

Abstract
Degradation Factor 1 was discovered 20 years ago as a yeast protein copurifying with Rad26, a helicase involved in transcription-coupled DNA repair. It was subsequently shown to control the ubiquitylation and destruction of the large subunit of DNA damage-arrested RNA Polymerase II. Since that time, much has been learned about Def1’s role in polymerase destruction and new functions of the protein have been revealed. We now understand that Def1 is involved in more than just RNA polymerase II regulation. Most of its known functions are associated with maintaining chromosome and genomic integrity, but other exciting activities outside this realm have been suggested. Here we review this fascinating protein, describe its regulation and present a hypothesis that Def1 is a central coordinator of ubiquitin signaling pathways in cells.
Keywords: Degradation Factor 1, DEF1, RNA polymerase II, ubiquitylation, DNA Damage Resistance, protein aggregation
Background:
DNA damage elicits cellular mechanisms which minimize transcription errors while also maintaining genomic integrity. One event occurring is the irreversible stalling or arrest of the transcriptional machinery comprising RNA polymerase II (RNAPII) and transcription elongation factors at the site of DNA damage, which must be removed from the lesion for the repair of the damaged DNA strand.[1, 2].
Early research efforts targeted at unraveling the pathways involved in yeast DNA damage response identified Rad26 as a key player in the transcription-coupled repair (TCR) [3]. TCR preferentially repairs the transcribed strand and is rapid compared to its counterpart global genomic repair (GGR). GGR requires the ubiquitylation and subsequent degradation of RNAPII[1, 2, 4]. The interplay between GGR and TCR was suggested by discovering a large protein that co-purified with the TCR factor Rad26 from chromatin preparations, which was subsequently named the RNAPII Degradation Factor 1 (DEF1) [5]. This finding pioneered the investigation of the function of Def1 in the ubiquitylation of RNAPII and revealed a role for polymerase degradation in GGR. Because Def1 is a yeast factor, the next section of the review will focus on the pathways leading to RNAPII degradation in yeast. However, later in this review, we will make a case that a functional homolog of Def1 exists in mammals.
Def1 mediates RNA polymerase II degradation
Three classes of enzymes work in series to ubiquitinate proteins, ubiquitin-activating E1, ubiquitin-conjugating E2 and ubiquitin-ligase E3 [6]. While yeast only has one E1 protein, there are multiple E2 and E3 enzymes in this organism. Using a combination of genetic analysis and in vitro reconstitution ubiquitylation assays, the enzymes required for the ubiquitylation of Rpb1, the largest subunit of RNAPII were identified. The E2 Ubc4/5, HECT-domain-containing E3 ligase Rsp5, and the cullin-RING domain E3 ligase, Elongin (Ela1/Elc1/Cul3), were shown to be required for UV-induced degradation of Rpb1, through the ubiquitin-proteasome pathway[2, 7, 8]. Biochemical assays characterized the roles of each of the two E3 ligases, Rsp5 and Elongin complex, in the polyubiquitylation of Rpb1 [9]. These studies led to a model where Rsp5 initially adds monoubiquitin to RNAPII, and this monoubiquitin unit is subsequently extended using lysine 48-linked chains by the Elongin complex [9]. For quite some time, it was not known how the Elongin complex targets RNAPII. In an elaborate set of experiments, Def1 was identified to be that factor [10]. Recruitment is mediated by a CUE domain contained in the N-terminus of Def1 (Figure 1). Instead of binding ubiquitin, the CUE domain of Def1 bound a ubiquitin-like domain (UBL) in the Elongin subunit Ela1. Thus, Def1 acts as a bridging factor connecting the Elongin complex to RNAPII.
Figure 1. The domain structure and known functions of Def1.
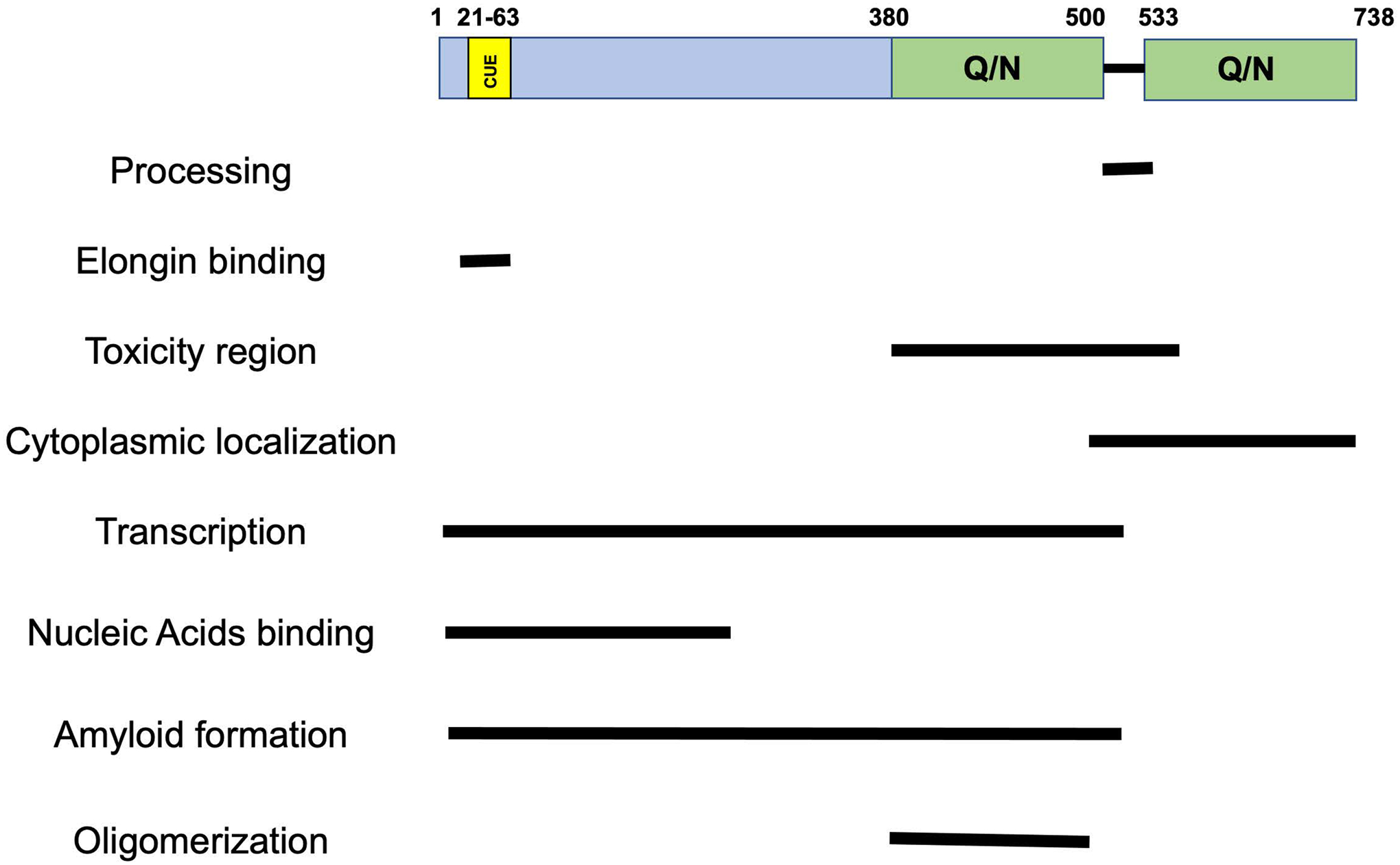
The amino acid numbers are located about the graphic. The functions attributted to each of the domains are based on publications described in the body of the review.
Nuclear localization of Def1 is dependent on its processing by the proteasome
A puzzling observation was that Def1 is a cytoplasmic protein, yet it regulates the ubiquitylation of chromatin-bound RNAPII. It is possible that a small fraction of Def1 was nuclear, which was sufficient to degrade RNAPII. However, it is much more interesting than that. It was later discovered that cytoplasmic Def1 is an inactive form of the protein (at least in regard to Rpb1 ubiquitylation), and Def1 is processed into a shorter form called processed Def1 (pr-Def1) when transcription stress is induced (Figure 2, right) [10]. The authors could not precisely map the cleavage site, but molecular genetic experiments estimated it occurs between amino acids 500–530 (Figure 1). This region contains a short interruption of the long, very Q/N-rich C-terminus. Cleavage of Def1 removes a large portion of the glutamine- and arginine-rich region (Q/N) located at the C-terminus (more discussion of the function of the Q/N-rich region is below). Def1 processing caused the relocation of pr-Def1 to the nucleus, where it can interact with RNAPII. The processing and nuclear accumulation of Def1 upon transcriptional stress is the major mechanism regulating its binding to RNAPII and initiation of the ubiquitin modification cascade. This mechanism was elegantly shown using an engineered version of Def1 that contained a tobacco-etch virus (TEV) protease site near the natural site of processing. Induction of processing by expression of the protease led to Rpb1 ubiquitylation independent of transcriptional stress. Interestingly though, the ubiquitylation of Rpb1 resulting from this mode of activation did not lead to its destruction, indicating transcriptional stress signals have additional roles in RNAPII degradation. Perhaps stress signals affect the removal of chromatin-bound RNAPII by Ccd48 or Ino80 complexes, a prerequisite for Rpb1 destruction [11, 12].
Figure 2. UVSSA and Def1 coordinate TCR and RNAPII degradation pathways through ubiquitin dependent signaling.
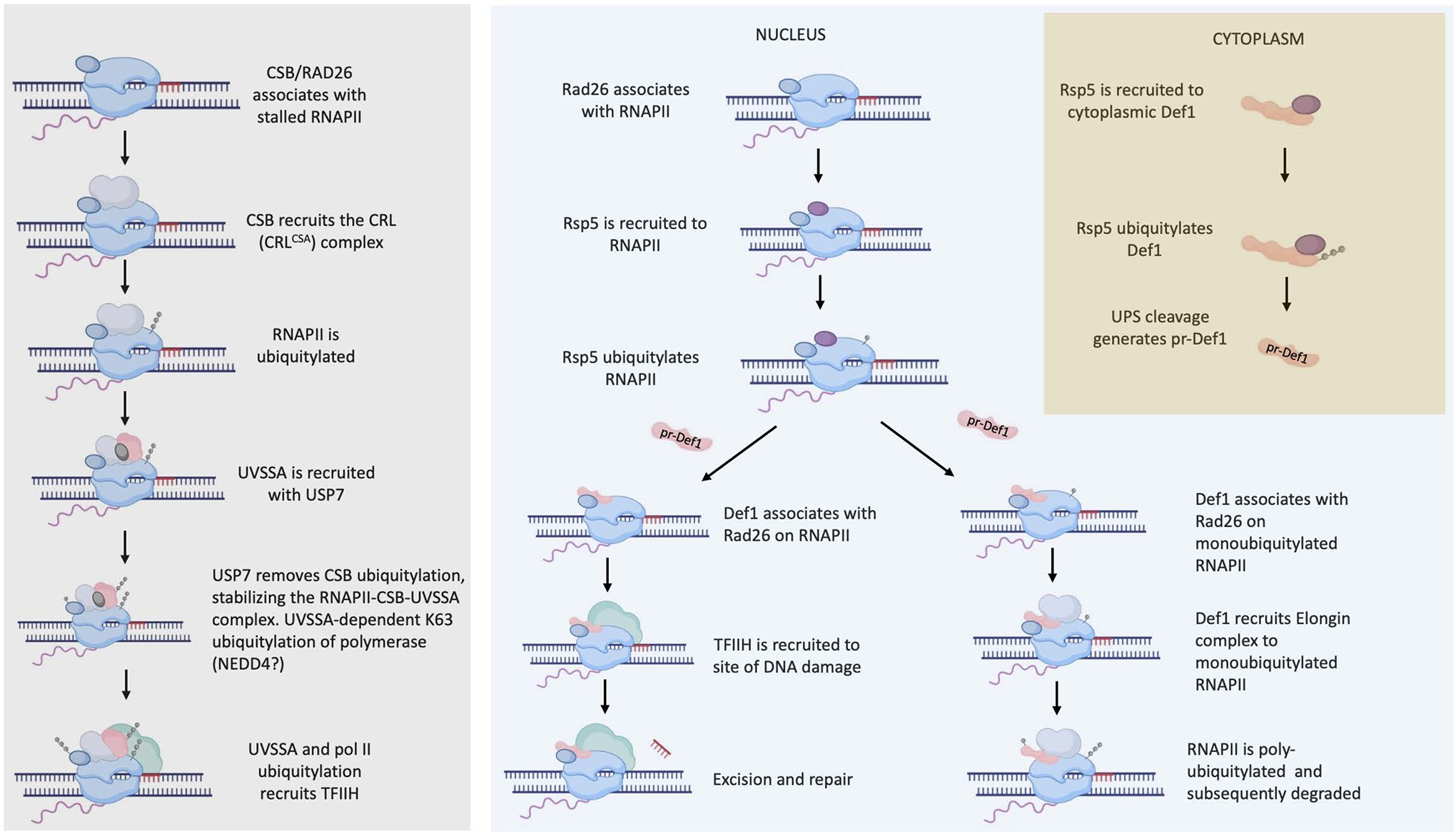
Comparison of the mechanism of action of UVSSA (left) and Def1 (right). The tan box highlights events in the cytoplasm. UPS: ubiquitin-proteosome system.
The processing of Def1 occurs by ubiquitin-dependent proteasome cleavage. The proteasome is widely known for its protein destruction function; however, it also can process proteins at a precise site, including transcription factors [13, 14]. Surprisingly, Rsp5 was identified as the E3 ubiquitin ligase that modified Def1 to target it to the proteasome for cleavage. As described above, in vitro ubiquitylation experiments determined that Rsp5 monoubiquitylates Rpb1. Therefore, Rsp5 has a dual function in Rpb1 ubiquitylation, the processing of Def1 in the cytoplasm and modification of Rpb1 in the nucleus. A few questions arise from the discovery of Def1 processing as a major regulatory step. First, what E2 is responsible for the processing event? It is likely to be Ubc4/5, but this must be proven experimentally. Second, and most importantly, how does transcriptional stress in the nucleus signal to the cytoplasm to initiate the processing of Def1? Def1 processing is an early point of control and a requisite for Rpb1 degradation.
Regulation of Def1 expression
The first hint that DEF1 is important for stress responses came from a study showing its expression is upregulated by 26 different cell-damaging conditions [15]. While a few stress-regulated transcription factors bind to the promoter of DEF1 [16, 17], expression is also controlled by an interesting transcription attenuation mechanism [18]. This was discovered by mapping alternative polyadenylation and cleavage sites within the open reading frames of genes during DNA damage. A promoter-proximal (less than 50nt away from the start codon) and a promoter-distal poly(A) site were identified in the DEF1 gene [18, 19]. In undamaged cells, the shorter, attenuated transcript predominates, which would not produce a full-length mRNA or protein. The shift in poly(A) site usage may be caused by reduced cleavage and polyadenylation factor (CPF-CF) levels in cells. DNA damage causes the destruction of multiple CPF-CF subunits, and reduced CPF-CF increases the by-pass of the first noncanonical terminator [18, 20]. The result of by-passing the attenuator is a 2-fold increase in full length transcript.
Contrary to the rule that transcription attenuation occurs by one of the two major pathways, Sen1-dependent (NNS) pathway or poly (A)-dependent (CPF-CF) pathway [21, 22], transcription attenuation of DEF1 at the proximal terminator requires elements of both pathways and was hence characterized as a hybrid terminator [19]. Based on the analysis of mutants in each of the pathway, the attenuator is more strongly influenced by the CPF-CF than the NNS termination pathway. This explains how reduced levels of CPF-CF in cells can cause the by-pass of the promoter-proximal termination site and production of more full-length mRNA. Transcription attenuation may be part of a mechanism of controlling the overexpression of DEF1, which can be toxic to cells (read below).
DEF1 in DNA damage resistance and repair pathways
DEF1 is required for the resistance of cells to multiple DNA damage agents, including those that cause replication fork collapse, oxidative damage to bases, bulky lesions, double-stranded breaks and chromosome rearrangements [5, 23–27]. The sensitivity of def1Δ cells to a wide variety of mutagens suggests it does not play a specific role in any one type of repair. This section of the review will describe its contributions to different repair pathways, and we provide a hypothesis for why DEF1 mutant cells are sensitive to many types of DNA damage.
As noted above, Def1 was first identified as a protein associating with the transcription-coupled repair (TCR) factor Rad26 [5]. Rad26 is an ATP-dependent helicase that moves RNA polymerase II along DNA, exposing the lesion previously buried in the active site of polymerase [28, 29]. Even though Def1 binds Rad26, it is not required for transcription-coupled repair [5, 27]. This was demonstrated by T4 endonuclease assays for the repair of UV damage within the RPB2 gene and genetic epistasis analysis using mutants in TCR and GGR. Thus, the importance of the Def1-Rad26 interaction in DNA repair is unclear, but Def1 appears to function in GGR indirectly by removing RNA polymerase. We know that Rpb1-degradation is not required for the repair of lesions[5, 8]; thus, Def1 does more in DNA repair than initiate the destruction of RNAPII.
Interestingly, deleting RAD26, which accelerates Rpb1 degradation, can reverse the need for Def1 to degrade RNA polymerase II [5]. The result was a striking reversal of the defect, not a partial suppression. Curiously, deleting RAD26 did not suppress the Rpb1 degradation defect of a not4Δ mutant (Ccr4-Not subunit) or a conditional mutant of CDC48 (removes polymerase from chromatin) [11, 30]. Genetic and biochemical data suggest that Ccr4-Not and CDC48 function upstream and downstream of Def1, respectively. Thus, the ability of Rad26 to modulate Rpb1 degradation is specific for Def1. A plausible model was proposed that Rad26 blocks Def1 from initiating Rpb1 degradation to allow for repair via TCR [5, 28]. Rad26 binds upstream of RNAPII at a location where DNA exits the polymerase and the transcript emerges [28]. Def1 specifically stimulates Rsp5-dependent ubiquitylation of Rpb1 incorporated into elongation complexes, suggesting it recognizes features of the EC absent in free RNA polymerase II [31]. Interestingly, the N-terminus of Def1 (amino acids 1–207, Figure 1) binds single- and double-stranded DNA [32], a feature of RNAPII elongation complexes. There is little known about how Def1 associates with RNAPII, other than its binding to RNAPII in cells is DNA damage dependent [33]. It cannot be ruled out that DNA damage-generated signals influence the interaction, but since initiating processing in the absence of DNA damage (above) leads to Rpb1 ubiquitylation and recent data showing recombinant Def1 binds to purified RNA polymerase II elongation complexes [34], the DNA damage -dependent binding of Def1 to RNAPII is most likely explained by the need to process Def1 and for nuclear translocation. Additional biochemical and structural characterization of the Def1-Rad26-RNAPII complex will be very valuable for understanding the complex relationship between these two factors.
Def1 mutants are mildly sensitive to oxidative stress [24]. Hydrogen peroxide does more than cause DNA damage, but there is evidence that Def1 may be important for repairing oxidative DNA damage during transcription. Owiti and colleagues used a system to measure transcription-associated mutagenesis (TAM) that relies on the mutation-induced reversion of a stop codon within a reporter gene in a strain accumulating unrepairable apurinic sites (AP) sites [25]. Failure to repair AP sites through the normal pathway leads to the accumulation of mutations due to error-prone repair DNA polymerase utilization. Deleting DEF1 elevated TAM rate about 15-fold, which was further enhanced in a rad26Δ mutant background. Furthermore, repair of the AP-sites was more affected on the transcribed strand. Since eliminating Rad26 from cells enhanced, rather than suppressed, TAM suggests that Def1’s Rpb1 degradation function is not required for repair of AP sites. The double def1Δ/rad26Δ has restored Rpb1 degradation [5]. Interestingly, the mutability phenotype of the def1Δ mutant was reported to depend on the carbon source used to grow the cells; mutability was not observed when grown in non-fermentable carbon sources [25]. The authors of that paper speculated that the metabolic state of the cells regulates Def1 by altering the ratio of reactive oxygen species in the cell. However, it should be noted that TAM was suppressed in mutants of other repair factors grown in non-fermentable carbon sources and Def1 is required for mitochondrial maintenance and function [32]. A trivial explanation for the carbon source dependence is that slowed growth of the def1Δ mutant, exacerbated by the growth in glycerol-ethanol due to impaired respiration, reduced transcription rates in the mutant, and thus, TAM.
Def1 mutants are also sensitive to X-rays and zeocin, agents that create double-stranded breaks (DSBs), and methyl methanesulfonate (MMS) and hydroxyurea that result in DSBs during replication [23, 24]. A proteomics study first suggested a mechanism for Def1’s involvement in managing DSBs. Wang and colleagues used a chromatin affinity purification-mass spectrometry procedure to identify more than 100 proteins, including Def1, recruited to an HO-endonuclease directed DSB [24]. Surprisingly, while def1Δ cells were shown to be sensitive to zeocin in this study, and x-rays in another [23], DEF1 was not required to repair an HO-induced break at the MAT locus or when the break was induced within a transcription unit. These results suggest that while Def1 is recruited to HO-induced DSB, it may not be directly involved in break repair. The sensitivity of the mutant strain to DSB-causing agents can be explained by another phenotype. Pulse-field gel electrophoresis of yeast chromosomes demonstrated that def1Δ cells displayed delayed repair of global DSBs and an increase in chromosome rearrangements [24]. These results suggest that Def1 plays a broad role in chromosome integrity. This hypothesis is further supported by two other chromosome maintenance functions of Def1. First, DEF1 is required for pre-meiotic DNA replication and homologous chromosome pairing during meiosis [23]. This is due partly to the reduced loading of Zip1. Interestingly, FACS analysis revealed that def1Δ cells are prone to aneuploidy and defective nuclear division events. Second, DEF1 mutant cells undergo self-diploidization stimulated by transformation-induced homologous recombination during mutant construction [26]. An interesting aspect of the latter study is that the authors correlated the DNA damage sensitivity and mutability phenotypes to the strain’s ploidy; isolates that underwent diploidization were UV immutable, while true haploids were mutable. This result suggests caution in working with def1Δ cells and the need to use conditional depletion strategies to identify immediate effects of the loss of Def1.
Can the broad role of DEF1 in the repair of DNA damage be linked to a common function?
The DNA damage sensitivity of DEF1 mutant cells is mild compared to that of mutants of factors directly involved in repair. At this time, evidence for a direct role of Def1 in repair is lacking. Def1 is particularly important when canonical, dedicated repair pathways are inactive or non-functional. For example, when TCR is impaired, Def1 is required as a last resort to destroy RNA polymerase II by ubiquitin-mediated proteolysis [4]. Unrepairable DNA damage is dealt with by error-prone replication pathways, translesion DNA synthesis (TLS) as a “last resort”. Utilizing TLS increases mutations and mutability [35, 36]. An explanation for the connection between Def1 and multiple DNA repair pathways may hinge upon its control of Pol3 destruction [37]. Pol3 is the catalytic subunit of DNA polymerase delta, which participates in lagging strand synthesis, DNA replication during meiosis, recombination, DSB repair, and nucleotide excision repair (NER) [38]. Other subunits of Pol3-containing complexes include Pol31, Pol32, Rev3, and Rev7. The discovery that Def1 controls the balance of replicative and TLS synthesis through Pol3 was based on the MMS-dependent association between it and Rad5, an ATPase that mediates the process of by-passing lesions through template switching [37]. Based on genetic analysis, RAD5 and DEF1 function in distinct aspects of DNA repair, with DEF1 belonging in the RAD6–RAD18-regulated DNA damage tolerance pathway that promotes the switch to error-prone TLS synthesis.
The authors of that paper showed that the ubiquitylation and destruction of the Pol3 after DNA damage requires Def1. A model was proposed that Def1-dependent destruction Pol3 causes its exchange with error-prone TLS catalytic subunits of DNA polymerase, Rev3 and Rev1. Blocking Pol3 destruction prevents switching to error-prone polymerases; thus, account for the reduced mutability in def1Δ mutants [25, 37]. Interestingly, Rev3 functions in DSB repair and Rev1 is used to by-pass abasic sites and guanosine adducts [39]. Def1’s function in regulating the switch to TLS DNA polymerases through Pol3 degradation would explain the broad DNA damage phenotypes and complex genetic interactions with multiple repair pathways of the def1Δ mutant.
Telomere silencing and maintenance
There is information on the role of DEF1 in telomere structure and function. The first evidence of Def1 regulating telomeres is based on its co-purification with Rrm3, a DNA helicase that promotes replication at telomeres and suppresses DNA damage at G4-quadruplex structures [32, 40]. The most striking result was that def1Δ cells displayed shortened telomeres, by ~200 bp. This shortening of telomere length caused a mild telomeric silencing defect of URA3-based reporters. The same work confirmed that telomere shortening was not observed in RAD26 or DST1 mutants, suggesting that this function of Def1 in telomere length control is distinct from its RNA polymerase II regulatory properties. Interestingly, the paper demonstrated that the first 207 amino acids of Def1 binds single-stranded and double-stranded DNA, and Def1 was recruited to telomeres. The recruitment was relatively weak, but as discussed above, the majority of Def1 is sequestered in the cytoplasm. Activating DNA damage pathways by UV or disrupted telomeres may be required to observe robust binding, and it would be interesting to examine if disrupting telomere structure causes the processing and localization of Def1 to the nucleus and telomeres specifically.
Rrm3 has a homolog, Pif1, that also promotes genome stability at telomeres by unwinding G4 structures at telomeres [41]. DEF1 and PIF1 mutants show negative genetic interactions, but the nature of this was not explored [42]. The fact that DEF1 shows genetic interactions with two helicases involved in suppressing DNA damage at telomeric structures reinforces its many roles in genomic maintenance. The loss of silencing is not unique to Saccharomyces cerevisiae DEF1 or telomeres. Schizosaccharomyces pombe DEF1 was identified in a screen for mutants disrupting heterochromatic silencing at the mating-type locus using several artificial boundary elements that replaced the natural IR-R+ sequences [43]. This result suggests that Def1’s role is not telomere specific but maybe a more general feature of heterochromatic silencing.
Def1 role in transcription
Def1 was first implicated in regulating transcription by its observed synthetic enhanced phenotypes with the gene encoding TFIIS, DST1, and elongation defective mutants of RNA polymerase II [5]. An explanation for these genetic interactions was that RNA polymerase arrested by non-DNA damage blockage requires Def1 processing and Def1-dependent Rpb1 degradation [4, 10, 31]. The failure to clear arrested RNAPII from genes synergized with the defects brought on by elongation factor mutations. However, there is more to the story than this. Additional lines of evidence point to a role for Def1 in promoting transcription, independent of its Rpb1 degrading activity. These are presented below.
The most direct evidence came from a more recent study that discovered that Def1 co-purifies with the general transcription factor TFIIH [34]. The same study also found subunits of the Ccr4-Not complex in these fractions (below). The interaction was mapped to the N-terminus of Def1 using crosslinking-mass spectrometry, which identified contacts between Def1 and the PH domain of Tfb1, Tfb2 and Rad3. Searching for a function of this interaction, the authors used a highly purified and well-defined transcription system to identify a role for Def1 in transcription. Adding Def1 alone to the reaction had no effect on initiation or elongation. However, when both TFIIS and Elongin were added, Def1 enhanced transcription initiation from a new initiation site on the SNR20 promoter. The transcription promoting activity of Def1 required the N-terminal half-of the polyQ region, since a version of the protein containing amino acids 1–380 was inactive, but one containing 1–530 was (see Figure 1). Furthermore, order of addition experiments suggests that Def1 (and Elongin) promote the reactivation of RNAPII after TFIIS-induced cleavage of the transcript displaced from the active-site of polymerase. This study also demonstrated that Def1 (1–530) can interact directly with purified elongation and pre-initiation complexes, indicating the direct involvement of Def1 in transcription. The interaction between Def1 and the EC, however, was insufficient to re-start transcription in the absence of Elongin and TFIIS, suggesting Def1 performs a recruitment function. Ela1, a subunit of the Elongin complex, contains a ubiquitin-like domain that binds to the CUE domain of Def1[10] to promote Rpb1 degradation. The role of Def1 in promoting elongation re-start in the absence of Rpb1 degradation may be to recruit Elongin to the EC. It would be very illuminating to conduct the transcription assays using a version of Def1 with a mutated CUE domain or Elongin subunit mutants to examine this possibility.
Another factor that connects Def1 function to elongation and Rpb1 degradation is Ccr4-Not. Ccr4-Not associates with elongation complexes, promotes elongation by preventing RNAPII arrest and functions with TFIIS [44–46]. Ccr4-Not, Not4 specifically, is required for Rpb1 destruction of arrested RNAPII[30]. Similar to Def1, Ccr4-Not subunit mutations display synthetic enhanced phenotypes with a dst1Δ mutation [46, 47]. Ccr4-Not subunits co-purified with TFIIH and Def1 and BioID proximity labeling studies indicate that Def1 and Ccr4-Not interact within cells [34]; Pfannenstein and Reese, unpublished). Ccr4-Not acts upstream of Def1 in the Rpb1 destruction pathway by promoting Rsp5-dependent ubiquitylation. Despite some similarities, two lines of evidence indicate that Ccr4-Not and Def1 have different functions in the cell. First, def1Δ and not4Δ mutations are synthetically lethal. Second, the Rpb1 degradation defect in the not4Δ mutant cannot be suppressed by deleting RAD26. We have suggested that Ccr4-Not and Def1 reactivate arrested RNAPII, which explains the synthetic lethality, and they may do so by acting as a scaffold to recruit and coordinate the actions of other elongation factor such as TFIIS and Elongin. The reciprocal interactions between Def1, Ccr4-Not, TFIIS and TFIIH suggest that further examination of the interplay among these factors would be a worthwhile pursuit and advances in cryo-EM may allow for a structure of these factors and elongation complexes to be solved.
Def1 in proteostasis
A distinguishing feature of Def1 is an extended glutamine- and asparagine-rich region (Q/N) at the C-terminus of the protein (Figure 1). One role of the C-terminus is to keep Def1 in the cytoplasm until its processing by the proteosome. Q/N-rich proteins have the propensity to aggregate and form amyloid-like fibrils[48, 49]. The first lines of evidence that Def1 regulates proteostasis, protein aggregation specifically, came from studies reporting that the toxicity of overexpressed mutant forms of Huntington’s disease proteins (Htt103Q) in yeast is suppressed by deleting DEF1 [50–52]. Overexpressed Def1 forms foci similar to other amyloid forming proteins and co-localized with over expressed human Huntington’s disease proteins in the cytoplasm [49, 51]. Furthermore, proteomics and genetic studies revealed that Def1 forms SDS-resistant aggregates in cells, a feature of amyloid-forming proteins [49, 53, 54]. Finally, Def1 protein forms amyloid-like aggregates in vitro, which has been mapped recently to the N-terminal portion of the Q-rich region (amino acids 371–530) [34, 49]. There are a few points to consider, however. At least one study suggested that the expression of the Huntingdon proteins, both disease causing (HttQ103) and control (HttQ25), was reduced in def1Δ cells [52]. So reduced accumulation of HttQ103 protein can explain the loss of toxicity in the mutant. Also, the toxicity of HttQ103 proteins requires ”seeding” of the aggregates by intercellular proteins prone to aggregation and prions, aggregation and toxicity does not always correlate[51, 55] and the degree of toxicity appears to be strain dependent [52].
So, what is the function of the Q-rich domain? Domain mapping studies revealed that expressing the shortened version of Def1 (1–500) is toxic to cells [10]. Toxicity was not observed when the wild type protein is overexpressed or a version with smaller portion of the Q-rich domain is removed (1–600), suggesting an extended C-terminus protects cells from Def1 toxicity [10]. Interestingly, driving the toxic versions of Def1 out of the nucleus by fusing a nuclear export signal to it, suppressed the toxicity. This suggests that accumulating processed protein in the nucleus is the cause of the toxicity and that the N-terminal half of the Q-rich domain contributes to this phenotype. Thus, the extended C-terminus is important for sequestering the protein in the cytoplasm and this mis-regulation interferes with nuclear functions. This is analogous to how cells contend with accumulation of other Q/N-rich domain proteins, sequester them in a form that reduces toxicity.
The ability of Def1 to form punctate, amyloid-like structures in cells requires overexpression. This may representant an experimentally visible, exaggerated state of a structure that forms to a lesser extent under natural conditions. The C-terminus of Def1 undergoes self-oligomerization [10, 32] and likely mediates its association with other Q/N rich proteins in cell. Indeed, amino acids 381–480 of Def1 forms oligomers in vitro [32]. Shortening of the Q/N-rich region by proteolytic processing of Def1 could shift the equilibrium from forming high-ordered aggregates to restrict its activity, to one capable of more dynamic interactions with like-structured proteins. Def1 may act as a scaffold for coordinating the recruitment of activities in RNA polymerase II degradation and transcription, and recruiting factors with similar Q/N-rich domains may be the mechanism of action. It is noteworthy that a version of Def1 completely lacking the Q/N-rich region (1–380) did not support transcription reactivation in transcription assays [34]. Thus, the first half of the Q/N-rich domain may make contacts with components of the transcription machinery.
Domain mapping experiments demonstrated that the Q/N-rich region is responsible for keeping Def1 in cytoplasm [10]. Furthermore, the C-terminus of Def1 (500–738) is sufficient to transport GFP into the cytoplasm, despite lacking any known nuclear export signal (NES). An interesting, but unproven theory is that the C-terminus of Def1 associates with nuclear pore complex (NPC) proteins Nup49, Nup100 and Nu116, which also has Q/N-rich regions. The propensity of Q/N regions to oligomerize with like domains may be the basis for how Def1 is recognized by the NPC and transported out of the nucleus.
Is UVSSA the mammalian equivalent of Def1?
Def1 has no identifiable homologs outside of yeast, which raises the question if Def1’s functions are fungal-specific. However, there are examples where proteins without detectable sequence homology perform analogous functions in their respective organisms. Here we make a case that the UVSSA protein is the Def1 equivalent.
UV-stimulated scaffold protein A (UVSSA) binds to arrested RNAPII, is required for the ubiquitylation of Rpb1 and recruits TFIIH to promote TCR [1, 56–59]. The sequential ubiquitination of RNAPII and UVSSA coordinates the recruitment of TFIIH to damaged sites [56, 58], which occurs via a physical interaction between UVSSA and the p62 subunit (TFB1 in yeast) of TFIIH [57]. Overall, UVSSA acts as a scaffold to coordinate ubiquitylation of arrested EC components and regulates TCR by the recruitment of TFIIH, functions similar to those of Def1 in yeast (Figure 2).
Let’s draw some parallels between UVSSA and Def1. First, both Def1 and UVSSA are required for Rpb1 ubiquitylation. Second, both are recruited to chromatin-bound RNAPII by DNA damage. Third, UVSSA binds the TFB1 homolog of human TFIIH, p62, and crosslinking mass spectrometry studies detected direct interactions between Def1 and TFB1[34, 57]. Fourth, both are regulated by monoubiquitylation. In the case of UVSAA, ubiquitylation causes TFIIH recruitment to damage sites, but the only known role of Def1 modification is its processing by the proteasome [10, 58]. However, it should be noted that global proteome analysis detected ubiquitylation sites in Def1 distinct from those required for its processing [10, 60]. It cannot be ruled out that Def1 ubiquitylation has roles in the TFIIH recruitment. Fifth, UVSSA recruits the ubiquitin-specific protease USP7 to protect ERCC6/CSB/yRAD26 from degradation [61]. Interestingly, the closest yeast homolog to USP7 is Ubp15, which co-purifies with Def1 and TFIIH [34]. Finally, both proteins act as a scaffold to coordinate the recruitment of activities that remove RNAPII or utilize Rad26/CSB-dependent TCR [5]. There are functions and activities that set them apart, however. UVSSA lacks a Q/N-rich motif, is nuclear and not processed like Def1. Sequestration of Def1 in the cytoplasm may be necessary in fungi to prevent premature Rpb1 degradation due to yeast’s more compact and transcriptionally active genome. Second, Def1 is required for Rpb1 degradation in yeast cells, while this appears not to be the case for UVSSA. Cells from patients with UVSSA mutations and cells depleted of UVSSA degrade the large subunit of RNAPII [59, 61, 62]. In fact, it has been suggested that UVSSA stabilizes the RNAPII-CSB complex by preventing CSB degradation[62, 63]. Finally, another major difference is that UVSSA is required for TCR, but Def1 is not[1, 2, 5]. This may reflect more of a difference in requirement of Rpb1 degradation for TCR between yeast and mammals, rather than a direct difference in the mode of action of Def1 and UVSSA. Rpb1 degradation is not necessary for TCR in yeast, but it is in mammals[1, 2]. Thus, while details differ, in a broad sense, both Def1 and UVSSA play a key role in balancing the use of TCR and RNAPII degradation pathways and this function centers around recruitment of factors through ubiquitin-dependent signaling.
Perspectives on Def1 function: looking forward
Def1 has many functions, but most are centered around maintaining genomic integrity and chromosome structure. One potential thread that ties Def1’s activities together is ubiquitylation. As discussed above, destruction of RNAPII and Pol3 to initiate DNA polymerase switching during damage responses links its regulation of transcription -and DNA damage-related stresses to protein destruction. The only other distinguishing feature of Def1, besides the Q/N-rich domain, is the CUE- ubiquitin-like binding domain. The CUE domain recruits Elongin to arrested RNAPII, but it is feasible that it binds other proteins with ubiquitin-like domains. Def1 can be a “universal scaffold “ that recruits ubiquitylation machinery to multiple targets throughout the genome. Directed proteomics screens using the CUE-domain as bait could provide evidence for this hypothesis. Additionally, global ubiquitylome students in def1Δ or conditional depletion strains may identify proteins whose ubiquitylation is dependent on Def1. Identifying novel targets of Def1 has the potential to link its many functions in genome integrity to ubiquitylation of proteins.
An interesting question is if Def1 has specific cytoplasmic functions or if the only reason its localized there is to prevent it from interfering with nuclear processes. Def1 associates with the Ccr4-Not complex, which is the major mRNA deadenylase. Furthermore, the list of protein binding partners cataloged in the Saccharomyces Genome Database (SGD) includes many mRNA binding proteins. Def1 binds nucleic acids, and this activity may suggest a role in mRNA regulation in the cytoplasm. However, the co-purification of RNA binding proteins with Def1 may be attributed to nucleic acids dependent interactions during isolation of the complexes. Nucleases are not used routinely in tandem-affinity purification (TAP) procedures. Most studies on Def1 thus far examined nuclear functions, and a more thorough search for cytoplasmic functions is needed. The identification of novel cytoplasmic functions for Def1 will open new exciting areas of research.
Acknowledgements:
This research was supported by funds from National Institutes of Health (R35 GM136353 to J.C.R)
References
- [1].Lans H, Hoeijmakers JHJ, Vermeulen W, Marteijn JA, The DNA damage response to transcription stress, Nat Rev Mol Cell Biol 20(12) (2019) 766–784. [DOI] [PubMed] [Google Scholar]
- [2].Gregersen LH, Svejstrup JQ, The Cellular Response to Transcription-Blocking DNA Damage, Trends Biochem Sci 43(5) (2018) 327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].van Gool AJ, Verhage R, Swagemakers SM, van de Putte P, Brouwer J, Troelstra C, Bootsma D, Hoeijmakers JH, RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6, EMBO J 13(22) (1994) 5361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wilson MD, Harreman M, Svejstrup JQ, Ubiquitylation and degradation of elongating RNA polymerase II: the last resort, Biochim Biophys Acta 1829(1) (2013) 151–7. [DOI] [PubMed] [Google Scholar]
- [5].Woudstra EC, Gilbert C, Fellows J, Jansen L, Brouwer J, Erdjument-Bromage H, Tempst P, Svejstrup JQ, A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage, Nature 415(6874) (2002) 929–933. [DOI] [PubMed] [Google Scholar]
- [6].Oh E, Akopian D, Rape M, Principles of Ubiquitin-Dependent Signaling, Annu Rev Cell Dev Biol 34 (2018) 137–162. [DOI] [PubMed] [Google Scholar]
- [7].Beaudenon SL, Huacani MR, Wang G, McDonnell DP, Huibregtse JM, Rsp5 ubiquitin-protein ligase mediates DNA damage-induced degradation of the large subunit of RNA polymerase II in Saccharomyces cerevisiae, Mol Cell Biol 19(10) (1999) 6972–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ribar B, Prakash L, Prakash S, ELA1 and CUL3 are required along with ELC1 for RNA polymerase II polyubiquitylation and degradation in DNA-damaged yeast cells, Mol Cell Biol 27(8) (2007) 3211–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Harreman M, Taschner M, Sigurdsson S, Anindya R, Reid J, Somesh B, Kong SE, Banks CA, Conaway RC, Conaway JW, Svejstrup JQ, Distinct ubiquitin ligases act sequentially for RNA polymerase II polyubiquitylation, Proc Natl Acad Sci U S A 106(49) (2009) 20705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wilson MD, Harreman M, Taschner M, Reid J, Walker J, Erdjument-Bromage H, Tempst P, Svejstrup JQ, Proteasome-Mediated Processing of Def1, a Critical Step in the Cellular Response to Transcription Stress, Cell 154(5) (2013) 983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Verma R, Oania R, Fang R, Smith GT, Deshaies RJ, Cdc48/p97 mediates UV-dependent turnover of RNA Pol II, Mol Cell 41(1) (2011) 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lafon A, Taranum S, Pietrocola F, Dingli F, Loew D, Brahma S, Bartholomew B, Papamichos-Chronakis M, INO80 Chromatin Remodeler Facilitates Release of RNA Polymerase II from Chromatin for Ubiquitin-Mediated Proteasomal Degradation, Mol Cell 60(5) (2015) 784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Piwko W, Jentsch S, Proteasome-mediated protein processing by bidirectional degradation initiated from an internal site, Nat Struct Mol Biol 13(8) (2006) 691–7. [DOI] [PubMed] [Google Scholar]
- [14].Rape M, Jentsch S, Taking a bite: proteasomal protein processing, Nat Cell Biol 4(5) (2002) E113–6. [DOI] [PubMed] [Google Scholar]
- [15].Jelinsky SA, Estep P, Church GM, Samson LD, Regulatory networks revealed by transcriptional profiling of damaged Saccharomyces cerevisiae cells: Rpn4 links base excision repair with proteasomes, Mol Cell Biol 20(21) (2000) 8157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E, An improved map of conserved regulatory sites for Saccharomyces cerevisiae, BMC Bioinformatics 7 (2006) 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Venters BJ, Wachi S, Mavrich TN, Andersen BE, Jena P, Sinnamon AJ, Jain P, Rolleri NS, Jiang C, Hemeryck-Walsh C, Pugh BF, A comprehensive genomic binding map of gene and chromatin regulatory proteins in Saccharomyces, Mol Cell 41(4) (2011) 480–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Graber JH, Nazeer FI, Yeh PC, Kuehner JN, Borikar S, Hoskinson D, Moore CL, DNA damage induces targeted, genome-wide variation of poly(A) sites in budding yeast, Genome Res 23(10) (2013) 1690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Whalen C, Tuohy C, Tallo T, Kaufman JW, Moore C, Kuehner JN, RNA Polymerase II Transcription Attenuation at the Yeast DNA Repair Gene, DEF1, Involves Sen1-Dependent and Polyadenylation Site-Dependent Termination, G3 (Bethesda) 8(6) (2018) 2043–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kuehner JN, Kaufman JW, Moore C, Stimulation of RNA Polymerase II ubiquitination and degradation by yeast mRNA 3’-end processing factors is a conserved DNA damage response in eukaryotes, DNA Repair (Amst) 57 (2017) 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rondon AG, Mischo HE, Proudfoot NJ, Terminating transcription in yeast: whether to be a ‘nerd’ or a ‘rat’, Nat Struct Mol Biol 15(8) (2008) 775–6. [DOI] [PubMed] [Google Scholar]
- [22].Kuehner JN, Pearson EL, Moore C, Unravelling the means to an end: RNA polymerase II transcription termination, Nat Rev Mol Cell Biol 12(5) (2011) 283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jordan PW, Klein F, Leach DR, Novel roles for selected genes in meiotic DNA processing, PLoS Genet 3(12) (2007) e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang P, Byrum S, Fowler FC, Pal S, Tackett AJ, Tyler JK, Proteomic identification of histone post-translational modifications and proteins enriched at a DNA double-strand break, Nucleic Acids Res 45(19) (2017) 10923–10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Owiti N, Lopez C, Singh S, Stephenson A, Kim N, Def1 and Dst1 play distinct roles in repair of AP lesions in highly transcribed genomic regions, DNA Repair (Amst) 55 (2017) 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stepchenkova EI, Shiriaeva AA, Pavlov YI, Deletion of the DEF1 gene does not confer UV-immutability but frequently leads to self-diploidization in yeast Saccharomyces cerevisiae, DNA Repair (Amst) 70 (2018) 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gaillard H, Tous C, Botet J, Gonzalez-Aguilera C, Quintero MJ, Viladevall L, Garcia-Rubio ML, Rodriguez-Gil A, Marin A, Arino J, Revuelta JL, Chavez S, Aguilera A, Genome-wide analysis of factors affecting transcription elongation and DNA repair: a new role for PAF and Ccr4-not in transcription-coupled repair, PLoS Genet 5(2) (2009) e1000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xu J, Wang W, Xu L, Chen JY, Chong J, Oh J, Leschziner AE, Fu XD, Wang D, Cockayne syndrome B protein acts as an ATP-dependent processivity factor that helps RNA polymerase II overcome nucleosome barriers, Proc Natl Acad Sci U S A 117(41) (2020) 25486–25493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Oh J, Xu J, Chong J, Wang D, Molecular basis of transcriptional pausing, stalling, and transcription-coupled repair initiation, Biochim Biophys Acta Gene Regul Mech 1864(1) (2021) 194659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jiang H, Wolgast M, Beebe LM, Reese JC, Ccr4-Not maintains genomic integrity by controlling the ubiquitylation and degradation of arrested RNAPII, Genes Dev 33(11–12) (2019) 705–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Somesh BP, Sigurdsson S, Saeki H, Erdjument-Bromage H, Tempst P, Svejstrup JQ, Communication between distant sites in RNA polymerase II through ubiquitylation factors and the polymerase CTD, Cell 129(1) (2007) 57–68. [DOI] [PubMed] [Google Scholar]
- [32].Chen YB, Yang CP, Li RX, Zeng R, Zhou JQ, Def1p is involved in telomere maintenance in budding yeast, J Biol Chem 280(26) (2005) 24784–91. [DOI] [PubMed] [Google Scholar]
- [33].Reid J, Svejstrup JQ, DNA damage-induced Def1-RNA polymerase II interaction and Def1 requirement for polymerase ubiquitylation in vitro, J Biol Chem 279(29) (2004) 29875–29878. [DOI] [PubMed] [Google Scholar]
- [34].Damodaren N, Van Eeuwen T, Zamel J, Lin-Shiao E, Kalisman N, Murakami K, Def1 interacts with TFIIH and modulates RNA polymerase II transcription, P Natl Acad Sci USA 114(50) (2017) 13230–13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhao L, Washington MT, Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases, Genes (Basel) 8(1) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Vaisman A, Woodgate R, Translesion DNA polymerases in eukaryotes: what makes them tick?, Crit Rev Biochem Mol Biol 52(3) (2017) 274–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Daraba A, Gali VK, Halmai M, Haracska L, Unk I, Def1 promotes the degradation of Pol3 for polymerase exchange to occur during DNA-damage--induced mutagenesis in Saccharomyces cerevisiae, PLoS Biol 12(1) (2014) e1001771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Giot L, Chanet R, Simon M, Facca C, Faye G, Involvement of the yeast DNA polymerase delta in DNA repair in vivo, Genetics 146(4) (1997) 1239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim N, Mudrak SV, Jinks-Robertson S, The dCMP transferase activity of yeast Rev1 is biologically relevant during the bypass of endogenously generated AP sites, DNA Repair (Amst) 10(12) (2011) 1262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Makovets S, Herskowitz I, Blackburn EH, Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions, Mol Cell Biol 24(9) (2004) 4019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Paeschke K, Bochman ML, Garcia PD, Cejka P, Friedman KL, Kowalczykowski SC, Zakian VA, Pif1 family helicases suppress genome instability at G-quadruplex motifs, Nature 497(7450) (2013) 458–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Stundon JL, Zakian VA, Identification of Saccharomyces cerevisiae Genes Whose Deletion Causes Synthetic Effects in Cells with Reduced Levels of the Nuclear Pif1 DNA Helicase, G3 (Bethesda) 5(12) (2015) 2913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jahn LJ, Mason B, Brogger P, Toteva T, Nielsen DK, Thon G, Dependency of Heterochromatin Domains on Replication Factors, G3 (Bethesda) 8(2) (2018) 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Reese JC, The control of elongation by the yeast Ccr4-not complex, Biochim Biophys Acta 1829(1) (2013) 127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kruk JA, Dutta A, Fu J, Gilmour DS, Reese JC, The multifunctional Ccr4-Not complex directly promotes transcription elongation, Genes Dev 25(6) (2011) 581–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dutta A, Babbarwal V, Fu J, Brunke-Reese D, Libert DM, Willis J, Reese JC, Ccr4-Not and TFIIS Function Cooperatively To Rescue Arrested RNA Polymerase II, Mol Cell Biol 35(11) (2015) 1915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Denis CL, Chiang YC, Cui Y, Chen J, Genetic evidence supports a role for the yeast CCR4-NOT complex in transcriptional elongation, Genetics 158(2) (2001) 627–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fiumara F, Fioriti L, Kandel ER, Hendrickson WA, Essential role of coiled coils for aggregation and activity of Q/N-rich prions and PolyQ proteins, Cell 143(7) (2010) 1121–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Alberti S, Halfmann R, King O, Kapila A, Lindquist S, A systematic survey identifies prions and illuminates sequence features of prionogenic proteins, Cell 137(1) (2009) 146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ, A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease, Nat Genet 37(5) (2005) 526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Duennwald ML, Jagadish S, Giorgini F, Muchowski PJ, Lindquist S, A network of protein interactions determines polyglutamine toxicity, Proc Natl Acad Sci U S A 103(29) (2006) 11051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Serpionov GV, Alexandrov AI, Ter-Avanesyan MD, Distinct mechanisms of mutant huntingtin toxicity in different yeast strains, FEMS Yeast Res 17(1) (2017). [DOI] [PubMed] [Google Scholar]
- [53].Kryndushkin D, Pripuzova N, Burnett BG, Shewmaker F, Non-targeted identification of prions and amyloid-forming proteins from yeast and mammalian cells, J Biol Chem 288(38) (2013) 27100–27111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Nizhnikov AA, Alexandrov AI, Ryzhova TA, Mitkevich OV, Dergalev AA, Ter-Avanesyan MD, Galkin AP, Proteomic screening for amyloid proteins, PLoS One 9(12) (2014) e116003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Duennwald ML, Jagadish S, Muchowski PJ, Lindquist S, Flanking sequences profoundly alter polyglutamine toxicity in yeast, Proc Natl Acad Sci U S A 103(29) (2006) 11045–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].van der Weegen Y, Golan-Berman H, Mevissen TET, Apelt K, Gonzalez-Prieto R, Goedhart J, Heilbrun EE, Vertegaal ACO, van den Heuvel D, Walter JC, Adar S, Luijsterburg MS, The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II, Nat Commun 11(1) (2020) 2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Okuda M, Nakazawa Y, Guo C, Ogi T, Nishimura Y, Common TFIIH recruitment mechanism in global genome and transcription-coupled repair subpathways, Nucleic Acids Res 45(22) (2017) 13043–13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nakazawa Y, Hara Y, Oka Y, Komine O, van den Heuvel D, Guo C, Daigaku Y, Isono M, He Y, Shimada M, Kato K, Jia N, Hashimoto S, Kotani Y, Miyoshi Y, Tanaka M, Sobue A, Mitsutake N, Suganami T, Masuda A, Ohno K, Nakada S, Mashimo T, Yamanaka K, Luijsterburg MS, Ogi T, Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair, Cell 180(6) (2020) 1228–1244 e24. [DOI] [PubMed] [Google Scholar]
- [59].Nakazawa Y, Sasaki K, Mitsutake N, Matsuse M, Shimada M, Nardo T, Takahashi Y, Ohyama K, Ito K, Mishima H, Nomura M, Kinoshita A, Ono S, Takenaka K, Masuyama R, Kudo T, Slor H, Utani A, Tateishi S, Yamashita S, Stefanini M, Lehmann AR, Yoshiura K, Ogi T, Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair, Nat Genet 44(5) (2012) 586–92. [DOI] [PubMed] [Google Scholar]
- [60].Swaney DL, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan NJ, Villen J, Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation, Nat Methods 10(7) (2013) 676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Schwertman P, Lagarou A, Dekkers DH, Raams A, van der Hoek AC, Laffeber C, Hoeijmakers JH, Demmers JA, Fousteri M, Vermeulen W, Marteijn JA, UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair, Nat Genet 44(5) (2012) 598–602. [DOI] [PubMed] [Google Scholar]
- [62].Zhang X, Horibata K, Saijo M, Ishigami C, Ukai A, Kanno S, Tahara H, Neilan EG, Honma M, Nohmi T, Yasui A, Tanaka K, Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair, Nat Genet 44(5) (2012) 593–7. [DOI] [PubMed] [Google Scholar]
- [63].Steurer B, Marteijn JA, Traveling Rocky Roads: The Consequences of Transcription-Blocking DNA Lesions on RNA Polymerase II, J Mol Biol 429(21) (2017) 3146–3155. [DOI] [PubMed] [Google Scholar]