

Abstract
Heart failure (HF) is a leading cause of mortality worldwide. The pathogenesis of HF is complex and has not yet been fully elucidated, which has slowed drug development and long-term treatments. Inflammasome-mediated responses occur during the progression of HF. It has been reported that energy metabolism and metabolites of intestinal flora are also involved in the process of HF, and they interact with each other to promote the progression of HF. NLR family pyrin domain containing 3 (NLRP3) inflammasome may be a key target in the relationship between inflammation-mediated energy metabolism and metabolites of intestinal flora. Elucidating the relationship among the above three factors may help to identify new molecular targets for the prevention and treatment of HF and ultimately affect the course of HF. In this study, we systematically summarize evidence regarding the relationship among NLRP3 inflammasome, energy metabolism, intestinal microflora metabolites, and inflammation, as well as highlight advantages of NLRP3 inflammasome in treating HF.
Keywords: heart failure, inflammation, NLRP3, metabolism, flora
INTRODUCTION
Heart failure (HF) is the end stage of a variety of heart diseases, which has the characteristics of high hospitalization rate and high mortality. Although the treatment of HF has made great progress, the re-hospitalization and mortality rates are still high. Therefore, there is an urgent need to identify new targets for delaying HF (1).
At present, the pathogenesis of HF has been demonstrated to include hemodynamic abnormalities, ventricular remodeling, nervous system activation, fibrosis, and inflammasome responses. However, the relationship among different pathogenic components have not been fully characterized, and most of the current treatment programs are not comprehensive in ameliorating all of these components. Patients with HF often have energy metabolism disorder, such that the reaction substrate changes from free fatty acids to glucose, which further aggravates the process of HF (2, 3). Energy metabolism is closely related to inflammasome reactions. Patients with HF often have intestinal congestion, higher permeability of the intestinal wall, and unstable composition and content of intestinal microflora (4), which can promote the release of inflammasome factors, induce inflammasome reactions (5), and ultimately accelerate the development of HF (6, 7).
Taken together, HF often consists of energy metiabolism disorder and intestinal microflora metabolite disorder, accompanied by inflammasome activation, namely NLR family pyrin domain containing 3 (NLRP3) inflammasome. Therefore, it is speculated that inhibition of NLRP3 inflammasome can prevent and treat HF through modulating inflammation-related energy metabolism and intestinal microflora metabolites.
Heart failure consists of energy metabolism disorder and intestinal flora metabolite disorder
Energy metabolism disorder in heart failure
Changes in energy metabolism pathways in HF mainly involve the metabolic disorder of myocardial lipids and glucose as well as other substances, which eventually leads to ventricular remodeling. Some researchers have referred to this metabolic abnormality as myocardial metabolic remodeling (8). Ventricular remodeling is the main pathological basis of HF. Myocardial metabolic remodeling occurs earlier than ventricular remodeling, which will accelerate the process of ventricular remodeling. Therefore, the effect of improving myocardial metabolism remodeling is better than that of single-intervention ventricular remodeling (9). Wenjing et al. (10) confirmed that the metabolic remodeling of myocardial substrate in the early stage of HF is an adaptive response, and the late stage will cause the disorder of internal environment and accelerate the process of HF. Improving myocardial substrate metabolism can better protect and improve myocardial function and provide new targets and strategies for the treatment of heart failure. Noordali et al. (11) confirmed that there were significant changes in cardiac energy metabolism in patients with HF, and severe metabolic imbalance was considered to be an overall feature of HF. Therefore, we believe that energy metabolism disorder runs through HF.
The energy metabolism disorder implicated in HF involves myocardial metabolism reconstruction. With the development of HF, a decrease in capillary density decreases oxygen supply to the heart, and an increase in the load before and after the heart in the circulation intensifies myocardial energy shortage. To meet the increased energy demand in HF, the metabolic substrate of the myocardium is transformed from fatty acids to glucose (12). For the same oxygen consumption, glucose oxidation produces more ATP than does fatty acid oxidation (13). However, at the end of HF, severe hypoxia leads to a decline in insulin resistance and mitochondrial oxidation capacity. Pyruvate enters tricarboxylic acid cycle (TCA) and is reduced to lactate, which leads to severe reduction of ATP production. Meanwhile, a decrease in fatty acid β oxidation by cardiomyocytes, increase of fatty acids in the myocardium, and accumulation of lipotoxic substances, such as ceramide, aggravate the progression of HF (14). Moreover, evidence shows that energy metabolism disorder occurs in chronic HF and is accompanied by inflammation (15).
Intestinal flora metabolite disorder in HF
In recent years, it has been confirmed that cardiovascular disease (CVD) is closely related to dysfunction of intestinal microbiota. The metabolites of intestinal flora directly and/or indirectly affect changes in cardiac energy metabolism in HF. At present, the following four metabolites are most implicated; trimetlylamine oxide (TMAO), short-chain fatty acids (SCFAs), bile acid, and endotoxins.
TMAO is a key metabolite of intestinal flora and is produced by dietary precursors containing TMA, such as phosphatidylcholine, choline, and L-carnitine. Clinical research shows that increased plasma TMAO is related to diastolic dysfunction, indicating that TMAO may affect myocardial tissue mechanics (16); this also confirms that plasma TMAO levels are more effective than are BNP levels in predicting the prognosis of HF and the rate of re-hospitalization (17).
SCFAs are another key metabolite of intestinal flora. The gut microbiota in the distal intestine promote fermentation, which usually produces SCFAs, which are the main nutrient source of the cecum and colon epithelium (18). SCFAs not only maintain stability of the intestinal barrier, but also regulate the immune system and play a defensive function against the invasion of pathogenic bacteria and harmful substances. In addition, results have shown that SCFAs can improve the prognosis of HF by inhibiting ventricular remodeling (19, 20).
Bile acid is also a key metabolite of intestinal flora. Bile acids are traditionally considered to act as emulsifiers for the gut to absorb fat and fat-soluble vitamins. Primary bile acids are usually synthesized in the liver (21). These major bile acids are usually reabsorbed (>95%). Intestinal microflora can further metabolize bile acids that have not been reabsorbed and produce secondary bile acids (22, 23). Relevant clinical studies have shown that the ratio of primary bile acid to secondary bile acid is decreased in patients with HF (24).
Endotoxins, also known as lipopolysaccharides (LPS), represent typical pathogen-related molecules, and mainly signal through toll-like receptor 4 (TLR4) to induce the expression of downstream inflammasome products (25). This effect occurs in cardiomyocytes and cardiac fibroblasts (26). There is also evidence that bacterial translocation increases during HF owing to one or more mechanisms, including changes in gastrointestinal structure and function caused by visceral congestion and host immune defense abnormalities (27). Furthermore, intestinal microbiome dysfunction, especially metabolite dysfunction, also accelerates the process of HF (28).
INTERACTION BETWEEN ENERGY METABOLISM DISORDER AND INTESTINAL METABOLITE DISORDER IN HEART FAILURE
Energy metabolism disorder induces dysfunction of intestinal flora and related metabolites
The pathogenesis of HF is complex and is accompanied by energy metabolism disorder, which causes different degrees of intestinal barrier dysfunction (25). Fatty acids can have antibacterial action or can be used as metabolic substrates by intestinal bacteria, which can affect the intestinal flora and its metabolites. The composition of intestinal flora in mice on a low-fat diet is different from that of other groups, indicating that the metabolism of fatty acids in vivo affects the composition of intestinal flora in mice (29). Studies have shown that a high-fat diet can increase intestinal permeability and induce barrier dysfunction by changing bile acid concentration (30). It can also induce intestinal flora disorder (31). Changes in microorganisms induced by a high-sugar diet, including a decrease in diversity, increase in Proteus bacilli, and a decrease in pseudobacilli have common characteristics with microbial group disorders related to metabolic diseases, inflammatory bowel disease, and other human diseases (32). Therefore, energy metabolism disorder in HF leads to dysfunction of intestinal flora and related metabolites (33).
Energy metabolism disorder caused by intestinal metabolite disorder
The main metabolite disorders in the intestine consist of dysfunction of TMAO, SCFAs, and bile acids. An increased TMAO concentration will damage the oxidation of pyruvate and fatty acids in myocardial mitochondria, thus leading to energy metabolism disorder and further aggravating HF (34). Acetate is a type of SCFA. In a clinical study of SCFAs, it was found that acetate has a protective effect on hypertrophic heart (19). Bile acid, as a signaling molecule, can activate different receptors to participate in energy homeostasis and participates in the process of HF (35). In conclusion, harmful metabolites of intestinal flora are increased, and beneficial metabolites are decreased in chronic HF, which leads to energy metabolism disorder (36).
DISTURBANCES IN ENERGY METABOLISM AND INTESTINAL FLORA METABOLITES CAN ACTIVATE NLRP3 INFLAMMASOME IN HEART FAILURE
Activation of inflammasome corpuscles in NLRP3 inflammasome
The activation of NLRP3 inflammasome corpuscles induces the binding of NLRP3 to the adaptor ASC through the interaction of pyd-pyd, and results in the aggregation of ASC into a large mottled structure (37). NLRP3 protein interacts with ASC through pyrin domain to form NLRP3-ASC-pro-caspase-1 complex, which is called NLRP3 inflammasome (38, 39). Currently, there are three commonly accepted activation pathways of NLRP3 inflammasome corpuscles:
Potassium outflow: Excessive ATP production can activate P2X7 receptors, which increase K+ outflow in cells; and pannexin-1 membrane channels, which are combined with P2X7 receptors, release stimulating factors intracellularly and further activate NLRP3 corpuscles (7).
Lysosomal damage: Macrophages exhibit endocytotic processes that sequester cholesterol crystals and silicon and can break or damage lysosomes, after which the released protease substances activate the NLRP3 inflammasome body.
Reactive oxygen species (ROS): When the body is injured, ROS levels generally increase, which will induce thioredoxin (TRX) to separate TRX from TXNIP, which subsequently activates the inflammasome body of NLRP3 (40). NLRP3-mediated inflammation activated by the above mechanism increases the secretion of downstream inflammasome factors, damages cells, and induces pyroptosis, which exacerbates inflammasome responses (41) (Fig. 1 and 2).
Figure 1.

Activation mechanism of NLRP3 in heart failure: PAMPs and DAMPS trigger the formation of polyinflammatory complex composed of NLRP3, ASC, and pro-caspase-1. The disturbance of intestinal metabolites SCFAs, LPS, TMAO, and bile acid can activate NLRP3 inflammasome through TLR4. Energy metabolism was disrupted, ATP production was decreased, and NLRP3 inflammasome was activated by P2X7. At the same time, glucose metabolism disorder, activation of AMPK, resulting in ROS production, through TXNIP activate NLRP3 inflammasome. The release of IL-1β and IL-18 after the activation of NLRP3 inflammasome causes heart failure, whereas intestinal metabolism disorders and energy metabolism disorders also cause heart failure; therefore, so NLRP3 inflammasome is the central link of heart failure.
Figure 2.
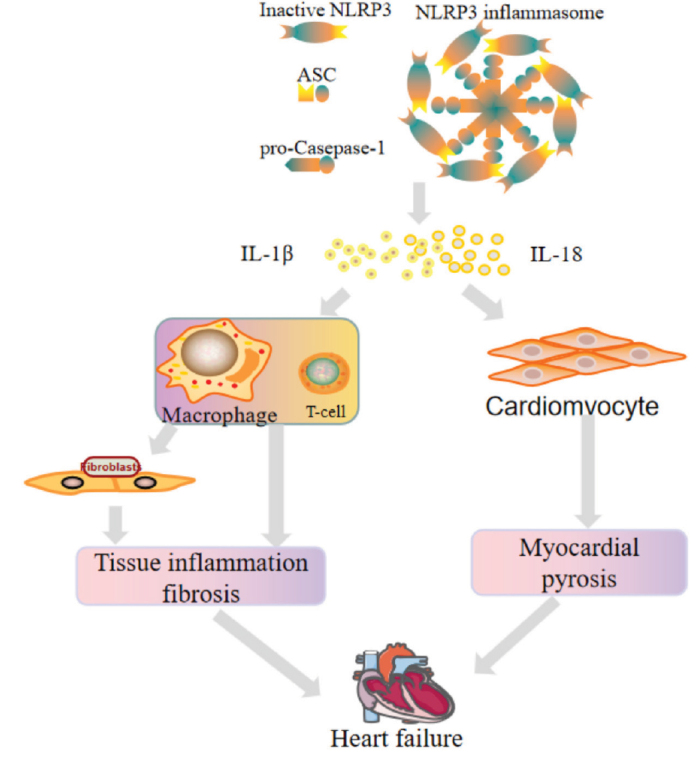
Heart failure caused by NLRP3 inflammasome corpuscle activation. Inflammatory factors produced by NLRP3 trigger cardiac inflammation by recruiting macrophages and subsequent T cells, leading to fibrosis, adverse cardiac remodeling, and heart failure. It can also induce cardiomyocytes scorch death and finally promote myocarditis and remodeling.
Energy metabolism disorder activates NLRP3 inflammasome corpuscles in heart failure
From the perspective of energy metabolism in HF, fatty acids and glycolysis as well as other energy metabolism pathways activate the NLRP3 inflammasome corpuscles through ATP and ROS (42). Saturated fatty acids, especially palmitic acid (PA), represent the most abundant fatty acid. When LPS is used to pretreat fibroblasts, it has been found that PA can induce the expression of both pro-interleukin (IL) 1β and NLRP3 mRNA in fibroblasts, which are necessary for activating the inflammasome body of NLRP3 (43). IL-1β is a cytokine from the IL-1 family. It has been found that pretreatment of mice with an IL-1 blocker retains systolic function and reduces contractions (44). In addition, in vitro studies have shown that IL-1 can affect myocardial diastolic pressure (45, 46).
AMP-activated protein kinase (AMPK) can regulate glycolipid metabolism to improve HF via classical pathways of energy metabolism. Specifically, AMPK can inhibit activation of the NF-κB pathway, activate the downstream NLRP3 inflammasome body, and participate in the regulation of inflammasome responses (47). This suggests that AMPK may affect HF by regulating the inflammasome corpuscles of NLRP3. ATP is the most direct energy source in an organism and can activate NLRP3 inflammasome corpuscles in cardiac fibroblasts (48). It is noteworthy that although ATP signaling can induce rapid activation of inflammasome cells in NLRP3 inflammasome, this may reflect the involvement of intermediate metabolites (49).
Energy metabolism of mitochondria: Mitochondria are important energy producing organelles that comprise the main energy sources of human cells and represent the common medium for activating NLRP3 inflammasome bodies by multiple channels (50). There is evidence that the assembly of mitochondrial DNA (mtDNA) by several mitochondrial-centered mechanisms in NLRP3 inflammasome corpuscles promotes the activation of inflammasome corpuscles (51). Mitochondrial damage induced by environmental or metabolic stress can induce mtDNA oxidation. Oxidized mtDNA can be released into the cytoplasm and combine with NLRP3 inflammasome, which leads to activation of inflammasome corpuscles and secretion of IL-1β (52). In conclusion, mitochondria play an important role in the activation of NLRP3 inflammasome.
Disorder of intestinal flora metabolites in heart failure initiates NLRP3 inflammasome corpuscles
Trimethylamine can activate the inflammasome body of NLRP3 (53). Lipid metabolism of intestinal cells is also an important upstream signal for activating NLRP3 inflammasome corpuscles. Moon et al. (54) have demonstrated that mitochondrial uncoupling protein can stimulate macrophages to regulate the expression of caspase-1 by inducing fatty acid synthesis. Other studies have shown that decreased fatty acid synthesis can lead to decreased activity of the NLRP3 inflammasome body in macrophages and can significantly increase the survival rate of mice (55).
The mechanism of SCFAs in protecting dysfunction of the intestinal barrier is complex. Acetic acid, propionic acid, and butyric acid are the main SCFAs near the large intestine in both humans and rodents. SCFAs can act as histone deacetylase (HDAC) inhibitor to affect the inflammasome body of NLRP3. Feng et al. (56) and other studies have shown that SCFAs inhibit the activation of NLRP3 inflammasome bodies and protect the function of the intestinal barrier. The same conclusion is obtained in dB/db mice. Yuan et al. (57) have suggested that SCFA may target the formation and activation of inflammasome corpuscles of NLRP3 to produce corresponding biological effects.
Bile acids are important mediators of intestinal microecology and NLRP3 activation. Bile acids do not activate NLRP3 inflammasome bodies under normal conditions. However, when bile acid deposits in the liver, NLRP3 inflammasome bodies are activated.
Similarly, whether dietary fiber supplementation improves intestinal inflammation also depends on changes in the axis of inflammasome corpuscles of microbial bile acid NLRP3 (58). Collectively, bile acid metabolism is related to activation of NLRP3 inflammasome.
NLRP3 INFLAMMASOME IS A KEY TARGET FOR HEART FAILURE TREATMENT
Targeted inhibition of assembly of the NLRP3 inflammasome body mitigates heart failure
Colchicine is a nonspecific inhibitor of NLRP3 inflammasome. Colchicine was initially thought to only inhibit microtubule polymerization and leukocyte exudation. As an anti-inflammasome drug, a large part of the efficacy of colchicine is related to inhibition of NLRP3 inflammasome (59). Colchicine alleviates pressure-overload HF in dogs (60) and rats (61, 62). In vivo and in vitro studies have shown that colchicine can effectively improve myocardial cell injury (63–65). Colchicine also increases calcium currents in rat cardiomyocytes (66, 67). All these results indicate that colchicine can be used to treat HF by inhibiting NLRP3. In one clinical trial, colchicine significantly reduced IL-6 concentrations compared with that of a placebo (63).
MCC950 selectively inhibits NLRP3 inflammasome (68, 69). Mcc950 can prevent diseases associated with NLRP3 inflammasome (70), such as hypertension (71) and CVD (72). Another NLRP3 inflammasome inhibitor, cy-09, can also prevent NLRP3 related diseases (73).
In HF, ASC methylation is increased, expression levels of ASC protein and mRNA are decreased, and the expression of IL-1 β is decreased (74). ASC methylation is a potential mechanism to reduce inflammation and ameliorate HF (75). Caspase-1 protein has been demonstrated to be significantly up-regulated in mice with HF, and deletion of endogenous caspase-1 can mitigate the development of HF after myocardial infarction (76). The application of ice/caspase-1 inhibitors has indicated that caspase-1 may be a therapeutic target for preventing the progression of cardiac remodeling in HF (77). Triptolide (TP) attenuates isoproterenol (ISO)-induced cardiac hypertrophy in mice (78). In addition, low dose TP can block NLRP3 inflammasome to improve myocardial fibrosis (79).
Inhibition of NLRP3 inflammasome release-effector factor mitigates heart failure
IL-1β is an important effector of NLRP3 activation. By treating mice with recombinant IL-1β, contractile myocardial damage can be reproduced in vivo. If a single injection of IL-1β causes myocardial contractile dysfunction, repeated injections of IL-1β will produce reversible non-ischemic cardiomyopathy. Anakinra is an IL-1 blocker. A clinical follow-up of anakinra in patients with HF has shown that the incidence of adverse remodeling and HF tends to decrease after three months (80). In clinical trials on canakinumab, inhibition of IL-1 expression can reduce the incidence of CVD (81, 82).
IL-18 is another important effector of NLRP3 activation. In patients with HF, IL-18 is released from the plasma by p38 MAPK, which leads to myocardial systolic dysfunction. Blocking IL-18 with a neutralizing antibody to IL-18bpa or via genetic manipulation can prevent the development of myocardial systolic dysfunction caused by IL-1β (83). These findings suggest that blockade of IL-18 may improve myocardial systolic function in patients with HF. Atorvastatin can reduce the expression of NLRP3 inflammasome, IL-1β, and IL-18. The effect of rosuvastatin, however, is unclear. Furthermore, statins can inhibit endothelial dysfunction (84) and ischemic reperfusion injury. However, further research is needed to confirm this finding (85).
CONCLUSION
HF is accompanied by energy metabolism disorder and intestinal microflora metabolite disorder, which can activate NLRP3 inflammasome and further aggravate the process of HF. Energy metabolism disorder and intestinal microflora metabolite disorder interact with each other, and a large number of relevant studies have reported that regulating NLRP3 inflammasome can ameliorate HF. Therefore, we propose NLRP3-inflammasome centered comprehensive prevention and treatment of HF. Future studies to determine validation of this perspective may provide new directions for the treatment of HF.
HIGHLIGHTS
NLRP3 is the new target for the treatment of heart failure (HF).
In this review, we investigate the role of energy metabolism disorder, inflammation, intestinal flora metabolites, and other aspects in HF.
This study serves to provide a theoretical basis for experimental research and clinical treatment.
Footnotes
Ethics approval and consent to participate: Ethics approval not applicable. All the authors provided consent for publication.
Institutional and financial support: National Natural Science Foundation of China (81774071); Tianjin Natural Science Foundation (19JCYBJC26400); Innovation Fund for graduate students of the School of integrated traditional Chinese and Western medicine of Tianjin University of Traditional Chinese Medicine in 2019 (ZXYCXLX201901); Research and innovation project funding for Postgraduates of Tianjin University of Traditional Chinese Medicine (YJSKC-20201034).
Conflict of interest: None declared.
Peer-review: Externally peer-reviewed.
Author contributions: Concept – S.W., J.Z., Y.W., X.J., M.G., Z.Y.; Design – S.W., J.Z., Y.W., X.J., M.G., Z.Y.; Supervision – S.W., J.Z., Y.W., X.J., M.G., Z.Y.; Fundings – S.W., J.Z., Y.W., X.J., M.G., Z.Y.; Materials – S.W., J.Z., Y.W., X.J., M.G., Z.Y.; Data collection &/or processing – S.W., J.Z., Y.W., X.J., M.G., Z.Y.; Analysis &/or interpretation – S.W., J.Z., Y.W., X.J., M.G., Z.Y.; Literature search – S.W., J.Z., Y.W., X.J., M.G., Z.Y.; Writing – S.W., J.Z., Y.W., X.J., M.G., Z.Y.; Critical review – S.W., J.Z., Y.W., X.J., M.G., Z.Y.
REFERENCES
- 1.Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016;13:368–78. doi: 10.1038/nrcardio.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kızılırmak P, Üresin Y, Özdemir O, Kılıçkıran Avcı B, Tokgözoğlu L, Öngen Z. Renin-angiotensin-aldosterone system blockers and cardiovascular outcomes: a meta-analysis of randomized clinical trials. Turk Kardiyol Dern Ars. 2017;45:49–66. doi: 10.5543/tkda.2016.78006. [DOI] [PubMed] [Google Scholar]
- 3.Li H, Ma Z, Zhai Y, Lv C, Yuan P, Zhu F, et al. Trimetazidine Ameliorates Myocardial Metabolic Remodeling in Isoproterenol-Induced Rats Through Regulating Ketone Body Metabolism via Activating AMPK and PPAR α. Front Pharmacol. 2020;11:1255. doi: 10.3389/fphar.2020.01255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui X, Ye L, Li J, Jin L, Wang W, Li S, et al. Metagenomic and metabolomic analyses unveil dysbiosis of gut microbiota in chronic heart failure patients. Sci Rep. 2018;8:635. doi: 10.1038/s41598-017-18756-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schiattarella GG, Sequeira V, Ameri P. Distinctive patterns of inflammation across the heart failure syndrome. Heart Fail Rev. 2021;26:1333–44. doi: 10.1007/s10741-020-09949-5. [DOI] [PubMed] [Google Scholar]
- 6.Tang WHW, Li DY, Hazen SL. Dietary metabolism, the gut microbiome, and heart failure. Nat Rev Cardiol. 2019;16:137–54. doi: 10.1038/s41569-018-0108-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sokolova M, Ranheim T, Louwe MC, Halvorsen B, Yndestad A, Aukrust P. NLRP3 Inflammasome: A Novel Player in Metabolically Induced Inflammation-Potential Influence on the Myocardium. J Cardiovasc Pharmacol. 2019;74:276–84. doi: 10.1097/FJC.0000000000000704. [DOI] [PubMed] [Google Scholar]
- 8.Wang SM, Ye LF, Wang LH. Shenmai Injection Improves Energy Metabolism in Patients With Heart Failure: A Randomized Controlled Trial. Front Pharmacol. 2020;11:459. doi: 10.3389/fphar.2020.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, Ji J, Yan XH. Cross-talk between AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr. 2012;52:373–81. doi: 10.1080/10408398.2010.500245. [DOI] [PubMed] [Google Scholar]
- 10.Wenjing G, Hong M, Meixiang X. Research progress on the relationship between myocardial energy substrate metabolic remodeling and heart failure. People’s Liberation Army Medical Journal. 2021:1–7.. [Google Scholar]
- 11.Noordali H, Loudon BL, Frenneaux MP, Madhani M. Cardiac metabolism - A promising therapeutic target for heart failure. Pharmacol Ther. 2018;182:95–114. doi: 10.1016/j.pharmthera.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Pasqua T, Rocca C, Giglio A, Angelone T. Cardiometabolism as an Interlocking Puzzle between the Healthy and Diseased Heart: New Frontiers in Therapeutic Applications. J Clin Med. 2021;10:721. doi: 10.3390/jcm10040721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karwi QG, Uddin GM, Ho KL, Lopaschuk GD. Loss of Metabolic Flexibility in the Failing Heart. Front Cardiovasc Med. 2018;5:68. doi: 10.3389/fcvm.2018.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao H, Feng XJ, Li ZM, Li M, Gao S, He YH, et al. Downregulation of adipose triglyceride lipase promotes cardiomyocyte hypertrophy by triggering the accumulation of ceramides. Arch Biochem Biophys. 2015;565:76–88. doi: 10.1016/j.abb.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 15.He L, Wang T, Chen BW, Lu FM, Xu J. Puerarin inhibits apoptosis and inflammation in myocardial cells via PPARα expression in rats with chronic heart failure. Exp Ther Med. 2019;18:3347–56. doi: 10.3892/etm.2019.7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang WH, Wang Z, Shrestha K, Borowski AG, Wu Y, Troughton RW, et al. Intestinal microbiota-dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J Card Fail. 2015;21:91–6. doi: 10.1016/j.cardfail.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuett K, Kleber ME, Scharnagl H, Lorkowski S, März W, Niessner A, et al. Trimethylamine-N-oxide and Heart Failure With Reduced Versus Preserved Ejection Fraction. J Am Coll Cardiol. 2017;70:3202–4. doi: 10.1016/j.jacc.2017.10.064. [DOI] [PubMed] [Google Scholar]
- 18.Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev. 2001;81:1031–64. doi: 10.1152/physrev.2001.81.3.1031. [DOI] [PubMed] [Google Scholar]
- 19.Marques FZ, Nelson E, Chu PY, Horlock D, Fiedler A, Ziemann M, et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation. 2017;135:964–77. doi: 10.1161/CIRCULATIONAHA.116.024545. [DOI] [PubMed] [Google Scholar]
- 20.Pluznick J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes. 2014;5:202–7. doi: 10.4161/gmic.27492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–91. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 22.Devlin AS, Fischbach MA. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat Chem Biol. 2015;11:685–90. doi: 10.1038/nchembio.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–59. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Mayerhofer CCK, Ueland T, Broch K, Vincent RP, Cross GF, Dahl CP, et al. Increased Secondary/Primary Bile Acid Ratio in Chronic Heart Failure. J Card Fail. 2017;23:666–71. doi: 10.1016/j.cardfail.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 25.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–51. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 26.Frangogiannis NG. The inflammasome response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255–65. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang WHW, Li DY, Hazen SL. Dietary metabolism, the gut microbiome, and heart failure. Nat Rev Cardiol. 2019;16:137–54. doi: 10.1038/s41569-018-0108-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makrecka-Kuka M, Volska K, Antone U, Vilskersts R, Grinberga S, Bandere D, et al. Trimethylamine N-oxide impairs pyruvate and fatty acid oxidation in cardiac mitochondria. Toxicol Lett. 2017;267:32–8. doi: 10.1016/j.toxlet.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 29.Moszak M, Szulińska M, Bogdański P. You Are What You Eat-The Relationship between Diet, Microbiota, and Metabolic Disorders-A Review. Nutrients. 2020;12:1096. doi: 10.3390/nu12041096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stenman LK, Holma R, Eggert A, Korpela R. A novel mechanism for gut barrier dysfunction by dietary fat: epithelial disruption by hydrophobic bile acids. Am J Physiol Gastrointest Liver Physiol. 2013;304:G227–34. doi: 10.1152/ajpgi.00267.2012. [DOI] [PubMed] [Google Scholar]
- 31.Daniel H, Gholami AM, Berry D, Desmarchelier C, Hahne H, Loh G, et al. High-fat diet alters gut microbiota physiology in mice. ISME J. 2014;8:295–308. doi: 10.1038/ismej.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Do MH, Lee E, Oh MJ, Kim Y, Park HY. High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change. Nutrients. 2018;10:761. doi: 10.3390/nu10060761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jakobsson HE, Rodríguez-Piñeiro AM, Schütte A, Ermund A, Boysen P, Bemark M, et al. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015;16:164–77. doi: 10.15252/embr.201439263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki T, Heaney LM, Bhandari SS, Jones DJ, Ng LL. Trimethylamine N-oxide and prognosis in acute heart failure. Heart. 2016;102:841–8. doi: 10.1136/heartjnl-2015-308826. [DOI] [PubMed] [Google Scholar]
- 35.Zhang R, Ma WQ, Fu MJ, Li J, Hu CH, Chen Y, et al. Overview of bile acid signaling in the cardiovascular system. World J Clin Cases. 2021;9:308–20. doi: 10.12998/wjcc.v9.i2.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jia Q. Mechanism of Yiqi Huoxue compound in treating chronic HF based on intestinal flora and its metabolites. Tianjin University of Traditional Chinese Medicine; 2020. [Google Scholar]
- 37.Steimle A, Michaelis L, Di Lorenzo F, Kliem T, Münzner T, Maerz JK, et al. Weak Agonistic LPS Restores Intestinal Immune Homeostasis. Mol Ther. 2019;27:1974–91. doi: 10.1016/j.ymthe.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Xu A, Lv J, Zhang Q, Ran Y, Wei C, et al. Development of small molecule inhibitors targeting NLRP3 inflammasome pathway for inflammatory diseases. Eur J Med Chem. 2020;185:111822. doi: 10.1016/j.ejmech.2019.111822. [DOI] [PubMed] [Google Scholar]
- 39.Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10:128. doi: 10.1038/s41419-019-1413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toldo S, Marchetti C, Abbate A. Re “NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective”. Biochem Biophys Res Commun. 2016;470:811–2. doi: 10.1016/j.bbrc.2016.01.088. [DOI] [PubMed] [Google Scholar]
- 41.Conforti-Andreoni C, Ricciardi-Castagnoli P, Mortellaro A. The inflammasomes in health and disease: from genetics to molecular mechanisms of autoinflammation and beyond. Cell Mol Immunol. 2011;8:135–45. doi: 10.1038/cmi.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Próchnicki T, Latz E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017;26:71–93. doi: 10.1016/j.cmet.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 43.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–15. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ikonomidis I, Lekakis JP, Nikolaou M, Paraskevaidis I, Andreadou I, Kaplanoglou T, et al. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. 2008;117:2662–9. doi: 10.1161/CIRCULATIONAHA.107.731877. [DOI] [PubMed] [Google Scholar]
- 45.Radin MJ, Holycross BJ, Dumitrescu C, Kelley R, Altschuld RA. Leptin modulates the negative inotropic effect of interleukin-1beta in cardiac myocytes. Mol Cell Biochem. 2008;315:179–84. doi: 10.1007/s11010-008-9805-6. [DOI] [PubMed] [Google Scholar]
- 46.Tsujino M, Hirata Y, Imai T, Kanno K, Eguchi S, Ito H, et al. Induction of nitric oxide synthase gene by interleukin-1 beta in cultured rat cardiocytes. Circulation. 1994;90:375–83. doi: 10.1161/01.CIR.90.1.375. [DOI] [PubMed] [Google Scholar]
- 47.Qi Y, Shang L, Liao Z, Su H, Jing H, Wu B, et al. Intracerebroventricular injection of resveratrol ameliorated Aβ-induced learning and cognitive decline in mice. Metab Brain Dis. 2019;34:257–66. doi: 10.1007/s11011-018-0348-6. [DOI] [PubMed] [Google Scholar]
- 48.Sandanger Ø, Ranheim T, Vinge LE, Bliks⊘en M, Alfsnes K, Finsen AV, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2013;99:164–74. doi: 10.1093/cvr/cvt091. [DOI] [PubMed] [Google Scholar]
- 49.Sokolova M, Vinge LE, Alfsnes K, Olsen MB, Eide L, Kaasb⊘ll OJ, et al. Palmitate promotes inflammatory responses and cellular senescence in cardiac fibroblasts. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:234–45. doi: 10.1016/j.bbalip.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 50.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203. doi: 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meyers AK, Zhu X. The NLRP3 Inflammasome: Metabolic Regulation and Contribution to Inflammaging. Cells. 2020;9:1808. doi: 10.3390/cells9081808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun. 2017;482:426–31. doi: 10.1016/j.bbrc.2016.11.088. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, Li Y, Yang P, Liu X, Lu L, Chen Y, et al. Trimethylamine-N-Oxide Promotes Vascular Calcification Through Activation of NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome and NF-κB (Nuclear Factor κB) Signals. Arterioscler Thromb Vasc Biol. 2020;40:751–65. doi: 10.1161/ATVBAHA.119.313414. [DOI] [PubMed] [Google Scholar]
- 54.Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee PJ, et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 2015;125:665–80. doi: 10.1172/JCI78253. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Moon JS, Nakahira K, Chung KP, DeNicola GM, Koo MJ, Pabón MA, et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med. 2016;22:1002–12. doi: 10.1038/nm.4153. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Feng Y, Wang Y, Wang P, Huang Y, Wang F. Short-Chain Fatty Acids Manifest Stimulative and Protective Effects on Intestinal Barrier Function Through the Inhibition of NLRP3 Inflammasome and Autophagy. Cell Physiol Biochem. 2018;49:190–205. doi: 10.1159/000492853. [DOI] [PubMed] [Google Scholar]
- 57.Yuan X, Wang L, Bhat OM, Lohner H, Li PL. Differential effects of short chain fatty acids on endothelial Nlrp3 inflammasome activation and neointima formation: Antioxidant action of butyrate. Redox Biol. 2018;16:21–31. doi: 10.1016/j.redox.2018.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh V, Yeoh BS, Walker RE, Xiao X, Saha P, Golonka RM, et al. Microbiota fermentation-NLRP3 axis shapes the impact of dietary fibres on intestinal inflammation. Gut. 2019;68:1801–2. doi: 10.1136/gutjnl-2018-316250. [DOI] [PubMed] [Google Scholar]
- 59.Mauro AG, Bonaventura A, Mezzaroma E, Quader M, Toldo S. NLRP3 Inflammasome in Acute Myocardial Infarction. J Cardiovasc Pharmacol. 2019;74:175–87. doi: 10.1097/FJC.0000000000000717. [DOI] [PubMed] [Google Scholar]
- 60.Koide M, Hamawaki M, Narishige T, Sato H, Nemoto S, DeFreyte G, et al. Microtubule depolymerization normalizes in vivo myocardial contractile function in dogs with pressure-overload left ventricular hypertrophy. Circulation. 2000;102:1045–52. doi: 10.1161/01.CIR.102.9.1045. [DOI] [PubMed] [Google Scholar]
- 61.Scopacasa BS, Teixeira VP, Franchini KG. Colchicine attenuates left ventricular hypertrophy but preserves cardiac function of aortic-constricted rats. J Appl Physiol (1985) 2003;94:1627–33. doi: 10.1152/japplphysiol.00744.2002. [DOI] [PubMed] [Google Scholar]
- 62.Tsutsui H, Ishibashi Y, Takahashi M, Namba T, Tagawa H, Imanaka-Yoshida K, et al. Chronic colchicine administration attenuates cardiac hypertrophy in spontaneously hypertensive rats. J Mol Cell Cardiol. 1999;31:1203–13. doi: 10.1006/jmcc.1999.0953. [DOI] [PubMed] [Google Scholar]
- 63.Deftereos S, Giannopoulos G, Papoutsidakis N, Panagopoulou V, Kossyvakis C, Raisakis K, et al. Colchicine and the heart: pushing the envelope. J Am Coll Cardiol. 2013;62:1817–25. doi: 10.1016/j.jacc.2013.08.726. [DOI] [PubMed] [Google Scholar]
- 64.Yamamoto S, Tsutsui H, Takahashi M, Ishibashi Y, Tagawa H, Imanaka-Yoshida K, et al. Role of microtubules in the viscoelastic properties of isolated cardiac muscle. J Mol Cell Cardiol. 1998;30:1841–53. doi: 10.1006/jmcc.1998.0747. [DOI] [PubMed] [Google Scholar]
- 65.Nishimura S, Nagai S, Katoh M, Yamashita H, Saeki Y, Okada J, et al. Microtubules modulate the stiffness of cardiomyocytes against shear stress. Circ Res. 2006;98:81–7. doi: 10.1161/01.RES.0000197785.51819.e8. [DOI] [PubMed] [Google Scholar]
- 66.Gómez AM, Kerfant BG, Vassort G. Microtubule disruption modulates Ca(2+) signaling in rat cardiac myocytes. Circ Res. 2000;86:30–6. doi: 10.1161/01.RES.86.1.30. [DOI] [PubMed] [Google Scholar]
- 67.Kerfant BG, Vassort G, Gómez AM. Microtubule disruption by colchicine reversibly enhances calcium signaling in intact rat cardiac myocytes. Circ Res. 2001;88:E59–65. doi: 10.1161/hh0701.090462. [DOI] [PubMed] [Google Scholar]
- 68.Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu D, Chen Y, Sun Y, Gao Q, Yu B, Jiang X, et al. Gasdermin family: a promising therapeutic target for cancers and inflammation-driven diseases. J Cell Commun Signal. 2020;14:293–301. doi: 10.1007/s12079-020-00564-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ward R, Li W, Abdul Y, Jackson L, Dong G, Jamil S, et al. NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol Res. 2019;142:237–50. doi: 10.1016/j.phrs.2019.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krishnan SM, Ling YH, Huuskes BM, Ferens DM, Saini N, Chan CT, et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc Res. 2019;115:776–87. doi: 10.1093/cvr/cvy252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pavillard LE, Cañadas-Lozano D, Alcocer-Gómez E, Marín-Aguilar F, Pereira S, Robertson AAB, et al. NLRP3-inflammasome inhibition prevents high fat and high sugar diets-induced heart damage through autophagy induction. Oncotarget. 2017;8:99740–56. doi: 10.18632/oncotarget.20763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. 2017;214:3219–38. doi: 10.1084/jem.20171419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Butts B, Gary RA, Dunbar SB, Butler J. Methylation of Apoptosis-Associated Speck-Like Protein With a Caspase Recruitment Domain and Outcomes in Heart Failure. J Card Fail. 2016;22:340–6. doi: 10.1016/j.cardfail.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Butts B, Butler J, Dunbar SB, Corwin EJ, Gary RA. ASC Methylation and Interleukin-1β Are Associated with Aerobic Capacity in Heart Failure. Med Sci Sports Exerc. 2017;49:1072–8. doi: 10.1249/MSS.0000000000001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Merkle S, Frantz S, Schön MP, Bauersachs J, Buitrago M, Frost RJ, et al. A role for caspase-1 in heart failure. Circ Res. 2007;100:645–53. doi: 10.1161/01.RES.0000260203.55077.61. [DOI] [PubMed] [Google Scholar]
- 77.Randle JC, Harding MW, Ku G, Schönharting M, Kurrle R. ICE/Caspase-1 inhibitors as novel anti-inflammatory drugs. Expert Opin Investig Drugs. 2001;10:1207–9. doi: 10.1517/13543784.10.7.1207. [DOI] [PubMed] [Google Scholar]
- 78.Ding YY, Li JM, Guo FJ, Liu Y, Tong YF, Pan XC, et al. Triptolide Upregulates Myocardial Forkhead Helix Transcription Factor p3 Expression and Attenuates Cardiac Hypertrophy. Front Pharmacol. 2016;7:471. doi: 10.3389/fphar.2016.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pan XC, Liu Y, Cen YY, Xiong YL, Li JM, Ding YY, et al. Dual Role of Triptolide in Interrupting the NLRP3 Inflammasome Pathway to Attenuate Cardiac Fibrosis. Int J Mol Sci. 2019;20:360. doi: 10.3390/ijms20020360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Van Tassell BW, Arena RA, Toldo S, Mezzaroma E, Azam T, Seropian IM, et al. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS One. 2012;7:e33438. doi: 10.1371/journal.pone.0033438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 82.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. CANTOS Trial Group. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–31. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 83.Toldo S, Mezzaroma E, O’Brien L, Marchetti C, Seropian IM, Voelkel NF, et al. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2014;306:H1025–31. doi: 10.1152/ajpheart.00795.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Satoh M, Tabuchi T, Itoh T, Nakamura M. NLRP3 inflammasome activation in coronary artery disease: results from prospective and randomized study of treatment with atorvastatin or rosuvastatin. Clin Sci (Lond) 2014;126:233–41. doi: 10.1042/CS20130043. [DOI] [PubMed] [Google Scholar]
- 85.Yu SY, Tang L, Zhao GJ, Zhou SH. Statin protects the heart against ischemia-reperfusion injury via inhibition of the NLRP3 inflammasome. Int J Cardiol. 2017;229:23–4. doi: 10.1016/j.ijcard.2016.11.219. [DOI] [PubMed] [Google Scholar]