mortem interval (PMI) range was 14–81 h for controls and 20–77 h for ALS. Both control and ALS groups were negative for β‐amyloid and α‐synuclein upon post‐mortem evaluation by MGH ADRC and VABBB, except for a single ALS case that revealed brain alterations likely due to AD (CERAD plaque stage: sparse; Braak neurofibrillary stage: III/IV; moderate amyloid angiopathy) but insufficient for a concomitant diagnosis of AD. Clinicopathological information for the control and all other ALS post‐mortem mCTX samples used in this study are summarized in Table 1. The AD cases were both male, aged 88 and 61 years old (PMI: unknown, and 24 h, respectively).
TABLE 1.
Post‐mortem motor cortex sample information
| Sex | Onset site | Genotype | fALS | Protein aggregates | FTD | Age at disease onset | Age of death | PMI | |
|---|---|---|---|---|---|---|---|---|---|
| Control 1 | M | N/A | N/A | N/A | N/A | N/A | N/A | 82 | 50 |
| Control 2 | M | N/A | N/A | N/A | N/A | N/A | N/A | 66 | 81 |
| Control 3 | M | N/A | N/A | N/A | N/A | N/A | N/A | 49 | 45 |
| Control 4 | F | N/A | N/A | N/A | N/A | N/A | N/A | 58 | 18 |
| Control 5 | M | N/A | N/A | N/A | N/A | N/A | N/A | 60 | 14 |
| Control 6 | F | N/A | N/A | N/A | N/A | N/A | N/A | 77 | 72 |
| Control 7 | F | N/A | N/A | N/A | N/A | N/A | N/A | 90 | 24 |
| Control 8 | F | N/A | N/A | N/A | N/A | N/A | N/A | 79 | Unknown |
| Control 9 | F | N/A | N/A | N/A | N/A | N/A | N/A | 57 | 13 |
| Control 10 | F | N/A | N/A | N/A | N/A | N/A | N/A | 60 | 15 |
| Control 11 | M | N/A | N/A | N/A | N/A | N/A | N/A | 63 | 16 |
| Control 12 | M | N/A | N/A | N/A | N/A | N/A | N/A | 86 | 10 |
| Control 13 | F | N/A | N/A | N/A | N/A | N/A | N/A | 73 | 20 |
| Control 14 | M | N/A | N/A | N/A | N/A | N/A | N/A | 92 | 23 |
| Control 15 | F | N/A | N/A | N/A | N/A | N/A | N/A | 52 | 10 |
| Control 16 | M | N/A | N/A | N/A | N/A | N/A | N/A | 55 | 19 |
| Control 17 | F | N/A | N/A | N/A | N/A | N/A | N/A | 85 | 24 |
| Control 18 | F | N/A | N/A | N/A | N/A | N/A | N/A | >90 | 45 |
| Control 19 | F | N/A | N/A | N/A | N/A | N/A | N/A | 79 | 9 |
| Control 20 | M | N/A | N/A | N/A | N/A | N/A | N/A | >90 | 23 |
| Control 21 | F | N/A | N/A | N/A | N/A | N/A | N/A | >90 | 24 |
| Control 2 | M | N/A | N/A | N/A | N/A | N/A | N/A | 89 | 36 |
| Control 23 | M | N/A | N/A | N/A | N/A | N/A | N/A | 70 | 56 |
| Control 24 | M | N/A | N/A | N/A | N/A | N/A | N/A | >90 | 86 |
| Control 25 | M | N/A | N/A | N/A | N/A | N/A | N/A | 66 | 71 |
| ALS 1 | M | Limb | Unknown | Unknown | TDP43 | No | 57 | 62 | 46 |
| ALS 2 | F | Limb | Unknown | Unknown | TDP43; Ubiquitin | No | 22 | 34 | 65 |
| ALS 3 | M | Bulbar | C9ORF72 | Yes | TDP43; Ubiquitin | No | 55 | 61 | 77 |
| ALS 5 | M | Limb | Unknown | No | TDP43 | No | 58 | 66 | 43 |
| ALS 6 | M | Limb | Unknown | No | TDP43 | No | 62 | 68 | 43 |
| ALS 7 | M | Limb | Unknown | No | TDP43 | No | 54 | 78 | 35 |
| ALS 8 | M | Limb | Unknown | Unknown | TDP43; tau | No | 74 | 85 | 38 |
| ALS 9 | F | Limb | Unknown | Unknown | TDP43 | No | 64 | 70 | 30 |
| ALS 10 | M | Unknown | Unknown | Unknown | TDP43 | No | Unknown | 58 | 30 |
| ALS 11 | M | Limb | Unknown | Unknown | TDP43 | No | Unknown | 62 | 24 |
| ALS 12 | M | Limb | C9ORF72 | Yes | TDP43 | No | 57 | 61 | 22 |
| ALS 13 | M | Unknown | Unknown | Unknown | TDP43 | No | Unknown | 64 | 20 |
| ALS 14 | M | Limb | Unknown | Unknown | TDP43 | No | 41 | 62 | 24 |
| ALS 15 | F | Limb | Unknown | Unknown | Ubiquitin | No | 65 | 66 | 24 |
| ALS 16 | F | Bulbar | Unknown | Unknown | TDP43 | No | 46 | 49 | 25 |
| ALS 17 | F | Limb | Unknown | Unknown | TDP43 | Yes | 52 | 53 | 30 |
| ALS 18 | M | Limb | Unknown | No | Unknown | Unknown | 75 | 79 | Unknown |
| ALS 19 | M | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | 69 | 24 |
| ALS 20 | M | Bulbar | Unknown | Unknown | Unknown | Yes | 58 | 61 | 24 |
| ALS 21 | M | Bulbar | Unknown | Unknown | Unknown | No | 79 | 81 | 4 |
| ALS 22 | M | Limb | Unknown | Unknown | Unknown | Unknown | 75 | 76 | 14 |
| ALS 23 | F | Bulbar | Unknown | No | Unknown | No | 76 | Unknown | 24 |
| ALS 24 | M | Unknown | Unknown | Unknown | TDP43 | Unknown | Unknown | 58 | 30 |
| ALS 25 | F | Unknown | C9ORF72 | Unknown | TDP43 | Unknown | Unknown | 67 | 19 |
| ALS 26 | M | Limb | Unknown | No | Unknown | No | 53 | 53 | 24 |
| ALS 27 | M | Limb | Unknown | Unknown | Unknown | No | 59 | 60 | 19 |
| ALS 28 | M | Limb | Unknown | Unknown | Unknown | No | 57 | 60 | 22 |
| ALS 29 | M | Limb | Unknown | No | TDP43 | No | 49 | 51 | 24 |
| ALS 30 | M | Limb | Unknown | No | TDP43; Ubiquitin | No | 58 | 62 | 24 |
| ALS 31 | F | Limb | Unknown | No | TDP43 | No | 90 | 91 | 33 |
| ALS 32 | F | Bulbar | Unknown | No | Unknown | No | 50 | 52 | 28 |
| ALS 33 | F | Bulbar | C9ORF72 | Unknown | TDP43 | No | 54 | 56 | Unknown |
| ALS 34 | F | Bulbar | C9ORF72 | Unknown | TDP43 | No | 49 | 58 | 48 |
| ALS 35 | M | Bulbar | Unknown | No | Unknown | No | 53 | 55 | 36 |
| ALS 36 | M | Limb | Unknown | No | Unknown | No | 50 | 61 | 8 |
| ALS 37 | F | Limb | Unknown | No | Unknown | No | 63 | 65 | 36 |
| ALS 38 | M | Limb | Unknown | No | Unknown | No | 59 | 61 | 12 |
| ALS 39 | F | Limb | Unknown | Unknown | Unknown | Unknown | 66 | 68 | 20 |
| ALS 40 | M | Bulbar | Unknown | Unknown | TDP43 | No | 54 | 57 | 24 |
| ALS 41 | F | Bulbar | Unknown | No | TDP43 | No | 47 | 54 | 24 |
| ALS 42 | F | Bulbar | C9ORF72 | Possible | TDP43 | Yes | Unknown | 59 | 20 |
| ALS 43 | F | Unknown | Unknown | Unknown | TDP43 | Unknown | Unknown | 70 | Unknown |
| ALS 44 | M | Bulbar | Unknown | Unknown | TDP43 | No | Unknown | 70 | 18 |
| ALS 45 | M | Limb | Unknown | No | TDP43 | No | 50 | 55 | 12 |
| ALS 46 | F | Bulbar | Unknown | No | TDP43 | No | 73 | 75 | 56 |
| ALS 47 | M | Bulbar | Unknown | No | TDP43 | No | 70 | 71 | 14 |
| ALS 48 | M | Bulbar | Unknown | No | TDP43 | No | 62 | 70 | 28 |
| ALS 49 | M | Limb | Unknown | No | TPD43; Ubiquitin | No | 68 | 73 | 28 |
| ALS 50 | M | Limb | C9ORF72 | No | TPD43; Ubiquitin | No | 58 | 65 | 80 |
| ALS 51 | M | Limb | C9ORF72 | Possible | TDP43 | No | 40 | 50 | 36 |
| ALS 52 | M | Bulbar | Unknown | Unknown | TDP43 | No | 69 | 73 | 45 |
2.2. Immunohistochemistry and image analysis
Paraffin‐embedded brain sections of 7‐µm‐thick were immunostained for pTau‐ S396 (1:100; #Ab109390, Abcam, MA) and pTau‐S396/S404 (PHF1; 1:250; Peter Davies) using a Bond Rx autostainer (Leica Biosystems, IL), according to the manufacturer's instructions. One set of sections was also incubated for 4 min in thionin to visualize neurons. [23] Briefly, slides were batch processed with the following settings: Bake and Dewax, IHC protocol F 60 min, HIER 20 min with ER1. Slides were then transferred into water and dehydrated by 1‐min incubations into baths of 70% ethanol, 95% ethanol, 100% ethanol, and xylene. Slides were then cover slipped using Permount Mounting Medium (Fisher Scientific) and left to dry overnight. All the sections were stained at the same time for either pTau‐S396, PHF1, or thionin using the same antibody cocktail. Slides were scanned using an Olympus VS120 virtual slide microscope at a magnification of 20X. Scanned slide images were analyzed in Olympus cellSens and OlyVIA analysis software.
2.3. Western blotting
Western blots were performed using previously described protocols [24, 25, 26]. Briefly, 50 μg of proteins was resuspended in sample buffer and fractionated on a 4%–12% bis–tris gel for 90 min at 120V. Proteins were then transferred to a PVDF membrane in an iBlot Dry Blotting System (Invitrogen, Thermo Fisher, MA), and the membrane was blocked with 5% bovine serum albumin (BSA) in tris‐buffered saline with Tween 20 (TBST) before immunodetection with the following primary antibodies: pTau‐S396 (1:500; #Ab109390, Abcam, MA), pTau‐S404 (1:500; #Ab92676, Abcam, MA), pTau‐T181 (1:500; #Ab75679, Abcam, MA), tau (1:1000; #A0024, DAKO, Denmark), and GAPDH (1:1000; #MAB374, Millipore Sigma, CA) overnight at 4°C. Primary antibody incubation was followed by four washes in TBST before incubation with the secondary antibody for 1 h (HRP‐conjugated goat anti‐rabbit IgG and HRP‐conjugated goat anti‐mouse IgG; Jackson ImmunoResearch Laboratories, West Grove, PA). After four washes in TBST, protein bands were visualized using the ECL detection system (Thermo Fisher Scientific, MA). Integrated density values (IDVs) for protein bands of interest were quantified in ImageJ 1.53a (National Institutes of Health, Bethesda, MD) and normalized to GAPDH IDVs.
2.4. Identifying MAPT variants in ALS patients
The genetic approach taken herein was previously described by Petrozziello et al. [27]. The genetic data were obtained from ALS Knowledge Portal (ALSKP) [28] and Project MinE data browser [29]. We included any variants annotated as missense, non‐synonymous, and splice altering variants within ALS cases (ALSKP, n = 3864 cases and n = 7,839 controls; Project MinE, n = 4366 cases and n = 1832 controls). We used the genome Aggregation Database (gnomAD, 125,748 exomes and 15,708 genomes, total n = 141,456) [30] to determine the global population frequency of each variant. For all the variants observed, we included the annotation outputs derived from CADD (Combined Annotation Dependent Depletion) and MPC (for Missense badness, PolyPhen‐2, and Constraint), which helped guide us during variant interpretation and prioritization. All variants are displayed in Table S1. Variant positions are based on reference genome assembly GRCh37/hg19. We also surveyed ClinVar, a repository of genetic variants reported in patients with disease for any MAPT pathogenic or likely pathogenic variants. Lastly, we used MAPT isoform NM_016835 to report the genetic variants in accordance with gnomAD and ClinVar.
2.5. Human CSF samples
After obtaining written informed consent, CSF samples were obtained from participants with ALS (n = 40) and controls (n = 10) at the Healey Center for ALS at Mass General (Table 2). Longitudinal CSF samples and accompanying clinical information from participants with ALS were collected between 2011 and 2016 as a part of a prospective, multicenter observational study. Control samples were obtained in a concurrently enrolling single‐center study at MGH using identical techniques to obtain, process, store, and share biofluid samples. In each study, participants were enrolled, and detailed clinical information and CSF samples were obtained at baseline and, for longitudinally collected samples, at follow‐up visits, approximately every 4 months. Cerebrospinal fluid was centrifuged, aliquoted, and frozen at −80°C. Processing was initiated within 15 min of collection. Clinical data collected from participants with ALS included timing and location of disease onset and progression, including the revised ALS Functional Rating Scale (ALSFRS‐R) and slow vital capacity (SVC). Raters for the ALSFRS‐R and VC were trained by the Northeast ALS Consortium (NEALS) Outcomes Training Center at the Barrow Neurological Institute. CSF sample information is provided in Table 3.
TABLE 2.
CSF sample information
| Sex | Onset site | Age at disease onset | ALSFRS‐R visit 1 | ALSFRS‐R visit 2 | ALSFRS‐R visit 3 | ALSFRS‐R visit 4 | ALSFRS‐R visit 5 | ALSFRS‐R visit 6 | ALSFRS‐R visit 7 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Control 26 | F | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Control 27 | F | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Control 28 | F | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Control 29 | M | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Control 30 | M | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Control 31 | M | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Control 32 | M | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Control 33 | F | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Control 34 | F | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Control 35 | M | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| ALS 53 | F | Limb | 68 | 36 | 34 | 33 | 27 | 26 | Unknown | Unknown |
| ALS 54 | F | Bulbar | 64 | 45 | 44 | 38 | 38 | 36 | 29 | 29 |
| ALS 55 | F | Limb | 39 | 43 | 43 | 41 | 39 | 37 | 36 | Unknown |
| ALS 56 | M | Limb | 54 | 41 | 45 | 42 | 36 | Unknown | Unknown | Unknown |
| ALS 57 | M | Limb | 46 | 44 | 43 | 40 | 42 | 39 | 35 | Unknown |
| ALS 58 | F | Bulbar | 60 | 31 | 32 | 28 | 25 | Unknown | Unknown | Unknown |
| ALS 59 | F | Limb | 49 | 41 | 40 | 36 | 38 | 33 | Unknown | Unknown |
| ALS 60 | F | Limb | 39 | 37 | 32 | 32 | 25 | 32 | Unknown | Unknown |
| ALS 61 | M | Limb | 63 | 41 | 37 | 37 | 41 | 36 | 37 | 36 |
| ALS 62 | M | Limb | 57 | 28 | 22 | 27 | 25 | Unknown | Unknown | Unknown |
| ALS 63 | M | Limb | 55 | 45 | 43 | 44 | 45 | Unknown | Unknown | Unknown |
| ALS 64 | F | Limb | 67 | 45 | 43 | 42 | Unknown | Unknown | Unknown | Unknown |
| ALS 65 | F | Limb | 65 | 32 | 30 | 23 | Unknown | Unknown | Unknown | Unknown |
| ALS 66 | M | Limb | 64 | 38 | 37 | 37 | 26 | 27 | 22 | 18 |
| ALS 67 | F | Limb | 36 | 36 | 31 | 33 | 33 | 24 | 24 | Unknown |
| ALS 68 | F | Bulbar | 57 | 42 | 35 | 30 | 27 | 25 | 19 | 15 |
| ALS 69 | M | Limb | 45 | 45 | 44 | 32 | 32 | 29 | 19 | Unknown |
| ALS 70 | M | Limb | 44 | 31 | 40 | 28 | 27 | 25 | 23 | Unknown |
| ALS 71 | M | Bulbar | 42 | 35 | 31 | 20 | 17 | Unknown | Unknown | Unknown |
| ALS 72 | M | Bulbar | 74 | 35 | 27 | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 73 | M | Limb | 67 | 42 | 34 | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 74 | M | Limb | 57 | 46 | 44 | 44 | 42 | Unknown | Unknown | Unknown |
| ALS 75 | M | Limb | 48 | 43 | 41 | 38 | Unknown | Unknown | Unknown | Unknown |
| ALS 76 | M | Limb | 47 | 34 | 25 | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 77 | M | Bulbar | 56 | 37 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 78 | M | Limb | 53 | 36 | 26 | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 79 | M | Limb | 25 | 42 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 80 | M | Limb | 36 | 37 | 33 | 29 | Unknown | Unknown | Unknown | Unknown |
| ALS 81 | M | Limb | 34 | 40 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 82 | M | Limb | 48 | 40 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 83 | F | Bulbar | 49 | 43 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 84 | M | Limb | 49 | 40 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 85 | F | Limb | 59 | 29 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 86 | M | Limb | 56 | 12 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 87 | M | Bulbar | 55 | 29 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 88 | M | Bulbar | 47 | 37 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 89 | M | Limb | 25 | 40 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 90 | M | Limb | 35 | 42 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 91 | M | Limb | 33 | 37 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
| ALS 92 | M | Limb | 46 | 40 | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown |
TABLE 3.
Demographics of ALS CSF samples
| Age at sample collection [mean (SD)] | Disease duration in months [mean (SD)] | ALSFRS‐R total score [mean (SD)] | Pre‐baseline ALSFRS‐R slope [mean (SD)] | |
|---|---|---|---|---|
| Visit 1 (n = 40) | 52.41 (12.32) | 25.32 (22.56) | 37.92 (6.41) | 0.55 (0.47) |
| Visit 2 (n = 26) | 56.52 (11.18) | 34.04 (26.27) | 36.00 (6.82) | 0.40 (0.25) |
| Visit 3 (n = 22) | 55.82 (10.81) | 40.06 (28.55) | 34.27 (6.74) | 0.37 (0.26) |
| Visit 4 (n = 16) | 58.25 (8.91) | 51.03 (32.85) | 32.94 (8.19) | 0.32 (0.24) |
| Visit 5 (n = 12) | 55.59 (11.32) | 53.41 (29.61) | 30.75 (5.40) | 0.30 (0.16) |
| Visit 6 (n = 10) | 54.87 (11.11) | 54.87 (11.11) | 27.70 (7.13) | 0.26 (0.14) |
| Visit 7 (n = 5) | 62.08 (11.47) | 56.88 (17.45) | 25.40 (8.68) | 0.30 (0.19) |
2.6. Quanterix Simoa assays
CSF tau and pTau‐T181 concentrations were measured using the Simoa Tau and pTau181 Advantage Kits on a fully automated Simoa HD‐X Analyzer using manufacturer's recommendations (Quanterix Corporation, Billerica, MA). CSF samples were centrifuged at 3,000g for 10 min, diluted 1:4 (pTau‐T181) or 1:10 (tau) in sample buffer, and run in duplicate. The coefficient of variance (CV) was 0%–8% (mean ± SD = 2.2 ± 1.9%) for pTau‐T181 and 0%–26% (mean ± SD = 5.4 ± 5.2%) for tau.
2.7. Statistics
Normal distribution of data was not assumed regardless of sample size or variance. The data are presented as bar graphs demonstrating individual values with the whiskers representing the standard error or as individual value plots with the central line representing the median and the whiskers representing the interquartile range. For CSF analysis, pre‐baseline ALSFRS‐R slope was calculated as the [(48 – ALSFRS‐R score)/(months since disease onset)] where ALSFRS‐R score is the first ALSFRS‐R total score recorded. Trajectory of ALSFRS‐R total score was estimated using a mixed effects model with a fixed effect for time and a random intercept and slope for each subject with an unstructured covariance. A covariate for each CSF measure was added separately to the ALSFRS‐R total score trajectory model to describe any effect on change in ALSFRS‐R total score. Changes in CSF measures were compared with changes in ALSFRS‐R total score by taking the first value for each subject subtracted from the last available visit with complete data for the given comparison. Comparisons between groups were performed using a non‐parametric Mann‐Whitney U test and one‐way ANOVA followed by Tukey's test. Correlations of CSF measures with clinical measures were performed as non‐parametric Spearman correlations. Comparisons for clinical measures were not corrected for multiple comparisons. All tests were two sided with a significance level of 0.05, and exact p values are reported. Analyses were performed using GraphPad Prism and R, a language and environment for statistical computing (https://www.R‐project.org/).
2.8. Study approval
The study was approved by the Partners Healthcare IRB. Written informed consent was obtained from all participants prior to study enrollment. Post‐mortem consent was obtained from the appropriate representative (next of kin or health care proxy) prior to autopsy.
3. RESULTS
3.1. pTau levels are not altered in ALS post‐mortem motor cortex
Our latest findings demonstrate a significant increase in synaptic pTau‐S396 levels in ALS mCTX [22], therefore, we used a large cohort of post‐mortem samples and assessed the overall levels of pTau‐S396 in ALS and control mCTX using immunohistochemistry (IHC). Nissl body marker thionin was used to visualize cells in the adjacent sections from the same samples (Figure 1A, top) and two AD EC were included as positive controls. pTau‐S396 immunosignal was detected in control, ALS, and AD samples (Figure 1A, bottom). As expected, extensive neuropil threads and NFTs were observed in AD brains (Figure 1Af). In mCTX, pTau‐S396 immunosignal was heterogenous in both control (Figure 1Ag,h) and ALS mCTX (Figure 1Ai,j). Given the heterogeneity of pTau‐S396 staining across all samples, we categorized the intensity of the signal as summarized in Table 4. Overall pTau‐S396 immunostaining in ALS mCTX was intense (+++) in 12.4%, moderate (++) in 43.8%, and weak (+) in 43.8% of cases. In controls, pTau‐S396 was intense in 28.6%, moderate in 42.8%, and weak in 14.3% of cases. Neuropil threads detected in ALS were extensive in 12.4%, moderate in 18.7%, and weak in 62.6% of cases. Neuropil threads in controls were extensive in 14.3%, moderate in 57.1%, and weak in 14.3% of cases. NFTs in ALS mCTX were detected in high numbers in 6.3%, moderate numbers in 18.7%, and few in 31.2% of cases. In controls, few NFTs were observed in 28.6% of cases. Lastly, pTau‐S396 overall staining was absent (−) in 14.3% of control cases, pTau‐S396 neuropil threads were absent in 6.3% of ALS and 14.3% of control mCTX, and no NFTs were detected in 73.8% of ALS and in 71.4% of control cases. Further quantitative analysis revealed no significant difference in pTau‐S396 levels between ALS and control mCTX (Figure 1B). To further delineate the heterogeneity of pTau‐S396 immunosignal among the ALS cases, pTau‐S396 levels were correlated with the known patient clinicopathological information. The analysis demonstrated an increase in pTau‐S396 levels in a case revealing brain alterations reminiscent of AD (Figure 1B, green dot) as well as in a case harboring a mutation in C9ORF72 and diagnosed with bulbar onset ALS (Figure 1B, red dot). Interestingly, the other C9ORF72‐ALS case used in this study, diagnosed with limb onset disease, had lower levels of pTau‐S396 (Figure 1B, blue dot).
FIGURE 1.
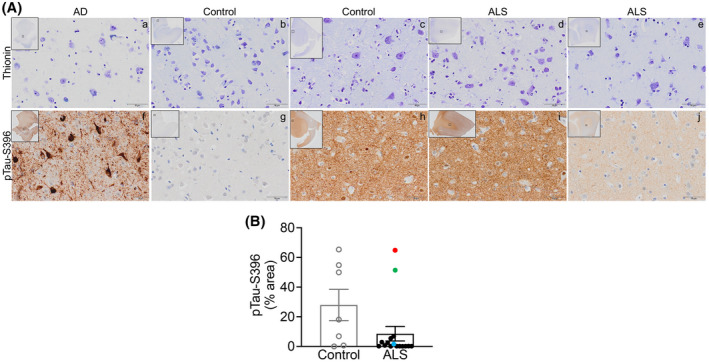
pTau‐S396 levels are not altered in ALS post‐mortem motor cortex. (A) Top. Representative thionin immunostaining in grey matter from (a) AD EC, (b, c) control mCTX, and (d, e) ALS mCTX. Bottom. Representative pTau‐S396 immunostaining in grey matter from (f) AD EC, (g, h) control mCTX, and (i, j) ALS mCTX. (B) There was no significant change in pTau‐S396 levels in ALS mCTX (n = 16) compared with controls (n = 7) (Mann–Whitney U test = 32, p = 0.1181). Bulbar onset C9ORF72‐ALS is indicated with a red dot, limb onset C9ORF72‐ALS with a blue dot, and a single ALS case revealing brain alterations likely due to AD is indicated with a green dot. Scale bar: 50 µm
TABLE 4.
pTau‐S396 and PHF1 immunostaining in control and ALS post‐mortem mCTX
| pTau‐S396 | PHF1 | |||||||
|---|---|---|---|---|---|---|---|---|
| Diffuse staining | Neuropil threads | NFTs | WM staining | Diffuse staining | Neuropil threads | NFTs | WM staining | |
| Control 1 | ++ | ++ | + | +++ | ++ | + | + | +++ |
| Control 2 | +++ | ++ | − | ++ | ++ | ++ | + | + |
| Control 3 | +++ | +++ | − | +++ | ++ | ++ | − | ++ |
| Control 4 | ++ | ++ | − | + | + | + | − | − |
| Control 5 | ++ | + | − | + | + | − | − | − |
| Control 6 | − | − | − | − | − | − | − | − |
| Control 7 | + | ++ | + | + | − | + | + | − |
| ALS 1 | ++ | + | − | + | + | − | − | − |
| ALS 2 | ++ | ++ | + | ++ | − | − | − | − |
| ALS 3 | +++ | +++ | + | +++ | ++ | ++ | + | +++ |
| ALS 4 | ++ | ++ | + | ++ | + | + | + | + |
| ALS 5 | + | + | − | − | + | − | − | − |
| ALS 6 | + | + | ++ | +++ | + | + | + | + |
| ALS 7 | +++ | +++ | +++ | +++ | ++ | +++ | +++ | ++ |
| ALS 8 | + | + | − | + | + | − | − | − |
| ALS 9 | + | ++ | ++ | + | + | ++ | ++ | − |
| ALS 10 | ++ | + | − | ++ | + | − | − | − |
| ALS 11 | ++ | + | + | + | + | − | − | − |
| ALS 12 | + | + | − | − | − | − | − | − |
| ALS 13 | + | + | ++ | + | ||||
| ALS 14 | ++ | + | − | + | ||||
| ALS 15 | + | − | − | − | ||||
| ALS 16 | ++ | + | + | + | ||||
Summary of pTau‐S396 and PHF1 IHC results for diffuse staining, neuropil threads, NFTs, and WM staining is given and indicated as follows: −, no staining; +, weak staining; ++, moderate staining; +++, intense staining.
Next, pTau‐S396 immunostaining was analyzed in white matter (WM) from the same AD EC and control and ALS mCTX (Figure 2A). Similar to the grey matter, the immunostaining in WM was heterogenous and, therefore, categorized as summarized in Table 4. pTau‐S396 immunostaining and neuropil threads detected in ALS were extensive (+++) in 18.7%, moderate (++) in 18.7%, and weak (+) in 43.8% of cases. In controls, neuropil threads were extensive in 28.6%, moderate in 14.3%, and weak in 42.8% of cases. Absence of neuropil threads (−) was observed in 18.7% of ALS and 14.3% of control cases. Further quantitative analysis revealed no significant difference in pTau‐S396 levels between ALS and control mCTX (Figure 2B). Correlation of known patient clinicopathological information with pTau‐S396 levels revealed that the absence or weak pTau‐S396 immunostaining in ALS WM corresponded with severe myelin loss at post‐mortem evaluation, while a moderate‐to‐intense staining was observed in the other ALS cases. Additionally, similar to grey matter, pTau‐S396 levels were increased in the case revealing brain alterations reminiscent of AD (Figure 2B, green dot) and in the bulbar onset C9ORF72‐ALS (Figure 2B, red dot), while lower levels were detected in the C9ORF72‐ALS case diagnosed with limb onset disease (Figure 2B, blue dot).
FIGURE 2.
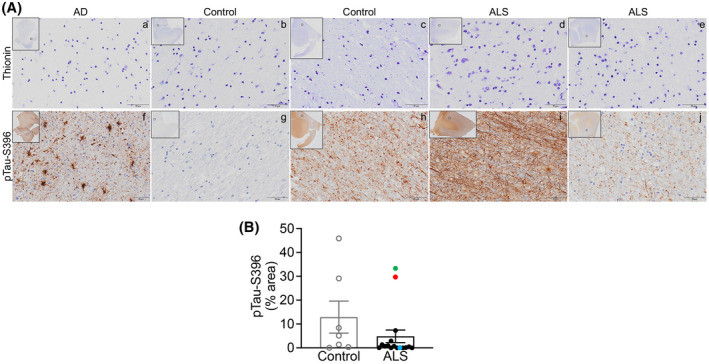
pTau‐S396 levels are not altered in ALS motor cortex white matter. (A) Top. Representative thionin immunostaining in white matter from (a) AD EC, (b, c) control mCTX, and (d, e) ALS mCTX. Bottom. Representative pTau‐S396 immunostaining in white matter from (f) AD EC, (g, h) control mCTX, and (i, j) ALS mCTX. (B) There was no significant change in pTau‐S396 levels in ALS mCTX WM (n = 16) compared with controls (n = 7) (Mann–Whitney U test = 34.50, p = 0.1574). Bulbar onset C9ORF72‐ALS is indicated with a red dot, limb onset C9ORF72‐ALS with a blue dot, and a single ALS case revealing brain alterations likely due to AD is indicated with a green dot. Scale bar: 50 µm
These results were further confirmed using PHF1 antibody which recognizes tau phosphorylation at both S396 and S404, in the same samples as those used for pTau‐S396 except for four ALS mCTX. Similar to pTau‐S396, PHF1 immunosignal was detected in control, ALS, and AD samples. As expected, extensive neuropil threads and NFTs were observed in AD brains (Figure 3Af). Both control (Figure 3Ag,h) and ALS mCTX (Figure 3Ai,j) demonstrated heterogeneity of PHF1 immunosignal, although the immunostaining was weaker compared with pTau‐S396 labeling. As outlined in Table 4, PHF1 overall immunostaining in ALS mCTX was moderate (++) in 16.7%, and weak (+) in 66.6% of cases. PHF1 staining in controls was moderate in 42.8% and weak in 28.6% of cases. Neuropil threads detected by PHF1 in ALS mCTX were extensive (+++) in 8.3%, moderate in 16.7%, and weak in 16.7% of cases. Neuropil threads in controls were moderate in 28.6% and weak in 42.8% of cases. NFTs in ALS mCTX were detected in high numbers in 8.3%, moderate numbers in 16.7%, and few in 16.7%. In controls, few and sparse NFTs were detected in 42.8% of cases. Lastly, PHF1 staining was absent (−) in 16.7% of ALS and 28.6% of controls, neuropil threads were absent in 58.3% of ALS and 28.6% of control cases, and no NFTs were detected in 58.3% of ALS and 57.2% of control cases. Similar to pTau‐S396, there were no significant alterations in PHF1 levels in ALS mCTX compared with controls (Figure 3B). PHF1 levels were then correlated with the known patient clinicopathological information and the results demonstrated an increase in PHF1 levels in the ALS case revealing histopathological alterations reminiscent of AD (Figure 3B, green dot) and in the bulbar onset ALS harboring mutation in C9ORF72 (Figure 3B, red dot), while immunosignal and neuropil threads were moderate for the limb onset C9ORF72‐ALS case (Figure 3B, blue dot).
FIGURE 3.
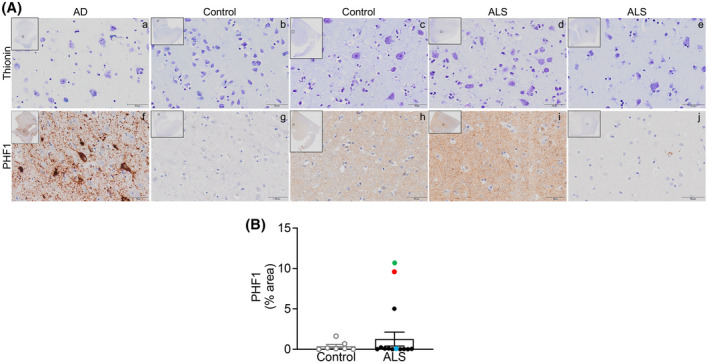
PHF1 levels are not altered in ALS post‐mortem motor cortex. (A) Top. Representative thionin immunostaining in grey matter from (a) AD EC, (b, c) control mCTX, and (d, e) ALS mCTX. Bottom. Representative PHF1 immunostaining in grey matter from (f) AD EC, (g, h) control mCTX, and (i, j) ALS mCTX. (B) There was no significant change in PHF1 levels in ALS mCTX (n = 12) compared with controls (n = 7) (Mann–Whitney U test = 34.50, p = 0.5483). Bulbar onset C9ORF72‐ALS is indicated with a red dot, limb onset C9ORF72‐ALS with a blue dot, and a single ALS case revealing brain alterations likely due to AD is indicated with a green dot. Scale bar: 50 µm
Similarly, PHF1 immunostaining in WM revealed a heterogenous signal in both control and ALS mCTX summarized in Table 4, and PHF1 immunosignal was also detected in the AD brains used as positive control (Figure 4A). Neuropil threads detected by PHF1 in ALS mCTX were extensive (+++) in 8.3%, moderate (++) in 8.3%, and weak (+) in 16.7% of cases. Neuropil threads in controls were extensive in 14.3%, moderate in 14.3%, and weak in 14.3% of cases. Absence of immunosignal (‐) was reported in 66.7% of ALS cases and in 57.1% of control cases. No significant alterations in PHF1 levels were found in ALS mCTX compared with controls in WM (Figure 4B). Similar to pTau‐S396, absence or weak PHF1 staining was observed in WM of ALS cases with severe myelin loss at post‐mortem evaluation. Furthermore, there was an increase in PHF1 in the single ALS case revealing alterations likely due to AD (Figure 4B, green dot) and in the bulbar onset C9ORF72‐ALS (Figure 4B, red dot); however, lower PHF1 levels were detected in the limb onset C9ORF72‐ALS (Figure 4B, blue dot).
FIGURE 4.
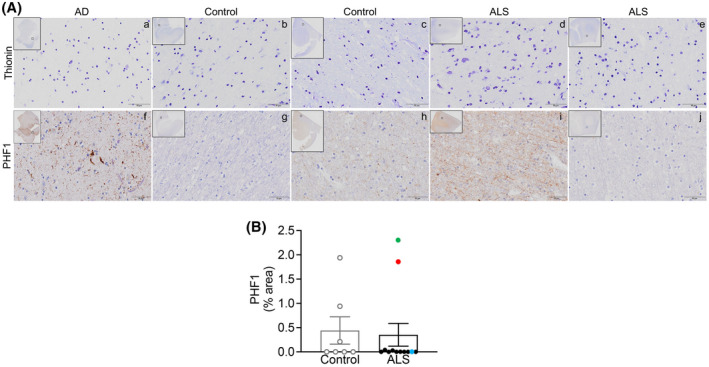
PHF1 levels are not altered in ALS motor cortex white matter. (A) Top. Representative thionin immunostaining in white matter from (a) AD EC, (b, c) control mCTX, and (d, e) ALS mCTX. Bottom. Representative PFH1 immunostaining in white matter from (f) AD EC, (g, h) control mCTX, and (i, j) ALS mCTX. (B) There was no significant change in PHF1 levels in WM from ALS mCTX (n = 12) compared with controls (n = 7) (Mann–Whitney U test = 39, p = 0.8068). Bulbar onset C9ORF72‐ALS is indicated with a red dot, limb onset C9ORF72‐ALS with a blue dot, and a single ALS case revealing brain alterations likely due to AD is indicated with a green dot. Scale bar: 50 µm
3.2. While pTau‐T181 levels are decreased across ALS, pTau‐S396 and total tau levels are increased in C9ORF72‐ALS
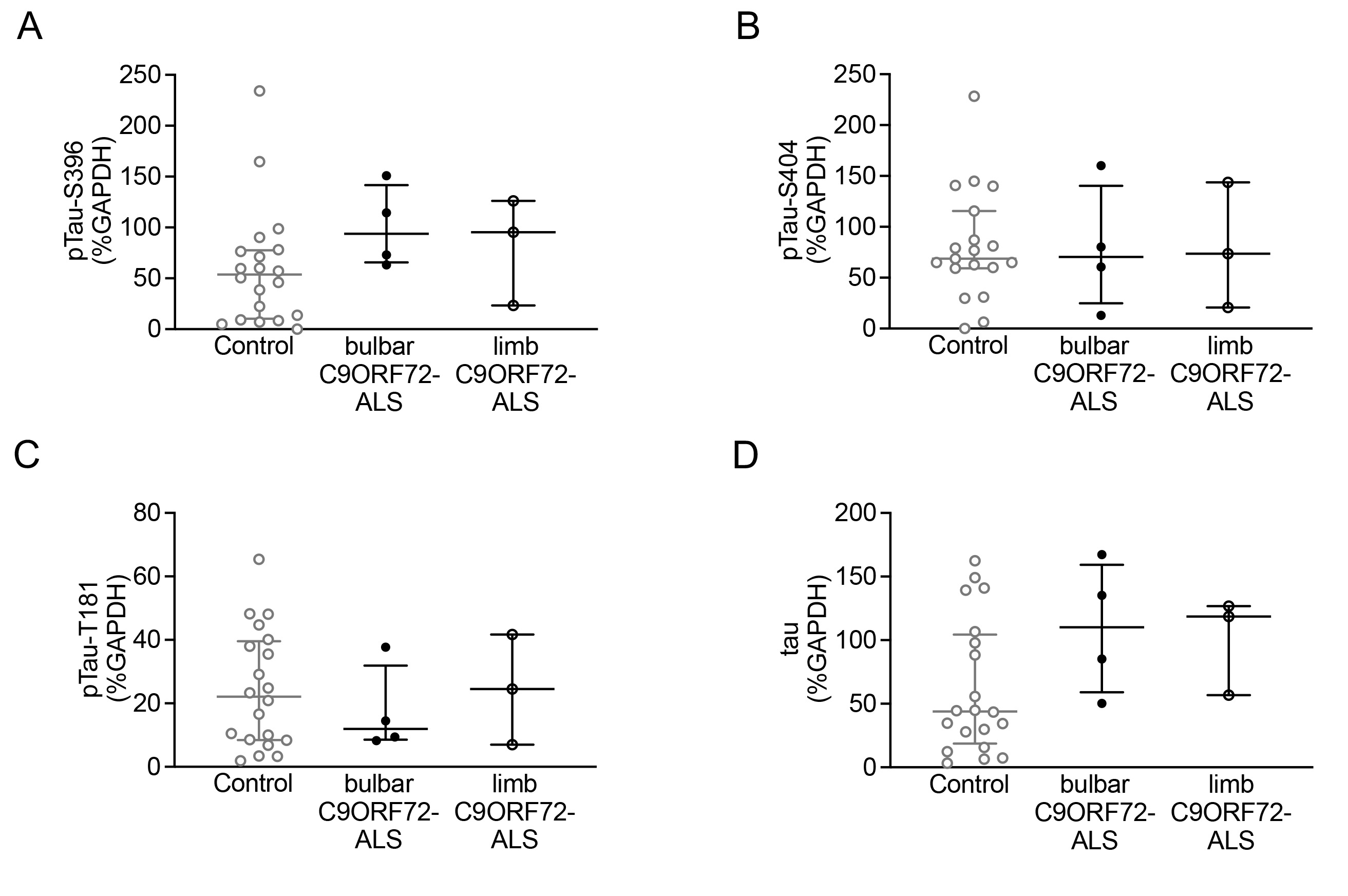
To verify the IHC findings and determine whether increases in tau phosphorylation are linked to C9ORF72 mutation and/or to bulbar onset disease, we assessed the levels of pTau‐S396, pTau‐S404, pTau‐T181, and total tau in a larger cohort of human post‐mortem mCTX using western blots. The analysis revealed that while there was no significant alteration in the overall levels of pTau‐S396, pTau‐S404, and total tau in ALS mCTX, there was a significant decrease in pTau‐T181 levels in ALS compared with controls (Figure 5A–E). In addition, while pTau‐S404 and pTau‐T181 levels were not altered, there was a significant increase in both pTau‐S396 and total tau levels in C9ORF72‐ALS (Figure 5F–J). Given that the IHC results revealed higher levels of pTau‐S396 and PHF1 in the bulbar onset C9ORF72‐ALS compared with the limb onset C9ORF72‐ALS, we next used the known patient clinicopathological information to correlate pTau and total tau levels to ALS region of onset. The analysis revealed that there were no significant alterations in pTau‐S396, pTau‐S404, pTau‐T181 or total tau levels in either bulbar or limb onset ALS (Figure 5K–O). Similarly, there were no alterations in pTau‐S396, pTau‐S404, pTau‐S181, and total tau in either bulbar or limb onset C9ORF72‐ALS (Figure S1).
FIGURE 5.
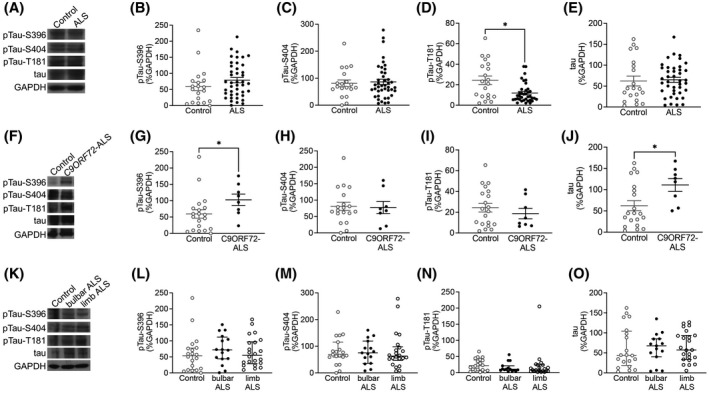
While pTau‐T181 levels are decreased in ALS motor cortex, pTau‐S396 and total tau levels are increased only in C9ORF72‐ALS. (A) Representative western blot images of pTau‐S396, pTau‐S404, pTau‐T181, total tau and GAPDH in control and ALS mCTX. There were no significant change in the levels of (B) pTau‐S396 (Mann–Whitney U test = 317, p = 0.1234) and (C) pTau‐S404 (Mann–Whitney U test = 396, p = 0.9665) between ALS (n = 43) and controls (n = 21). (D) pTau‐T181 levels were significantly decreased in ALS mCTX (n = 43) compared with controls (n = 21) (Mann–Whitney U test = 226.5, p = 0.0157). (E) There was no significant change in total tau levels between ALS (n = 43) and control mCTX (n = 21) (Mann–Whitney U test = 366, p = 0.5071). (F) Representative western blot images of pTau‐S396, pTau‐S404, pTau‐T181, total tau and GAPDH in control, and C9ORF72‐ALS mCTX. (G) There was a significant increase in pTau‐S396 levels in C9ORF72‐ALS (n = 8) compared with control mCTX (n = 21) (Mann–Whitney U test = 36, p = 0.0247). There were no significant changes in (H) pTau‐S404 (Mann–Whitney U test = 76, p > 0.999) and (I) pTau‐T181 levels (Mann–Whitney U test = 66, p = 0.5002) in C9ORF72‐ALS (n = 8) compared with control mCTX (n = 21). (J) There was a significant increase in total tau levels in C9ORF72‐ALS (n = 8) compared with controls (n = 21) (Mann–Whitney U test = 61, p = 0.0211). (K) Representative western blot images of pTau‐S396, pTau‐S404, pTau‐T181, total tau, and GAPDH in mCTX from control, bulbar, and limb onset ALS. There were no significant alterations in the levels of (L) pTau‐S396 (one‐way ANOVA [F(2,55) = 0.3652, p = 0.6958], (M) pTau‐S404 (one‐way ANOVA [F(2,53) = 0.02583, p = 0.9745], (N) pTau‐T181 (one‐way ANOVA [F(2,53) = 0.2965, p = 0.7447], and (O) total tau (one‐way ANOVA [F(2,55) = 0.04461, p = 0.9564] between controls (n = 21), bulbar onset ALS (n = 16), or limb onset ALS (n = 23). *p < 0.05
3.3. Identification of novel MAPT genetic variants in ALS
To further investigate the potential pathogenic role exerted by tau in ALS, we leveraged ALSKP and Project MinE data sets to verify whether there were genetic variants in MAPT in people living with ALS. We observed a total of 36 heterozygous variants in the MAPT gene in a total of 42 ALS cases (Table S1) and no homozygous variants aggregated in ALSKP and Project MinE. Among the variants, 33 were missense (Figure 6A) in the canonical MAPT transcript NM_016835 and 27 in the post‐spliced functional MAPT transcript NM_005910 (Figure 6B). Next, we evaluated the population frequency of each MAPT variant using large datasets aggregating exomes and genomes from non‐ALS individuals such as gnomAD. Among the 36 variants, 15 variants were unique to ALS and absent in gnomAD and were accordingly classified as “ALS unique variants.” Twenty‐one variants were classified as “ALS rare variants” as they were observed in gnomAD at a very low frequency (MAF < 4.71E‐05). Following variant annotation with CADD and MPC, we divided the missense variants into five distinct categories (M1‐M5) based on the predicted severity of their impact on protein function with M1 (MPC > 2 and CADD > 25) being the most probable to induce a pathogenic effect and M5 being the least probable category (MPC < 1.5 and CADD < 20). The variants in categories M1 and M2, which are within the upper limit of predicted pathogenicity, cluster near the C‐terminus of the protein transcript within or around the microtubule‐binding domain (Figure 6B). The addition of known pathogenic or likely pathogenic MAPT variants reported further supports the predicted pathogenicity of M1 and M2 variants as these ClinVar variants also cluster at the C‐terminus within or neighboring the microtubule‐binding domain (Figure 6B). Of the four ALS variants that are harbored within the microtubule‐binding domain namely, p.Leu583Val (NM_005910 p.Leu266Val), p.Lys616del (NM_005910 p.Lys281del), p.Lys652Met (NM_005910 p.Lys317Met), and p.Val698Ile (NM_005910 p.Val363Ile), two variants were previously reported in ClinVar. One variant, p.Leu266Val, was also reported in a patient with frontotemporal dementia (FTD), classified as a pathogenic variant [31], and also observed in two unrelated ALS cases in the Project MinE browser. Another variant, p.Lys317Met, which is unique to ALS and was also observed in two unrelated ALS patients, has been previously reported in ClinVar in two pedigrees displaying FTD and ALS [32]. For the other variants within and outside of the microtubule‐binding domain, despite their rarity and even several being unique to ALS, their pathogenicity and contribution to neurodegeneration remains elusive, and their effect would need to be validated. Interestingly, all the likely pathogenic variants identified localized adjacent to the phosphorylation sites that appear to be involved in ALS (Figure 6C).
FIGURE 6.
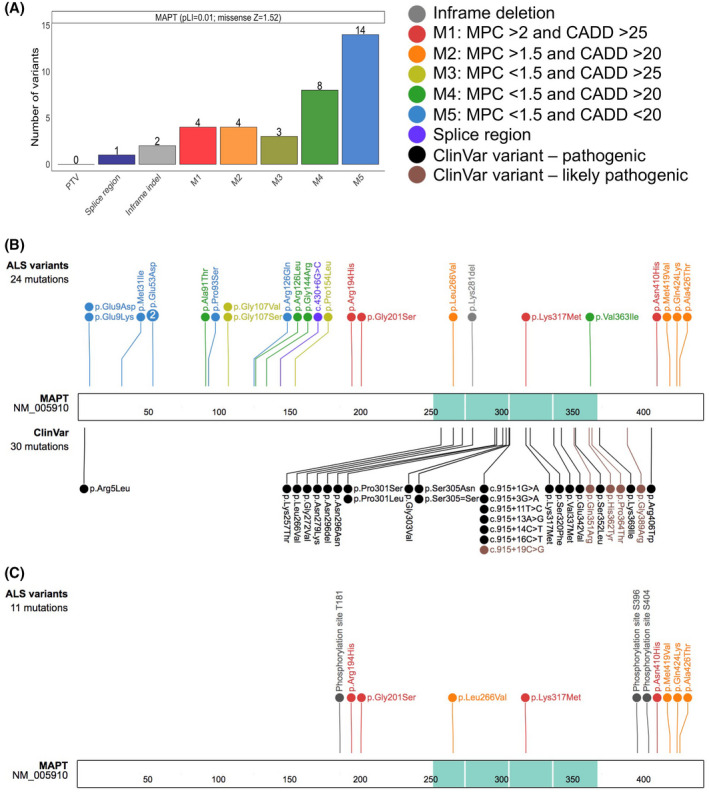
Protein schematic of MAPT variants observed in ALS. (A) Variant types are displayed on the X‐axis with their respective counts on the Y‐axis for the MAPT gene. The colors represent the type of non‐synonymous changes observed, and the number next to the variants, noted on top of each bar, depicts the number of individuals observed to carry the corresponding variant. The probability of loss of function (pLI) and the missense constraint Z scores for MAPT are shown adjacent to the gene label. PTV, protein truncating variant; indel, insertion/deletion; M in M1‐M5, missense. Missense variants were divided into five classes depending on their MPC (Missense badness, PolyPhen‐2, and Constraint) and CADD (Combined Annotation Dependent Depletion) deleteriousness scores. (B) Schematic representation highlighting the novel ALS MAPT variants displayed on top. Variants identified only in ALS cases were classified as “ALS unique variants,” while the variants observed in ALS cases and also at a very low allele frequency in gnomAD were classified as “ALS rare variants.” The variants displayed on the bottom were ClinVar pathogenic (black) and likely pathogenic (brown) variants. The microtubule‐binding domain is shown in turquoise. The numbers within the protein sequence depicts the amino acid position. (C) Schematic representation of the newly identified pathogenic or likely pathogenic MAPT variants in ALS together with the known phosphorylation sites at T181, S396 and S404 across tau protein
3.4. pTau‐T181:tau ratio is decreased in ALS CSF
Recent studies have suggested that alterations in tau and pTau‐T181 may be viable biomarkers for AD [33, 34], therefore, we assessed the levels of total tau and pTau‐T181 in CSF from healthy controls and people living with ALS using Quanterix Simoa assays (Table S2). Mann–Whitney U test indicated that while there was no significant difference in total tau or pTau‐T181 levels in ALS CSF compared to control CSF, there was a significant decrease in pTau‐T181:tau ratio in ALS CSF (Figure 7A–C). Next, we correlated alterations in tau and pTau in ALS CSF with the known patient clinical information when available. The analysis revealed that although CSF tau levels were significantly increased in bulbar onset ALS, there was no change in pTau‐T181, and pTau‐T181:tau ratio was significantly decreased in bulbar onset ALS (Figure 7D–F). Similarly, while tau and pTau‐T181 levels were not altered, pTau‐T181:tau ratio was significantly decreased in CSF from limb onset ALS (Figure 7G–I).
FIGURE 7.
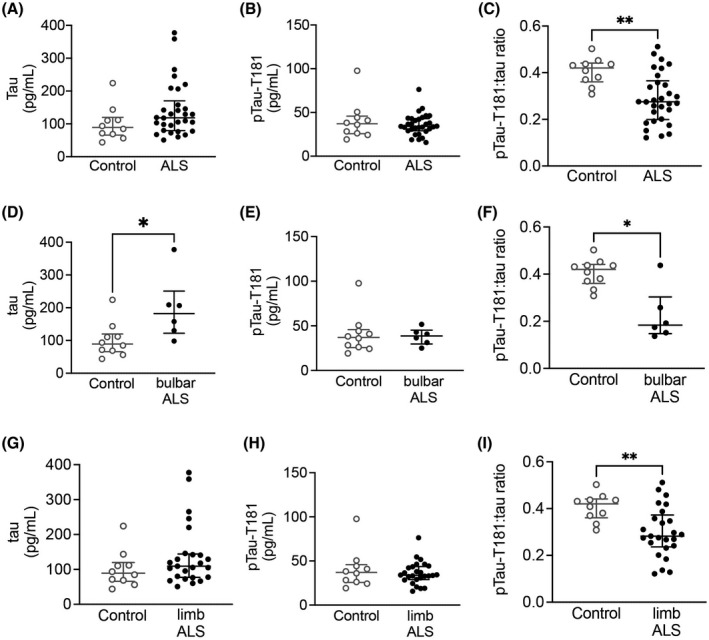
CSF pTau‐T181:tau ratio is decreased in ALS. While there was no difference in CSF (A) tau (Mann–Whitney U test = 100, p = 0.1234) and (B) pTau‐T181 (Mann–Whitney U test = 147, p = 0.8227), there was a significant decrease in (C) pTau‐T181:tau ratio (Mann–Whitney U test = 56, p = 0.0025) in ALS CSF (n = 40) compared with healthy controls (n = 10). (D) There was a significant increase in CSF tau levels in bulbar onset ALS (n = 6) (Mann–Whitney U test = 9, p = 0.0225). (E) CSF pTau‐T181 levels were not altered in bulbar onset ALS (Mann–Whitney U test = 27, p = 0.7925). (F) CSF pTau‐T181:tau ratio was significantly decreased in bulbar onset ALS (Mann–Whitney U test = 7, p = 0.0110). (G) CSF tau (Mann–Whitney U test = 91, p = 0.2253) and (H) pTau‐T181 levels (Mann–Whitney U test = 123, p = 0.8214) were not altered in limb onset ALS (n = 25). (I) There was a significant decrease in pTau‐T181:tau ratio in limb onset ALS (Mann–Whitney U test = 49, p = 0.0045). *p < 0.05; **p < 0.01
Further analysis revealed no correlation between tau, pTau‐T181, or pTau‐T181:tau ratio and age at time of sample collection (Figure 8A–C). Similarly, there was no correlation between tau or pTau‐T181 levels and disease duration; however, there was a trend toward a significant correlation between pTau‐T181:tau ratio and disease duration (Figure 8D–F). There was also a trend toward a significant correlation between the ALSFRS‐R at time of first visit with tau levels; however, ALSFRS‐R did not correlate with pTau‐T181 or pTau‐T181:tau ratio levels (Figure 8G–I). While there was no correlation between pTau‐T181 levels and pre‐baseline ALSFRS‐R slope, there was a trend toward a significant correlation between tau and pre‐baseline ALSFRS‐R slope and a significant correlation between pre‐baseline ALSFRS‐R slope and pTau‐T181:tau ratio (Figure 8J–L).
FIGURE 8.
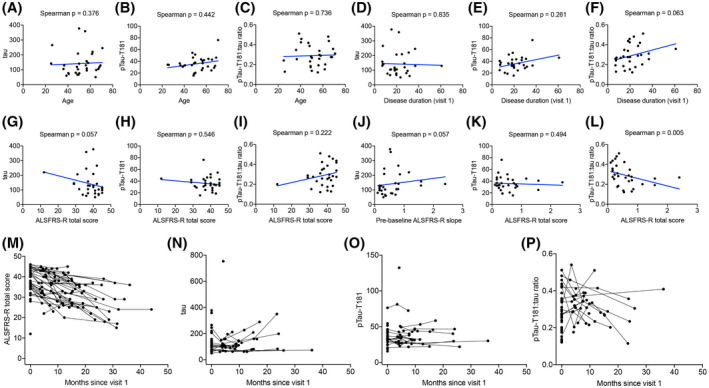
CSF pTau‐T181:tau ratio correlates with ALS duration and progression. There was no correlation between CSF (A) tau (Spearman correlation, p = 0.376), (B) pTau‐T181 (Spearman correlation, p = 0.442), or (C) pTau‐T181:tau ratio (Spearman correlation, p = 0.736) and the age at first visit. (D) CSF tau (Spearman correlation, p = 0.835) and (E) pTau‐T181 levels (Spearman test, p = 0.261) did not correlate with ALS disease duration. (F) There was a trend toward a significant correlation between CSF pTau‐T181:tau ratio and ALS disease duration (Spearman correlation, p = 0.063). (G) There was a trend toward a significant inverse correlation between CSF tau and ALSFRS‐R (Spearman correlation, p = 0.057). (H) There was no correlation between CSF pTau‐T181 and ALSFRS‐R (Spearman correlation, p = 0.546). (I) There was no correlation with CSF pTau‐T181:tau ratio and ALSFRS‐R (Spearman correlation, p = 0.222). (J) There was a trend toward a negative correlation between CSF tau and ALSFRS‐R slope (Spearman correlation, p = 0.057). (K) There was no correlation between CSF pTau‐T181 and ALSFRS‐R slope (Spearman correlation, p = 0.494). (L) There was a positive correlation between CSF pTau‐T181:tau ratio and ALSFRS‐R slope (Spearman correlation, p = 0.005). (M) There was a significant decline in ALSFRS‐R over time (p < 0.001). There was no significant alterations in CSF (N) tau (p = 0.907), (O) pTau‐T181 (p = 0.222), and (P) pTau‐T181:tau ratio (p = 0.578)
Lastly, longitudinal analysis of ALS CSF revealed that there was high variability in both tau and pTau‐T181 trajectory. Specifically, there was a significant decline over time based on the ALSFRS‐R assessed for each patient at each visit; however, there were no significant alterations in tau, pTau‐T181, or pTau‐T181:tau ratio in ALS over time (Figure 8M–P) (Table 5). When tau was added to the ALSFRS‐R trajectory model, there was a significant effect of tau on the change in ALSFRS‐R over time. This model estimated a baseline ALSFRS‐R total score of 37.9 points and a slope of −0.56 point/month and a 10‐point increase in tau at baseline resulted in a further −0.026 point/month change in ALSFRS‐R. While pTau‐T181 showed no significant effects, pTau‐T181:tau ratio revealed a trend toward a significant effect (Table 6).
TABLE 5.
Trajectories of ALSFRS‐R, tau, pTau‐T181, and pTau‐T181: tau ratio in ALS CSF
| Intercept | Slope | Slope SE | Slope p | |
|---|---|---|---|---|
| ALSFRS‐R | 37.8 | −0.47 | 0.06 | <0.001 |
| Tau | 147.9 | 0.16 | 1.38 | 0.907 |
| pTau‐T181 | 39.07 | −0.09 | 0.07 | 0.222 |
| pTau‐T181:tau ratio | 0.3 | 0 | 0 | 0.578 |
TABLE 6.
Effect of baseline tau, pTau‐T181, and pTau‐T181:tau ratio on ALSFRS‐R trajectory
| Value | SE | DF | t value | p value | |
|---|---|---|---|---|---|
| tau | |||||
| Intercept | 37.933 | 1.224 | 60 | 30.998 | <0.001 |
| Disease duration since visit 1 | −0.557 | 0.08 | 60 | −6.93 | <0.001 |
| Baseline tau | −0.188 | 0.151 | 28 | −1.245 | 0.224 |
| Disease duration: baseline tau | −0.026 | 0.011 | 60 | −2.31 | 0.024 |
| pTau‐T181 | |||||
| Intercept | 38.111 | 1.229 | 65 | 30.999 | <0.001 |
| Disease duration since visit 1 | −0.493 | 0.075 | 65 | −6.608 | <0.001 |
| Baseline pTau‐T181 | −0.612 | 1.034 | 29 | −0.592 | 0.558 |
| Disease duration: baseline pTau‐T181 | −0.066 | 0.055 | 65 | −1.186 | 0.240 |
| pTau‐T181:tau ratio | |||||
| Intercept | 37.964 | 1.227 | 60 | 30.929 | <0.001 |
| Disease duration since visit 1 | −0.571 | 0.09 | 60 | −6.36 | <0.001 |
| Baseline pTau‐T181:tau ratio | 13.89 | 11.492 | 28 | 1.209 | 0.237 |
| Disease duration: baseline pTau‐T181:tau ratio | 1.423 | 0.715 | 60 | 1.989 | 0.051 |
4. DISCUSSION
In this study, we demonstrated that while tau and its phosphorylation at S396 and S404 are not altered in ALS post‐mortem mCTX, pTau‐T181 levels were significantly decreased in ALS. Furthermore, we report a significant increase in tau and pTau‐S396 levels in C9ORF72‐ALS. These findings highlight a differential pattern of phosphorylation at specific amino acid residues across tau protein in ALS. We also identified several pathogenic or likely pathogenic variants in MAPT, clustered within the microtubule‐binding domain and proline‐rich region (PRR), which were unique to ALS cases. Consistent with previous reports, our findings also revealed a significant decrease in pTau‐T181:tau ratio in ALS CSF [16, 17, 18, 19]. Although CSF total tau levels were not altered across all ALS samples, they were significantly increased in ALS patients with bulbar onset disease and were inversely correlated with the ALSFRS‐R score. Furthermore, pTau‐T181:tau ratio was significantly decreased in ALS and positively correlated with ALSFRS‐R score. Total tau and pTau‐T181:tau ratio did not function as disease‐monitoring biomarkers. Instead, they functioned as predictive biomarkers given that the rate of ALS progression correlated with increases in total tau and decreases in pTau‐T181:tau ratio. Collectively, our findings revealed similar trends in pTau‐T181 levels across post‐mortem mCTX and CSF in ALS.
Tau pathology has been described recently in both sporadic and familial forms of ALS [4, 5], as increases in total tau and cytoplasmic inclusions of hyperphosphorylated tau have been reported in post‐mortem mCTX and spinal cord from ALS patients [13, 14, 15]. Furthermore, we demonstrated that pTau‐S396 mislocalizes to synapses in human post‐mortem mCTX across ALS subtypes and contributes to mitochondrial fragmentation and dysfunction via interaction with DRP1 [22], providing additional support for tau's pathogenic role in ALS. However, our findings here demonstrate that there is not an overall increase in tau phosphorylation in ALS. Specifically, our findings reveal that pTau immunostaining was heterogenous in post‐mortem mCTX and that neuropil threads and sparse NFTs were observed in both ALS and controls, a finding that was confirmed by western blots. The intense pTau staining detected in a single ALS case was likely due to AD pathology (Braak neurofibrillary stage: III/IV; CERAD plaque stage: sparse as reported by VABBB). Yet, pTau levels were increased in the C9ORF72‐ALS mCTX with bulbar onset ALS. Western blot analysis in a larger cohort of post‐mortem mCTX confirmed that alterations in tau phosphorylation were due to the presence of C9ORF72 mutation and not associated with region of ALS onset. This finding suggests that altered protein homeostasis or misfolding, previously reported in C9ORF72‐ALS [35, 36], may contribute to tau accumulation. Specifically, it has been suggested that C9ORF72 expansion leads to the disruption of protein degradation pathways, further increasing the accumulation of several proteins, including tau, ubiquitin, and p62 [37]. Similarly, previous findings demonstrated that the repeat expansion G4C2 in C9ORF72 increases tau phosphorylation in both ALS and ALS/FTD [38, 39, 40]. Additionally, a recent positron emission tomography (PET) imaging study has demonstrated an increase in tau PET signal that correlated with an increase in aggregated tau in the brains of people carrying C9ORF72 expansions [41], further suggesting that tau phosphorylation may increase in the presence of mutations linked with protein misfolding and aggregation. A more in‐depth analysis in a larger cohort of ALS cases with various mutations causing protein aggregation [1, 2] is required. Such a study would also help determine whether increases in tau phosphorylation are more closely linked to genetic ALS cases associated with alteration in protein homeostasis.
We identified 36 MAPT genetic variants in 42 ALS cases from ALSKP and Project MinE data browsers. Of the 36 variants, 15 were unique to ALS and the remaining 21 were observed at a very low frequency in gnomAD. Thirty‐three of 36 were missense variants, which we subsequently annotated using the scores of pathogenicity CADD and MPC and classified into five pathogenicity categories. Interestingly, the eight ALS variants associated with a high predicted pathogenicity cluster near the C‐terminus of the protein transcript within or around the microtubule‐binding domain consistent with the other pathogenic or likely pathogenic variants in patients with FTD and motor neuron dysfunction previously reported in ClinVar. Specifically, two variants, p.Leu583Val and p.Lys652Met, which were collectively observed in four unrelated ALS patients in ALSKP and Project MinE, were also observed in pedigrees displaying FTD, parkinsonism, and motor neuron degeneration [31, 32]. The brain of the patient carrying the p.Leu583Val showed Pick body‐like inclusions and unique tau‐positive argyrophilic astrocytes with stout filaments and naked, round, or irregular argyrophilic inclusions with deposits of tau. Additionally, recombinant tau with a L266V mutation revealed a reduced ability to promote microtubule assembly, which may be the primary effect of the mutation [31]. Furthermore, two apparently unrelated pedigrees with an autosomal dominant FTD with parkinsonism and ALS were shown to carry the p.Lys652Met mutation. These people revealed a massive degeneration of the substantia nigra without Lewy bodies. A variable degree of frontotemporal atrophy was found and extensive deposition of abnormal tau protein in a mixed pattern (neuronal, glial) was observed [32]. The recurrence of these variants may suggest either a common haplotype among the four ALS patients and these pedigrees or a mutation hotspot. Given the evidence of variant pathogenicity clustering near or within the microtubule‐binding domain in multiple pedigrees with neurodegeneration, it is likely that these eight variants observed in ALS cases explain or significantly contribute to neurodegeneration in these patients. However, in the absence of brain samples from these specific ALS cases harboring these newly identified variants, future studies will focus on sequencing the available post‐mortem mCTX cohort to validate these findings and determine the functional consequences of these mutations. Here, we reported alterations in tau phosphorylation at T181 and S396 in ALS mCTX, and both of these phosphorylation sites are adjacent to the likely and newly described pathogenic variants identified in MAPT. Although the variants are located near the known pathogenic sites, alterations in tau phosphorylation are post‐translational and may be caused by different molecular mechanisms which remain to be investigated.
Recent studies suggest that alterations in tau levels in biofluids, such as CSF, may serve as a biomarker for the diagnosis of tauopathies [42, 43]. Whether tau levels could serve as a viable biomarker in ALS remains contradictory, with studies to date reporting both increases and no change in CSF tau in ALS [16, 17, 18, 19, 44, 45]. While all studies demonstrate no significant difference in CSF pTau‐T181 levels in ALS, several reports demonstrate significant decreases in CSF pTau‐T181:tau ratio in ALS [16, 19, 46]. Similarly, contradictory data have been reported regarding the potential prognostic validity of CSF tau in ALS [47, 48], with studies suggesting a positive correlation between tau or pTau:tau ratio and disease progression [16, 19, 49]. Our findings here demonstrate that while there are no alterations in tau and pTau‐T181 in ALS CSF, there is a significant decrease in the pTau‐T181:tau ratio, supporting previously published reports [16, 19, 44, 45]. Importantly, we reported a significant increase in tau levels in ALS CSF in patients diagnosed with bulbar onset ALS, suggesting that tau levels may increase in the CSF depending on the region of disease onset and, therefore, may serve as a biomarker for a subset of ALS patients. Additionally, increases in tau may be associated with more rapid progression, while decreases in pTau‐T181:tau ratio may be associated with slower progression. Therefore, CSF tau levels and pTau‐T181:tau ratio may serve as a predictive biomarker for ALS. It should be noted that decreases in CSF pTau‐T181:tau ratio may also result from increases in CSF total tau levels, thus indicating that CSF pTau‐T181 levels are reduced in relation to an overall increase in CSF tau. However, our findings in CSF align with post‐mortem findings, highlighting a potential pathogenic role for pTau‐T181 in ALS. This residue is located within the PRR of tau which plays a role in tau's tubulin and actin binding [50, 51] and more recently, liquid–liquid phase separation (LLPS), a mechanism that may link tau to ALS pathology, where aggregates form via LLPS [52, 53, 54]. Tau's PRR drives LLPS and does so under the control of its phosphorylation state, contributing to the formation of non‐filamentous pathogenic tau species [52, 53]. Therefore, it is possible that decreases in pTau‐T181 may be critical for these events given that this site is localized within the PRR [55]. Future studies will focus on determining the functional role of pTau‐T181 in ALS and whether it plays a role in LLPS.
One limitation of our study is that we used three different cohorts of ALS samples for biochemical and bioinformatics evaluation. Future studies will aim to assess pTau levels in post‐mortem mCTX and CSF from the same patients when possible. Ongoing efforts are concentrated on sequencing available post‐mortem mCTX to identify pathogenetic variants in MAPT. Furthermore, our results revealed heterogenous levels of tau phosphorylation in control mCTX likely due to other unknown causes including differences in age, as the control cases in this cohort were generally older than the ALS cases. In addition, our study focused on phosphorylation of three specific amino acid residues across tau protein, therefore, further investigation is required to determine the involvement of other tau phosphorylation sites, such as T175, whose involvement in ALS has been recently described [14].
Collectively, our results in a large cohort of human post‐mortem mCTX suggest that pTau‐S396 and total tau levels are increased in C9ORF72‐ALS, suggesting that alterations in tau may be due to altered protein homoeostasis in this ALS subtype. Furthermore, the identification of specific variants in MAPT in ALS suggests that those mutations may act as disease modifiers that may alter the onset and duration of disease. Our data also provide additional support for the use of pTau‐T181:tau ratio as a potential biomarker for ALS and highlight a potential role for pTau‐T181 in ALS pathogenesis, given that CSF levels reflect the significant decrease in pTau‐T181 across ALS.
CONFLICT OF INTEREST
T.G.I. serves as member of a Lilly Monitoring Committee (DMC). B.T.H. is a member of Novartis, Dewpoint, and Cell Signaling Scientific Advisory Board (SAB), and of Biogen DMC, and acts as consultant for US DoJ, Takeda, Virgil, W20, and Seer; he receives grants from Abbvie, F prime, NIH, Tau consortium, Cure Alzheimer's fund, Brightfocus, and JPB foundations. S.E.A. has received honoraria and/or travel expenses for lectures from Abbvie, Eisai, and Biogen and has served on SAB of Cortexyme and vTv, and as consultant for Athira, Cassava, Cognito Therapeutics, EIP Pharma and Orthogonal Neuroscience, and has received research grant support from NIH, Alzheimer's Association, Alzheimer's Drug Discovery Foundation, Abbvie, Amylyx, EIP Pharma, Merck, Janssen/Johnson & Johnson, Novartis, and vTv. T.S.J. is on the scientific advisory board of Cognition Therapeutics and receives collaborative grant funding from European Research Council, UK Dementia Research Institute, and Autifony. M.E.C. acts as consultant for Aclipse, Mt Pharma, Immunity Pharma Ltd., Orion, Anelixis, Cytokinetics, Biohaven, Wave, Takeda, Avexis, Revelasio, Pontifax, Biogen, Denali, Helixsmith, Sunovian, Disarm, ALS Pharma, RRD, Transposon, and Quralis, and as DSBM Chair for Lilly. J.D.B. has received personal fees from Biogen, Clene Nanomedicine, and MT Pharma Holdings of America, and grant support from Alexion, Biogen, MT Pharma of America, Anelixis Therapeutics, Brainstorm Cell Therapeutics, Genentech, nQ Medical, NINDS, Muscular Dystrophy Association, ALS One, Amylyx Therapeutics, ALS Association, and ALS Finding a Cure. G.S‐V. is a consultant for MarvelBiome. None of these had any influence over the current paper.
AUTHOR CONTRIBUTIONS
Tiziana Petrozziello contributed to the study design, data collection, data analysis, and drafting of the manuscript. Ana C. Amaral, Simon Dujardin, Sali M. K. Farhan, James Chan, Bianca A. Trombetta, Pia Kivisäkk, Alexandra N. Mills, Evan A. Bordt, Spencer E. Kim, Patrick M. Dooley, and Anubrata Ghosal contributed to the data collection, data analysis, and editing of the manuscript. Teresa Gomez‐Isla, Bradley T. Hyman, Steven E. Arnold, Tara Spires‐Jones, Merit E. Cudkowicz, James D. Berry contributed to the study design and editing of the manuscript. Ghazaleh Sadri‐Vakili contributed to the study design, data analysis, and drafting of the manuscript.
ETHICS APPROVAL
The study was approved by the Mass General Brigham Healthcare Institutional Review Board (IRB). Written informed consent was obtained from all participants prior to study enrollment. Post‐mortem consent was obtained from the appropriate representative (next of kin or health care proxy) prior to autopsy.
PATIENT CONSENT STATEMENT
Not applicable.
PERMISSION TO REPRODUCE MATERIAL FROM OTHER SOURCES
Not applicable.
Supporting information
Fig S1
FIGURE S1 Tau phosphorylation is not altered based on region of onset in C9ORF72‐ALS. There were no significant alterations in the levels of (A) pTau‐S396 (one‐way ANOVA [F(2,24) = 1.003], p = 0.3816), (B) pTau‐S404 (one‐way ANOVA [F(2,23) = 0.004261, p = 0.9957], (C) pTau‐T181 (one‐way ANOVA [F(2,24) = 0.2557, p = 0.7764], and (D) total tau (one‐way ANOVA [F(2,24) = 1.870, p = 0.1759] between controls (n = 21), bulbar onset ALS (n = 4), or limb onset ALS (n = 3) mCTX as measured by western blots
{kind=link}
Table S1‐S2
TABLE S1 MAPT variants in ALS
TABLE S2 Tau, pTau‐T181, and pTau‐T181:tau ratio levels in cerebrospinal fluid from ALS patients
ACKNOWLEDGMENTS
The authors thank the patients and their families for sample donations and the Veterans Affairs Biorepository Brain Bank for sample collection.
Petrozziello T, Amaral AC, Dujardin S, Farhan SMK, Chan J, Trombetta BA, et al. Novel genetic variants in MAPT and alterations in tau phosphorylation in amyotrophic lateral sclerosis post‐mortem motor cortex and cerebrospinal fluid. Brain Pathol. 2022;32:e13035. 10.1111/bpa.13035
Funding information
T.P. was supported by an award from the Judith and Jean Pape Adams Charitable Foundation and Byrne Family Endowed Fellowship in ALS Research. SD was supported by the Alzheimer's Association (2018‐AARF‐591935) and the Jack Satter Foundation. S.M.K.F. was supported by the ALS Canada Tim E. Noël Postdoctoral Fellowship. The Massachusetts Alzheimer's Disease Research Center is supported by the National Institute on Aging NIA (Grant P30AG062421)
DATA AVAILABILITY STATEMENT
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Brown RH Jr, Al‐Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:1602. [DOI] [PubMed] [Google Scholar]
- 2. Kim G, Gautier O, Tassoni‐Tsuchida E, Ma XR, Gitler AD. ALS genetics: gains, losses, and implications for future therapies. Neuron. 2020;108(5):822–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakamura S, Wate R, Kaneko S, Ito H, Oki M, Tsuge A, et al. An autopsy case of sporadic amyotrophic lateral sclerosis associated with the I113T SOD1 mutation. Neuropathology. 2014;34:58–63. [DOI] [PubMed] [Google Scholar]
- 4. Takeuchi R, Toyoshima Y, Tada M, Tanaka H, Shimizu H, Shiga A, et al. Globular glial mixed four repeat tau and TDP‐43 proteinopathy with motor neuron disease and frontotemporal dementia. Brain Pathol. 2016;26:82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bodakuntla S, Jijumon AS, Villablanca C, Gonzalez‐Billault C, Janke C, et al. Microtubule‐associated proteins: structuring the cytoskeleton. Trends Cell Biol. 2019;29:804–19. [DOI] [PubMed] [Google Scholar]
- 6. Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wolfe MS. The role of tau in neurodegenerative diseases and its potential as a therapeutic target. Scientifica (Cairo). 2012;2012:796024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–22. [DOI] [PubMed] [Google Scholar]
- 9. Pirscoveanu DFV, Pirici I, Tudorica V, Balseanu TA, Albu VC, Bondari S, et al. Tau protein in neurodegenerative diseases—a review. Rom J Morphol Embryol. 2017;58:1141–50. [PubMed] [Google Scholar]
- 10. Combs B, Mueller RL, Morfini G, Brady ST, Kanaan NM. Tau and axonal transport misregulation in tauopathies. Adv Exp Med Biol. 2019;1184:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gotz J, Halliday G, Nisbet RM. Molecular pathogenesis of the tauopathies. Annu Rev Pathol. 2019;14:239–61. [DOI] [PubMed] [Google Scholar]
- 12. Henstridge CM, Sideris DI, Carroll E, Rotariu S, Salomonsson S, Tzioras M, et al. Synapse loss in the prefrontal cortex is associated with cognitive decline in amyotrophic lateral sclerosis. Acta Neuropathol. 2018;135:213–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ayaki T, Ito H, Komure O, Kamada M, Nakamura M, Wate R, et al. Multiple proteinopathies in familial ALS cases with optineurin mutations. J Neuropathol Exp Neurol. 2018;77:128–38. [DOI] [PubMed] [Google Scholar]
- 14. Moszczynski AJ, Hintermayer MA, Strong MJ. Phosphorylation of threonine 175 tau in the induction of tau pathology in amyotrophic lateral sclerosis‐frontotemporal spectrum disorder (ALS‐FTSD). A review. Front Neurosci. 2018;12:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stevens CH, Guthrie NJ, van Roijen M, Halliday GM, Ooi L. Increased tau phosphorylation in motor neurons from clinically pure sporadic amyotrophic lateral sclerosis patients. J Neuropathol Exp Neurol. 2019;78:605–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grossman M, Elman L, McCluskey L, McMillan CT, Boller A, Powers J, et al. Phosphorylated tau as a candidate biomarker for amyotrophic lateral sclerosis. JAMA Neurol. 2014;71:442–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilke C, Deuschle C, Rattay TW, Maetzler W, Synofzik M. Total tau is increased, but phosphorylated tau not decreased, in cerebrospinal fluid in amyotrophic lateral sclerosis. Neurobiol Aging. 2015;36:1072–4. [DOI] [PubMed] [Google Scholar]
- 18. Bourbouli M, Rentzos M, Bougea A, Zouvelou V, Constantinides VC, Zaganas I, et al. Cerebrospinal fluid TAR DNA‐binding protein 43 combined with tau proteins as a candidate biomarker for amyotrophic lateral sclerosis and frontotemporal dementia spectrum disorders. Dement Geriatr Cogn Disord. 2017;44:144–52. [DOI] [PubMed] [Google Scholar]
- 19. Schreiber S, Spotorno N, Schreiber F, Acosta‐Cabronero J, Kaufmann J, Machts J, et al. Significance of CSF NfL and tau in ALS. J Neurol. 2018;265:2633–45. 10.1007/s00415-018-9043-0 [DOI] [PubMed] [Google Scholar]
- 20. Hardiman O, Al‐Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071. [DOI] [PubMed] [Google Scholar]
- 21. Kim HJ, Taylor JP. Lost in transportation: nucleocytoplasmic transport defects in als and other neurodegenerative diseases. Neuron. 2017;96:285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Petrozziello T, Bordt EA, Mills AN, Kim SE, Sapp E, Devlin BA, et al. Targeting tau mitigates mitochondrial fragmentation and oxidative stress in amyotrophic lateral sclerosis. Mol Neurobiol. 2021. 10.1101/2021.03.22.436505 [DOI] [PubMed] [Google Scholar]
- 23. Lister JP, Blatt GJ, DeBassio WA, Kemper TL, Tonkiss J, Galler JR, et al. Effect of prenatal protein malnutrition on numbers of neurons in the principal cell layers of the adult rat hippocampal formation. Hippocampus. 2005;15:393–403. [DOI] [PubMed] [Google Scholar]
- 24. Mueller KA, Glajch KE, Huizenga MN, Wilson RA, Granucci EJ, Dios AM, et al. Hippo signaling pathway dysregulation in human huntington's disease brain and neuronal stem cells. Sci Rep. 2018;8:11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tai H‐C, Serrano‐Pozo A, Hashimoto T, Frosch MP, Spires‐Jones TL, Hyman BT, et al. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin‐proteasome system. Am J Pathol. 2012;181:1426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petrozziello T, Mills AN, Vaine CA, Penney EB, Fernandez‐Cerado C, Legarda GPA, et al. Neuroinflammation and histone H3 citrullination are increased in X‐linked Dystonia Parkinsonism post‐mortem prefrontal cortex. Neurobiol Dis. 2020;144:105032. [DOI] [PubMed] [Google Scholar]
- 27. Petrozziello T, Mills AN, Farhan SMK, Mueller KA, Granucci EJ, Glajch KE, et al. Lipocalin‐2 is increased in amyotrophic lateral sclerosis. Muscle Nerve. 2020;62:272–83. [DOI] [PubMed] [Google Scholar]
- 28. Farhan SMK, Howrigan DP, Abbott LE, Klim JR, Topp SD, Byrnes AE, et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat‐shock protein. Nat Neurosci. 2019;22:1966–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van der Spek RAA, van Rheenen W, Pulit SL, Kenna KP, van den Berg LH, Veldink JH, et al. The project MinE databrowser: bringing large‐scale whole‐genome sequencing in ALS to researchers and the public. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kobayashi T, Ota S, Tanaka K, Ito Y, Hasegawa M, Umeda Y, et al. A novel L266V mutation of the tau gene causes frontotemporal dementia with a unique tau pathology. Ann Neurol. 2003;53:133–7. [DOI] [PubMed] [Google Scholar]
- 32. Zarranz JJ, Ferrer I, Lezcano E, Forcadas MI, Eizaguirre B, Atares B, et al. A novel mutation (K317M) in the MAPT gene causes FTDP and motor neuron disease. Neurology. 2005;64:1578–85. [DOI] [PubMed] [Google Scholar]
- 33. Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19:422–33. [DOI] [PubMed] [Google Scholar]
- 34. Lee JC, Kim SJ, Hong S, Kim Y. Diagnosis of Alzheimer's disease utilizing amyloid and tau as fluid biomarkers. Exp Mol Med. 2019;51:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parakh S, Atkin JD. Protein folding alterations in amyotrophic lateral sclerosis. Brain Res. 2016;1648:633–49. [DOI] [PubMed] [Google Scholar]
- 36. Blokhuis AM, Groen EJN, Koppers M, van den Berg LH, Pasterkamp RJ. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125:777–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bieniek KF, Murray ME, Rutherford NJ, Castanedes‐Casey M, DeJesus‐Hernandez M, Liesinger AM, et al. Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathol. 2013;125(2):289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Strong MJ, Donison NS, Volkening K. Alterations in tau metabolism in ALS and ALS‐FTSD. Front Neurol. 2020;11:598907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He H, Huang W, Wang R, Lin Y, Guo Y, Deng J, et al. Amyotrophic lateral sclerosis‐associated GGGGCC repeat expansion promotes tau phosphorylation and toxicity. Neurobiol Dis. 2019;130:104493. [DOI] [PubMed] [Google Scholar]
- 40. King A, Al‐Sarraj S, Troakes C, Smith BN, Maekawa S, Iovino M, et al. Mixed tau, TDP‐43 and p62 pathology in FTLD associated with a C9ORF72 repeat expansion and pAla239Thr MAPT (tau) variant. Acta Neuropathol. 2013;125(2):303–10. [DOI] [PubMed] [Google Scholar]
- 41. Ly CV, Koenig L, Christensen J, Gordon B, Beaumont H, Dahiya S, et al. Tau positron emission tomography imaging in C9orf72 repeat expansion carriers. Eur J Neurol. 2019;26(9):1235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shoji M. Cerebrospinal fluid and plasma tau as a biomarker for brain tauopathy. Adv Exp Med Biol. 2019;1184:393–405. [DOI] [PubMed] [Google Scholar]
- 43. Schraen‐Maschke S, Sergeant N, Dhaenens C‐M, Bombois S, Deramecourt V, Caillet‐Boudin M‐L, et al. Tau as a biomarker of neurodegenerative diseases. Biomark Med. 2008;2:363–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scarafino A, D’Errico E, Introna A, Fraddosio A, Distaso E, Tempesta I, et al. Diagnostic and prognostic power of CSF Tau in amyotrophic lateral sclerosis. J Neurol. 2018;265:2353–62. [DOI] [PubMed] [Google Scholar]
- 45. Lanznaster D, Hergesheimer RC, Bakkouche SE, Beltran S, Vourc'h P, Andres CR, et al. Abeta1‐42 and tau as potential biomarkers for diagnosis and prognosis of amyotrophic lateral sclerosis. Int J Mol Sci. 2020;21:2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abu‐Rumeileh S, Steinacker P, Polischi B, Mammana A, Bartoletti‐Stella A, Oeckl P, et al. CSF biomarkers of neuroinflammation in distinct forms and subtypes of neurodegenerative dementia. Alzheimers Res Ther. 2019;12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brettschneider J, Petzold A, Sussmuth SD, Ludolph AC, Tumani H. Axonal damage markers in cerebrospinal fluid are increased in ALS. Neurology. 2006;66:852–6. [DOI] [PubMed] [Google Scholar]
- 48. Paladino P, Valentino F, Piccoli T, Piccoli F, La Bella V. Cerebrospinal fluid tau protein is not a biological marker in amyotrophic lateral sclerosis. Eur J Neurol. 2009;16:257–61. [DOI] [PubMed] [Google Scholar]
- 49. Steinacker P, Feneberg E, Weishaupt J, Brettschneider J, Tumani H, Andersen PM, et al. Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry. 2016;87:12–20. [DOI] [PubMed] [Google Scholar]
- 50. He HJ, Wang XS, Pan RM, Wang DL, Liu MN, He RQ. The proline‐rich domain of tau plays a role in interactions with actin. BMC Cell Biology 2009;10:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McKibben KM, Rhoades E. Independent tubulin binding and polymerization by the proline‐rich region of tau is regulated by tau’s N‐terminal domain. J Biol Chem. 2019;294(50):19381–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kanaan NM, Hamel C, Grabinski T, Combs B. Liquid‐liquid phase separation induces pathogenic tau conformations in vitro. Nat Commun. 2020;11(1):2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang X, Vigers M, McCarty J, Rauch JN, Fredrickson GH, Wilson MZ, et al. The proline‐rich domain promotes tau liquid‐liquid phase separation in cells. J Cell Biol. 2020;219(11):e202006054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Watanabe S, Inami H, Oiwa K, Murata Y, Sakai S, Komine O, et al. Aggresome formation and liquid‐liquid phase separation independently induce cytoplasmic aggregation of TAR DNA‐binding protein 43. Cell Death Dis. 2020;11(10):909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pakravan D, Michiels E, Bratek‐Skicki A, De Decker M, Van Lindt J, Alsteens D, et al. Liquid‐liquid phase separation enhances TDP‐43 LCD aggregation but delays seeded aggregation. Biomolecules. 2021;11(4):548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
FIGURE S1 Tau phosphorylation is not altered based on region of onset in C9ORF72‐ALS. There were no significant alterations in the levels of (A) pTau‐S396 (one‐way ANOVA [F(2,24) = 1.003], p = 0.3816), (B) pTau‐S404 (one‐way ANOVA [F(2,23) = 0.004261, p = 0.9957], (C) pTau‐T181 (one‐way ANOVA [F(2,24) = 0.2557, p = 0.7764], and (D) total tau (one‐way ANOVA [F(2,24) = 1.870, p = 0.1759] between controls (n = 21), bulbar onset ALS (n = 4), or limb onset ALS (n = 3) mCTX as measured by western blots
Table S1‐S2
TABLE S1 MAPT variants in ALS
TABLE S2 Tau, pTau‐T181, and pTau‐T181:tau ratio levels in cerebrospinal fluid from ALS patients
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.