

Abstract
BACKGROUND:
Acute traumatic coagulopathy often accompanies traumatic brain injury (TBI) and may impair cognitive recovery. Antithrombin III (AT-III) reduces the hypercoagulability of TBI. Antithrombin III and heparinoids such as enoxaparin (ENX) demonstrate potent anti-inflammatory activity, reducing organ injury and modulating leukocyte (LEU) activation, independent of their anticoagulant effect. It is unknown what impact AT-III exerts on cerebral LEU activation and blood-brain barrier (BBB) permeability after TBI. We hypothesized that AT-III reduces live microcirculatory LEU-endothelial cell (EC) interactions and leakage at the BBB following TBI.
METHODS:
CD1 mice (n = 71) underwent either severe TBI (controlled cortical impact (CCI), 6-m/s velocity, 1-mm depth, and 4-mm diameter) or sham craniotomy and then received either AT-III (250 IU/kg), ENX (1.5 mg/kg), or vehicle (saline) every 24 hours. Forty-eight hours post-TBI, cerebral intravital microscopy visualized in vivo penumbral microvascular LEU-EC interactions and microvascular leakage to assess BBB inflammation/permeability. Body weight loss and the Garcia neurological test (motor, sensory, reflex, balance) served as surrogates of clinical recovery.
RESULTS:
Both AT-III and ENX similarly reduced in vivo penumbral LEU rolling and adhesion (p < 0.05). Antithrombin III also reduced live BBB leakage (p < 0.05). Antithrombin III animals demonstrated the least 48-hour body weight loss (8.4 ± 1%) versus controlled cortical impact and vehicle (11.4 ± 0.5%, p < 0.01). Garcia neurological test scores were similar among groups.
CONCLUSION:
Antithrombin III reduces post-TBI penumbral LEU-EC interactions in the BBB leading to reduced neuromicrovascular permeability. Antithrombin III further reduced body weight loss compared with no therapy. Further study is needed to determine if these AT-III effects on neuroinflammation affect longer-term neurocognitive recovery after TBI.
Keywords: Antithrombin III, traumatic brain injury, neutrophil, blood brain barrier, murine
Traumatic brain injury (TBI) is a principal source of mortality and a leading etiology of morbidity after injury in the developed world.1 There were approximately 2.87 million TBI-related emergency department visits, acute care hospitalizations, and deaths in the United States in 2014. Despite decades of intense laboratory and bedside research, effective neuroprotection after TBI remains elusive. Moreover, impacted individuals, overrepresented by young males, may require lifelong care.2,3 Better understanding of the pathophysiology of both primary and secondary brain injury may help guide development of neurotherapeutic agents.
Primary brain injury encompasses the immediate mechanical damage incurred upon the neurons, supporting glia and cerebral blood vessels as a result of the direct external blunt force trauma to the skull and the brain parenchyma. This is followed by secondary injury, a phase that can last up to weeks and is primarily driven by a cascade of neuroimmune responses believed to increase the initial scope of the injury thereby worsening neurological outcome through glial cell activation, leukocyte (LEU) recruitment, and upregulation of inflammatory mediators as well as excitotoxic and apoptotic processes.4,5
An early occurrence after primary injury is disruption of the blood-brain barrier (BBB). This key event enables interchange between the plasma space and injured cerebral parenchyma. Importantly, adhesion of circulating LEUs and trafficking of more remote inflammatory cells to areas of injury may worsen the primary tissue damage and promote leakage of plasma and circulating cells to the interstitium of the injured brain penumbra. Locally curated LEUs release inflammatory mediators that further mobilize glia and other immune cells to the site of injury and aggravate neuroinflammation.5,6
The influx of plasma and blood components combined with cytotoxic cell membrane dysfunction exacerbates local edema, increases intracranial pressure, and promotes local and global cerebral ischemia.7 We have demonstrated that certain anticoagulants possess anti-inflammatory properties that can reduce LEU-mediated cerebral inflammation and swelling. In particular, unfractionated or low–molecular weight heparins, such as enoxaparin (ENX), blunt post-TBI LEU interactions with the BBB and improve neurological recovery independent of their anticoagulant properties.8,9 Antithrombin III (AT-III), another anticoagulant plasma protein, protects against endothelial injury in animal models of sepsis, ischemia-reperfusion, and acute lung injury.10-13 Such effects are attributed to AT-III’s ability to reduce peri-injury neutrophil accumulation and local vascular permeability in a fashion similar to that of ENX.14 A putative role for AT-III in mitigating against blunt TBI induced cerebral microinflammation is therefore plausible and is reasonable to explore.
Accordingly, we hypothesized that AT-III administered after blunt TBI would reduce LEU-mediated cerebrovascular inflammation and decrease microvascular permeability, thereby reducing secondary brain injury and improving neurological recovery.
MATERIALS AND METHODS
Experimental Design and Study Groups
Procedures received approval from the University of Pennsylvania Institutional Animal Care and Use Committee. CD1 adult male mice (25–30 g) (Charles River Laboratories, Wilmington, MA) were acclimated in standard housing with ad libitum water and chow, for 5 days before experiments and immediately after recovery from brain injury. Mice underwent controlled cortical impact (CCI) or sham craniotomy and then received one of three agents: (1) subcutaneous ENX (1.5 mg/kg) (Winthrop; Sanofi-Aventis, Bridgewater, NJ), (2) intravenous (femoral vein) pharmaceutical-grade AT-III (250 IU/kg) (donated by Grifols S.A., Durham, NC), or (3) an equal volume of intravenous normal saline (0.9%, NS; Baxter Healthcare Corporation, Deerfield, IL). Doses were administered 0.5 and 24 hours after the CCI (Fig. 1). Antithrombin III dosing was determined with discussion with the manufacturer and previous reports,14,15 while ENX doses were equivalent to therapeutic doses in humans and reflected previous literature demonstrating beneficial effects in reducing brain inflammation after TBI.9
Figure 1.
Timeline of experimental procedures. NS, normal saline; wt, weight.
Seventy-one (71) mice were randomly assigned to one of six groups: (1) sham craniotomy and vehicle (VEH; 0.9% NS) (SHAM-VEH, n = 9), (2) sham craniotomy and ENX (SHAM-ENX, n = 8), (3) sham craniotomy and AT-III (SHAM–AT-III, n = 9), (4) TBI and vehicle (CCI-VEH, n = 19), (5) TBI and ENX (CCI-ENX, n = 13), and (6) TBI and AT-III (CCI–AT- III, n= 13).
Severe TBI Model
Controlled cortical impact was used to replicate TBI through a well-validated rodent model.16,17 Briefly, on day 1, mice were anesthetized with intraperitoneal ketamine (Hospira, Lake Forest, IL), xylazine (Akorn, Decatur, IL), and acepromazine (Boehringer Ingelheim, St. Joseph, MO) (KXA, 100, 10, and 1 mg/kg, respectively) and placed prone in a stereotactic device. After scalp exposure, a left-sided 4-mm craniotomy was created between bregma and lambda sutures using a dental drill (Henry Schein, Melville, NY) without violating the dura. The left parietotemporal cortex was then injured via a controlled cortical impactor (AMS201; AmScien Instruments, Richmond, VA), which resulted in a reproducible injury previously correlated to severe TBI (3-mm-diameter impactor tip, impact velocity of 6 m/s, and cortical deformation depth of 1 mm).
Pial Intravital Microscopy
In vivo assessment of cerebral microcirculation using pial intravital microscopy was performed 48 hours after CCI as described previously because this is the optimal time to observe peak LEU mobilization to the BBB penumbra after brain injury.16,18 Under KXA anesthesia, the right jugular vein was cannulated and a second 2.5-mm craniotomy was created immediately anterior to the first and covered with a 5-mm coverslip (Fisher Scientific, Waltham, MA). Secured in the stereotactic device, mice were transferred to an intravital microscope (ECLIPSE FN1; Nikon Instruments, Melville, NY) and received a 50-μL intrajugular injection of 0.3% rhodamine 6G (Sigma-Aldrich, St. Louis, MO) to fluorescently label circulating LEUs visualized at a 590-nm epiillumination emission exposure. A randomly selected area of nonbranching pial venules measuring 25 to 50 μm in diameter was selected for a 1-minute digital video recording (QuantEM camera; Photometrics, Tucson, AR). Intravenous bovine fluorescein isothiocyanate (FITC)–labeled albumin (100 mg/kg) (Sigma-Aldrich) was then administrated to visualize albumin leakage in same venule as a surrogate for microvascular permeability. Permeability was observed under a 488-nm fluorescent filter. A 10-second digital video recording was captured for offline assessment of microvascular permeability.
Offline Quantification of LEU-EC Interactions and Albumin Leakage
Video data captured at a 590-nm λ were imported into analysis software (NIS-Elements; Nikon Instruments, Melville, NY), and LEU–endothelial cell (EC) interactions were documented by a blinded observer quantifying the following parameters: (1) LEU transit, mean number of labeled LEU crossing a 100-μm-long venular segment in 60 seconds; (2) LEU adhesion, number of LEU stationary for at least 30 seconds during the recording period. Fluorescently labeled spherical cells measuring 7 to 12 μm were counted as LEU, and interactions were reported as the number of cells/100 μm per minute (Fig. 2).
Figure 2.
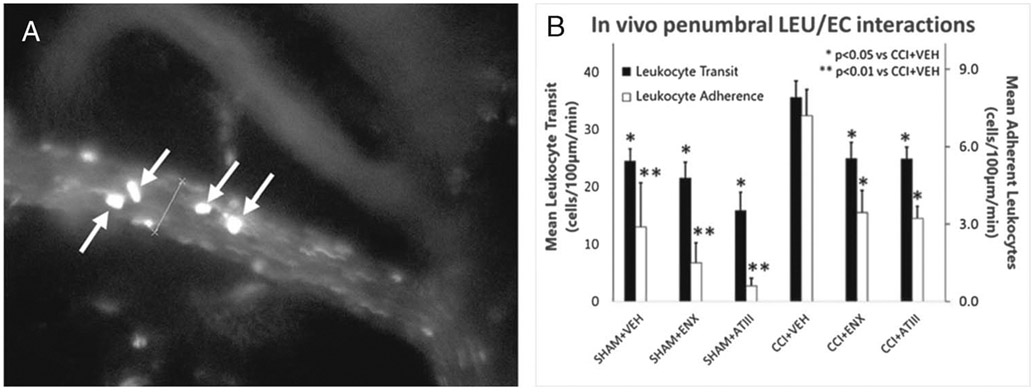
In vivo LEU/endothelial interactions. (A) Representative image showing in vivo LEUs interacting with pial microvascular endothelium. White arrows indicate fluorescently labeled LEU transiting on the endothelium. (B) Leukocyte transit and adherence in the pial penumbral microcirculation 48 hours after CCI. Compared with untreated injured animals (CCI-VEH), both AT-III and ENX reduced post-TBI LEU transit and adherence (*p < 0.05, **p < 0.01 vs. CCI-VEH).
Footage captured under the 488-nm filter was evaluated for fluorescence intensity from FITC-labeled albumin measured in three distinct regions within the vessel (venular intensity) and outside the vessel wall (perivenular intensity) (Fig. 3). The ratio of mean venular intensity to mean perivenular intensity was averaged to determine the permeability index for a given vessel indicating the degree of vessel macromolecular leakage.
Figure 3.

In vivo cerebral microvascular permeability. (A) Representative image of FITC-albumin leakage in the cerebral microcirculation. The microvascular permeability index equals the ratio of mean fluorescence of three separate locations outside the vessel wall (IP) tomean fluorescence of three separate locations within venule (IV). (B) Fluorescein isothiocyanate–albumin leakage in the pial penumbral microcirculation 48 hours after CCI. Compared with untreated injured animals (CCI-VEH), only AT-III reduced post-TBI cerebrovascular albumin leakage (*p < 0.05 vs. CCI-VEH). The box plot contains the median (−) and themean (X). IV, venular intensity;IP, perivenular intensity.
Body Weight Loss and Neurologic Recovery
Animal body weights were obtained just before (W0h) as well as 24 hours (W24h) and 48 hours (W48h) after CCI with weight loss expressed as a ratio ([W0h – W24h or W48h]/W0h × 100%). Murine neurologic function was assessed by the modified stroke Garcia neurological test (GNT) assessing motor, sensory, reflex, and balance ability at 24 hours and 48 hours after TBI; 18 points is the maximum achievable score.19
Brain Water Content
Forty-eight hours after CCI, mice were sacrificed and excised brains were separated into injured (ipsilateral) and uninjured (contralateral) hemispheres. Wet weight (WW) was immediately assessed. Dry weight (DW) was assessed after 72 hours of dehydration at 70°C. Tissue water content was calculated using wet-to-dry ratios (% water content = 100 × [WW × DW]/WW).
Statistical Analysis
All data are presented as mean or median (as marked) ± standard error of the mean. Statistical analyses were performed using SPSS (SPSS, Chicago, IL; 2019). Differences between group means were compared using the Kruskal-Wallis test; significance was assumed for p < 0.05.
RESULTS
In Vivo LEU-EC Interactions and Microvascular Permeability (48 Hours)
Forty-eight hours after CCI, injured animals demonstrated great numbers of LEU-EC interactions and microvascular leakage; uninjured control mice demonstrated the lowest levels of both parameters. Leukocyte transit in CCI-VEH mice (35.6 ± 2.9 LEUs/100 μm per minute) was reduced by administration of both ENX (24.9 ± 2.8 LEUs/100 μm per minute, p = 0.016) and AT-III (24.8 ± 2.1 LEUs/100 μm per minute, p = 0.026) (Fig. 2). Both treatments (ENX, AT-III) resulted in similar LEU transit levels. Specifically, LEU adhesion in CCI-VEH mice (7.2 ± 1 LEUs/100 μm per minute) was reduced by ENX (3.5 ± 0.8 LEUs/100 μm per minute, p = 0.03) and AT-III (3.2 ± 0.5 LEUs/100 μm per minute, p = 0.048).
Cerebrovascular FITC-albumin leakage (Fig. 3) was greater in CCI-VEH (52.8 ± 1.3%) compared with CCI–AT-III mice (41.9 ± 2.8%, p = 0.002) but similar to CCI-ENX mice (49.1 ±2.3%, p = 0.3).
Brain Water Content (48 Hours)
Uninjured animals receiving no treatment demonstrated less ipsilateral cerebral hemisphere edema compared with injured counterparts (Table 1). Treatment with AT-III or with ENX did not significantly reduce cerebral edema as compared with CCI-VEH (p > 0.05). Contralateral cerebral hemisphere edema was similar in all groups (p > 0.05).
TABLE 1.
Cerebral Hemisphere Water Content
| Sham-VEH | Sham-ENX | Sham–AT-III | CCI-VEH | CCI-ENX | CCI–AT-III | |
|---|---|---|---|---|---|---|
| Ipsilateral, mean, % | 76.64 | 77.4716 | 77.66745 | 78.51845 | 78.3674 | 77.64315 |
| Ipsilateral, median, % | 77.66 | 77.69902 | 77.68606 | 78.68589 | 79.02885 | 78.6192 |
| Ipsilateral, SEM | 0.74 | 0.184223 | 0.224232 | 0.215833 | 0.546356 | 1.10583 |
| Contralateral, mean, % | 76.65 | 77.5164 | 77.77869 | 77.44365 | 77.26 | 75.11578 |
| Contralateral, median, % | 77.47 | 77.67654 | 77.83964 | 77.56415 | 77.37609 | 77.69782 |
| Contralateral, SEM | 0.65 | 0.173227 | 0.195306 | 0.184909 | 0.403147 | 2.104034 |
| Ratio (ipsi/contra) | 0.999 | 0.999 | 0.998 | 1.014 | 1.014 | 1.033 |
Contra, contralateral; ipsi, ipsilateral; SEM, standard error of the mean.
Body Weight Loss and Neurologic Function (24 and 48 Hours)
Body weight loss was greatest 48 hours after TBI particularly in injured untreated animals (CCI-VEH, 11.4 ± 0.5%) (Fig. 4A). Among injured animals, only AT-III treatment reduced weight loss (8.4% ± 1%, p < 0.01) observed in CCI-VEH animals. All uninjured groups (SHAM) also demonstrated less body weight loss than CCI-VEH animals (p < 0.01).
Figure 4.
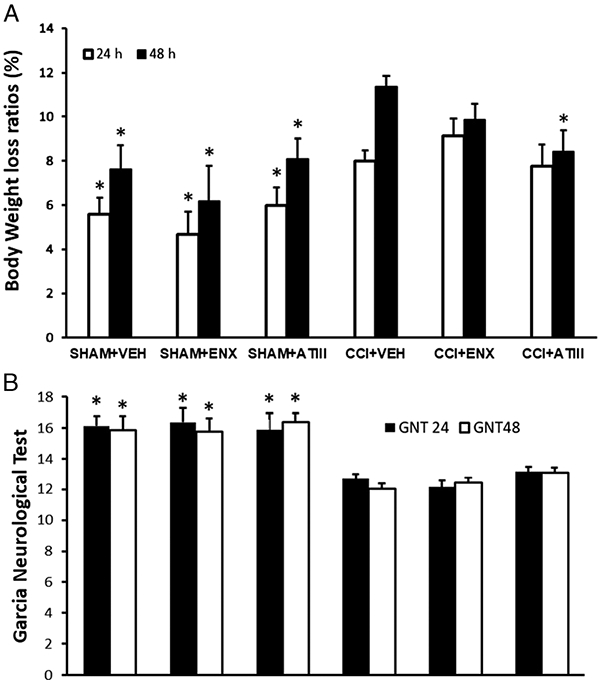
Body weight and GNT scores. (A) Body weight assessment: all the groups scored similarly at 24 hours, but at 48 hours, AT-III treatment significantly reduced weight loss as compared with CCI-VEH (*p < 0.01 vs. CCI-VEH for given time frame). The mean initial body weight of each group was as follows: SHAM-VEH, 33.4 ± 0.7 g; SHAM-ENX, 34.2 ± 0.6 g; SHAM–AT-III, 32.1 ± 1.2 g; CCI-VEH, 31.5 ± 0.8 g; CCI-ENX, 33.0 ± 0.5 g; and CCI–AT-III, 30.9 ± 1.3 g. (B) Garcia neurological test score: all uninjured animals scored significantly better than injured counterparts (*p < 0.01 vs. CCI-VEH for given time frame).Neither AT-III nor ENX treatment injured groups were significantly different from the injured untreated group. Garcia neurological test score: all negative controls were significantly better than the positive control (*p < 0.01 vs. CCI-VEH). Neither AT-III nor ENX treatment groups were significantly different from the positive control group.
The stroke functional recovery score (GNT) was highest in uninjured animals compared with all other groups (Fig. 4B). All injured mice demonstrated similar 24- or 48-hour post-TBI GNT scores (untreated, 12 ± 0.3; ENX, 12.4 ± 0.4; AT-III, 13.1 ± 0.3; p > 0.05) with highest scores in the AT-III but failing to reach significance.
DISCUSSION
In the present study, we provide what we believe is the first in vivo evidence of the ability of AT-III to reduce LEU activation in the injured pericontusional neurovasculature, blocking its interactions with the blood brain barrier. We further demonstrate how in the same pial vessels AT-III preserves BBB integrity, decreasing macromolecular leakage by more than 25%. Antithrombin III animals appeared to clinically benefit, with reduced weight loss.
The economic and human burden of TBI is enormous, yet few interventions discovered in the last decades have led to significant curbing of TBI-related mortality. Key to finding a beneficial therapy is the deeper understanding of the underpinnings of brain injury pathophysiology. Traumatic brain injury can be described as having two separate but interconnected injury components: a primary injury, which occurs upon external impact to the skull and immediately causes mechanical damage to neurons, axons, glia and cerebral blood vessels, and a delayed secondary injury, which lasts for the subsequent hours and days and which is primarily driven by the host response to tissue injury. The latter is characterized by an internal subsequent cascade of neuroimmune mechanisms believed to cause further tissue damage, which can, in some cases, ultimately lead to brain herniation and death.4
Local penumbral activation of the innate immune response and, specifically, circulating LEU and neurovascular endothelium is believed to initiate and sustain the initial BBB disruption, increasing microvascular permeability and leading to worsening cerebral edema.20-22 Activated LEUs marginate out of laminar circulatory flow interacting with and adhering to concurrently activated ECs and resulting in transmigration through the microvasculature into the brain parenchyma.23 These LEUs are mostly polymorphonuclear neutrophils (PMNs) but also include macrophages and T lymphocytes.24 Local PMN accumulation increases in the first 24 hours after injury and peaks at 48 to 72 hours when critical loss of BBB integrity is observed.7 Specifically, peripheral LEU infiltration into brain tissue activates resident immune cells such as microglia and astrocytes, which has been linked to an imbalance between local concentrations of proinflammatory and anti-inflammatory cytokines.22,25 Locally activated LEUs release metalloproteinases, proteases, reactive oxygen species, tumor necrosis factor α, interleukin 1-ß, and interleukin 626 within hours from injury further aggravating BBB disruption.4,27 Tissue LEU infiltration is further facilitated by a sustained endotheliopathy and upregulation of endothelial surface adhesion molecules such as E-selectin, intercellular adhesion molecules 1, and vascular adhesion molecules 1.28 In normal conditions, this process has a physiologic purpose to destroy and dispose of injured tissue and debris, ultimately allowing astrocytes to produce microfilaments to synthesize scar tissue.29 However, with severe cerebral tissue disruption, this process becomes dysregulated and injurious to the host. There is a close relationship between the magnitude of posttraumatic leukocytic mobilization and the extent of brain edema and brain tissue loss in secondary brain injury.30
Antithrombin III is a plasma glycoprotein synthesized in the liver and belonging to the family of serine protease inhibitors (serpins).31 It is the predominant, naturally occurring inhibitor of coagulation, which mainly, but not exclusively, targets activated factor II (thrombin) and factor Xa. The clinical use of AT-III is currently limited to hereditary or acquired AT-III deficiency, which is associated with inadequate anticoagulation and increased risk for thromboembolic events.32 Independent of AT-III’s anticoagulant activity, various studies have demonstrated evidence of potent anti-inflammatory effects, particularly when AT-III plasma activity ranges from 150% ± 200% of its normal plasma levels.14,33,34 These anti-inflammatory effects appear to be mediated through interactions between AT-III and endothelium,35 producing a profound increase in endothelial production of prostacyclin,36 which blocks PMN adhesion to ECs.37 Antithrombin III has been shown to inhibit LEU rolling and adhesion to EC in a feline intestinal mesentery ischemia-reperfusion model.38 In a similar model, Nevière et al.39 used small intestine intravital videomicroscopy to also demonstrate blunted LEU-EC adhesion by AT-III and reduced intestinal injury after endotoxemia. The authors additionally showed how the addition of indomethacin, a prostacyclin inhibitor, reversed the blockade of LEU-EC interactions observed with AT-III.40 Antithrombin III also binds to other surface LEU receptors (i.e., syndecan-4) on neutrophils, monocytes, and lymphocytes also directly inhibiting their interaction with ECs.13,41
Enoxaparin, a low–molecular weight heparin has been shown to reduce brain edema, lesion volume, and neurological impairment, specifically blocking LEU-EC interactions in the BBB and reducing tissue edema while enhancing neurological recovery.42 Our group previously investigated how this effect could be mediated through high mobility group box 1 (HMGB1) protein inhibition.9 In the current experiments, both ENX and AT-III blunted in vivo LEU-EC interactions and pericontusional LEU mobilization, but only AT-III reduced local microvascular permeability. The ENX administration in the current study was at lower doses and wider intervals, which may explain why it failed to also reduce in vivo permeability. These data thus suggest that, similar to heparins, early administration of AT-III after TBI may block LEU activation at the injured BBB and mitigate against worsening cerebral swelling and secondary brain injury. Antithrombin III has been associated with reductions in tissue edema and microvascular permeability in other studies as well. For example, in animal lung injury models, separate authors have demonstrated how AT-III attenuated vascular leakage via inhibiting neutrophil activation, also proposing an effect mediated by high mobility group box 1 protein inhibition.43 Relatedly, AT-III has been shown to reduce spinal cord edema and improve incomplete spinal cord injury recovery through facilitation of the spinal cord-evoked potentials and motor functions.44
To our knowledge, the current study is the first report of AT-III modulation of LEU-EC interactions in the cerebral microcirculation following TBI. In agreement with previous reports, our study linked live reductions in pericontusional LEU-EC interactions and reduced microvascular leakiness. Nonetheless, the current report has important limitations. First, it was conducted in mice, and there is no assurance that the pathophysiology of TBI recovery or modulation by AT-III is directly translatable to mechanisms involved in human TBI. Second, only one dose of 250 IU/kg of AT-III was administered after injury based on multiple reports of AT-III in other disease processes and on the manufacturer’s recommendations.14,43, 44 A dose-response curve and measurement of serum AT-III activity in both ENX and AT-III animals but also untreated animals would have been optimal. Nonetheless, this first pilot dosage in TBI will serve to inform future dose/frequency clarifications in brain injury. Third, despite observing less weight loss in injured animals treated with AT-III, their GNT evaluation at 24 and 48 hours tended to demonstrate a trend of improved scores, but this failed to reach significance. Perhaps this reflects a small sample size or could be explained by the GNT scale not being the optimal clinical tool for rodent TBI recovery because it was designed primarily for stroke models.19 Alternatively, our 48-hour animal observation period was insufficiently long to manifest small differences between groups, and a longer observation of several days to weeks would have revealed such differences. The 48-hour time frame was nonetheless necessary to conduct the terminal surgery of intravital microscopy, which needs to occur 2 days after injury to capture the optimal time of LEU mobilization to the penumbra. Finally, AT-III and ENX are anticoagulants and may have caused unobserved bleeding. Although our previous study9 did not grossly demonstrate greater cerebral hematoma size with ENX, this was not similarly confirmed in the current with AT-III. Furthermore, the administration of any anticoagulant within 30 minutes of TBI would not follow in any standard of care algorithm in humans. In our model, worsening cerebral hemorrhage could have affected animal neurological recovery and ultimately canceled any benefit on cerebral swelling, potentially explaining the lack of GNT score improvement by AT-III.
CONCLUSIONS
In conclusion, the current study demonstrates novel findings indicating that early post-TBI administration of AT-III reduces in vivo penumbral recruitment of LEUs and local microvascular permeability, resulting in greater animal weight recovery. Additional exploration of AT-III safety and efficacy in brain-injured large animals and eventually humans will be necessary to determine if such AT-III preparations can improve outcomes after TBI.
ACKNOWLEDGMENTS
We thank Ms. Robin Armstrong for her technical and organizational assistance.
Footnotes
DISCLOSURE
The authors declare no conflicts of interests. This work was supported by an investigator-sponsored research grant from GRIFOLS International.
This study was presented at the 79th Annual Meeting of AAST and Clinical Congress of Acute Care Surgery, September 9–12, 2020, in Waikoloa, Hawaii.
Contributor Information
Mohamed ElSaadani, Division of Traumatology, Surgical Critical Care and Emergency Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania..
Syed M. Ahmed, Division of Traumatology, Surgical Critical Care and Emergency Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania..
Christina Jacovides, Division of Traumatology, Surgical Critical Care and Emergency Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania..
Alfonso Lopez, Division of Traumatology, Surgical Critical Care and Emergency Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania..
Victoria E. Johnson, Department of Neurosurgery, Center for Brain Injury and Repair, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania..
Lewis J. Kaplan, Division of Traumatology, Surgical Critical Care and Emergency Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania..
C. William Schwab, Division of Traumatology, Surgical Critical Care and Emergency Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania..
Douglas H. Smith, Department of Neurosurgery, Center for Brain Injury and Repair, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania..
Jose L. Pascual, Division of Traumatology, Surgical Critical Care and Emergency Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania.; Department of Neurosurgery, Center for Brain Injury and Repair, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania..
REFERENCES
- 1.Gardner AJ, Zafonte R. Neuroepidemiology of traumatic brain injury. In: Rosano C, Ikram MA, Ganguli M, eds. Handbook of Clinical Neurology, Vol. 138: Neuroepidemiology. New York, NY: Elsevier; 2016. [DOI] [PubMed] [Google Scholar]
- 2.Campbell-Sills L, Stein MB, Liu H, Agtarap S, Heeringa SG, Nock MK, Ursano RJ, Kessler RC. Associations of lifetime traumatic brain injury characteristics with prospective suicide attempt among deployed US army soldiers. J Head Trauma Rehabil. 2020;35:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harrison-Felix C, Whiteneck G, DeVivo M, Hammond FM, Jha A. Mortality following rehabilitation in the traumatic brain injury model Systems of Care. NeuroRehabilitation. 2004;19(1):45–54. [PubMed] [Google Scholar]
- 4.Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99(1):4–9. [DOI] [PubMed] [Google Scholar]
- 5.Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16(3):165–177. [DOI] [PubMed] [Google Scholar]
- 6.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26(8):1191–1201. [DOI] [PubMed] [Google Scholar]
- 7.Morganti-Kossmann MC, Semple BD, Hellewell SC, Bye N, Ziebell JM. The complexity of neuroinflammation consequent to traumatic brain injury: from research evidence to potential treatments. Acta Neuropathol. 2019;137(5):731–755. [DOI] [PubMed] [Google Scholar]
- 8.Nagata K, Kumasaka K, Browne KD, Li S, St-Pierre J, Cognetti J, Marks J, Johnson VE, Smith DH, Pascual JL. Unfractionated heparin after TBI reduces in vivo cerebrovascular inflammation, brain edema and accelerates cognitive recovery. J Trauma Acute Care Surg. 2016;81(6):1088–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li S, Marks JA, Eisenstadt R, et al. Enoxaparin ameliorates post-traumatic brain injury edema and neurologic recovery, reducing cerebral leukocyte endothelial interactions and vessel permeability in vivo. J Trauma Acute Care Surg. 2015;79(1):78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maeda A, Ohta K, Ohta K, Nakayama Y, Hashida Y, Toma T, Saito T, Maruhashi K, Yachie A. Effects of antithrombin III treatment in vascular injury model of mice. Pediatr Int. 2011;53(5):747–753. [DOI] [PubMed] [Google Scholar]
- 11.Rehberg S, Yamamoto Y, Sousse LE, Jonkam C, Zhu Y, Traber LD, Cox RA, Prough DS, Traber DL, Enkhbaatar P. Antithrombin attenuates vascular leakage via inhibiting neutrophil activation in acute lung injury. Crit Care Med. 2013;41(12):e439–e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iba T, Levy JH, Hirota T, Hiki M, Sato K, Murakami T, Nagaoka I. Protection of the endothelial glycocalyx by antithrombin in an endotoxin-induced rat model of sepsis. Thromb Res. 2018;171:1–6. [DOI] [PubMed] [Google Scholar]
- 13.Mizutani A, Okajima K, Uchiba M, Isobe H, Harada N, Mizutani S, Noguchi T. Antithrombin reduces ischemia/reperfusion-induced renal injury in rats by inhibiting leukocyte activation through promotion of prostacyclin production. Blood. 2003;101(8):3029–3036. [DOI] [PubMed] [Google Scholar]
- 14.Uchiba M, Okajima K, Murakami K. Effects of various doses of antithrombin III on endotoxin-induced endothelial cell injury and coagulation abnormalities in rats. Thromb Res. 1998;89(5):233–241. [DOI] [PubMed] [Google Scholar]
- 15.Uchiba M, Okajima K. Antithrombin III (AT III) prevents LPS-induced pulmonary vascular injury: novel biological activity of AT III. Semin Thromb Hemost. 1997;23(6):583–590. [DOI] [PubMed] [Google Scholar]
- 16.Pascual JL, Murcy MA, Li S, et al. Neuroprotective effects of progesterone in traumatic brain injury: blunted in vivo neutrophil activation at the blood-brain barrier. Am J Surg. 2013;206(6):840–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, Mcintosh TK. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma. 1995;12(2):169–178. [DOI] [PubMed] [Google Scholar]
- 18.Marks JA, Li S, Gong W, Sanati P, Eisenstadt R, Sims C, Smith DH, Reilly PM, Pascual JL. Similar effects of hypertonic saline and mannitol on the inflammation of the blood-brain barrier microcirculation after brain injury in a mouse model. J Trauma Acute Care Surg. 2012;73(2):351–357. [DOI] [PubMed] [Google Scholar]
- 19.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats: statistical validation. Stroke. 1995;26(4):627–635. [DOI] [PubMed] [Google Scholar]
- 20.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6(7):393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukaszewicz AC, Soyer B, Payen D. Water, water, everywhere: sodium and water balance and the injured brain. Curr Opin Anaesthesiol. 2011;24(2):138–143. [DOI] [PubMed] [Google Scholar]
- 22.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147 Suppl 1(Suppl 1):S232–S240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Leden RE, Parker KN, Bates AA, Noble-Haeusslein LJ, Donovan MH. The emerging role of neutrophils as modifiers of recovery after traumatic injury to the developing brain. Exp Neurol. 2019;317:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, Artelt M, Burnet M, Trautmann K, Schluesener HJ. Early infiltration of CD8+ macrophages/microglia to lesions of rat traumatic brain injury. Neuroscience. 2006;141(2):637–644. [DOI] [PubMed] [Google Scholar]
- 25.Kubes P, Ward PA. Leukocyte recruitment and the acute inflammatory response. Brain Pathol. 2000;10(1):127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DiStasi MR, Ley K. Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends Immunol. 2009;30(11):547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen HX, O’Barr TJ, Anderson AJ. Polymorphonuclear leukocytes promote neurotoxicity through release of matrix metalloproteinases, reactive oxygen species, and TNF-α. J Neurochem. 2007;102(3):900–912. [DOI] [PubMed] [Google Scholar]
- 28.Liu YW, Li S, Dai SS. Neutrophils in traumatic brain injury (TBI): friend or foe? J Neuroinflammation. 2018;15(1):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ, Lauritzen M. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129(3):778–790. [DOI] [PubMed] [Google Scholar]
- 30.Carlos TM, Clark RS, Franicola-Higgins D, Schiding JK, Kochanek PM. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J Leukoc Biol. 1997;61(3):279–285. [DOI] [PubMed] [Google Scholar]
- 31.Bucur SZ, Levy JH, Despotis GJ, Spiess BD, Hillyer CD. Uses of antithrombin III concentrate in congenital and acquired deficiency states. Transfusion. 1998;38(5):481–98. [DOI] [PubMed] [Google Scholar]
- 32.Allingstrup M, Wetterslev J, Ravn FB, Møller AM, Afshari A. Antithrombin III for critically ill patients. Cochrane Database Syst Rev. 2016;2(2):CD005370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uchiba M, Okajima K, Murakami K, Okabe H, Takatsuki K. Attenuation of endotoxin-induced pulmonary vascular injury by antithrombin III. Am J Physiol. 1996;270(6 Pt 1):L921–L930. [DOI] [PubMed] [Google Scholar]
- 34.Wiedermann CJ, Römisch J. The anti-inflammatory actions of antithrombin —a review. Acta Med Austriaca. 2002;29(3):89–92. [DOI] [PubMed] [Google Scholar]
- 35.Hoffmann JN, Vollmar B, Römisch J, Inthorn D, Schildberg FW, Menger MD. Antithrombin effects on endotoxin-induced microcirculatory disorders are mediated mainly by its interaction with microvascular endothelium. Crit Care Med. 2002;30(1):218–225. [DOI] [PubMed] [Google Scholar]
- 36.Yamauchi T, Umeda F, Inoguchi T, Nawata H. Antithrombin III stimulates prostacyclin production by cultured aortic endothelial cells. Biochem Biophys Res Commun. 1989;163(3):1404–1411. [DOI] [PubMed] [Google Scholar]
- 37.Lindemann S, Gierer C, Darius H. Prostacyclin inhibits adhesion of polymorphonuclear leukocytes to human vascular endothelial cells due to adhesion molecule independent regulatory mechanisms. Basic Res Cardiol. 2003;98(1):8–15. [DOI] [PubMed] [Google Scholar]
- 38.Ostrovsky L, Woodman RC, Payne D, Teoh D, Kubes P. Antithrombin III prevents and rapidly reverses leukocyte recruitment in ischemia/reperfusion. Circulation. 1997;96(7):2302–2310. [DOI] [PubMed] [Google Scholar]
- 39.Nevière R, Tournoys A, Mordon S, Maréchal X, Song FL, Jourdain M, Fourrier F. Antithrombin reduces mesenteric venular leukocyte interactions and small intestine injury in endotoxemic rats. Shock. 2001;15(3):220–225. [DOI] [PubMed] [Google Scholar]
- 40.Hoffmann JN, Vollmar B, Inthorn D, Schildberg FW, Menger MD. Antithrombin reduces leukocyte adhesion during chronic endotoxemia by modulation of the cyclooxygenase pathway. Am J Physiol Cell Physiol. 2000;279(1):C98–C107. [DOI] [PubMed] [Google Scholar]
- 41.Horie S, Ishii H, Kazama M. Heparin-like glycosaminoglycan is a receptor for antithrombin III-dependent but not for thrombin-dependent prostacyclin production in human endothelial cells. Thromb Res. 1990;59(6):895–904. [DOI] [PubMed] [Google Scholar]
- 42.Wahl F, Grosjean-Piot O, Bareyre F, Uzan A, Stutzmann JM. Enoxaparin reduces brain edema, cerebral lesions, and improves motor and cognitive impairments induced by a traumatic brain injury in rats. J Neurotrauma. 2000;17(11):1055–1065. [DOI] [PubMed] [Google Scholar]
- 43.Hagiwara S, Iwasaka H, Matsumoto S, Noguchi T. High dose antithrombin III inhibits HMGB1 and improves endotoxin-induced acute lung injury in rats. Intensive Care Med. 2008;34(2):361–367. [DOI] [PubMed] [Google Scholar]
- 44.Arai M, Goto T, Seichi A, Nakamura K. Effects of antithrombin III on spinal cord-evoked potentials and functional recovery after spinal cord injury in rats. Spine. 2004;29(4):405–412. [DOI] [PubMed] [Google Scholar]