

Abstract
Synthetic biology enables microbial hosts to produce complex molecules that are otherwise produced by organisms that are rare or difficult to cultivate, but the structures of these molecules are limited to those formed by chemical reactions catalyzed by natural enzymes. The integration of artificial metalloenzymes (ArMs) that catalyze unnatural reactions into metabolic networks could broaden the cache of molecules produced biosynthetically by microorganisms. We report an engineered microbial cell expressing a heterologous biosynthetic pathway, which contains both natural enzymes and ArMs, that produces an unnatural product with high diastereoselectivity. To create this hybrid biosynthetic organism, we engineered Escherichia coli (E. coli) with a heterologous terpene biosynthetic pathway and an ArM containing an iridium-porphyrin complex that was transported into the cell with a heterologous transport system. We improved the diastereoselectivity and product titer of the unnatural product by evolving the ArM and selecting the appropriate gene induction and cultivation conditions. This work shows that synthetic biology and synthetic chemistry can produce, together with natural and artificial enzymes in whole cells, molecules that were previously inaccessible to nature.
Keywords: artificial metalloenzymes, biosynthesis, engineered microorganism
Introduction
Molecules produced by natural biosynthetic pathways are the inspiration or the actual matter in many products, ranging from medicines and agrochemicals to commodity chemicals and biofuels. Although many molecules with structures that are the envy of chemists are produced by these natural or engineered biosynthetic pathways, the structures remain limited by the available chemical reactions catalyzed by natural enzymes. If artificial enzymes could be incorporated into such biosynthetic pathways, then unnatural products could be produced from a combination of nature’s reactions and chemists’ imaginations.
Recently, artificial metalloenzymes (ArMs) have been created that catalyze a series of unnatural reactions2–13. However, most of these reactions do not occur on the types of molecules that could be produced by biosynthesis. In contrast, an iridium-containing analog of a heme enzyme catalyzes the cyclopropanation of unconjugated, hindered, terminal alkenes that are present in natural products, such as terpenes4. This reactivity provides the potential to create an artificial biosynthetic pathway that combines natural pathways manipulated by synthetic biology with the catalysis created by ArMs.
The assembly of artificial metalloenzymes has advanced from in vitro reconstitution of purified proteins with organometallic complexes to assembly of ArMs in the periplasm13,14 or on the outer-membrane15,16 of living cells. However, methods to assemble ArMs in more complex intracellular environments are lacking17,18. We hypothesized that artificial cytochrome P450s containing synthetic heme derivatives could be generated in the cytoplasm by employing the natural heme transport machinery19 and the affinity of hemoproteins to heme derivatives. Thus, artificial cytochrome P450s are potential ArMs for conducting unnatural biotransformations in vivo in a fashion that enables the assembly of artificial biosynthetic pathways.
Terpenes are the largest and most structurally diverse family of natural products20. Their core skeletons are assembled in one step and modified by a suite of tailoring enzymes including cytochrome P450s21. While native P450s typically catalyze oxidations of terpenes, extensively engineered P450s, such as P450BM322, and artificial P450s substituted with abiotic metalloporphyrins, could catalyze unnatural transformations3–5, 23,24 that would introduce an artificial dimension to the diversification of terpene structures. Here, we report the combination of limonene biosynthesis by a series of heterologously expressed natural enzymes and the catalysis by an artificial metalloenzyme Ir-CYP119 (CYP119 containing Ir(Me)MPIX as cofactor; CYP119, a P450 enzyme from Sulfolobus solfataricus; MPIX, mesoporphyrin IX) to produce a terpenoid that was previously inaccessible in a microbial host. This unnatural combination of synthetic biology and synthetic chemistry mimics the production of a terpene and subsequent conversion of this hydrocarbon to a terpenoid by P450 enzymes during natural biosynthesis.
Our overall strategy for production of the unnatural terpenoid is depicted in Fig. 1. The artificial metalloenzyme is assembled in E. coli with the aid of a cofactor transport system and overexpression of the apo-enzyme. In concert, the same E. coli is engineered with the pathway for production of a natural product that serves as the substrate for the artificial metalloenzyme. This combination of natural and unnatural reactions in the same cell would produce an unnatural derivative of a classic natural product.
Figure 1.

A schematic representation of a reaction sequence to produce an unnatural terpenoid by combining a heterologously expressed natural biosynthetic pathway and an ArM in E. coli.
Results And Discussion
Assembling the ArM
in vivo The first major challenge facing this integration of an artificial metalloenzyme with a cellular biosynthetic process is to assemble such enzymes in the cytoplasm25. Previously, we reported the in vitro construction of Ir-CYP119 by incorporation of Ir(Me)MPIX into the heme binding site of the purified apo-form of CYP119. To construct this artificial enzyme in a cell, the cofactor must enter into the cytoplasm from the medium through the cell membranes. Metal-substituted heme derivatives have been reported to be transported across cell membranes when the outer-membrane receptor ChuA is overexpressed26–28. Thus, we first considered whether the co-expression of such a heme transporter and CYP119 would enable the artificial cofactor Ir(Me)MPIX to be transported into E. coli and incorporated into the apo-CYP119 mutant in the cytoplasm.
To test the feasibility of assembling Ir-CYP119 and catalyzing a reaction in E. coli, we studied the cyclopropanation of the monoterpenoid (−)-carvone. We used the diastereoselectivity of the reaction as a parameter to distinguish any background reaction or reaction catalyzed by free cofactor from the reaction catalyzed by the assembled artificial metalloenzyme. During our previous study, Ir-CYP119 generated from in vitro reconstitution of Ir(Me)MPIX and purified apo-CYP119 mutants catalyzed the cyclopropanation of (−)-carvone with EDA (ethyl diazoacetate) to give an 8 : 1 : 1 : 1 ratio of diastereomeric products. The major diastereomer of this reaction is a minor diastereomer of the 1 : 1 : 3 : 3 ratio of diastereomers formed from the analogous reaction catalyzed by the free iridium porphyrin complex4. Thus, a reaction that forms a mixture of diastereomers resembling the ratio produced by the pure ArM in vitro would denote reaction by the assembled ArM in the cell.
To this end, E. coli BL21(DE3) cells co-expressing the heme transporter ChuA and the CYP119 mutant were supplemented with 10 µM Ir(Me)MPIX, and the cell pellets containing in vivo-assembled Ir-CYP119 were directly employed for catalyzing the reaction of (−)-carvone with EDA after re-suspension in reaction buffer. However, the diastereomeric ratio (dr) of the whole-cell reaction was 1.7 : 1.0 : 2.7 : 3.3 (Supplementary Fig. 1a), indicating that the majority of the product of the whole-cell system was formed from a reaction catalyzed by the free iridium porphyrin complex, rather than by an assembled ArM. In addition, no product was detected when E. coli cultures were incubated in the absence of Ir(Me)MPIX (Supplementary Fig. 1a).
To reduce interference from the background reactivity of the free iridium porphyrin, we evaluated the reaction of (−)-carvone with EDA catalyzed by cells grown in the presence of Ir(Me)MPIX in concentrations ranging from 10 µM to 0.02 µM. These experiments showed that the dr of the cyclopropane product was much higher (23 : 3.5 : 1.0 : 1.2) when the concentration of Ir(Me)MPIX was 0.02 µM (Fig. 2a). We surmised that the lower concentrations of Ir(Me)MPIX reduced the amount of product formed by a background reaction catalyzed by the cofactor unbound to CYP119. Control experiments showed that the dr was low for products formed in E. coli expressing ChuA without co-expression of CYP119 (Supplementary Fig. 1b). Under the conditions of this control experiment, the cyclopropanation would be catalyzed by the free iridium cofactor.
Figure 2.

The whole cells containing Ir-CYP119 catalyze the cyclopropanation of (−)-carvone with high diastereoselectivity. a, Decreasing the concentration of Ir(Me)MPIX increased the diastereoselectivity of whole-cell reactions. The identity of the major diastereomer has been reported previously4, the conformations of minor diastereomers are not known. b, E. coli cells co-expressing CYP119 (with lacUV5 promoter) and the hug operon could maintain the high diastereoselectivity under low expression level of CYP119. E. coli cells containing CYP119 (with lacUV5 promoter) and an RFP-coding plasmid are shown as control (RFP, Red Fluorescent Protein). Both strains were cultivated in the presence of 0.1 µM Ir(Me)MPIX. c, The percentages of added iridium (0.1 µM) that were recovered from the subcellular fractions of E. coli cells. Hug strain: E. coli co-expressing the HUG system and CYP119 (with lacUV5 promoter). Control strain: E. coli co-expressing RFP and CYP119 (with lacUV5 promoter). P1-P4 are the four diastereomeric products numbered in the order of elution by GC (gas chromatography). Selectivity in the plots corresponds to the ratio of each diastereomer versus the total of the four diastereomeric products. All data are shown as the average from three biological replicates, with error bars indicating 1 standard deviation.
Next, we sought to combine the Ir-CYP119 with a natural biosynthetic pathway to produce an unnatural terpenoid. The analysis of Ir(Me)MPIX uptake for cyclopropanation in whole cells described above was conducted with carvone because we had shown previously that cyclopropanation of this terpenoid occurred in high yield and high diastereoselectivity4. However, the biosynthesis of (−)-limonene, which has the same carbon skeleton as (−)-carvone, occurs with much higher titer29,30, so we sought to assess the potential to combine heterologous biosynthesis with artificial metalloenzymes by generating unnatural derivatives of limonene.
Initial attempts to express a heterologous biosynthetic pathway to form limonene in concert with expression of CYP119 regulated by a T7 promoter caused the titer of limonene to decrease drastically from 188 mg/L without co-expression of CYP119 to 28 mg/L with co-expression (Supplementary Fig. 2a). To restore the production of limonene, we expressed CYP119 with a weaker promoter. However, when the reaction of exogenously added (−)-carvone with EDA was conducted with cells co-expressing ChuA and CYP119 with the weaker lacUV5 promoter, the cyclopropane was formed with low diastereoselectivity, again implying that assembly of the artificial metalloenzyme was inefficient (Supplementary Fig. 3). Thus, we sought conditions that would assemble the artificial metalloenzyme more efficiently at low expression levels of CYP119 to generate product with high diastereoselectivity.
To increase the efficiency of the cellular uptake of Ir(Me)MPIX, we replaced the ChuA transporter with a heme transport system encoded by the hug operon from Plesiomonas shigelloides, which has been used less frequently than ChuA, but has been shown previously to increase expression yields of hemoglobin to unusually high levels with heme supplementation31,32. Reactions of (−)-carvone with EDA catalyzed by E. coli cells expressing the HUG transport system, in concert with CYP119 (expressed from the lacUV5 promoter), formed the cyclopropane product with a high diastereoselectivity of 82% (the % diastereoselectivity in this case is the percentage of the major diastereomer in the 24.0 : 3.1 : 1.0 : 1.2 ratio of diastereomeric products) under conditions that are identical to those of the reactions with cells expressing the ChuA transporter (Fig. 2b).
To elucidate the origin of the higher efficiency of the assembly of ArMs in cells expressing the HUG transport system, the distribution of iridium was analyzed by ICP-MS (inductively coupled plasma mass spectrometry) (Fig. 2c). In cells harboring CYP119 in the presence of 0.1 µM Ir(Me)MPIX, 65% of the iridium was located in the cytoplasm of the cells expressing the HUG transport system, while only 10% of the iridium was located in the cytoplasm of the cells expressing no transporter. Further, the cytoplasmic fraction isolated from the cells co-expressing the HUG transport system catalyzed the cyclopropanation of (−)-carvone with a diastereoselectivity of 80%, and the yield of cyclopropane was more than 13 times higher than that of the reaction catalyzed by the cytoplasmic fraction from cells expressing no transporter (Supplementary Fig. 4). These results indicate that the HUG system significantly enhances the uptake of Ir(Me)MPIX into the cytoplasm.
Cyclopropyl Limonene Biosynthesis
Having achieved the diastereoselective cyclopropanation of (−)- carvone with EDA in cells expressing CYP119, even at a relatively low level, we sought to integrate such reactivity with the biosynthesis of limonene from a simple sugar (Fig. 3a). Again, we assessed whether the expression of the artificial metalloenzyme would affect the production of limonene. The production of limonene was unaffected by the expression level of CYP119 achieved with the pJHA135 plasmid (Fig. 3b). Moreover, the limonene titer was unaffected by the presence of 0.1 µM Ir(Me)MPIX (Supplementary Fig. 2b). Finally, both Ir(Me)MPIX and cyclopropyl limonene did not inhibit cell growth within the concentration range relevant for our study (Supplementary Fig. 5).
Figure 3.

Combining limonene biosynthesis with ArM. a, A schematic diagram of E. coli strain that combines the limonene biosynthetic pathway and the ArM that catalyzes cyclopropanation of limonene. The string of genes at the top represents the plasmid harboring the mevalonate-based limonene biosynthetic pathway (pJBEI6410)29. The bottom one represents the plasmid harboring the HUG system and CYP119 mutant (pJHA135). b, (−)-Limonene was produced with high titer in E. coli strains expressing the limonene pathway/hug operon (pJBEI6410+pJHA047) and limonene pathway/hug operon/CYP119 (pJBEI6410+pJHA135), respectively. c, E. coli cells expressing Ir-CYP119 catalyzed the cyclopropanation of exogenously added (−)-limonene with diastereoselectivity distinct from that of the free cofactor and enhanced over that of the reaction in vitro. d, The cyclopropyl limonene was produced diastereoselectively from glucose in E. coli expressing the limonene pathway/hug operon/CYP119. P1-P4 are the four diastereomeric products numbered in the order of elution by GC. The selectivity of P1-P4 corresponds to the ratio of each diastereomer versus the total of the four diastereomeric products. Ptotal is the sum of four diastereomeric products. All data from whole-cell reactions are shown as the average from three biological replicates, with error bars indicating 1 standard deviation.
The cyclopropanation of limonene added exogenously to the whole-cell system catalyzed by E. coli expressing the hug operon and CYP119 in the M9-rich medium was then evaluated. These cells produced the cyclopropyl limonene with a diastereoselectivity of 52% (1.0 : 3.3 : 1.1 : 1.0 dr) (Fig. 3c). This ratio of diastereomers is distinct from that of the reaction catalyzed by free Ir(Me)MPIX (1.0 : 1.2 : 3.5 : 4.2 dr) and the diastereoselectivity was fortuitously higher than that of the reaction catalyzed by the same Ir-CYP119 mutant in vitro (1.0 : 2.4 : 2.3 : 2.9 dr) (Fig. 3c). We suggest that the higher diastereoselectivity in the whole-cell system results from a greater stability of the enzyme in vivo than in vitro.
To fully combine terpene production and artificial catalysis within the same cell, gene expression for limonene biosynthesis and CYP119 was induced concurrently with addition of Ir(Me)MPIX (Fig. 3d). After overnight incubation, the synthesis of cyclopropyl limonene was initiated by addition of EDA. Under these conditions, 250 µg/L of cyclopropyl limonene was produced after 4.5 h with diastereoselectivity (43%, 1.0 : 3.2 : 1.8 : 1.5 dr) that was within 10% of that of the whole-cell reaction with exogenously added limonene. This similarity in diastereoselectivity indicates that the cyclopropyl limonene is formed from the combination of the heterologous biosynthesis of limonene and cyclopropanation catalyzed by the ArM. To assess the possibility that formation of the cyclopropane was catalyzed by the Ir-CYP119 released from dead cells, the cells were separated from the culture by centrifugation. The catalytic activity of the supernatant and of resuspended cells was evaluated by the reaction of (−)-carvone and EDA. The formation of cyclopropyl carvone with resuspended cells occurred with high diastereoselectivity and a yield more than 10 times that of the reaction conducted with the supernatant (Supplementary Fig. 6). These data show that the product was produced primarily by intact cells.
Increasing diastereoselectivity and titer
To increase the diastereoselectivity and titer of the cyclopropyl limonene from the artificial pathway, we evolved the CYP119 mutant. We constructed a small library of CYP119 mutants containing additional changes to the amino acids around the binding site of the cofactor and tested the diastereoselectivity of these mutants for the cyclopropanation of limonene in whole cells (Supplementary Fig. 7). After three rounds of screening, mutant P/R256W/V254A (P designates the parent) catalyzed the cyclopropanation of limonene with a higher diastereoselectivity of 72% (1.0 : 9.6 : 1.7 : 1.1 dr) and a higher yield (Fig. 4a,b). This evolved mutant was then introduced into the E. coli strain containing the limonene pathway and hug operon to perform artificial production of cyclopropyl limonene. With the procedures previously developed (Fig. 3d), the titer and diastereoselectivity of the products formed from this new pathway were 265 µg/L and 59% (1.0 : 7.0 : 2.3 : 1.5 dr) respectively (Supplementary Fig. 8).
Figure 4.
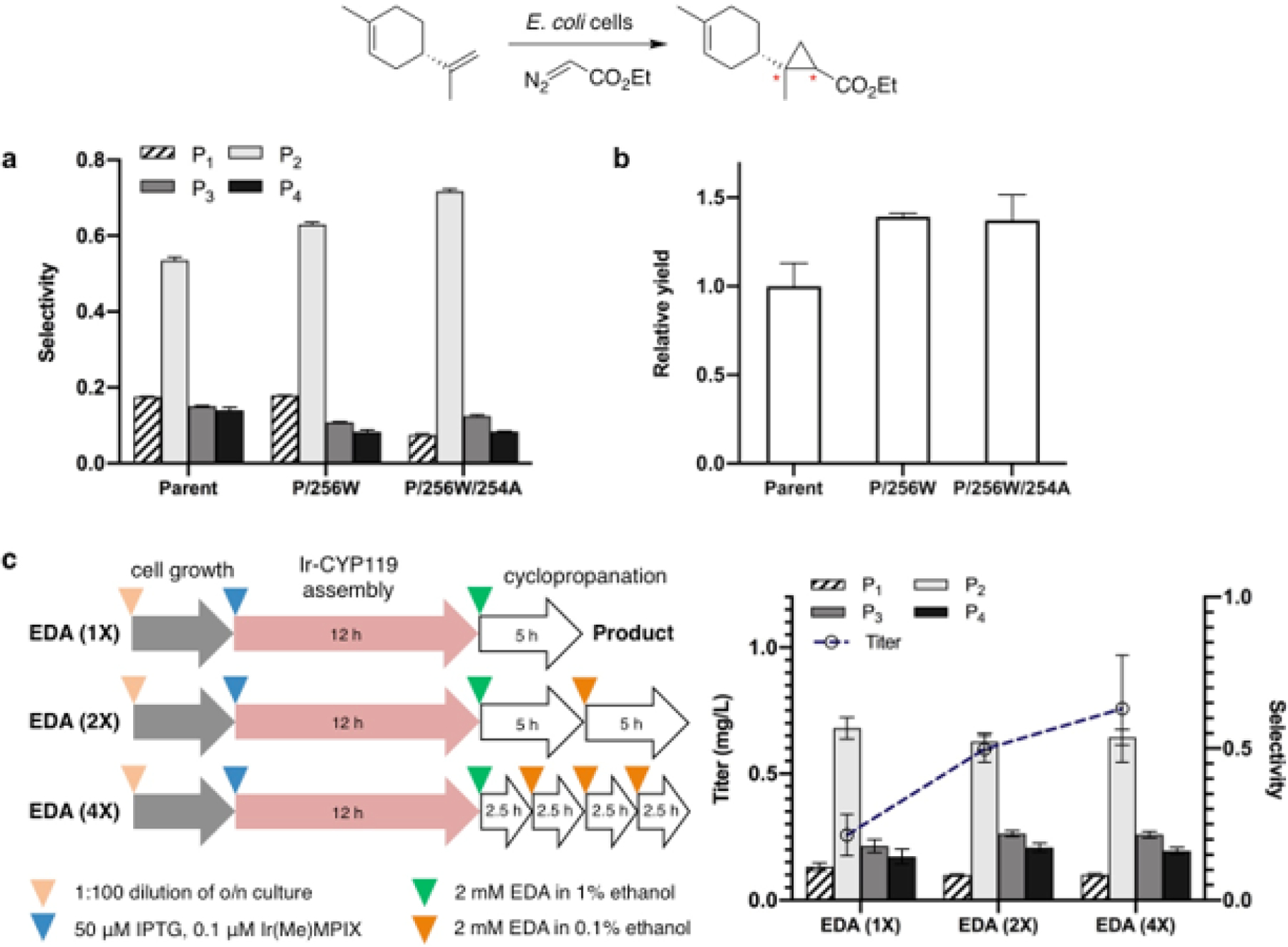
Increasing the diastereoselectivity and titer of cyclopropyl limonene by directed evolution and process optimization. a, Directed evolution of CYP119 mutants with higher diastereoselectivity. b, The evolved mutants also catalyzed the cyclopropanation in higher yields. The relative yield is the amount of cyclopropyl limonene formed from the whole-cell reaction catalyzed by evolved CYP119 mutants relative to that of the reaction with the parent mutant. c, The improved titer of cyclopropyl limonene with evolved CYP119 mutant (C317G, T213G, V254A, L155W, R256W) and batch-wise addition of EDA. The diagram shows different conditions used for the production of cyclopropyl limonene by the artificial biosynthetic pathway. The concentrations of reagents listed in the diagram are the final concentrations in culture media. All data are shown as the average from three biological replicates, with error bars indicating 1 standard deviation.
The conversion efficiency of limonene by natural P450s in heterologous hosts is known to be low30,33, and the titer of cyclopropyl limonene is limited, in part, by the volatility of limonene. The titer of limonene is just 2 mg/L when the cells are grown without an organic overlay, but is 145 mg/L when the cells are grown with a dodecane overlay (Supplementary Fig. 9). However, an overlay is used only if limonene is the final product; it would be incompatible with our system in which we must preserve limonene intracellularly as a substrate for the ArM. Thus, we sought to optimize the titer of cyclopropyl limonene by alternate methods. To do so, we investigated the potential toxicity of EDA to E. coli in the process of producing cyclopropyl limonene and investigated the effect of adding EDA portion-wise (Supplementary Fig. 10). We found that addition of EDA at an initial concentration of 2 mM and refeeding EDA three times over 10 h led to a titer of cyclopropyl limonene that was almost 3 times its initial value (from 258 ug/L to 760 µg/L) with little change in diastereoselectivity (54%, 1.0 : 6.4 : 2.6 : 1.9 dr) (Fig. 4c). Additional increases in product titer would likely be gained by further optimization of the protocol for feeding EDA, investigation of additional enzyme mutants, and manipulating the expression levels of proteins in the pathway.
Conclusions
Production of an unnatural product using a microorganism engineered with a heterologous natural product biosynthetic pathway, an ArM, and a heterologous cofactor transport system demonstrates the feasibility of creating organic molecules by artificial biosynthesis comprising synthetic biology and synthetic chemistry. One can envision this combination of biosynthesis and artificial metalloenzymes being applied to engineered pathways that produce unusual core structures, with enzymes that catalyze functionalization of C-H bonds to install abiotic groups at positions where P450s might operate naturally, or with the artificial metalloenzyme operating in the middle of a pathway followed by diversification of the abiotic product with evolved downstream enzymes. One can even envision creating reagents, such as diazoesters, biosynthetically based on known pathways to these species34. Such goals could be achieved in E. coli or in a range of organisms that produce diverse classes of natural products, such as polyketides or various alkaloids, thus opening a realm of biosynthesis beyond that limited by the scope of natural reactions.
Methods
General procedure for the artificial biosynthesis of cyclopropyl limonene
BL21(DE3) cells were transformed with a plasmid (pJBEI6410) coding for enzymes in limonene biosynthetic pathway and a plasmid (pJHA135 or pJHA170) encoding both the heme transport system HUG and the corresponding CYP119 mutant. Individual colonies from freshly transformed plates (incubated at 30 °C) were inoculated into 2 mL M9-rich medium supplemented with 100 μg/mL carbenicillin and 25 μg/mL kanamycin. The cultures were grown overnight at 30 °C, 200 rpm for 18 h. In a 50 mL glass culture tube, 10 mL of M9-rich medium was inoculated with 100 μL of overnight starter culture and incubated at 37 °C, 200 rpm to an OD600 of ~1. The cultures were then induced by adding 50 μM IPTG, at which time 2 μL of a 0.5 mM solution of Ir(Me)MPIX in DMSO was added to give a final Ir(Me)MPIX concentration of 0.1 μM. Upon induction, the incubation temperature was reduced to 30 °C, and expression was allowed to continue for 18 h at 200 rpm. To the 10 mL culture was added 21.2 μL EDA dissolved in 100 μL ethanol to give a final EDA concentration of 20 mM and a final ethanol concentration of 1% (v/v), and the mixture was further incubated at 30 °C, 200 rpm for 4.5 h. To analyze the titer of cyclopropyl limonene, aliquots of cell culture (1.4 mL) were removed from the tube and extracted with an equal volume of ethyl acetate. The organic layer (1 mL) was concentrated to 90 μL with a Speedvac, and to this concentrate was added 10 μL of ethyl acetate containing 1 g/L (−)-carvone (carvone used as internal standard). The final solution was analyzed by GC-MS.
Supplementary Material
Acknowledgements:
We thank D. P. Henderson for kindly sharing pHug21 plasmid. This work was supported by the Department of Energy, Laboratory Directed Research and Development funding, under contract DE-AC01–05CH11231 and National Science Foundation grant 2027943. ZL is an A*Star predoctoral fellow. We thank the College of Chemistry’s NMR facility for resources provided and the staff for their assistance. Instruments in CoC-NMR are supported in part by the NIH (S10OD024998). ICP-MS measurements were performed in the OHSU Elemental Analysis Core with partial support from NIH (S10RR025512).
Footnotes
Declarations
Competing interests: JDK has a financial interest in Amyris, Lygos, Demetrix, Maple Bio, Napigen, Apertor Pharma, Ansa Biotechnologies, Berkeley Yeast, and Zero Acre Farms.
Contributor Information
Jing Huang, Lawrence Berkeley National Laboratory.
Zhennan Liu, University of California, Berkeley.
Brandon Bloomer, University of California, Berkeley.
Douglas Clark, UC Berkeley.
Aindrila Mukhopadhyay, Lawrence Berkeley National Laboratory.
Jay Keasling, University of California, Berkeley.
John Hartwig, University of California, Berkeley.
Data availability:
All data are available in the main text or the Supplementary Information.
References
- 1.Cravens A, Payne J & Smolke CD Synthetic biology strategies for microbial biosynthesis of plant natural products. Nat. Commun 10, 2142 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwizer F et al. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev 118, 142–231 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Gu Y, Natoli SN, Liu Z, Clark DS & Hartwig JF Site-Selective Functionalization of (sp3)C−H Bonds Catalyzed by Artificial Metalloenzymes Containing an Iridium-Porphyrin Cofactor. Angew. Chem. Int. Ed 58, 13954–13960 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Key HM et al. Beyond Iron: Iridium-Containing P450 Enzymes for Selective Cyclo-propanations of Structurally Diverse Alkenes. ACS Cent. Sci 3, 302–308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dydio P, Key HM, Hayashi H, Clark DS & Hartwig JF Chemoselective, Enzymatic C–H Bond Amination Catalyzed by a Cytochrome P450 Containing an Ir(Me)-PIX Cofactor. J. Am. Chem. Soc 139, 1750–1753 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee A et al. An Enantioselective Artificial Suzukiase Based on the Biotin-Streptavidin Technology. Chem. Sci 7, 673 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abe S et al. Control of the Coordination Structure of Organometallic Palladium Complexes in an apo-Ferritin Cage. J. Am. Chem. Soc 130, 10512–10514 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Letondor C et al. Artificial Transfer Hydrogenases Based on the Biotin-(Strept)Avidin Technology: Fine Tuning the Selectivity by Saturation Mutagenesis of the Host Protein. J. Am. Chem. Soc 128, 8320 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Skander M et al. Artificial Metalloenzymes: (Strept)Avidin as Host for Enantioselective Hydrogenation by Achiral Biotinylated Rhodium-Diphosphine Complexes. J. Am. Chem. Soc 126, 14411 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Lin CC, Lin CW & Chan ASC Catalytic Hydrogenation of Itaconic Acid in a Biotinylated Pyrphos–Rhodium(I) System in a Protein Cavity. Tetrahedron: Asymmetry 10, 1887 (1999). [Google Scholar]
- 11.Philippart F et al. A Hybrid Ring-Opening Metathesis Polymerization Catalyst Based on an Engineered Variant of the Beta-Barrel Protein Fhua. Chem. - Eur. J 19, 13865 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Lo C, Ringenberg MR, Gnandt D, Wilson Y & Ward TR Artificial Metalloenzymes for Olefin Metathesis Based on the Biotin-(Strept)Avidin Technology. Chem. Commun 47, 12065 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Jeschek M et al. Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 537, 661–665 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Zhao J et al. Genetic Engineering of an Artificial Metalloenzyme for Transfer Hydrogenation of a Self-Immolative Substrate in Escherichia coli’s Periplasm. J. Am. Chem. Soc 140, 13171–13175 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Grimm AR et al. A Whole Cell E. coli Display Platform for Artificial Metalloenzymes: Poly(phenylacetylene) Production with a Rhodium–Nitrobindin Metalloprotein. ACS Catal 8, 2611–2614 (2018). [Google Scholar]
- 16.Heinisch T et al. E. coli surface display of streptavidin for directed evolution of an allylic deallylase. Chem. Sci 9, 5383–5388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khanna N, Esmieu C, Mészáros LS, Lindblad P & Berggren G In vivo activation of an [FeFe] hydrogenase using synthetic cofactors. Energy Environ. Sci 10, 1563–1567 (2017). [Google Scholar]
- 18.Song WJ & Tezcan FA A designed supramolecular protein assembly with in vivo enzymatic activity. Science 346, 1525 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Huang W & Wilks A Extracellular Heme Uptake and the Challenge of Bacterial Cell Membranes. Annu. Rev. Biochem 86, 799–823 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Brahmkshatriya PP & Brahmkshatriya PS in Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes (eds Ramawat Kishan Gopal & Mérillon Jean-Michel) 2665–2691 (Springer Berlin Heidelberg, 2013). [Google Scholar]
- 21.Helfrich EJN, Lin G-M, Voigt CA & Clardy J Bacterial terpene biosynthesis: challenges and opportunities for pathway engineering. Beilstein J. Org. Chem 15, 2889–2906 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coelho PS, Brustad EM, Kannan A & Arnold FH Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 339, 307–310 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Key HM, Dydio P, Clark DS & Hartwig JF Abiological Catalysis by Artificial Haem Proteins Containing Noble Metals in Place of Iron. Nature 534, 534 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dydio P et al. An Artificial Metalloenzyme with the Kinetics of Native Enzymes. Science 354, 102 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeschek M, Panke S & Ward TR Artificial Metalloenzymes on the Verge of New-to-Nature Metabolism. Trends Biotechnol 36, 60–72 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Lelyveld VS, Brustad E, Arnold FH & Jasanoff A Metal-Substituted Protein MRI Contrast Agents Engineered for Enhanced Relaxivity and Ligand Sensitivity. J. Am. Chem. Soc 133, 649–651 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bordeaux M, Singh R & Fasan R Intramolecular C(sp3)H amination of arylsulfonyl azides with engineered and artificial myoglobin-based catalysts. Biorg. Med. Chem 22, 5697–5704 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reynolds EW, Schwochert TD, McHenry MW, Watters JW & Brustad EM Orthogonal Expression of an Artificial Metalloenzyme for Abiotic Catalysis. ChemBioChem 18, 2380–2384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alonso-Gutierrez J et al. Metabolic engineering of Escherichia coli for limonene and perillyl alcohol production. Metab. Eng 19, 33–41 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Ascue Avalos GA, Toogood HS, Tait S, Messiha HL & Scrutton NS From Bugs to Bioplastics: Total (+)-Dihydrocarvide Biosynthesis by Engineered Escherichia coli. ChemBioChem 20, 785–792 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henderson DP, Wyckoff EE, Rashidi CE, Verlei H & Oldham AL Characterization of the Plesiomonas shigelloides Genes Encoding the Heme Iron Utilization System. J. Bacteriol 183, 2715–2723 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith BJZ, Gutierrez P, Guerrero E, Brewer CJ & Henderson DP Development of a Method To Produce Hemoglobin in a Bioreactor Culture of Escherichia coli BL21(DE3) Transformed with a Plasmid Containing Plesiomonas shigelloides Heme Transport Genes and Modified Human Hemoglobin Genes. Appl. Environ. Microbiol 77, 6703 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carter OA, Peters RJ & Croteau R Monoterpene biosynthesis pathway construction in Escherichia coli. Phytochemistry 64, 425–433 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Waldman AJ & Balskus EP Discovery of a Diazo-Forming Enzyme in Cremeomycin Biosynthesis. J. Org. Chem 83, 7539–7546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the Supplementary Information.