

Abstract
Alzheimer's disease and Parkinson's disease are two of the most prevalent and disabling neurodegenerative diseases globally. Both are proteinopathic conditions and while occasionally inherited, are largely sporadic in nature. Although the advances in our understanding of the two have been significant, they are far from complete and neither diagnosis nor the current practices in treatment and rehabilitation is adequately helpful. Animal models have historically found application as testing beds for novel therapeutics and continue to be valuable aids in pharmacological research. This review chronicles the development of those models in the context of Alzheimer's and Parkinson's disease, and highlights the shifting paradigms in studying two human‐specific conditions in non‐human organisms.
Keywords: Alzheimer's disease, animal models, neurodegeneration, Parkinson's disease
Cellular and molecular changes in neuronal cells may induce proteinopathies, leading to Alzheimer's and/or Parkinson's diseases (AD and PD respectively). Both AD and PD are marked by the loss of neurons due to cell death and result in the loss of cognition and impaired movement of patients. This review discusses the etiology of AD and PD in terms of historical perspectives known for these diseases.

1. INTRODUCTION
Alzheimer's disease (AD) and Parkinson's disease (PD), first described in 1906 and 1817, respectively, are the two most prevalent neurodegenerative diseases in the world, with a rising trend in the number of people that are affected and ultimately die because of them. Alzheimer's disease remained among the top ten non‐communicable diseases affecting persons over the age of 50 while Parkinson's disease climbed the ranks, with a 6% greater rate of incidence in 2019 than in 1990. 1 The etiologies of these disorders are still largely unknown and diagnosis of PD and, until recently, AD is entirely clinical. Diagnostic criteria for the two, as well as other neurological disorders, are regularly updated to better guide the delineation of idiopathic conditions from those that present with similar clinical symptoms but have distinct pathogeneses. Even with advances in testing that now enable the detection of amyloid‐β in suspected AD 2 and dopaminergic deficit in suspected PD, 3 definitive diagnoses for both require postmortem evaluation of brain tissue.
As biological samples, postmortem tissues are inadequate sources of information for the pre‐ clinical and prodromal stages of a disease. 4 , 5 Sample collection in such cases also becomes ethically and practically challenging. However, the growing burden of neurodegenerative disease demonstrates the need for a system which can model the intricacies of complex diseases like AD and PD, and help in the identification of biomarkers, risk factors and drug targets to improve diagnosis and treatment. Animal models have long been the answer to the problems of replicability and reliability inherent in samples collected directly from the patient. In addition, they provide a more holistic view of multifactorial diseases that even the current technology in human cellular models of neurodegeneration 6 fails to match. The discovery of genetic mutations and pathological proteins such as amyloid‐β, Tau, and α‐synuclein (αS) that define AD and PD as proteinopathies has also driven the engineering of more robust animal models of these diseases, although no animal model has completely phenocopied either of the two. Most of them exhibit key parts of the neurodegenerative cascade, some to a greater extent than others, but whether the entire pathophysiology has or could be modelled in non‐human subjects remains unclear to this day. Even so, animal models have undeniably granted us important insights into brain function, and continue to serve as platforms for the testing of drugs and novel therapeutics.
Mounting evidence suggests a clinical and pathological overlap between AD and PD 7 , 8 , 9 (Figure 1) and transgenic models of synucleinopathy that simultaneously reflect AD‐reminiscent cognitive impairment 10 , 11 have even prompted genome‐wide association studies (GWAS) into establishing a possible genetic link between the two diseases. 12 , 13 However, being ultimately unique to human beings, modelling AD and PD in lesser animals is a steep learning curve that is the subject of this review. Through the discussion that follows, we aim to highlight not only how animal models have been instrumental in the explication of disease pathology but also how modelling practices have evolved over the years.
FIGURE 1.
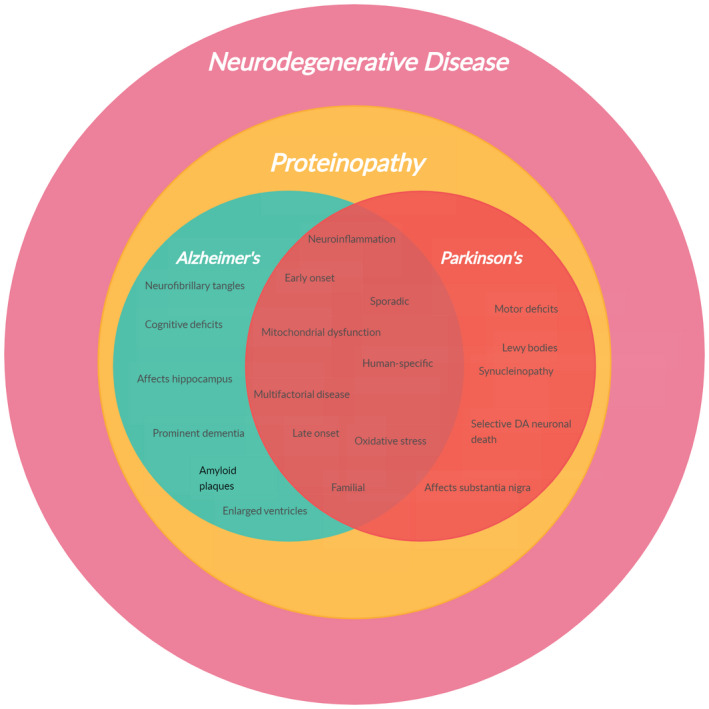
Clinical and pathological overlap between AD and PD
2. ANIMAL MODELS OF ALZHEIMER'S DISEASE
AD most commonly affects people above 60 years of age in what is called late‐onset (LOAD) or sporadic AD. Early‐onset (EOAD), familial AD is a much rarer occurrence although most of the animal models in AD research carry genetic mutations identified in EOAD patients. Over the last few years, however, the validity of data from these transgenic models with respect to the more prevalent sporadic disease has been increasingly questioned, and rodent models that bypass the APP gene mutation to develop age‐dependent behavioral changes are currently being investigated. 14
2.1. APP models
The APP gene encodes the amyloid precursor protein (APP) from which amyloid‐β (Aβ), the primary component of senile plaques, is derived. The amyloid cascade hypothesis that places Aβ at the center of AD pathology and upon which much of the present work on AD and animal models of AD rests, was established through a series of findings between 1984 and 1987. 15 , 16 , 17 , 18 , 19 At that time, genetic engineering techniques were still relatively novel and animal models of AD were scarce to none because no proof of this disease could be found in species other than humans. Subsequently, neuritic plaques and cerebrovascular amyloid deposits found in non‐human aged mammals began to be reported, 20 but in 1989 Rapoport theorized that AD was phylogenetically novel and specific to human beings. 21 Indeed, there exists a body of evidence separating amyloid and tauopathies found in primates from the neurodegenerative manifestation of classical AD, 22 and primate participation in AD research has received insufficient attention. The 1990s welcomed a surge in scientific intrigue about this disease and three pathogenic mutations on the APP gene were reported within the same year. 23 , 24 , 25 Then, in 1995, the first mouse model carrying the third v717F mutation and recapitulating many of the pathological hallmarks, including thioflavin‐S‐positive Aβ deposits, neuritic plaques, astrocytosis, microgliosis and even synaptic loss, was developed, 26 blazing a trail for transgenic mice in the study of neurodegenerative diseases. Other mouse models of mutant APP quickly followed the first (called PD‐APP mice after the PDGF promoter used to express the transgene), utilized different promoters 27 and even carried multiple mutations 28 to display elevated levels of Aβ, amyloid plaques, neuritic dystrophy, and gliosis. Behavioral deficits and other neuropathological and electrophysiological aspects related to AD were also commonly observed in these models. 29 Despite such similarities, plaque deposition in these models varied both temporally and spatially. The TgCRND8 mice which expressed multiple APP mutations, for example, showed amyloid deposition in the parenchyma by 3 months of age. 28 PD‐APP mice and Tg2576 mice, both carrying single mutations, differed in the composition of their plaques in that Tg2576 plaques were larger and denser and had fewer diffuse deposits than PD‐APP mice. 29 Furthermore, Aβ40 and Aβ42 (truncations of the Aβ peptide) were more or less equally elevated in Tg2576 models as opposed to a disproportionate elevation of Aβ42 in the PD‐APP model. 29
2.2. PSEN models
Linkage analysis and cloning studies gained momentum in the 1990s and led to the identification of a second novel gene in 1995, bearing missense mutations found in EOAD patients. 30 That same year, the third EOAD gene was found 31 and shown to be homologous to the second gene. Mutations on these genes, since named PSEN1 and PSEN2 respectively, were subsequently reported and more than 380 combined PSEN mutations are known today, making PSEN variants the leading cause of familial AD, with APP variants coming up second. 32 PSEN directs the expression of presenilins which comprise the catalytic subunit of the γ‐secretase enzyme complex that catalyzes intramembranous APP proteolysis, the site of which differs for EOAD‐associated presenilin variants, giving rise to the longer and more amyloidogenic Aβ42 peptide species. 33 Mutant PSEN1 transgenic lines were developed a year later in 1996, using many of the same promoters that were used in APP mice. 34 , 35 These mice produced elevated levels of Aβ42 with little to no change in Aβ40 levels, although single mutant lines were found without plaque formation. 36 Crossed with APP lines, however, plaque deposition was expedited and more extensive 37 , 38 with significant neuronal loss in the hippocampus. 39
By the 1990s, invertebrates were already being investigated as potential models of AD, with the introduction of nematodes (Caenorhabditis elegans) expressing the Aβ42 peptide in 1994. 40 This, of course, was 4 years before the completion of its genome sequencing led to the discovery of the C. elegans APP homolog, apl‐1, which is expressed in multiple tissue types and is now known to be essential for viability of the organism. 41 Loss of apl‐1, as well as its overexpression, produces adverse developmental effects, including larval lethality, which may be rescued by the neuronal expression of this protein's extracellular domain. 41 A reduction in the activity of sel‐12, a presenilin homolog discovered in 1995 as a suppressor of a lin‐12 gain‐of‐function mutation, 42 has also been reported to partially rescue apl‐1 lethality, generally mimicking the regulatory relationship between human PSEN and APP. 41 More recently, the effects of age‐altered metal homeostasis on the deposition of Aβ, as well as other late‐onset symptoms like paralysis, have been studied in C. elegans models of AD. 43 Directed expression of the human Aβ peptide in Drosophila, despite the presence of the APP‐homolog Appl, has been used to study Aβ toxicity in vivo, where it causes locomotive and cognitive defects in the flies. 44
2.3. MAPT models
Reports of Aβ‐dependent synaptic function impairment, synaptic loss, and dystrophic neurites in mouse APP and APP/PSEN1 models notwithstanding, the lack of neuronal loss and neurodegeneration seen in human AD could not be adequately explained in these animals. In the mid‐1980s, even before amyloid cascade was the leading hypothesis in the modelling of AD, families diagnosed with heritable fronto‐ temporal dementia (FTD; earlier more popular as Pick's disease 45 ) had revealed evidence of Tau inclusions containing hyperphosphorylated Tau. 46 In 1997, this pathological feature was termed ‘tauopathy’ 47 and the following year mutations in the microtubule‐associated protein tau (MAPT) gene were reported. 48 , 49 , 50 These mutations in FTD patients seemed to manifest clinically in a variety of ways that included changes in behavior and impairment of speech, memory and motor functions, and pathologically as the neurodegeneration of multiple neuronal systems. 51 Six isoforms of Tau are known to be formed through alternate splicing 52 —three containing 3 microtubule‐binding repeats (3R Tau) and three containing 4 of those repeats (4R Tau). The ratio between these isoforms is essential to prevent neurodegeneration and dementia in adult humans. Tauopathy‐related MAPT mutations alter the splicing of exon 10, causing an imbalance in this ratio with the overproduction of 4R Tau. 49 Functionally, Tau was characterized in 1977 as a contributor to axonal stability where only the phosphorylated protein could bind the axonal microtubules 53 and in 1986, hyperphosphorylated Tau was found to be abundantly present in the neurofibrillary tangles (NFTs) that had been established as a pathological hallmark of AD. 54 However, no causal relationship between the state of its phosphorylation and its aggregation in AD patients has been sufficiently described to date. The first transgenic mice to model a tauopathy expressed 4R, the largest tau isoform and the most natural substrate for hyperphosphorylation but showed little to no NFT formation. 55 Contrastingly, mice expressing the mutated P301L protein developed behavioral and motor deficits in addition to the age‐ and dose‐dependent formation of NFTs across multiple regions of the brain and spinal cord. 56 It is important to note, however, that while Tau clearly participates in the pathogenesis of AD, Tau mutations that could explain its aberrant behavior were never explicitly associated with this disease. Nevertheless, the FTD mutation, P301L, was extensively used to study MAPT pathology and its role in neurodegeneration. Many important discoveries were made in this process—the onset of memory loss in mice models before the development of significant NFT pathology and the success of transgene suppression in halting neuronal loss but not the formation of NFTs suggested that not NFTs but another toxic MAPT intermediate could be responsible for the neurodegeneration and memory deficit seen in AD. 57 In general, NFT pathology is seen to progress hierarchically, spreading gradually from the transentorhinal cortex to the hippocampus and then to other cortical regions in a cell‐to‐cell transfer that is reminiscent of AD. 58 , 59 , 60 Compared with Aβ deposition, Tau pathology is believed to occur at a later stage but correlates better with cognitive decline in AD patients. 61 , 62 In Drosophila, overexpression of mutant or even wild‐type human Tau causes age‐dependent neurodegeneration and early death. Several determinants of Tau toxicity like kinases, phosphatases, apoptotic regulators, and cytoskeleton proteins have been identified in vivo using large‐scale screens based on such model neurodegenerative phenotypes. 63 Although there exist certain structural differences between human Tau and fly Tau, many phosphorylation sites that are present in MAPT are conserved throughout the Tau family.
2.4. Neuroinflammation in AD
For a long time, neuroinflammation as seen in APP/Aβ or Tau models was thought to be a secondary effect of plaque and NFT accumulation, although neither pathology, either independently or in complex, could adequately explain the progression of AD in humans. 64 Eventually, growing evidence of inflammatory responses in patient brains, postmortem tissues, and even preclinical models of AD 65 , 66 have suggested that neuroinflammation plays a more central role in AD pathogenesis. Microglial activation and cytokine release is thought to be responsible for neuroinflammation, and therefore it cannot be assumed that this response is exclusive to AD; in fact, numerous studies have revealed an increase in inflammation markers in the brains of those diagnosed with PD, 67 Huntington's disease, 68 traumatic brain injuries, 69 and amyotrophic lateral sclerosis. 70 However, further research into this phenotype has shown that not only is this inflammation associated with neuronal loss but it also exacerbates both amyloid and NFT pathologies and could potentially be what links the early signs of Aβ accumulation and the late‐stage development of Tau tangles. 71 It has been hypothesized that microglial activation is, in reality, driven by the building levels of insoluble Aβ. 72 In the early 2000s, the induction of gliosis resulted in at least partial clearance of Aβ deposits and rescue of some of the symptoms in model systems 73 but prolonged activation was shown to reverse those effects with a gradual decline in microglial capacity to bind and process Aβ 74 but not its capacity to produce cytokines. 75 These pro‐inflammatory cytokines, along with other neurotoxins of microglial origin, are thought to contribute to neuroinflammation and eventually neurodegeneration.
Whatever the transgene or peptide, the establishment of disease or disease phenotypes in animal models is fundamentally dependent on their overexpression. This in itself contradicts the development of AD in humans which, so far, has not been linked to increases in genetic expression. How far this difference affects what we know about the disease is still a matter of debate.
3. ANIMAL MODELS OF PARKINSON'S DISEASE
PD manifests pathologically as the progressive loss of nigrostriatal dopaminergic neurons and formation of αS‐rich aggregates called Lewy bodies and Lewy neurites. Like with AD, a very low percentage (15%) of patients globally report a family history of PD‐related symptoms, while the rest are classified as sporadic. However, no single gene mutation has been shown to have complete penetrance in PD, and there's a real possibility that multiple genetic factors synergistically increase the risk of both familial and sporadic PD. 76 The physiological manifestations are heterogenous but clinically apparent as motor—including bradykinesia, akinesia, tremors and postural difficulties—and non‐motor symptoms like disturbed sleep, anxiety, depression, and cognitive deficits, which often occur prior to the motor symptoms. 76
3.1. Neurotoxin models
3.1.1. Reserpine
PD was first described in 1817, almost a century before AD, and until the implication of the SNCA gene in the late‐90s, the risk factors for this disease were believed to be mostly, if not entirely, environmental. The discovery of reserpine, a plant extract traditionally used in the treatment of insanity, associated fever, and snakebites, 77 in the 1950s paved the way for the development of the earliest rodent models of PD. In ‘reserpinized’ animals, transmitter depletion, chiefly that of 5‐hydroxytryptamine among other catechol amines, 78 occurs in the adrenergic system, knocking them into a ‘tranquilized state’ characterized by the slowing of movement and other parkinsonian symptoms. The rescue of these symptoms, for the first time in 1957, by the administration of 3,4‐dihydroxyphenylalanine (commercially, L‐DOPA), a natural precursor of the transmitter amines that could cross the blood‐brain‐ barrier (BBB), was a breakthrough in pharmacological research into PD. 79 Soon after, this effect was replicated in human subjects 80 and established reserpine rats as robust screens for the symptomatic efficacy of novel drugs. Despite this, arguments built against the suitability of reserpine models in the study of PD, a disorder then thought to be orchestrated largely and selectively by the loss of dopamine which was only one among the class of neurotransmitters that were blocked by reserpine. Subsequently noradrenergic and serotonergic systems also began to be implicated in PD pathogenesis 81 ; however, since it was later elucidated that reserpine causes over 95% loss of striatal dopamine within hours of injection, much of the research focus remained on dopaminergic degeneration, with reserpine rats the subject of choice. Besides neurochemical deficits, the compound also affected basal ganglia structures 82 , 83 and induced akinesia and hind limb rigidity, but these effects were unfortunately transient, and not much can be said beyond the pre‐clinical validity of these models. 84
3.1.2. 6‐OHDA
The hydroxylated dopamine analogue 6‐OHDA was first identified as a toxin capable of inducing the degeneration of nigrostriatal dopaminergic neurons in 1968 85 and although it is now widely used in the study of Parkinsonian disorders, it is unable to cross the BBB and requires to be administered via direct injection into the brain. The site of injection varies, however, and is usually determined by the requirements of the study. Usually, to avoid inadvertent injury to the substantia nigra pars compacta (SNpc), where A9 dopaminergic neurons are found in large numbers, the toxin is injected into the median forebrain bundle (MFB) or the terminal striatum. For purposes of nigral drug target analysis, a single in‐dwelling cannula for both toxin and drug administration is often more practical. 86 , 87 Although its specifics remain poorly understood, the general mechanism of 6‐OHDA action involves the inhibition of the mitochondrial respiratory complex and the generation of reactive oxygen species (ROS) which create oxidative stress. 88 These events, catalyzed by iron and antagonized by iron chelators and antioxidants, closely mimic those of human PD 89 and lend considerable construct validity to 6‐OHDA models. Injected into the SNpc, 6‐OHDA produces rapid, but dose‐dependent neurodegeneration, marked by full or partial lesions 84 and accompanied by microglial activation, 90 and drug‐induced rotational behavior. 91 However, this pathology is still incomplete with respect to PD in that it does not noticeably affect other regions of the brain besides the basal ganglia. No proteinaceous aggregates resembling Lewy bodies can be detected in 6‐OHDA models either, and the neurodegeneration is much more rapid than it would be in PD patients. Nevertheless, the 6‐OHDA continues to facilitate preclinical drug discovery and validate their symptomatic efficacy in PD.
3.1.3. MPTP
In 1982, ‘synthetic heroin’ was found to cause detectable signs of advanced PD almost overnight in San Francisco patients. 92 Subsequently identified as 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP), this compound is not toxic by itself but is able to cross the BBB and is rapidly converted to 1‐methyl‐4‐phenylpyridinium (MPP+) 93 in the brain via a reaction catalyzed by monoamine oxidase B (MAO‐B), an enzyme that has been shown to inhibit 6‐OHDA pathology. 88 MPP+ is selectively taken up by dopaminergic neurons in the substantia nigra 94 , 95 and, in toxic concentrations, inhibits the mitochondrial complex I 96 and results in the production of ROS. Interestingly, MPTP‐induced parkinsonism had first been observed in humans and is perhaps why—coupled with the fact that rats were found to be resistant to MPTP 97 —closely‐related primate monkeys arose as the earliest non‐human models of this disease. 98 , 99 , 100 In primates, even upon moderate exposure, MPTP damages vesicular monoamine transporter type‐2 (VMAT‐2), which normally serves to store dopamine. 101 Cytosolic dopamine levels are thereby increased which, in turn, generate ROS and could be implicated in the retrograde degeneration of dopaminergic neurons. 101 A similar mechanism has, in fact, already been identified in humans, 102 and even though monkeys recapitulate almost the entire spectrum of motor symptoms seen in humans, respond to levodopa, and present with significant dopaminergic neurodegeneration in the SNpc, 103 not many research facilities are normally equipped to conduct studies on primates, and thus the mouse MPTP model has become a more affordable and accessible option. MPTP mice can be both acute or subacute models depending on the dose and frequency of injection and display acute or subacute loss of dopaminergic neurons in the SNpc. Subacute MPTP models even carry αS inclusions, which acute models do not, 104 but unlike the former, acute models develop certain motor deficits; however, those are transient and have limited applicability in behavioral studies. 105
3.2. Genetic models
Traditionally, PD has been characterized more in clinical terms than pathological. Since the first implication of a genetic culprit in the etiology of this disease, several PD risk loci have been identified and some pathological features common to both idiopathic and genetic PD (mainly Lewy inclusions and dystrophic neurites) are now considered hallmarks. To date, 23 genes—called PARK genes and numbered in the order of their identification—have been linked to PD; however, their penetrance and actual relevance in the context of both clinical and pathological disease are still burning questions. In truth, only a few of the PARK genes have reasonably confirmed inheritance patterns and pathogenic roles, while the involvement of the rest is largely unclear. Moreover, two or more of these genes have been mapped to the same molecular pathway, leading to the likelihood of oligogenic involvement in determining PD risk. 106 We will discuss only the most popular genetic models in this review.
3.2.1. SNCA (PARK1‐4) models
αS was originally isolated in 1993 from plaques found in AD brains and called NACP (non‐ amyloid component of plaque). 107 Its gene, SNCA, was later mapped to chromosome 4q21 108 but it was not until 1996 that SNCA was first found to cosegregate with familial, autosomal dominant PD. 109 The following year the first SNCA mutation, A53T, was reported 110 and αS was also independently identified as the principal component of Lewy bodies. 111 Other missense mutations were subsequently reported and today eight pathogenic αS mutations are known. Functionally, αS, a small 140aa protein, has been characterized as playing important roles in vesicle trafficking, docking, priming and fusion, neurotransmitter release and axonal transport, although the exact nature of these roles requires further elucidation. 112 The first transgenic mice to model αS‐mediated neurodegeneration expressed the wild‐type human protein and progressively developed both αS‐ and ubiquitin‐immunoreactive inclusions in multiple regions of the brain including the substantia nigra. 113 These changes were accompanied by some motor deficits resembling those of PD, but the inclusions observed were not of the fibrillar structure that is typical of Lewy inclusions and no significant loss of dopaminergic neurons was reported either. 113 Likewise, models carrying single autosomal dominant mutations like A53T, A30P and E46K also fail to recapitulate dopaminergic neurodegeneration, although they do form αS aggregates. 114 However, between 2000 and 2011, varying patterns and severity of αS and Lewy body pathology were observed in different models of mutated or wild‐type SNCA. Expression of truncated protein and the overexpression of wild‐type αS both resulted in some degree of nigro‐striatal dopaminergic neuron loss, 115 , 116 while in knock‐in/knock‐out models, only double knock‐out mice displayed dopaminergic degeneration. Replacement of endogenous αS with wild‐type or mutated human αS produced some striatal pathology, as made evident by increased levels of striatal catecholamines and metabolites, whereas doubly mutant transgenes displayed additional neurite dysptrophy and motor deficits. 117 Then in 2012, a new mouse model that displayed progressive formation of fibrillar αS inclusions with striking resemblance to Lewy bodies, nigral neurodegeneration, decrease in striatal dopamine, neuroinflammation as well as motor deficits entered the scene, carrying pre‐formed fibrils (PFF) that were produced in vitro and then injected directly into their brains. 118 Since then, a large number of PFF models have been generated using both rodents and non‐human primates, with varying sites (striatum, substantia nigra, and cortex), strains and doses of injection. 119 Evidence of retrograde transport templating endogenous αS has been observed in these models. 118
The presence of an endogenous SNCA gives mice the edge over other model candidates like C. elegans and Drosophila melanogaster, which require an artificial introduction of the gene. Human SNCA may be overexpressed in Drosophila to mimic to some degree human PD pathophysiology, including the development of proteinase‐K‐resistant and ubiquitin‐positive α‐synuclein aggregates. Most synucleinopathy symptoms are, in fact, more intense and develop quicker in flies than in other animals and all SNCA mutations currently known to us are reported to have been modelled in Drosophila. 120
3.2.2. LRRK2 (PARK8) models
The PARK8 locus was first linked to autosomal dominant PD in 2002 but characterized with low penetrance. 121 The gene was later identified as LRRK2 and a series of mutations in multiple domains of the protein were reported in 2004. 122 , 123 These included R1441C, Y1699C, and I2020T, which are among the seven pathogenic LRRK2 mutations known today. 124 LRRK2 function is not yet well understood, although putative roles in protein translation, axonal growth and brain aging have been associated with this protein, along with putative kinase activity based on its domain structure and sequence homology with existing kinases. 125 The G2019S mutation was reported a few years later and estimated to be the most common PD‐linked mutation, 126 followed by R1441C/G/H. Like SNCA, rodents carry an endogenous LRRK2 homolog which encodes a protein similar in structure and function to human LRRK2, and also have conserved dopaminergic neuroanatomical pathways. This has set the stage for a multitude of knock in/knock out models, R1441C knock in mice being some of the earliest animals to ever model the LRRK2 pathology. 127 However, no signs of αS aggregation were detected and the nigrostriatal tract also remained largely unaffected. 127 Contrastingly, a BAC‐transgenic model carrying the R1441G mutation showed neuritic inclusions, but no neurodegeneration. 128 Similar Tau pathology was observed in other BAC models expressing G2019S and wild‐type LRRK2 but without αS inclusions or overt neurodegeneration. 129 Adenovector‐based delivery of mutant and wild‐type LRRK2 also resulted in limited neurofilamental developments and little to no synucleinopathy. Thereafter, in a multi‐hit approach, viral vectors delivered human wild‐type αS to the substantia nigra of adult LRRK2 rats, inducing a significant dopaminergic neuron loss that is further increased in BAC G2019S LRRK2 rodents. 129 A53T‐mutant αS also induced a massive neurodegeneration of the substantia nigra but the copresence of the G2019S mutation seemed to have no synergistic effect on the pathophysiology.
Instead, G2019S knock‐in mice exhibited nearly a twofold increase in the severity of nigral synucleinopathy. 129
3.2.3. Parkin and PINK1 (PARK2, PARK6) models
In 1998, Kitada and colleagues became the first to identify mutations in Parkin and further characterize them as causes of an autosomal recessive form of juvenile parkinsonism (AR‐JP). 130 The Parkin protein is analogous in function to E3 ubiquitin‐protein ligases. The amino‐terminal domain of Parkin is required for substrate recognition, while the RING‐finger domain interacts with its corresponding E2 ubiquitin‐conjugating enzyme, UbcH7. Numerous studies in Drosophila, mice, and human cells suggest that Parkin, together with another pathologically significant protein, PINK1, is part of a common pathway that protects against the effects of damaged mitochondria. 131 The phosphate and tensin homolog‐induced putative kinase 1 gene (Pink1) is located on the short arm of chromosome 1. Valente et al. first identified two Pink1 homozygous mutations in 2004 in Sicilian subjects, which were subsequently associated with the autosomal recessive form of PD (AR‐PD). 132 More than 10 different mutations in this gene have since been reported to cause PD. Other than missense mutations, combinations of homozygous and heterozygous loss‐of‐function mutations commonly facilitate the onset of AR‐PD. Knocking out Pink1 has been the most common transgenic approach to engineering models of this gene. In the year 2006, Drosophila PINK1−/− models were used to establish that Pink1 knockouts have degenerated flight muscles in addition to mitochondrial damage and result in apoptosis, muscle degeneration, and sterility, specially in males. 133 In 2009, C. elegans models were explored to establish the antagonistic role of LRRK2, and PINK1; axon swelling and neurodegeneration were observed, which were rescued by the expression of either WT‐LRRK2 or WT‐PINK1, suggesting that PINK1 acts downstream of LRRK2. 134 In 2014, a study was funded by The Michael J. Fox Foundation for Parkinson's Research (MJFF) to generate novel rat models with targeted disruption of Pink1, which manifests mainly as DA neuronal loss in the substantia nigra, motor defects, proteinase K‐resistant LB formation, and mitochondrial damage. 135
4. iPSC MODELS AND FUTURE DIRECTIONS
The discovery and use of induced pluripotent stem cells (iPSCs) can be traced back to the early 2000s. Their greatest impact to date has been on pharmacological research, disease modelling, and drug screening. It is true that iPSCs present attractive advantages over animal models in terms of their ability to provide a complete or nearly‐complete replica of endogenous cellular systems, but given the high skill requirements, poor reproducibility, and large costs, it is still too soon to say whether they could replace animal models altogether. iPSCs have provided unique insights into the biochemical functions of pathogenic proteins in AD and PD. SNCA iPSC models have recapitulated αS inclusion pathology in dopaminergic neurons. iPSC models of LRRK2 have suggested that the protein plays a role in αS upregulation, regulates the expression of oxidative stress‐response genes, and contributes to mitochondrial dysfunction during the progression of PD. 136 It is also through iPSCs that the role of Parkin and PINK1 in mitochondrial regulation, and dopamine uptake, as well as release, were elucidated. 136 Brain organoids were developed from human midbrain tissues in 2017 but neuroblastoma cell lines and catecholaminergic cell lines like PC‐12 are more popularly used to model PD. 137 More recently, iPSCs derived from postmortem tissues are being used to directly model a diseased cell, and libraries of such iPSC models are being developed as preclinical testing resources. 138 iPSCs that model the effects of mutant APOE4 have been used to study its importance as a risk factor for AD. These cells display increased levels of Aβ and Tau, and induce GABA transmission. 139 iPSC technology has also made possible the development of neuronal precursors and, subsequently, astrocytes which can be used to study Aβ homeostasis, PSEN mutations, and cytokine release, among others. 136 iPSC products in combination with CRISPR‐Cas9 gene editing have started to find some applicability as therapeutic agents in the treatment of diseases. However, certain inherent risks and limitations, including the risk of tumorigenicity and the tendency to accumulate karyotypic aberrations over time, makes their transplantation into living humans a risky venture. 140 Nevertheless, iPSCs are more than a promising aid in the study of disease pathologies and if their challenges are appropriately overcome, may well be the key to understanding, preventing and curing many complex diseases.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
RB wrote the paper, AR, SN, SMI, contributed information, MT compiled and edited the review.
ACKNOWLEDGEMENTS
We thank all our peers at the Department of Biological Sciences at the Birla Institute of Science and Technology, who have been supportive throughout the entire process of putting this review together. We also sincerely apologize to the authors whose work we could not include due to limitations of the space.
Banerjee R, Rai A, Iyer SM, Narwal S, Tare M. Animal models in the study of Alzheimer's disease and Parkinson's disease: A historical perspective. Anim Models Exp Med. 2022;5:27–37. doi: 10.1002/ame2.12209
Funding information
SMI is funded by Department of Science and Technology, Science and Engineering Research Board Early Career grant to MT. SN is supported from Birla Institute of Technology and Science PhD student fellowship at Biological Sciences.
REFERENCES
- 1. GBD 2019 Diseases and Injuries Collaborators . Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396:1204‐1222. doi: 10.1016/S0140-6736(20)30925-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weller J, Budson A. Current understanding of Alzheimer's disease diagnosis and treatment. F1000Res. 2018;7:F1000 Faculty Rev‐1161. doi: 10.12688/f1000research.14506.1 [DOI] [Google Scholar]
- 3. Rizek P, Kumar N, Jog MS. An update on the diagnosis and treatment of Parkinson disease. CMAJ. 2016;188(16):1157‐1165. doi: 10.1503/cmaj.151179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McDade E, Bateman RJ. Stop Alzheimer's before it starts. Nature. 2017;547(7662):153‐155. doi: 10.1038/547153a [DOI] [PubMed] [Google Scholar]
- 5. Lang AE, Espay AJ. Disease modification in Parkinson's disease: current approaches, challenges, and future considerations. Mov Disord. 2018;33(5):660‐677. doi: 10.1002/mds.27360 [DOI] [PubMed] [Google Scholar]
- 6. Liu C, Oikonomopoulos A, Sayed N, Wu JC. Modeling human diseases with induced pluripotent stem cells: from 2D to 3D and beyond. Development. 2018;145(5):dev156166. doi: 10.1242/dev.156166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Han Z, Tian R, Ren P, et al. Parkinson's disease and Alzheimer's disease: a Mendelian randomization study. BMC Med Genet. 2018;19:215. doi: 10.1186/s12881-018-0721-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aarsland D, Batzu L, Halliday GM, et al. Parkinson disease‐associated cognitive impairment [published correction appears in Nat Rev Dis Primers. 2021 Jul 13;7(1):53]. Nat Rev Dis Primers. 2021;7(1):47. doi: 10.1038/s41572-021-00280-3 [DOI] [PubMed] [Google Scholar]
- 9. Caruana M, Cauchi R, Vassallo N. Putative role of red wine polyphenols against brain pathology in Alzheimer's and Parkinson's disease. Front Nutr. 2016;3:31. doi: 10.3389/fnut.2016.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clinton LK, Blurton‐Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Abeta, tau, and alpha‐synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30(21):7281‐7289. doi: 10.1523/JNEUROSCI.0490-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gallardo G, Schlüter OM, Südhof TC. A molecular pathway of neurodegeneration linking alpha‐synuclein to ApoE and Abeta peptides. Nat Neurosci. 2008;11(3):301‐308. doi: 10.1038/nn2058 [DOI] [PubMed] [Google Scholar]
- 12. Emon MA, Heinson A, Wu P, et al. Clustering of Alzheimer's and Parkinson's disease based on genetic burden of shared molecular mechanisms. Sci Rep. 2020;10:19097. doi: 10.1038/s41598-020-76200-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu XC, Cao L, Tan MS, et al. Association of Parkinson's disease GWAS‐linked loci with Alzheimer's disease in Han Chinese. Mol Neurobiol. 2017;54:308‐318. doi: 10.1007/s12035-015-9649-5 [DOI] [PubMed] [Google Scholar]
- 14. Le Bras A. A new mouse model to study late‐onset Alzheimer's disease. Lab Anim. 2021;50:151. doi: 10.1038/s41684-021-00780-5 [DOI] [Google Scholar]
- 15. Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120(3):885‐890. doi: 10.1016/s0006-291x(84)80190-4 [DOI] [PubMed] [Google Scholar]
- 16. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82(12):4245‐4249. doi: 10.1073/pnas.82.12.4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kang J, Lemaire HG, Unterbeck A, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell‐surface receptor. Nature. 1987;325(6106):733‐736. doi: 10.1038/325733a0 [DOI] [PubMed] [Google Scholar]
- 18. Tanzi RE, Gusella JF, Watkins PC, et al. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235(4791):880‐884. doi: 10.1126/science.2949367 [DOI] [PubMed] [Google Scholar]
- 19. Delabar JM, Goldgaber D, Lamour Y, et al. Beta amyloid gene duplication in Alzheimer's disease and karyotypically normal Down syndrome. Science. 1987;235(4794):1390‐1392. doi: 10.1126/science.2950593 [DOI] [PubMed] [Google Scholar]
- 20. Selkoe DJ, Bell DS, Podlisny MB, Price DL, Cork LC. Conservation of brain amyloid proteins in aged mammals and humans with Alzheimer's disease. Science. 1987;235(4791):873‐877. doi: 10.1126/science.3544219 [DOI] [PubMed] [Google Scholar]
- 21. Rapoport SI. Hypothesis: Alzheimer's disease is a phylogenetic disease. Med Hypotheses. 1989;29(3):147‐150. doi: 10.1016/0306-9877(89)90185-0 [DOI] [PubMed] [Google Scholar]
- 22. Finch CE, Austad SN. Commentary: is Alzheimer's disease uniquely human? Neurobiol Aging. 2015;36(2):553‐555. doi: 10.1016/j.neurobiolaging.2014.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goate A, Chartier‐Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349(6311):704‐706. doi: 10.1038/349704a0 [DOI] [PubMed] [Google Scholar]
- 24. Chartier‐Harlin MC, Crawford F, Houlden H, et al. Early‐onset Alzheimer's disease caused by mutations at codon 717 of the beta‐amyloid precursor protein gene. Nature. 1991;353(6347):844‐846. doi: 10.1038/353844a0 [DOI] [PubMed] [Google Scholar]
- 25. Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science. 1991;254(5028):97‐99. doi: 10.1126/science.1925564 [DOI] [PubMed] [Google Scholar]
- 26. Games D, Adams D, Alessandrini R, et al. Alzheimer‐type neuropathology in transgenic mice overexpressing V717F beta‐amyloid precursor protein. Nature. 1995;373(6514):523‐527. doi: 10.1038/373523a0 [DOI] [PubMed] [Google Scholar]
- 27. Hsiao K, Chapman P, Nilsen S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274(5284):99‐102. doi: 10.1126/science.274.5284.99 [DOI] [PubMed] [Google Scholar]
- 28. Chishti MA, Yang D‐S, Janus C, et al. Early‐onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276(24):21562‐21570. doi: 10.1074/jbc.M100710200 [DOI] [PubMed] [Google Scholar]
- 29. Games D, Buttini M, Kobayashi D, Schenk D, Seubert P. Mice as models: transgenic approaches and Alzheimer's disease. J Alzheimers Dis. 2006;9(3 Suppl):133‐149. doi: 10.3233/jad-2006-9s316 [DOI] [PubMed] [Google Scholar]
- 30. Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early‐onset familial Alzheimer's disease. Nature. 1995;375(6534):754‐760. doi: 10.1038/375754a0 [DOI] [PubMed] [Google Scholar]
- 31. Levy‐Lahad E, Wijsman EM, Nemens E, et al. A familial Alzheimer's disease locus on chromosome 1. Science. 1995;269(5226):970‐973. doi: 10.1126/science.7638621 [DOI] [PubMed] [Google Scholar]
- 32. Xiao X, Liu H, Liu X, Zhang W, Zhang S, Jiao B. APP, PSEN1, and PSEN2 variants in Alzheimer's disease: systematic re‐evaluation according to ACMG guidelines. Front Aging Neurosci. 2021;13:695808. doi: 10.3389/fnagi.2021.695808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kabir MT, Uddin MS, Setu JR, Ashraf GM, Bin‐Jumah MN, Abdel‐Daim MM. Exploring the role of PSEN mutations in the pathogenesis of Alzheimer's disease. Neurotox Res. 2020;38(4):833‐849. doi: 10.1007/s12640-020-00232-x [DOI] [PubMed] [Google Scholar]
- 34. Duff K, Eckman C, Zehr C, et al. Increased amyloid‐beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383(6602):710‐713. doi: 10.1038/383710a0 [DOI] [PubMed] [Google Scholar]
- 35. Borchelt DR, Thinakaran G, Eckman CB, et al. Familial Alzheimer's disease‐linked presenilin 1 variants elevate Abeta1‐42/1‐40 ratio in vitro and in vivo. Neuron. 1996;17(5):1005‐1013. doi: 10.1016/s0896-6273(00)80230-5 [DOI] [PubMed] [Google Scholar]
- 36. Nakano Y, Kondoh G, Kudo T, et al. Accumulation of murine amyloidbeta42 in a gene‐ dosage‐dependent manner in PS1 ‘knock‐in’ mice. Eur J Neurosci. 1999;11(7):2577‐2581. doi: 10.1046/j.1460-9568.1999.00698.x [DOI] [PubMed] [Google Scholar]
- 37. Holcomb L, Gordon MN, McGowan E, et al. Accelerated Alzheimer‐type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4(1):97‐100. doi: 10.1038/nm0198-097 [DOI] [PubMed] [Google Scholar]
- 38. Schmitz C, Rutten BPF, Pielen A, et al. Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer's disease. Am J Pathol. 2004;164(4):1495‐1502. doi: 10.1016/S0002-9440(10)63235-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Casas C, Sergeant N, Itier JM, et al. Massive CA1/2 neuronal loss with intraneuronal and N‐ terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165(4):1289‐1300. doi: 10.1016/s0002-9440(10)63388-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Link CD. Expression of human beta‐amyloid peptide in transgenic Caenorhabditis elegans . Proc Natl Acad Sci USA. 1995;92(20):9368‐9372. doi: 10.1073/pnas.92.20.9368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hornsten A, Lieberthal J, Fadia S, et al. APL‐1, a Caenorhabditis elegans protein related to the human beta‐amyloid precursor protein, is essential for viability. Proc Natl Acad Sci USA. 2007;104(6):1971‐1976. doi: 10.1073/pnas.0603997104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Levitan D, Greenwald I. Facilitation of lin‐12‐mediated signalling by sel‐12, a Caenorhabditis elegans S182 Alzheimer's disease gene. Nature. 1995;377(6547):351‐354. doi: 10.1038/377351a0 [DOI] [PubMed] [Google Scholar]
- 43. Jing Y. Regulation of Caenorhabditis elegans model in Alzheimer's disease. E3S Web Conf. 2020;185(03043):1‐10. doi: 10.1051/e3sconf/202018503043 [DOI] [Google Scholar]
- 44. Tsuda L, Lim YM. Alzheimer's disease model system using Drosophila . Adv Exp Med Biol. 2018;1076:25‐40. doi: 10.1007/978-981-13-0529-0_3 [DOI] [PubMed] [Google Scholar]
- 45. Groen JJ, Endtz LJ. Hereditary Pick's disease: second re‐examination of the large family and discussion of other hereditary cases, with particular reference to electroencephalography, a computerized tomography. Brain. 1982;105(Pt 3):443‐459. doi: 10.1093/brain/105.3.443 [DOI] [PubMed] [Google Scholar]
- 46. Grundke‐Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule‐associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83(13):4913‐4917. doi: 10.1073/pnas.83.13.4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci USA. 1997;94(8):4113‐4118. doi: 10.1073/pnas.94.8.4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia [published correction appears in Ann Neurol 1998 Sep; 44(3):428]. Ann Neurol. 1998;43(6):815‐825. doi: 10.1002/ana.410430617 [DOI] [PubMed] [Google Scholar]
- 49. Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5'‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature. 1998;393(6686):702‐705. doi: 10.1038/31508 [DOI] [PubMed] [Google Scholar]
- 50. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95(13):7737‐7741. doi: 10.1073/pnas.95.13.7737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: frontotemporal dementia caused by microtubule‐associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging [published correction appears in Neuropathol Appl Neurobiol. 2015 Jun;41(4):571] [published correction appears in Neuropathol Appl Neurobiol. 2015 Jun;41(4):571]. Neuropathol Appl Neurobiol. 2015;41(1):24‐46. doi: 10.1111/nan.12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule‐associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3(4):519‐526. doi: 10.1016/0896-6273(89)90210-9 [DOI] [PubMed] [Google Scholar]
- 53. Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol. 1977;116(2):227‐247. doi: 10.1016/0022-2836(77)90214-5 [DOI] [PubMed] [Google Scholar]
- 54. Grundke‐Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule‐associated protein tau (tau) in Alzheimer cytoskeletal pathology. PNAS. 1986;83(13):4913‐4917. doi: 10.1073/pnas.83.13.4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Probst A, Götz J, Wiederhold KH, et al. Axonopathy and amyotrophy in mice transgenic for human four‐repeat tau protein. Acta Neuropathol. 2000;99(5):469‐481. doi: 10.1007/s004010051148 [DOI] [PubMed] [Google Scholar]
- 56. Lewis J, McGowan E, Rockwood J, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein [published correction appears in Nat Genet 2000 Sep; 26(1):127]. Nat Genet. 2000;25(4):402‐405. doi: 10.1038/78078 [DOI] [PubMed] [Google Scholar]
- 57. SantaCruz K, Lewis J, Spires T, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476‐481. doi: 10.1126/science.1113694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jankowsky JL, Zheng H. Practical considerations for choosing a mouse model of Alzheimer's disease. Mol Neurodegeneration. 2017;12:89. doi: 10.1186/s13024-017-0231-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Braak H, Braak E. Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiol Aging. 1995;16(3):271‐284. doi: 10.1016/0197-4580(95)00021-6 [DOI] [PubMed] [Google Scholar]
- 60. Liu LI, Drouet V, Wu JW, et al. Trans‐synaptic spread of tau pathology in vivo. PLoS One. 2012;7(2):e31302. doi: 10.1371/journal.pone.0031302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79(1):110‐119. doi: 10.1002/ana.24546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bischof GN, Endepols H, van Eimeren T, Drzezga A. Tau‐imaging in neurodegeneration. Methods. 2017;130:114‐123. doi: 10.1016/j.ymeth.2017.08.003 [DOI] [PubMed] [Google Scholar]
- 63. Prüßing K, Voigt A, Schulz JB. Drosophila melanogaster as a model organism for Alzheimer's disease. Mol Neurodegener. 2013;8:35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement. 2018;4:575‐590. doi: 10.1016/j.trci.2018.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lagarde J, Sarazin M, Bottlaender M. In vivo PET imaging of neuroinflammation in Alzheimer's disease. J Neural Transm. 2018;125(5):847‐867. doi: 10.1007/s00702-017-1731-x [DOI] [PubMed] [Google Scholar]
- 66. Gomez‐Nicola D, Boche D. Post‐mortem analysis of neuroinflammatory changes in human Alzheimer's disease. Alzheimers Res Ther. 2015;7(1):42. doi: 10.1186/s13195-015-0126-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson's disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19. doi: 10.1186/s40035-015-0042-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Crotti A, Glass CK. The choreography of neuroinflammation in Huntington's disease. Trends Immunol. 2015;36(6):364‐373. doi: 10.1016/j.it.2015.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Faden AI, Loane DJ. Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics. 2015;12(1):143‐150. doi: 10.1007/s13311-014-0319-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McCombe PA, Henderson RD. The Role of immune and inflammatory mechanisms in ALS. Curr Mol Med. 2011;11(3):246‐254. doi: 10.2174/156652411795243450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ismail R, Parbo P, Madsen LS, et al. The relationships between neuroinflammation, beta‐ amyloid and tau deposition in Alzheimer's disease: a longitudinal PET study. J Neuroinflammation. 2020;17:151. doi: 10.1186/s12974-020-01820-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Baik SH, Kang S, Son SM, Mook‐Jung I. Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer's disease mouse model. Glia. 2016;64(12):2274‐2290. doi: 10.1002/glia.23074 [DOI] [PubMed] [Google Scholar]
- 73. Chakrabarty P, Jansen‐West K, Beccard A, et al. Massive gliosis induced by interleukin‐6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24(2):548‐559. doi: 10.1096/fj.09-141754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Krabbe G, Halle A, Matyash V, et al. Functional impairment of microglia coincides with Beta‐amyloid deposition in mice with Alzheimer‐like pathology. PLoS One. 2013;8(4):e60921. doi: 10.1371/journal.pone.0060921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta‐amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28(33):8354‐8360. doi: 10.1523/JNEUROSCI.0616-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tran J, Anastacio H, Bardy C. Genetic predispositions of Parkinson's disease revealed in patient‐derived brain cells. NPJ Parkinsons Dis. 2020;6(8): doi: 10.1038/s41531-020-0110-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Britannica, The Editors of Encyclopedia . Reserpine. Encyclopedia Britannica. 2019. https://www.britannica.com/science/reserpine. Accessed 14 December 2021. [Google Scholar]
- 78. Shore PA, Pletscher A, Tomich EG, Carlsson A, Kuntzman R, Brodie BB. Role of brain serotonin in reserpine action. Ann N Y Acad Sci. 1957;66:609‐617. doi: 10.1111/j.1749-6632.1957.tb40751.x [DOI] [PubMed] [Google Scholar]
- 79. Carlsson A, Lindqvist M, Magnusson T. 3,4‐Dihydroxyphenylalanine and 5‐hydroxytryptophan as reserpine antagonists. Nature. 1957;180(4596):1200. doi: 10.1038/1801200a0 [DOI] [PubMed] [Google Scholar]
- 80. Degkwitz R, Frowein R, Kulenkampff C, Mohs U. [On the effects of L‐dopa in man and their modification by reserpine, chlorpromazine, iproniazid and vitamin B6]. Klin Wochenschr. 1960;38:120‐123. doi: 10.1007/BF02189076 [DOI] [PubMed] [Google Scholar]
- 81. Jellinger KA. Pathology of Parkinson's disease. Changes other than the nigrostriatal pathway. Mol Chem Neuropathol. 1991;14(3):153‐197. doi: 10.1007/BF03159935 [DOI] [PubMed] [Google Scholar]
- 82. Robledo P, Feger J. Acute monoaminergic depletion in the rat potentiates the excitatory effect of the subthalamic nucleus in the substantia nigra pars reticulata but not in the pallidal complex. J Neural Transm Gen Sect. 1991;86(2):115‐126. doi: 10.1007/BF01250572 [DOI] [PubMed] [Google Scholar]
- 83. Biggs CS, Fowler LJ, Whitton PS, Starr MS. Extracellular levels of glutamate and aspartate in the entopeduncular nucleus of the rat determined by microdialysis: regulation by striatal dopamine D2 receptors via the indirect striatal output pathway? Brain Res. 1997;753(1):163‐175. doi: 10.1016/s0006-8993(97)00033-4 [DOI] [PubMed] [Google Scholar]
- 84. Duty S, Jenner P. Animal models of Parkinson's disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol. 2011;164(4):1357‐1391. doi: 10.1111/j.1476-5381.2011.01426.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ungerstedt U. 6‐Hydroxy‐dopamine induced degeneration of central monoamine neurons. Eur J Pharmacol. 1968;5(1):107‐110. doi: 10.1016/0014-2999(68)90164-7 [DOI] [PubMed] [Google Scholar]
- 86. Vernon AC, Croucher MJ, Dexter DT. Additive neuroprotection by metabotropic glutamate receptor subtype‐selective ligands in a rat Parkinson's model. NeuroReport. 2008;19(4):475‐478. doi: 10.1097/WNR.0b013e3282f602df [DOI] [PubMed] [Google Scholar]
- 87. Austin PJ, Betts MJ, Broadstock M, O'Neill MJ, Mitchell SN, Duty S. Symptomatic and neuroprotective effects following activation of nigral group III metabotropic glutamate receptors in rodent models of Parkinson's disease. Br J Pharmacol. 2010;160(7):1741‐1753. doi: 10.1111/j.1476-5381.2010.00820.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schober A. Classic toxin‐induced animal models of Parkinson's disease: 6‐OHDA and MPTP. Cell Tissue Res. 2004;318(1):215‐224. doi: 10.1007/s00441-004-0938-y [DOI] [PubMed] [Google Scholar]
- 89. Jenner P. Clues to the mechanism underlying dopamine cell death in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1989;52(Suppl):22‐28. doi: 10.1136/jnnp.52.suppl.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6‐OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci. 2002;15(6):991‐998. doi: 10.1046/j.1460-9568.2002.01938.x [DOI] [PubMed] [Google Scholar]
- 91. Barnéoud P, Parmentier S, Mazadier M, et al. Effects of complete and partial lesions of the dopaminergic mesotelencephalic system on skilled forelimb use in the rat. Neuroscience. 1995;67(4):837‐848. doi: 10.1016/0306-4522(95)00112-v [DOI] [PubMed] [Google Scholar]
- 92. Langston WJ. The MPTP story. J Parkinsons Dis. 2017;7(1):11‐19. doi: 10.3233/JPD-179006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Langston JW, Irwin I, Langston EB, Forno LS. 1‐Methyl‐4‐phenylpyridinium ion (MPP+): identification of a metabolite of MPTP, a toxin selective to the substantia nigra. Neurosci Lett. 1984;48(1):87‐92. doi: 10.1016/0304-3940(84)90293-3 [DOI] [PubMed] [Google Scholar]
- 94. Shen RS, Abell CW, Gessner W, Brossi A. Serotonergic conversion of MPTP and dopaminergic accumulation of MPP+. FEBS Lett. 1985;189(2):225‐230. doi: 10.1016/0014-5793(85)81028-0 [DOI] [PubMed] [Google Scholar]
- 95. Blanchard V, Raisman‐Vozari R, Vyas S, et al. Differential expression of tyrosine hydroxylase and membrane dopamine transporter genes in subpopulations of dopaminergic neurons of the rat mesencephalon. Brain Res Mol Brain Res. 1994;22(1–4):29‐38. doi: 10.1016/0169-328x(94)90029-9 [DOI] [PubMed] [Google Scholar]
- 96. Ramsay RR, Salach JI, Singer TP. Uptake of the neurotoxin 1‐methyl‐4‐phenylpyridine (MPP+) by mitochondria and its relation to the inhibition of the mitochondrial oxidation of NAD+‐linked substrates by MPP+. Biochem Biophys Res Commun. 1986;134(2):743‐748. doi: 10.1016/s0006-291x(86)80483-1 [DOI] [PubMed] [Google Scholar]
- 97. Johannessen JN, Chiueh CC, Burns RS, Markey SP. Differences in the metabolism of MPTP in the rodent and primate parallel differences in sensitivity to its neurotoxic effects. Life Sci. 1985;36(3):219‐224. doi: 10.1016/0024-3205(85)90062-1 [DOI] [PubMed] [Google Scholar]
- 98. Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine. Proc Natl Acad Sci USA. 1983;80(14):4546‐4550. doi: 10.1073/pnas.80.14.4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jenner P, Rupniak NMJ, Rose S, et al. 1‐Methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐induced parkinsonism in the common marmoset. Neurosci Lett. 1984;50(1–3):85‐90. doi: 10.1016/0304-3940(84)90467-1 [DOI] [PubMed] [Google Scholar]
- 100. Langston JW, Forno LS, Rebert CS, Irwin I. Selective nigral toxicity after systemic administration of 1‐methyl‐4‐phenyl‐1,2,5,6‐tetrahydropyrine (MPTP) in the squirrel monkey. Brain Res. 1984;292(2):390‐394. doi: 10.1016/0006-8993(84)90777-7 [DOI] [PubMed] [Google Scholar]
- 101. Chen MK, Kuwabara H, Zhou Y, et al. VMAT2 and dopamine neuron loss in a primate model of Parkinson's disease. J Neurochem. 2008;105(1):78‐90. doi: 10.1111/j.1471-4159.2007.05108.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lotharius J, Brundin P. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha‐synuclein. Nat Rev Neurosci. 2002;3(12):932‐942. doi: 10.1038/nrn983 [DOI] [PubMed] [Google Scholar]
- 103. Langston JW, Langston EB, Irwin I. MPTP‐induced parkinsonism in human and non‐ human primates–clinical and experimental aspects. Acta Neurol Scand Suppl. 1984;100:49‐54. [PubMed] [Google Scholar]
- 104. Vermilyea SC, Emborg ME. α‐Synuclein and nonhuman primate models of Parkinson's disease. J Neurosci Methods. 2015;255:38‐51. doi: 10.1016/j.jneumeth.2015.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sedelis M, Schwarting RK, Huston JP. Behavioral phenotyping of the MPTP mouse model of Parkinson's disease. Behav Brain Res. 2001;125(1–2):109‐125. doi: 10.1016/s0166-4328(01)00309-6 [DOI] [PubMed] [Google Scholar]
- 106. Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68(2):201‐206. doi: 10.1016/j.neuron.2010.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Uéda K, Fukushima H, Masliah E, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1993;90(23):11282‐11286. doi: 10.1073/pnas.90.23.11282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Spillantini MG, Divane A, Goedert M. Assignment of human alpha‐synuclein (SNCA) and beta‐synuclein (SNCB) genes to chromosomes 4q21 and 5q35. Genomics. 1995;27(2):379‐381. doi: 10.1006/geno.1995.1063 [DOI] [PubMed] [Google Scholar]
- 109. Golbe LI, Lazzarini AM, Duvoisin RC, et al. Clinical genetic analysis of Parkinson's disease in the Contursi kindred. Ann Neurol. 1996;40:767‐775. doi: 10.1002/ana.410400513 [DOI] [PubMed] [Google Scholar]
- 110. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the ‐synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045‐2047. doi: 10.1126/science.276.5321.2045 [DOI] [PubMed] [Google Scholar]
- 111. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha‐ synuclein in Lewy bodies. Nature. 1997;388(6645):839‐840. doi: 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- 112. Jankovic J, Tan EK. Parkinson's disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry. 2020;91(8):795‐808. doi: 10.1136/jnnp-2019-322338 [DOI] [PubMed] [Google Scholar]
- 113. Masliah E, Rockenstein E, Veinbergs I, et al. Dopaminergic loss and inclusion body formation in alpha‐synuclein mice: implications for neurodegenerative disorders. Science. 2000;287(5456):1265‐1269. doi: 10.1126/science.287.5456.1265 [DOI] [PubMed] [Google Scholar]
- 114. Konnova EA, Swanberg M. Animal models of Parkinson's disease. In: Stoker TB, Greenland JC, eds. Parkinson's disease: pathogenesis and clinical aspects [Internet]. Codon Publications; 2018. Chapter 5. Available from: https://www.ncbi.nlm.nih.gov/books/NBK536725/. doi: 10.15586/codonpublications.parkinsonsdisease.2018.ch5 [DOI] [Google Scholar]
- 115. Wakamatsu M, Ishii A, Iwata S, et al. Selective loss of nigral dopamine neurons induced by overexpression of truncated human alpha‐synuclein in mice. Neurobiol Aging. 2008;29(4):574‐585. doi: 10.1016/j.neurobiolaging.2006.11.017 [DOI] [PubMed] [Google Scholar]
- 116. Chesselet MF, Fleming S, Mortazavi F, Meurers B. Strengths and limitations of genetic mouse models of Parkinson's disease. Parkinsonism Relat Disord. 2008;14(Suppl 2);S84‐S87. doi: 10.1016/j.parkreldis.2008.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Prasad K, Tarasewicz E, Ohman Strickland PA, et al. Biochemical and morphological consequences of human α‐synuclein expression in a mouse α‐synuclein null background. Eur J Neurosci. 2011;33(4):642‐656. doi: 10.1111/j.1460-9568.2010.07558.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Luk KC, Kehm V, Carroll J, et al. Pathological α‐synuclein transmission initiates Parkinson‐like neurodegeneration in nontransgenic mice. Science. 2012;338(6109):949‐953. doi: 10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gómez‐Benito M, Granado N, García‐Sanz P, Michel A, Dumoulin M, Moratalla R. Modeling Parkinson's disease with the alpha‐synuclein protein. Front Pharmacol. 2020;11:356. doi: 10.3389/fphar.2020.00356. Published 2020 Apr 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Whitworth AJ. Drosophila models of Parkinson's disease. Adv Genet. 2011;73:1‐50. doi: 10.1016/B978-0-12-380860-8.00001-X [DOI] [PubMed] [Google Scholar]
- 121. Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2‐q13.1. Ann Neurol. 2002;51(3):296‐301. doi: 10.1002/ana.10113 [DOI] [PubMed] [Google Scholar]
- 122. Paisán‐Ruíz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8‐linked Parkinson's disease. Neuron. 2004;44(4):595‐600. doi: 10.1016/j.neuron.2004.10.023 [DOI] [PubMed] [Google Scholar]
- 123. Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal‐ dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601‐607. doi: 10.1016/j.neuron.2004.11.005 [DOI] [PubMed] [Google Scholar]
- 124. Rui Q, Ni H, Li D, Gao R, Chen G. The role of LRRK2 in neurodegeneration of Parkinson disease. Curr Neuropharmacol. 2018;16(9):1348‐1357. doi: 10.2174/1570159X16666180222165418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ren C, Ding Y, Wei S, et al. G2019S variation in LRRK2: an ideal model for the study of Parkinson's disease? Front Hum Neurosci. 2019;13:306. doi: 10.3389/fnhum.2019.00306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bouhouche A, Tibar H, Ben El Haj R, et al. LRRK2 G2019S mutation: prevalence and clinical features in Moroccans with Parkinson's disease. Parkinsons Dis. 2017;2017:2412486. doi: 10.1155/2017/2412486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tong Y, Pisani A, Martella G, et al. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci USA. 2009;106(34):14622‐14627. doi: 10.1073/pnas.0906334106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Li Y, Liu W, Oo TF, et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci. 2009;12(7):826‐828. doi: 10.1038/nn.2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Dues DJ, Moore DJ. LRRK2 and protein aggregation in Parkinson's disease: insights from animal models. Front Neurosci. 2020;14:719. doi: 10.3389/fnins.2020.00719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605‐608. doi: 10.1038/33416 [DOI] [PubMed] [Google Scholar]
- 131. Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. 2015;85(2):257‐273. doi: 10.1016/j.neuron.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Valente EM, Abou‐Sleiman PM, Caputo V, et al. Hereditary early‐onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304(5674):1158‐1160. doi: 10.1126/science.1096284 [DOI] [PubMed] [Google Scholar]
- 133. Grant LM, Kelm‐Nelson CA, Hilby BL, et al. Evidence for early and progressive ultrasonic vocalization and oromotor deficits in a PINK1 gene knockout rat model of Parkinson's disease. J Neurosci Res. 2015;93(11):1713‐1727. doi: 10.1002/jnr.23625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Sämann J, Hegermann J, von Gromoff E, Eimer S, Baumeister R, Schmidt E. Caenorhabditits elegans LRK‐1 and PINK‐1 act antagonistically in stress response and neurite outgrowth. J Biol Chem. 2009;284(24):16482‐16491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Cai X, Qiao JU, Knox T, et al. In search of early neuroradiological biomarkers for Parkinson's disease: alterations in resting state functional connectivity and gray matter microarchitecture in PINK1 −/− rats. Brain Res. 2019;1706:58‐67. doi: 10.1016/j.brainres.2018.10.033 [DOI] [PubMed] [Google Scholar]
- 136. Valadez‐Barba V, Cota‐Coronado A, Hernández‐Pérez OR, et al. iPSC for modeling neurodegenerative disorders. Regen Ther. 2020;15:332‐339. doi: 10.1016/j.reth.2020.11.006. Published 2020 Dec 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Jang W, Kim HJ, Li H, Jo KD, Lee MK, Yang HO. The neuroprotective effect of erythropoietin on rotenone‐induced neurotoxicity in SH‐SY5Y cells through the induction of autophagy. Mol Neurobiol. 2016;53(6):3812‐3821. doi: 10.1007/s12035-015-9316-x [DOI] [PubMed] [Google Scholar]
- 138. Smith HL, Freeman OJ, Butcher AJ, et al. Astrocyte unfolded protein response induces a specific reactivity state that causes non‐cell‐autonomous neuronal degeneration. Neuron. 2020;105(5):855‐866.e5. doi: 10.1016/j.neuron.2019.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Israel MA, Yuan SH, Bardy C, et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012;482(7384):216‐220. doi: 10.1038/nature10821. Published 2012 Jan 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Doss MX, Sachinidis A. Current challenges of iPSC‐based disease modeling and therapeutic implications. Cells. 2019;8(5):403. doi: 10.3390/cells8050403. Published 2019 Apr 30. [DOI] [PMC free article] [PubMed] [Google Scholar]