

Abstract
Pretargeted PET imaging is an emerging and fast-developing method to monitor immuno-oncology strategies. Currently, tetrazine ligation is considered the most promising bioorthogonal reaction for pretargeting in vivo. Recently, we have developed a method to 18F-label ultrareactive tetrazines by copper-mediated fluorinations. However, bispyridyl tetrazines—one of the most promising structures for in vivo pretargeted applications—were inaccessible using this strategy. We believed that our successful efforts to 18F-label H-tetrazines using low basic labeling conditions could also be used to label bispyridyl tetrazines via aliphatic nucleophilic substitution. Here, we report the first direct 18F-labeling of bispyridyl tetrazines, their optimization for in vivo use, as well as their successful application in pretargeted PET imaging. This strategy resulted in the design of [18F]45, which could be labeled in a satisfactorily radiochemical yield (RCY = 16%), molar activity (Am = 57 GBq/µmol), and high radiochemical purity (RCP > 98%). The [18F]45 displayed a target-to-background ratio comparable to previously successfully applied tracers for pretargeted imaging. This study showed that bispyridyl tetrazines can be developed into pretargeted imaging agents. These structures allow an easy chemical modification of 18F-labeled tetrazines, paving the road toward highly functionalized pretargeting tools. Moreover, bispyridyl tetrazines led to near-instant drug release of iTCO-tetrazine-based ‘click-to-release’ reactions. Consequently, 18F-labeled bispyridyl tetrazines bear the possibility to quantify such release in vivo in the future.
Keywords: bioorthogonal chemistry, tetrazine ligation, bispyridyl tetrazines, pretargeted imaging, PET, fluorine-18, molecular imaging
1. Introduction
Radioimmunoconjugates have emerged as important tools; e.g., in the diagnosis and treatment of cancer [1,2]. A subclass within these important substances are monoclonal antibodies (mAbs). They can be designed and produced with exquisite target affinity and selectivity. From a nuclear molecular imaging point of view, mAb-based agents can result in high target-to-background ratios with a low nondisplaceable binding component, which make them almost ideal tracers. Unfortunately, at the same time they possess slow pharmacokinetic properties; i.e., target accumulation and blood clearance takes days rather than hours [3,4]. Consequently, long-lived radionuclides must be used to match the pharmacokinetic profile of these vectors [3]. This results in unnecessary radiation burden for patients (Figure 1A), and sometimes even at a magnitude prohibitive for clinical studies [5]. An elegant way to overcome these limitations is a relatively new technique based on a pretargeted approach. In this approach, the accumulation timeframe of mAbs is temporally decoupled from the actual imaging process. In particular, a tagged mAb is administered days before a small molecule imaging agent is used to bioorthogonally react with the tag of the mAb. In this way, the accumulation of the mAb can be imaged even after days with the aid of a short-lived radionuclide (Figure 1B) [6].
Figure 1.

(A) Conventional immunoimaging vs. (B) pretargeted immunoimaging based on tetrazine ligation [3].
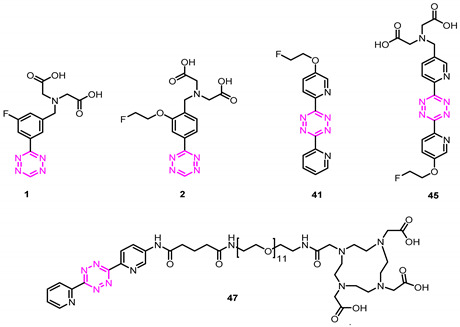
Pretargeted imaging consequently exploits the unique targeting properties of mAbs and the rapid pharmacokinetics properties of small molecules, enabling exceptional target-to-background ratios within hours [3]. Currently, the most promising reaction for pretargeted imaging is the tetrazine ligation between a tetrazine (Tz) and a trans-cyclooctene (TCO) (Figure 1B). Several preclinical examples have been reported that demonstrated the potential of this reaction [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22]. More importantly, in late 2020, the first clinical phase I trial based on tetrazine ligation was initiated (NCT04106492) [23]. From a clinical point of view, a fluorine-18 (18F)-labeled Tz would be ideal for positron emission tomography (PET) applications, since 18F possesses almost perfect physical characteristics for molecular imaging [24,25,26]. However, due to the intrinsic instability of the tetrazine scaffold to basic conditions—typically employed in direct 18F-fluorination approaches—no 18F-tetrazine was synthesized until few years ago [10,27,28,29,30,31]. Recently, we reported the first direct 18F-fluorination of highly reactive tetrazines, which could be labeled via copper-mediated oxidative 18F-fluorination [32]. This methodology allowed us to develop a low-lipophilicity, fast-clearing, and highly reactive tetrazine PET tracer (1) (Figure 2). In a similar way, we were able to develop the first direct aliphatic 18F-radiolabeled Tz (2) (Figure 2). This was possible using ultralow basic conditions [33,34]. Both compounds (1 and 2) showed high imaging contrast during in vivo experiments, and are currently subjects in further studies to evaluate their potential to be translated into the clinic.
Figure 2.

Compound evaluation and rationale—from previous work to this project: The 18F-labeling of H-Tz has already been established (previous work); labeling of bispyridyl Tzs succeeded in this study. Bispyridyl Tzs have intrinsic high-reaction kinetics and allow higher chemical flexibility. Pyridyl unit A can be used to introduce the necessary polarity for in vivo pretargeting, whereas pyridyl unit B can be used for labeling purposes.
However, both labeling methods (Cu-mediated 18F-fluorinartion and direct aliphatic 18F-labeling) have thus far only been successful for H-Tzs. Bispyridyl scaffolds could not be labeled with Cu-mediated approaches due to the chelating effect of the pyridyl nitrogen [32,35]. Aliphatic labeling was only applied on monosubstituted Tzs [34]. Bispyridyl Tzs are, however, very appealing subjects to be used for pretargeted experiments (Figure 2). Beside H-Tzs, these structures are the only other structural class that have been successfully applied in vivo [6,35,36,37,38]. Bispyridyl Tzs have intrinsic high-reaction kinetics and allow for greater flexibility with respect to structural modifications compared to H-Tzs. For example, one pyridyl unit can be used for labeling purposes, whereas the other can be used to introduce polarity or other functional scaffolds as NIR turn-on dyes or additional chelators for pretargeted radiotherapy [3,17,39,40]. Recently, bispyridyl Tzs also were shown to be superior for instant and near-quantitative ’click-to-release’ approaches [41]. Radiolabeled bispyridyl Tzs could be used as such to quantify drug release in vivo.
In this work, we describe the design, synthesis, and in vivo evaluation of the first 18F-labeled bispyridyl Tz. In order to develop such a Tz, we exploited one pyridyl ring to introduce polarity. We recently showed that a clogD7.4 of at least -3 is a necessary parameter for successful in vivo pretargeting [17]. The other pyridyl ring was used for labeling purposes using a set of different linkers. The selection of the linker for the 18F-labeling was initially performed using H-Tzs due to their more accessible chemistry. Promising structures in term of radiochemical conversions (RCC) were translated into their bispyridyl Tz counterparts. Finally, the best candidate was applied for pretargeted PET experiments.
2. Results and Discussion
2.1. Evaluation of Different Fluoroethyl Linkers Using H-Tz as Model Structures
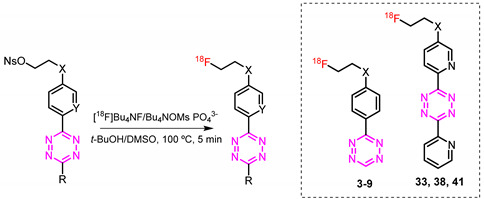
We have previously reported that fluoroethyl moieties can be used to introduce 18F-fluoride into H-Tz employing non-nucleophilic bases [33,34]. We believed that these conditions could also be suitable to label bispyridyl-based Tzs. In order to test the influence on the labeling step of different conjugation strategies between the fluoroethyl moiety and the Tz, we synthesized a set of structures possessing various linkers such as esters, amines, ethers and amides (Figure 3). We decided to use H-Tzs for this study, as they are easier to access. Seven different molecules (3–9) and their corresponding precursors were designed.
Figure 3.

Compound design strategy. (A) Screening of fluoroethyl linkers. H-Tzs were initially selected to evaluate different types of linkers due to their easier chemical accessibility. (B) The best linkers in terms of RCC were translated on the bispyridyl Tz scaffold. (C) The most promising structure was developed into a potential PET tracer [17].
2.2. Synthesis of H-Tz Derivatives
Compound 8 and its precursor 8a were synthesized as previously reported [33]. Compounds 3 and 4 and their corresponding precursor were obtained as reported in Scheme 1.
Scheme 1.

Reagents and conditions: (i) (a) NH2NH2.H2O, CH2Cl2, S8, EtOH, 50 °C, 24 h; (b) NaNO2, AcOH, 0 °C to rt, 30 min; (ii) 1-fluoro-2-iodoethane, DIPEA, DMF, 70 °C, 4 h; (iii) 2-bromoethanol, DIPEA, DMF, 70 °C, 4 h; (iv) nosyl chloride, DIPEA, DMAP, CH2Cl2, rt, 1 h; (v) 2-fluoroethylamine hydrochloride or ethanolamine, Et3N, CDI, CH3CN, 0 °C to rt, 12 h.
Briefly, Tz 10 was synthesized from 4-cyanobenzoic acid using a Pinner-like, sulfur-mediated procedure in modest yields [42]. Subsequent alkylation with 1-fluoro-2-iodoethane or 2-bromoethanol respectively gave compounds 3 and 11. The latter was then reacted with nosyl chloride to yield precursor 3a. Coupling of 4-cyanobenzoic acid with 2-fluoroethylamine or ethanolamine afforded 12 and 13; these were then converted into the corresponding Tz derivatives 4 and 14. These intermediates were further nosylated under basic conditions. However, we were not able to isolate 4a, since an intramolecular substitution occurred that could not be prevented. Tzs 5 and 6 and their respective precursors were obtained in a similar manner as reported in Scheme 2. The yields obtained were comparable to those of 3 and 4. However, the amide precursor 6a could be isolated in a yield of 83%. This most likely was possible due the lower reactivity of the tetrazine core compared to that of 4a.
Scheme 2.

Reagents and conditions:(i) (a) NH2NH2.H2O, CH2Cl2, S8, EtOH, 50 °C, 24 h; (b) NaNO2, AcOH, 0 °C to rt, 30 min; (ii) 1-fluoro-2-iodoethane, DIPEA, DMF, 70 °C, 4 h; (iii) 2-bromoethanol, DIPEA, DMF, 70 °C, 4 h; (iv) nosyl chloride, DIPEA, DMAP, CH2Cl2, rt, 1 h; (v) 2-fluoroethylamine or ethanolamine, CDI, CH3CN, 0 °C to rt, 12 h.
Compounds 7 and 9 were obtained using a different synthesis strategy (Scheme 3). 4-Hydroxybenzonitrile was reacted with 1-fluoro-2-iodoethane or 2-bromoethanol to respectively give 20 and 21 in 71% and 92% yields. These were converted into the corresponding Tzs 7 and 22 in modest yields (37% and 54%, respectively). The latter was further modified to give the nosylate precursor 7a. Intermediates 17 and 18 were obtained from 4-(bromomethyl)benzonitrile and the corresponding amines. N-Boc protection and reaction with hydrazine gave Tzs 27 and 28. Deprotection of 27 afforded compound 9 in an almost quantitative yield. Reaction of nosyl chloride with 28 resulted in an intramolecular reaction, and 9a could not be obtained. For this reason, a different protective group was selected. Then, 28 was deprotected and reacted with trityl chloride to give 31. In order to have the corresponding reference compound, 9 was converted to 30 as well. Finally, reaction with nosyl chloride gave 9b in a yield of 56%.
Scheme 3.

Reagents and conditions:(i) 1-fluoro-2-iodoethane or 2-bromoethanol, K2CO3, CH3CN, 70 °C, 12 h; (ii) (a) NH2NH2.H2O, CH2Cl2, S8, EtOH, 50 °C, 24 h; (b) NaNO2, AcOH, 0 °C to rt, 30 min; (iii) nosyl chloride, DIPEA, DMAP, CH2Cl2, rt, 1 h; (iv) 2-fluoroethylamine or ethanolamine, CH3CN, 0 °C to rt, 12 h; (v) Boc2O, Et3N, CH2Cl2, 0 °C to rt, 12 h; (vi) HCl, dioxane, rt, 4 h; (vii) TrCl, Et3N, CH2Cl2, 0 °C to rt, 12 h.
2.3. Labeling of H-Tz Derivatives
As we recently reported, nosyl leaving groups are optimally suited to aliphatically 18F-label high-reactive Tzs. A mesylate or tosylate base precursor resulted in low radiochemical yields (RCY) [33,43]. Radiolabeling only succeeded using low basicity conditions, since H-Tzs are too sensitive for standard—rather, basic—aliphatic 18F-fluorination approaches [29,33]. Within this study, we applied the same labeling conditions. Furthermore, we also investigated to what extent the precursor concentration influenced the RCC. Two amounts (3.1 and 9.3 nmol) were selected in this respect. With the exception of compound 3, both concentrations resulted in the same RCC range. Labeling attempts revealed further that the linkers influenced RCCs strongly. Only tetrazines 3, 7, and 8 could be synthesized. RCCs were in the range of 23–53% (based on radio-HPLC, n = 3). No or only minimal product formation could be observed with a different substitution profile. In light of that, bispyridyl Tzs 33, 38, and 41 were designed, synthesized, and then radiolabeled.
2.4. Synthesis of the Bispyridyl Tzs
Bispyridyl analogues of 3, 7, and 8 were synthesized as described in Scheme 4. 6-Cyanonicotinic acid was converted to the corresponding bispyridyl Tz 32 following a previously reported procedure [44]. The latter was alkylated with 1-fluoro-2-iodoethane or 2-bromoethanol to respectively give compounds 33 and 34. The alcohol derivative was then transformed to the nosylate precursor 33a. Differently, 5-methylpicolinonitrile was brominated and reacted with ethylene glycol to afford 36. Formation of the Tz ring and conversion to the fluorine analogue gave 38 [45]. Nosylation of the same alcohol intermediate yielded 38a. The last compounds, 41 and 41a, were obtained starting with 5-hydroxypicolinonitrile and using the same procedure employed for Tz 7.
Scheme 4.

Reagents and conditions: (i) 2-pyridinecarbonitrile, (a) NH2NH2.H2O, S8, EtOH, 90 °C, 4 h; (b) NaNO2, AcOH, 0 °C to rt, 30 min; (ii) 1-fluoro-2-iodoethane or 2-bromoethanol, DIPEA, DMF, 70 °C, 4 h; (iii) nosyl chloride, DIPEA, DMAP, CH2Cl2, rt, 1 h; (iv) NBS, AIBN, CH3CN, reflux, 12 h; (v) ethylene glycol, NaH, THF, 0 °C to reflux, 12 h; (vi) CF3(CF2)3SO2F, Et3N.3HF, THF, rt, 18 h; (vii) 1-fluoro-2-iodoethane or 2-bromoethanol, K2CO3, CH3CN, 70 °C, 12 h.
2.5. Labeling of Bispyridyl Tz Derivatives
Compounds [18F]33, [18F]38, and [18F]41 were labeled from their corresponding precursors 33a, 38a, and 41a following the same procedure reported in Section 2.3. The results are reported in Table 1. As expected from previous results, radiolabeling was successful in all cases. RCCs were in the range of 20–30%. However, only compounds [18F]33 and [18F]38 could be radiolabeled in a similar conversion when lower precursor concentrations were used. Due to that and its easier synthetic access compared to 33, we decided to use 38 as a template for our next steps, aiming to develop a pretargeted imaging agent.
Table 1.
18F-labeling of tetrazines developed in this study.
| ||||||
|---|---|---|---|---|---|---|
| Tetrazine | X | R | RCC (%) * Low Precursor Amount (3.1 nmol) |
RCC (%) * High Precursor Amount (9.3 nmol) |
||
| HPLC | TLC | HPLC | TLC | |||
| 3 | -COO- | -H | 0 | 0 | 18 ± 8 | 16 ± 6 |
| 5 | -CH2COO- | -H | - b | - b | - b | - b |
| 6 | -CH2CONH- | -H | - b | - b | - b | - b |
| 7 | -O- | -H | 45 ± 6 | 52 ± 4 | 53 ± 5 | 55 ± 4 |
| 8 | -CH2O- | -H | 23 ± 4 | 24 ± 3 | 26 ± 4 | 28 ± 3 |
| 9 c | -CH2NH- | -H | - d | - d | - d | - d |
| 33 | -COO- | -2-Pyr | 24 ± 3 | 22 ± 3 | 27 ± 4 | 25 ± 5 |
| 38 | -O- | -2-Pyr | 20 ± 11 | 21 ± 8 | 29 ± 10 | 28 ± 6 |
| 41 | -CH2O- | -2-Pyr | 9 ± 1 | 8 ± 1 | 23 ± 3 | 26 ± 6 |
a Precursor was not obtained. b No reaction observed. c Precursor protected with trityl. d Precursor decompose during reaction. n.d. = not determined. * Radiochemical conversions (RCC) were calculated for each compound by radio-HPLC and radio-TLCs as recently reported [43]. All results were based on n = 3 experiments.
2.6. Synthesis and Evaluation of 45 by Ex Vivo Blocking Assay
Compound 45 was designed based on a fluorothexy moiety identified to yield in the highest RCCs of previous results in this study. To decrease lipophilicity, we further conjugated a diacetic acid moiety to the structure. This combination should have resulted in an easy-to-label compound, as well as an imaging agent with the necessary polarity to be applied for in vivo pretargeted imaging. The pathway to synthesize 45 is depicted in Scheme 5. Alkylation of di-tert-butyl iminodiacetate by compound 35 under basic conditions yielded 43. The latter was reacted with an excess of 39 and hydrazine hydrate to give 44. Deprotection and purification by preparative-HPLC afforded 45 as a TFA salt.
Scheme 5.

Synthesis of 45 and its precursor. Reagents and conditions: (i) di-tert-butyl iminodiacetate, K2CO3, CH3CN, reflux, 12 h; (ii) 39, (a) NH2NH2.H2O, S8, EtOH, 90 °C, 4 h; (b) NaNO2, AcOH, 0 °C to rt, 30 min; (iii) TFA, CH2Cl2, rt, 4 h; (iv) 40, (a) NH2NH2.H2O, S8, EtOH, 90 °C, 4 h; (b) NaNO2, AcOH, 0 °C to rt, 30 min; (v) nosyl chloride, DIPEA, DMAP, CH2Cl2, rt, 1 h.
Next, 45 was evaluated for its abilities in pretargeting. We recently developed a blocking assay that allowed us to assess the in vivo ligation performance of unlabeled tetrazine derivatives, omitting the time-consuming development of radiolabeled tetrazines for each tested ligand. It was based on the ability of Tzs to block the binding of the literature-known pretargeted imaging agent [111In]47 to the pretargeting vector CC49-TCO (administered 72 h prior) in tumor-bearing mice [17,32,34]. In this setup, 45 showed almost complete blocking. This blocking effect was as good as the ones observed with currently successfully applied ‘state-of-the-art’ pretargeted imaging agents (Table 2). This result allowed us to assume that 45 could indeed be a suitable candidate for pretargeted imaging.
Table 2.
Structural scaffolds, calculated physicochemical properties (TPSA, clogD7.4), and blocking efficiencies of all investigated tetrazine derivatives. Compound 45 displayed a suitable profile to be translated into preclinical studies.
| ||||
|---|---|---|---|---|
| Tetrazine | clogD7.4 b | TPSA c | Blocking Effect f | % Tumor Uptake of [111In]47 after Blocking |
| 1 a | −6.93 | 129 | 90 ± 5 g | 10 ± 5 g |
| 2 a | −6.83 | 138 | 98 ± 0.3 h | 2 ± 0.3 h |
| 41 * | 3.65 | 35 | 22 ± 21 | 78 ± 21 |
| 45 a | −6.36 | 164 | 99 ± 0.7 | 1 ± 0.7 |
| 47 d | −4.13 e | 358 | 99 ± 0.5 | 1 ± 0.5 |
a The compounds were obtained as trifluoroacetate salt. b Calculated distribution coefficient at physiological pH (7.4) using Chemicalize software. c Calculated using Chemicalize software. d The compound was employed as a reference. e Calculated as chelated to a trivalent cation. f The blocking effect of nonradiolabeled Tz was determined as the change in tumor uptake of [111In]47 22 h p.i. Each Tz was administered 1 h prior to [111In]47, and the uptake was normalized to a group of animals in which no blocking was performed (control). Data represent mean from n = 3 mice/group; detailed information can be found in the Materials and Methods section. g Blocking data from [26]. h Blocking data from [27]. * Tz 41 was tested to demonstrate that the blocking effect was related to clogD7.4 and not the bispyridyl scaffold. TPSA = topological polar surface area.
2.7. Synthesis and Labeling of [18F]45
The precursor (45a) was synthesized over 4 synthesis steps (Scheme 5). Briefly, 40 was reacted with an excess of 43 and hydrazine hydrate to give, after oxidation, 46. This was then converted to the corresponding nosylated derivative 45a. Radiolabeling of [18F]45 was carried out in a one-pot, two-step reaction sequence (Figure 4A,B). The conditions were similar to those we recently reported and applied for all former labeling attempts in this study [29,32,33,46]. A protection/deprotection strategy was needed to successfully label [18F]45 in a radiochemical yield (RCY) of 16 ± 4% (n = 4), a radiochemical purity (RCP) of >98%, and a molar activity (AM) of 57 ± 15 GBq/µmol. The total synthesis was approximately 90 min, including labeling, purification, and formulation of the final product. The maximum isolated amount was 1.2–2 GBq (EOS) starting from ~17 GBq of fluoride-18. The [18F]45 rapidly reacted with TCO-PNP carbonate, and was stable in PBS at room temperature for minimum of 4 h, as confirmed by radio-HPLC (Figure 4C).
Figure 4.

Radiolabeling of [18F]45. (A) Reaction sequence. Reagents and conditions: (i) [18F] Bu4NF/Bu4NOMs PO43-, t-BuOH/DMSO, 100 °C, 5 min; (ii) TFA, CH3CN, 80 °C, 10 min. (B) Analytical chromatograms of formulated [18F]45 in PBS (pH = 7.4) spiked with nonradioactive 45 for identification (rt: 5.20 min). (C) Analytical chromatogram of the reaction between [18F]45 showing that the tetrazine core was intact. Analytical chromatogram of [18F]15 formulated before TCO-PNP addition (red) and after TCO-PNP addition (black).
2.8. In Vivo Pretargeting PET Imaging with [18F]45
The ability of [18F]45 to act as a pretargeting imaging agent was evaluated in LS174T tumor-bearing mice. Mice were grouped and administered with either monoclonal anti-TAG72 antibody, CC49, or TCO-conjugated CC49 (CC49-TCO) 72 h prior to injection of [18F]45. PET/CT scans were acquired 1 h later, as well as image-derived uptake values from tumor, heart, and muscle tissue extracted by manually created regions of interests (Figure 5).
Figure 5.

PET/CT imaging of [18F]45. (A) Images from PET/CT scans of [18F]45 1 h p.i. from mice pretreated with either CC49-TCO (left) or CC49 (right) 72 h earlier. Scale bar indicates mean percent of injected dose per gram (%ID/mL). White arrows indicate LS174T tumor xenografts. (B) Image-derived tissue uptake (mean %ID/mL) of [18F]45 1 h p.i. in mice administered with either CC49-TCO or CC49 72 h prior to [18F]45 injection. (C) Image-derived tissue uptake of [18F]45 in mean %ID/mL. All values are reported as mean ± S.E.M, n = 5/group. * Uptake in heart was considered as a surrogate for blood. ** p < 0.01 (Welch’s t-test).
The tumors of the mice in the group receiving CC49-TCO had a significant higher uptake of [18F]45 (1.8 ± 0.3%ID/mL, mean ± SEM, n = 5) compared to the animals in the control group (0.3 ± 0.1%ID/mL, n = 5). Uptake in muscle tissue was generally low, resulting in a tumor-to-muscle ratio of 8.3 in animals pretargeted with CC49-TCO. In contrast, the uptake in blood (heart used as a surrogate) in this group of animals was high, resulting in a tumor-to-blood (T/B) ratio of 0.8. The tumor-to-liver (T/L) ratio was 0.3. Therefore, the uptake and target-to-background contrast found for [18F]45 were at comparable levels to previous findings [17,32,34]. However, there is a general need to improve the target-to-background contrast before clinical translation. This could either be achieved by developing primary and secondary vectors with improved performance, thereby increasing the tumor uptake. Further, the use of a clearing agent could also potentially accelerate clearance of the primary vector and thus improve the T/B ratio in the future [14].
3. Materials and Methods
3.1. Chemistry
3.1.1. General
All reagents and solvents were purchased from commercial suppliers and used without further purification. Anhydrous tetrahydrofuran (THF) was obtained from an SG water solvent purification system (Pure Process Technology). Anhydrous dimethyl sulfoxide (DMSO), N,N-dimethylacetamide (DMA), MeCN, and pyridine were purchased from commercially suppliers and stored under argon. Reactions requiring anhydrous conditions were carried out under an inert atmosphere (nitrogen or argon) and using oven-dried glassware (152 °C). Syringes used to transfer anhydrous solvents or reagents were purged with argon prior to use. Other solvents were analytical or HPLC grade and were used as received. NMR spectra were acquired on a 600 MHz Bruker Avance III HD (600 MHz for 1H and 151 MHz for 13C), a 400 MHz Bruker Avance II (400 MHz for 1H, 376 MHz for 19F, and 101 MHz for 13C), and a 400 MHz Bruker Avance UltraShield (400 MHz for 1H, 376 MHz for 19F, and 101 MHz for 13C) using chloroform-d, MeOD, or DMSO-d6 as a deuterated solvent, and with the residual solvent as the internal reference. For all NMR experiments, the deuterated solvent signal was used as the internal lock. Coupling constants (J values) are given in Hertz (Hz). Multiplicities of 1H NMR signals are reported as follows: s, singlet; d, doublet; dd, doublet of doublets; ddd, doublet of doublets of doublets; dt, doublet of triplets; t, triplet; q, quartet; m, multiplet; br, broad signal. NMR spectra of all compounds were reprocessed in MestReNova software (version 12.0.22023) from the original FID files. Yields refer to isolated compounds estimated to be >90% pure as determined by 1H NMR (25 °C) and analytical HPLC (Please refer to the Supplementary Materials). Analytical HPLC method: Thermo Fisher UltiMate 3000 with a C-18 column (Luna 5 μm C18(2) 100 Å, 150 mm × 4.6 mm). Eluents: A, H2O with 0.1% TFA; B, MeCN with 0.1% TFA. Gradient from 100% A to 100% B for 12 min, back to 100% A for 3 min, flow rate 2 mL/min. Detection by UV absorption at λ = 254 nm on a UVD 170U detector. Thin-layer chromatography (TLC) was carried out on silica gel 60 F254 plates from Merck (Germany). Visualization was accomplished by UV lamp (254 nm). Preparative HPLC was carried out on an UltiMate HPLC system (Thermo Scientific) consisting of an LPG-3200BX pump (10 mL/min), a Rheodyne 9725i injector, a 10 mL loop, a MWD-3000SD detector (254 nm), and an AFC-3000SD automated fraction collector, using a Gemini-NX C18 column (21.2 × 250 mm, 5 µm, 110Å) (Phenomenex) equipped with a guard. Purifications were performed using linear gradients of 0.1% TFA in MilliQ-H2O (A) and 0.1% TFA, 10% MilliQ-H2O in MeCN (B). Data were acquired and processed using Chromeleon software v. 6.80. Semipreparative HPLC was performed on the same system using a Luna 5µ C18 column (250 × 10 mm) with a flow rate of 3 mL/min. Automated flash column chromatography was performed on a CombiFlash NextGen 300+ system supplied by TeleDyne ISCO, equipped with RediSep silica-packed columns. Detection of the compounds was carried out by means of a UV–vis variable wavelength detector operating at 200 to 800 nm and by an evaporative light-scattering detector (ELSD). Solvent systems for separation were particular for each compound, but consisted of various mixtures of heptane, EtOAc, CH2Cl2, and MeOH. Microwave-assisted synthesis was carried out in a Biotage Initiator apparatus operating in single mode; the microwave cavity produced controlled irradiation at 2.45 GHz (Biotage AB, Uppsala, Sweden). The reactions were run in sealed vessels. These experiments were performed by employing magnetic stirring and a fixed hold time using variable power to reach (during 1–2 min) and then maintain the desired temperature in the vessel for the programmed time period. The temperature was monitored by an IR sensor focused on a point on the reactor vial glass. The IR sensor was calibrated to the internal solution’s reaction temperature by the manufacturer. Mass spectra analysis was completed using MS-Acquity-A: Waters Acquity UPLC with QDa-detector. CC49-TCO was kindly provided by Tagworks Pharmaceuticals, and it was obtained as previously described [14].
3.1.2. 2-Fluoroethyl 4-(1,2,4,5-tetrazin-3-yl)benzoate (3)
4-(1,2,4,5-Tetrazin-3-yl)benzoic acid (10)
The compound was synthesized as previously reported [42]. 4-Cyanobenzoic acid (0.3 g, 2.00 mmol), CH2Cl2 (0.12 mL, 2.00 mmol), sulfur (0.12 g, 0.5 mmol), and ethanol (4.0 mL) were mixed together in a microwave reaction vial. Hydrazine monohydrate (0.82 mL, 16.00 mmol) was added dropwise with stirring. The vessel was sealed, and the reaction mixture was heated to 50 °C for 24 h. The reaction was diluted with 3 mL of CH2Cl2, and sodium nitrite (1.44 g, 20.00 mmol) in 20 mL of H2O was added dropwise to the mixture under cooling. Excess acetic acid (7 mL) was then added slowly, during which the solution turned bright red in color. The reaction mixture was extracted with CH2CL2 (3 × 20 mL). The organic phase was dried over MgSO4 and concentrated under reduced pressure. The resulting residue was purified using flash chromatography (CH2Cl2/MeOH 98/2) to give 0.08 g (20%) of the desired product as a pink solid. Rf = 0.31 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, DMSO) δ 13.32 (s, 1H), 10.66 (s, 1H), 8.62 (d, J = 8.5 Hz, 2H), 8.22 (d, J = 8.5 Hz, 2H); 13C NMR (101 MHz, DMSO) δ 166.67, 165.08, 158.24, 135.70, 134.32, 130.20, 127.97.
2-Fluoroethyl 4-(1,2,4,5-tetrazin-3-yl)benzoate (3)
Compound 10 (0.04 g, 0.20 mmol) and 1-fluoro-2-iodoethane (0.05 g, 0.30 mmol) were dissolved in 2 mL dry DMF, and DIPEA (0.10 mL, 0.60 mmol) was added. The reaction was left at 70 °C overnight. The reaction was diluted with CH2Cl2 (15 mL) and washed with a saturated aqueous solution of NH4Cl (10 mL) and H2O (2 × 10 mL). The organic phase was dried over MgSO4 and concentrated under reduced pressure. Purification by flash chromatography (85/15 heptane/EtOAc) afforded 0.04 g (81%) of 3 as a red solid. Rf = 0.23 (heptane/EtOAc 80/20); 1H NMR (600 MHz, CDCl3) δ 10.21 (s, 1H), 8.65 (d, J = 8.6 Hz, 2H), 8.23 (d, J = 8.5 Hz, 2H), 4.89–4.72 (m, 1H), 4.69–4.64 (m, 1H), 4.63–4.57 (m, 1H), 4.57–4.50 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 165.92, 165.50, 157.99, 135.75, 133.63, 130.58, 128.27, 81.26 (d, J = 171.1 Hz), 64.30 (d, J = 20.1 Hz).
3.1.3. 2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 4-(1,2,4,5-tetrazin-3-yl)benzoate (3a)
2-Hydroxyethyl 4-(1,2,4,5-tetrazin-3-yl)benzoate (11)
Compound 10 (0.03 g, 0.15 mmol) and 2-bromoethanol (0.02 mL, 0.22 mmol) were dissolved in 3 mL dry DMF, DIPEA (0.08 mL, 0.44 mmol) was dissolved in 1 mL DMF and added dropwise, and the reaction was left at 70 °C. After the reaction was completed, it was cooled down, diluted with water (30 mL) and extracted with CH2Cl2 (3 × 20 mL). The crude was purified by flash chromatography (60/40 heptane/EtOAc) to give 0.03 g (82%) of the desired product as a red solid. Rf = 0.24 (heptane/EtOAc 70/30); 1H NMR (600 MHz, CDCl3) δ 10.28 (s, 1H), 8.72 (d, J = 8.1 Hz, 2H), 8.29 (d, J = 8.1 Hz, 2H), 4.54 (t, J = 4.6 Hz, 2H), 4.02 (t, J = 4.7 Hz, 2H), 2.56–1.97 (m, 1H), 13C NMR (151 MHz, CDCl3) δ 166.04, 165.93, 158.00, 135.68, 133.87, 130.55, 128.28, 67.10, 61.31.
2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 4-(1,2,4,5-tetrazin-3-yl)benzoate (3a)
To a solution of compound 11 (0.03 g, 0.12 mmol) and DIPEA (0.04 mL, 0.24 mmol) in CH2Cl2 (10 mL) were added nosyl chloride (0.041 g, 0.18 mmol) and DMAP (0.001 g, 0.01 mmol). The reaction was stirred at room temperature for 1 h. The solvent was removed under reduced pressure. Purification by flash chromatography (80/20 heptane/EtOAc) afforded 0.035 (67%) of 3a as a red solid. Rf = 0.38 (heptane/EtOAc 70/30); 1H NMR (600 MHz, CDCl3) δ 10.31 (s, 1H), 8.71 (d, J = 8.5 Hz, 2H), 8.31 (d, J = 8.8 Hz, 2H), 8.16 (d, J = 8.5 Hz, 2H), 8.14–8.09 (m, 2H), 4.64–4.58 (m, 2H), 4.58–4.53 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 165.77, 165.07, 158.06, 150.73, 141.65, 136.14, 132.92, 130.48, 129.17, 128.27, 124.50, 68.72, 62.28.
3.1.4. N-(2-Fluoroethyl)-4-(1,2,4,5-tetrazin-3-yl)benzamide (4)
4-Cyano-N-(2-fluoroethyl)benzamide (12)
To a solution of 4-cyanobenzoic acid (0.73 g, 5.00 mmol) in CH3CN (20 mL) was added CDI (1.21 g, 7.50 mmol). The mixture was stirred at room temperature for 45 min, before addition of 2-fluoroethylamine hydrochloride (0.55 g, 5.50 mmol) and Et3N (2.09 mL, 15.00 mmol). The reaction mixture was stirred for 2 h. Water (30 mL) was added, and the mixture was extracted with EtOAc (3 × 20 mL). The organic phase was dried over MgSO4 and concentrated under reduced pressure to give 0.65 g (68%) of the desire compound as a white solid. Rf = 0.26 (heptane/EtOAc 60/40); 1H NMR (400 MHz, DMSO) δ 8.96 (s, 1H), 8.02 (d, J = 8.7 Hz, 2H), 7.97 (d, J = 8.6 Hz, 2H), 4.61 (t, J = 5.1 Hz, 1H), 4.50 (t, J = 5.1 Hz, 1H), 3.62 (q, J = 5.2 Hz, 1H), 3.56 (q, J = 5.2 Hz, 1H); 13C NMR (101 MHz, DMSO) δ 165.61, 138.57, 132.92, 128.55, 114.17, 82.49 (d, J = 165.8 Hz), 40.57 (d, J = 20.9 Hz).
N-(2-Fluoroethyl)-4-(1,2,4,5-tetrazin-3-yl)benzamide (4)
The compound was obtained from 12 (0.60 g, 3.12 mmol) as described for compound 10. Purification by flash chromatography (60/40 heptane/EtOAc) afforded 0.36 g (47%) of 4 as a red solid. Rf = 0.38 (heptane/EtOAc 50/50); 1H NMR (400 MHz, DMSO) δ 10.65 (s, 1H), 8.95 (t, J = 5.5 Hz, 1H), 8.60 (d, J = 8.6 Hz, 2H), 8.15 (d, J = 8.6 Hz, 2H), 4.65 (t, J = 5.1 Hz, 1H), 4.53 (t, J = 5.1 Hz, 1H), 3.66 (q, J = 5.3 Hz, 1H), 3.59 (q, J = 5.2 Hz, 1H); 13C NMR (101 MHz, DMSO) δ 166.24, 165.59, 158.70, 138.21, 134.81, 128.75, 128.23, 82.57 (d, J = 165.8 Hz), 40.56 (d, J = 16.2 Hz).
3.1.5. 2-(4-(1,2,4,5-Tetrazin-3-yl)benzamido)ethyl 4-nitrobenzenesulfonate (4a)
4-Cyano-N-(2-hydroxyethyl)benzamide (13)
The compound was obtained from 4-cyanobenzoic acid (1.17 g, 8.00 mmol) and ethanolamine (4.82 mL, 80.00 mmol) as described for compound 12. Purification by flash chromatography (CH2Cl2/MeOH 95/5) afforded 1.37 g (90%) of the desired compound as a colorless oil. Rf = 0.22 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, DMSO) δ 8.70 (t, J = 5.6 Hz, 1H), 8.01 (d, J = 8.6 Hz, 2H), 7.96 (d, J = 8.6 Hz, 2H), 4.75 (t, J = 5.6 Hz, 1H), 3.53 (q, J = 5.9 Hz, 2H), 3.39–3.32 (m, 2H); 13C NMR (101 MHz, DMSO) δ 165.38, 139.03, 132.83, 128.54, 118.83, 113.94, 60.01, 42.82.
N-(2-Hydroxyethyl)-4-(1,2,4,5-tetrazin-3-yl)benzamide (14)
The compound was obtained from 13 (1.00 g, 5.25 mmol) as described for compound 10. Purification by flash chromatography (CH2Cl2/MeOH 97/3) afforded 0.39 g (30%) of the desired compound as a colorless oil. Rf = 0.31 (CH2Cl2/MeOH 95/5); 1H NMR (600 MHz, DMSO) δ 10.64 (s, 1H), 8.68 (t, J = 5.6 Hz, 1H), 8.61–8.56 (m, 2H), 8.16–8.10 (m, 2H), 4.75 (t, J = 5.6 Hz, 1H), 3.56 (q, J = 6.1 Hz, 2H), 3.38 (q, J = 6.0 Hz, 2H); 13C NMR (151 MHz, DMSO) δ 166.00, 165.61, 158.71, 158.68, 138.66, 128.72, 128.16, 60.13, 42.80.
2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)-4,5-dihydrooxazole
The compound was obtained from 14 (0.05 g, 0.21 mmol) as described for compound 3a. An intramolecular substitution occurred during the reaction, giving this side product. Purification by flash chromatography (60/40 heptane/EtOAc) afforded 0.35 g (75%) of this side product as a red solid. Rf = 0.41 (heptane/EtOAc 50/50); 1H NMR (600 MHz, CDCl3) δ 10.18 (s, 1H), 9.09–8.44 (m, 2H), 8.11 (d, J = 8.1 Hz, 2H), 4.43 (t, J = 9.5 Hz, 2H), 4.06 (t, J = 9.5 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 166.07, 163.83, 157.88, 133.94, 132.05, 129.06, 128.22, 67.91, 55.17.
3.1.6. 2-Fluoroethyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (5)
2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)acetic acid (15)
The compound was obtained from 2-(4-cyanophenyl)acetic acid (0.97 g, 6.00 mmol) as described for compound 10. Purification by flash chromatography (98/2 CH2Cl2/MeOH) to afford 0.36 g (28%) of the desired compound as a pink solid. Rf = 0.19 (95/5 CH2Cl2/MeOH); 1H NMR (400 MHz, MeOD) δ 10.33 (s, 1H), 8.56 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 8.3 Hz, 2H), 3.78 (s, 2H); 13C NMR (101 MHz, MeOD) δ 173.27, 166.24, 157.83, 140.15, 130.69, 130.13, 127.78, 40.39.
2-Fluoroethyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (5)
The compound was obtained from 15 (0.07 g, 0.32 mmol) as described for compound 3. Purification by flash chromatography (80/20 heptane/EtOAc) afforded 0.042 g (49%) of 5 as a red solid. Rf = 0.20 (80/20 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 10.14 (s, 1H), 8.53 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.1 Hz, 2H), 4.63–4.57 (m, 1H), 4.51–4.46 (m, 1H), 4.37–4.32 (m, 1H), 4.30–4.25 (m, 1H), 3.74 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 170.58, 166.25, 157.79, 139.06, 130.62, 130.38, 128.56, 81.14 (d, J = 170.9 Hz), 63.96 (d, J = 20.2 Hz), 41.01.
3.1.7. 2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (5a)
2-Hydroxyethyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (16)
The compound was obtained from 15 (0.07 g, 0.32 mmol) as described for compound 11. Purification by flash chromatography (80/20 heptane/EtOAc) afforded 0.05 g (59%) of the desired product as a red solid. Rf = 0.17 (60/40 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 10.14 (s, 1H), 8.51 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.3 Hz, 2H), 4.24–4.17 (m, 2H), 3.81–3.74 (m, 2H), 3.73 (s, 2H), 1.91 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 171.09, 166.22, 157.78, 139.23, 130.58, 130.36, 128.55, 66.68, 61.06, 41.13.
2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (5a)
The compound was obtained from 16 (0.50 g, 0.19 mmol) as described for compound 3a. Purification by flash chromatography (75/25 heptane/EtOAc) afforded 0.05 g (58%) of 5a as a red solid. Rf = 0.27 (heptane/EtOAc 60/40); 1H NMR (400 MHz, CDCl3) δ 10.16 (s, 1H), 8.51 (d, J = 8.2 Hz, 2H), 8.30 (d, J = 8.8 Hz, 2H), 8.02 (d, J = 8.8 Hz, 2H), 7.43 (d, J = 8.2 Hz, 2H), 4.30 (s, 4H), 3.69 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 170.30, 157.84, 150.86, 141.58, 138.66, 130.76, 130.35, 129.20, 128.56, 124.52, 68.35, 62.00, 40.86.
3.1.8. 2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)-N-(2-fluoroethyl)acetamide (6)
2-(4-Cyanophenyl)-N-(2-fluoroethyl)acetamide (17)
The compound was obtained from 2-(4-cyanophenyl)acetic acid (0.5 g, 3.10 mmol) as described for compound 12 [17]. Concentration under reduced pressure afforded 0.46 g (72%) of the desired product as a white solid. Rf: 0.35 (heptane/EtOAc 30/70); 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 5.88 (s, 1H), 4.47 (t, J = 4.8 Hz, 1H), 4.35 (t, J = 4.8 Hz, 1H), 3.59–3.49 (m, 3H), 3.46 (dt, J = 5.8, 4.7 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 169.58, 140.02, 132.56, 130.10, 118.58, 111.32, 82.48 (d, J = 166.7 Hz), 43.36, 40.23 (d, J = 19.5 Hz).
2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)-N-(2-fluoroethyl)acetamide (6)
The compound was obtained from 17 (0.40 g, 1.94 mmol) as described for compound 3 [17]. Purification by flash chromatography (30/70 heptane/EtOAc) afforded 0.21 g (41%) of 6 as a red solid. Rf = 0.22 (heptane/EtOAc 40/60); 1H NMR (400 MHz, DMSO) δ 10.58 (s, 1H), 8.48–8.40 (m, 3H), 7.57 (d, J = 8.2 Hz, 2H), 4.51 (t, J = 5.0 Hz, 1H), 4.39 (t, J = 5.0 Hz, 1H), 3.62 (s, 2H), 3.43 (q, J = 5.3 Hz, 1H), 3.36 (q, J = 5.2 Hz, 1H); 13C NMR (101 MHz, DMSO) δ 170.27, 165.92, 158.54, 142.02, 130.61, 130.53, 128.15, 82.90 (d, J = 165.0 Hz), 42.56, 40.56 (d, J = 15.8 Hz).
3.1.9. 2-(2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)acetamido)ethyl 4-nitrobenzenesulfonate (6a)
2-(4-Cyanophenyl)-N-(2-hydroxyethyl)acetamide (18)
The compound was obtained from 2-(4-cyanophenyl)acetic acid (2.1 g, 13.03 mmol) as described for compound 13. Recrystallization from EtOAc afforded 1.60 g (60%) of the desired product as a white solid. Rf = 0.15 (heptane/EtOAc 20/80); 1H NMR (400 MHz, DMSO) δ 8.15 (s, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 4.68 (t, J = 5.4 Hz, 1H), 3.54 (s, 2H), 3.40 (q, J = 5.8 Hz, 2H), 3.12 (q, J = 5.9 Hz, 2H); 1H NMR (400 MHz, DMSO) δ 8.15 (s, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 4.68 (t, J = 5.4 Hz, 1H), 3.54 (s, 2H), 3.40 (q, J = 5.8 Hz, 2H), 3.12 (q, J = 5.9 Hz, 2H).
2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)-N-(2-hydroxyethyl)acetamide (19)
The compound was obtained from 18 (0.82 g, 4.00 mmol) as described for compound 3. Purification by flash chromatography (98/2 CH2Cl2/MeOH) and recrystallization from EtOAc afforded 0.33 g (32%) of the desired product as a pink solid. Rf = 0.31 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, DMSO) δ 10.31 (s, 1H), 8.54 (d, J = 8.2 Hz, 2H), 7.58 (d, J = 8.1 Hz, 2H), 3.67 (s, 2H), 3.62 (t, J = 5.8 Hz, 2H), 3.36–3.32 (m, 2H); 13C NMR (101 MHz, DMSO) δ 171.23, 165.43, 157.02, 140.25, 129.84, 129.01, 127.04, 59.30, 41.46, 40.99.
2-(2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)acetamido)ethyl 4-nitrobenzenesulfonate (6a)
The compound was obtained from 19 (0.06 g, 0.23 mmol) as described for compound 3a. Purification by flash chromatography (99/1 CH2Cl2/MeOH) afforded 0.08 g (83%) of 3a as a red solid. Rf = 0.31 (heptane/EtOAc 60/40); 1H NMR (400 MHz, DMSO) δ 10.51 (s, 1H), 8.46 (d, J = 8.9 Hz, 2H), 8.39 (d, J = 8.6 Hz, 2H), 8.29 (d, J = 8.9 Hz, 2H), 7.69 (d, J = 8.6 Hz, 2H), 5.83 (s, 1H), 4.29 (t, J = 7.0 Hz, 2H), 4.10 (t, J = 7.0 Hz, 2H), 3.31 (s, 2H); 13C NMR (101 MHz, DMSO) δ 165.77, 158.22, 151.32, 149.36, 141.57, 140.68, 129.72, 128.22, 128.11, 128.07, 125.43, 86.21, 67.35, 47.47.
3.1.10. 3-(4-(2-Fluoroethoxy)phenyl)-1,2,4,5-tetrazine (7)
4-(2-Fluoroethoxy)benzonitrile (20)
To a solution of 4-hydroxybenzonitrile (0.6 g, 5.00 mmol) and K2CO3 (1.38 g, 10.00 mmol) in CH3CN (20 mL) was added 1-fluoro-2-iodoethane (1.04 g, 6.00 mmol). The reaction was refluxed for 12 h and then concentrated under reduced pressure. The resulting mixture was diluted with water (50 mL), extracted with EtOAc (3 × 50 mL), washed with brine (50 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (heptane/EtOAc 80/20) afforded 0.8 g (97%) of the desired compound as a yellow oil. Rf = 0.37 (heptane/EtOAc 70/30); 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 8.9 Hz, 1H), 7.01 (d, J = 8.9 Hz, 1H), 5.05–4.79 (m, 1H), 4.77–4.71 (m, 1H), 4.36–4.29 (m, 1H), 4.29–4.22 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 161.69, 134.06, 119.03, 115.33, 104.59, 81.48 (d, J = 171.7 Hz), 67.37 (d, J = 20.5 Hz).
3-(4-(2-Fluoroethoxy)phenyl)-1,2,4,5-tetrazine (7)
The compound was obtained from 20 (0.73 g, 4.42 mmol) as described for compound 3. Purification by flash chromatography (heptane/EtOAc 90/10) afforded 0.36 g (37%) of the desired compound as a red oil. Rf = 0.33 (heptane/EtOAc 80/20); 1H NMR (400 MHz, CDCl3) δ 10.07 (s, 1H), 8.53 (d, J = 8.9 Hz, 2H), 7.06 (d, J = 8.9 Hz, 2H), 4.89–4.78 (m, 1H), 4.74–4.62 (m, 1H), 4.43–4.29 (m, 1H), 4.27–4.16 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 166.06, 162.56, 157.40, 130.26, 124.56, 115.38, 81.63 (d, J = 171.5 Hz), 67.29 (d, J = 20.6 Hz).
3.1.11. 2-(4-(1,2,4,5-Tetrazin-3-yl)phenoxy)ethyl 4-nitrobenzenesulfonate (7a)
4-(2-Hydroxyethoxy)benzonitrile (21)
To a solution of 4-hydroxybenzonitrile (1.42 g, 12.00 mmol) and K2CO3 (8.29 g, 60.00 mmol) in CH3CN (20 mL) was added 2-bromoethanol (2.55 mL, 36.00 mmol). The reaction was refluxed for 12 h and then concentrated under reduced pressure. The resulting mixture was diluted with water (50 mL), extracted with EtOAc (3 × 50 mL), washed with brine (50 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (heptane/EtOAc 40/60) afforded 1.41 g (72%) of the desired compound as a yellow oil. Rf = 0.18 (heptane/EtOAc 50/50); 1H NMR (400 MHz, DMSO) δ 7.76 (d, J = 8.8 Hz, 2H), 7.11 (d, J = 8.8 Hz, 2H), 4.92 (t, J = 5.5 Hz, 1H), 4.09 (t, J = 5.0 Hz, 2H), 3.73 (q, J = 5.0 Hz, 2H), 13C NMR (101 MHz, DMSO) δ 162.70, 134.63, 119.64, 116.05, 103.15, 70.56, 59.79.
2-(4-(1,2,4,5-Tetrazin-3-yl)phenoxy)ethan-1-ol (22)
The compound was obtained from 21 (0.98 g, 6.00 mmol) as described for compound 3. Purification by flash chromatography (heptane/EtOAc 50/50) afforded 0.71 g (54%) of the desired product as a red solid. Rf = 0.27 (heptane/EtOAc 50/50); 1H NMR (400 MHz, DMSO) δ 10.50 (s, 1H), 8.47 (d, J = 9.0 Hz, 2H), 7.23 (d, J = 9.0 Hz, 2H), 4.94 (t, J = 5.5 Hz, 1H), 4.15 (t, J = 4.9 Hz, 2H), 3.78 (q, J = 5.2 Hz, 2H); 13C NMR (101 MHz, DMSO) δ 165.65, 163.10, 158.16, 130.10, 124.35, 115.95, 70.44, 59.92.
2-(4-(1,2,4,5-Tetrazin-3-yl)phenoxy)ethyl 4-nitrobenzenesulfonate (7a)
Compound 21 (0.10 g, 0.46 mmol) was mixed with nitrobenzenesulfonyl chloride (0.15 g, 0.69 mmol) and dissolved in 4 mL dry CH2Cl2 under argon. A mixture of DIPEA (0.33 mL, 1.83 mmol) and DMAP (5 mg, 0.04 mmol) in 1 mL dry CH2Cl2 was added at 0 °C under argon. The reaction was slowly heated to room temperature and left for 1 h. The reaction was diluted with 10 mL CH2Cl2 and washed with 20 mL NH4Cl (sat.) and H2O (2 × 20 mL). The organic phase was dried over N2SO4 and concentrated under reduced pressure. Purification by flash chromatography (heptane/EtOAc 60/40) afforded 0.12 g (65%) of the desired product as a red solid. Rf = 0.41 (heptane/EtOAc 50/50); 1H NMR (400 MHz, DMSO) δ 10.52 (s, 1H), 8.97–8.31 (m, 5H), 8.33–7.87 (m, 2H), 7.11 (d, J = 8.9 Hz, 2H), 4.97–4.50 (m, 2H), 4.48–4.06 (m, 2H); 13C NMR (101 MHz, DMSO) δ 165.57, 161.88, 158.28, 154.85, 147.71, 130.25, 127.38, 125.26, 123.79, 116.05, 75.01, 67.33.
3.1.12. N-(4-(1,2,4,5-Tetrazin-3-yl)benzyl)-2-fluoroethan-1-amine (9)
4-(((2-Fluoroethyl)amino)methyl)benzonitrile (23)
To a solution of 4-(bromomethyl)benzonitrile (0.78 g, 4.00 mmol) in CH3CN (40 mL) was added K2CO3 (0.33 g, 24.0 mmol) and 2-fluoroethylamine hydrochloride (0.16 g, 16.0 mmol). The mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the residue was diluted with water (20 mL) and extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using EtOAc (heptane/EtOAc 50/50) in heptane to afford 0.54 g (76%) of 23 as a colorless oil. Rf = 0.24 (heptane/EtOAc 40/60). 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 8.2 Hz, 2H), 7.40 (d, J = 8.0 Hz, 2H), 4.63–4.48 (m, 1H), 4.47–4.37 (m, 1H), 3.84 (s, 2H), 2.93–2.84 (m, 1H), 2.84–2.72 (m, 1H), 1.65 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 145.6, 132.3, 128.6, 118.9, 110.9, 83.5 (d, J = 165.5 Hz), 53.1, 49.1 (d, J = 19.7 Hz)
Tert-butyl 4-cyanobenzyl(2-fluoroethyl)carbamate (25)
To a solution of 23 (540 mg, 3.03 mmol) and Et3N (1.27 mL, 9.09 mmol) in CH2Cl2 (40 mL) was added Boc2O (790 mg, 3.63 mmol), and the mixture was stirred at room temperature for 12 h. The solution was washed with water and saturated K2CO3 solution, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using (heptane/EtOAc 70/30) to afford 0.710 g (84%) of the desired product as a colorless oil (mixture of rotamers). Rf = 0.42 (heptane/EtOAc 80/20). 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 7.8 Hz, 2H), 7.27 (d, J = 7.8 Hz, 2H), 4.79–4.10 (m, 4H), 3.62–3.28 (m, 2H), 1.96–1.05 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 155.4, 144.2, 143.8, 132.4, 128.1, 127.5, 118.7, 111.1, 83.2 (d, J = 168.2 Hz), 82.7 (d, J = 170.5 Hz), 52.1, 51.2, 47.7, 28.3.
Tert-butyl 4-(1,2,4,5-tetrazin-3-yl)benzyl(2-fluoroethyl)carbamate (27)
The compound was obtained from 25 (0.68 g, 2.44 mmol) as described for compound 3. Purification by flash chromatography (heptane/EtOAc 80/20) afforded 0.18 g (22%) of the desired product as a red solid (mixture of rotamers). Rf = 0.21 (heptane/EtOAc 80/20); 1H NMR (400 MHz, CDCl3) δ 10.23 (s, 1H), 8.62 (d, J = 7.8 Hz, 2H), 7.49 (d, J = 7.8 Hz, 2H), 4.76–4.42 (m, 4H), 3.83–3.38 (m, 2H), 1.64–1.38 (m, 9H); 13C NMR (101 MHz, CDCl3) δ 166.32, 157.77, 155.51, 144.25, 132.37, 130.58, 128.55, 127.86, 83.46, 83.17 (d, J = 165.4 Hz), 82.67 (d, J = 170.4 Hz), 80.63, 52.07, 51.07, 47.49, 28.36.
N-(4-(1,2,4,5-tetrazin-3-yl)benzyl)-2-fluoroethan-1-amine (9)
To a solution of 27 (0.10 mg, 0.30 mmol) in dioxane (10 mL) was added a solution of HCl in dioxane (4.0 M, 3.0 mL). The mixture was stirred at room temperature for 2 h and then concentrated under reduced pressure. The obtained solid was washed with Et2O to afford 0.07 g (86%) of 9 as hydrochloride salt. 1H NMR (400 MHz, DMSO-d6) δ 10.64 (s, 1H), 9.87 (s, 2H), 8.55 (d, J = 8.3 Hz, 2H), 7.89 (d, J = 8.3 Hz, 2H), 4.89 (t, J = 4.6 Hz, 1H), 4.77 (t, J = 4.6 Hz, 1H), 4.35 (s, 2H), 3.38 (t, J = 4.7 Hz, 1H), 3.31 (t, J = 4.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 165.7, 158.7, 137.0, 132.8, 131.6, 128.4, 80.0 (d, J = 165.3 Hz), 50.1, 47.1 (d, J = 20.0 Hz).
3.1.13. 2-((4-(1,2,4,5-Tetrazin-3-yl)benzyl)(tert-butoxycarbonyl)amino)ethyl 4-nitro Benzenesulfonate (9a)
4-(((2-Hydroxyethyl)amino)methyl)benzonitrile (24)
The compound was obtained from 4-(bromomethyl)benzonitrile (2.00 g, 10.20 mmol) and ethanolamine (12.30 mL, 200.00 mmol) as described for compound 23. Concentration under reduced pressure afforded 1.65 g (92%) of the desired compound as a white solid. Rf = 0.15 (90/10 CH2Cl2/MeOH); 1H NMR (400 MHz, CDCl3) δ 7.62–7.52 (m, 2H), 7.46–7.37 (m, 2H), 3.88–3.77 (m, 2H), 3.69–3.59 (m, 2H), 2.79–2.70 (m, 2H), 2.58–2.23 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 145.76, 132.22, 128.67, 118.88, 110.71, 60.91, 53.07, 50.78.
Tert-butyl (4-cyanobenzyl)(2-hydroxyethyl)carbamate (26)
The compound was obtained from 24 (1.62 g, 9.19 mmol) as described for compound 25. Purification by flash chromatography (heptane/EtOAc 60/40) afforded 2.36 g (93%) of the desired product as a colorless oil (rotamers mixture). Rf = 0.4 (heptane/EtOAc 50/50); 1H NMR (600 MHz, CDCl3) δ 7.63 (d, J = 7.9 Hz, 2H), 4.55–4.52 (m, 2H), 3.74 (s, 2H), 3.51–2.94 (m, 2H), 2.69 (s, 1H), 1.76–0.53 (m, 9H); 13C NMR (151 MHz, CDCl3) δ 156.91, 144.11, 132.43, 128.02, 127.50, 118.72, 111.19, 81.02, 62.17, 61.46, 52.10, 51.17, 50.37, 49.51, 28.32.
Tert-butyl (4-(1,2,4,5-tetrazin-3-yl)benzyl)(2-hydroxyethyl)carbamate (28)
The compound was obtained from 26 (2.32 g, 8.32 mmol) as described for compound 3. Purification by flash chromatography (80/20 heptane/EtOAc) to give 1.07 g (39%) of the desired product as a red oil (rotamers mixture). Rf. = 0.21 (heptane/EtOAc 70/30); 1H NMR (400 MHz, CDCl3) δ 10.17 (s, 1H), 8.53 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.1 Hz, 2H), 4.57 (s, 2H), 3.71 (d, J = 5.8 Hz, 2H), 3.46–3.41 (m, 2H), 3.23 (q, J = 5.4 Hz, 1H), 1.39 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 166.23, 157.75, 156.89, 144.17, 132.36, 130.55, 128.52, 127.89, 80.75, 62.36, 61.76, 51.98, 51.12, 50.13, 49.44, 28.34.
3-(4-(1,2,4,5-Tetrazin-3-yl)benzyl)oxazolidin-2-one
The compound was obtained from 28 (0.06 g, 0.18 mmol) as described for compound 3a. An intramolecular substitution occurred during the reaction giving this side product. Purification by flash chromatography (50/50 heptane/EtOAc) afforded 0.035 (67%) of the side product as a red solid. Rf = 0.21 (heptane/EtOAc 50/50); 1H NMR (400 MHz, CDCl3) δ 10.16 (s, 1H), 8.55 (d, J = 8.3 Hz, 2H), 7.47 (d, J = 8.3 Hz, 2H), 4.46–3.83 (m, 2H), 3.45 (dd, J = 8.7, 7.3 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 166.16, 158.59, 157.88, 141.30, 131.38, 128.96, 128.83, 61.87, 48.23, 44.21.
3.1.14. N-(4-(1,2,4,5-Tetrazin-3-yl)benzyl)-2-fluoro-N-tritylethan-1-amine (30)
Compound 9 (0.05 g, 0.18 mmol) was mixed with trityl chloride (0.05 g, 0.20 mmol) and dissolved in 4 mL dry CH2Cl2 under argon. A mixture of Et3N (0.08 mL, 0.55 mmol) in 1 mL dry CH2Cl2 was added at 0 °C under argon. The reaction was slowly heated to room temperature and left for 1 h. The reaction was diluted with 10 mL CH2Cl2 and washed with NH4Cl (sat.) 20 mL and H2O (2 × 20 mL). The organic phase was dried over N2SO4 and concentrated under reduced pressure. Purification by flash chromatography (heptane/EtOAc 80/20) afforded 0.08 g (78%) as a red solid. Rf = 0.41 (heptane/EtOAc 80/20); 1H NMR (600 MHz, CDCl3) δ 10.13 (s, 1H), 8.57 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.4 Hz, 2H), 7.74–7.55 (m, 6H), 7.32–7.23 (m, 6H), 7.16–7.10 (m, 3H), 3.99 (t, J = 6.0 Hz, 1H), 3.91 (t, J = 6.1 Hz, 1H), 3.69 (s, 2H), 2.92–2.55 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 166.46, 157.73, 147.36, 143.10, 130.32, 129.32, 128.56, 128.43, 127.93, 127.87, 126.46, 81.94 (d, J = 168.6 Hz), 79.00, 57.49, 54.66 (d, J = 22.8 Hz).
3.1.15. 2-((4-(1,2,4,5-Tetrazin-3-yl)benzyl)(trityl)amino)ethyl 4-nitrobenzenesulfonate (9b)
2-((4-(1,2,4,5-Tetrazin-3-yl)benzyl)amino)ethan-1-ol (29)
To a solution of 28 (0.99 g, 2.98 mmol) in dioxane (10 mL) was added a solution of HCl in dioxane (4.0 M, 7.7 mL). The mixture was stirred at room temperature for 2 h and then concentrated under reduced pressure. The obtained solid was washed with Et2O to afford 0.72 g (90%) of 29 as hydrochloride salt. 1H NMR (400 MHz, DMSO) δ 10.64 (s, 1H), 9.50 (s, 3H), 8.96–8.31 (m, 2H), 8.09–7.60 (m, 2H), 4.32 (t, J = 5.7 Hz, 2H), 3.73 (t, J = 5.4 Hz, 2H), 3.01 (q, J = 5.6 Hz, 2H); 13C NMR (101 MHz, DMSO) δ 165.74, 158.72, 137.22, 132.77, 131.62, 128.37, 66.82, 56.83, 49.93, 49.16.
2-((4-(1,2,4,5-Tetrazin-3-yl)benzyl)(trityl)amino)ethan-1-ol (31)
The compound was obtained from 29 (0.05 g, 0.19 mmol) as described for compound 30. Purification by flash chromatography (60/40 heptane/EtOAc) to give 0.04 g (48%) of the desired product as a red solid. Rf = 0.53 (60/40 heptane/EtOAc); 1H NMR (600 MHz, CDCl3) δ 10.22 (s, 1H), 8.65 (d, J = 8.2 Hz, 2H), 7.86 (d, J = 8.1 Hz, 2H), 7.69 (d, J = 7.9 Hz, 6H), 7.33 (t, J = 7.7 Hz, 6H), 7.22 (t, J = 7.3 Hz, 3H), 3.76 (s, 2H), 3.29 (t, J = 6.8 Hz, 2H), 2.56 (d, J = 7.0 Hz, 2H), 1.63 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 166.40, 157.73, 147.86, 143.28, 130.35, 129.37, 128.55, 128.49, 126.38, 79.05, 61.76, 57.79, 57.12.
2-((4-(1,2,4,5-Tetrazin-3-yl)benzyl)(trityl)amino)ethyl 4-nitrobenzenesulfonate (9b)
The compound was obtained from 31 (0.04 g, 0.08 mmol) as described for compound 3a. Purification by flash chromatography (90/10 heptane/EtOAc) to give 0.04 g (48%) of the desired product as a red solid. Rf = 0.25 (80/20 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 10.19 (s, 1H), 8.46 (d, J = 8.3 Hz, 2H), 8.08 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 8.8 Hz, 2H), 7.64 (d, J = 8.2 Hz, 2H), 7.61–7.43 (m, 6H), 7.23 (t, J = 7.6 Hz, 6H), 7.16–7.09 (m, 3H), 4.32–3.39 (m, 4H), 2.57 (t, J = 7.2 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 166.20, 157.92, 150.45, 146.36, 142.60, 141.64, 130.79, 129.11, 128.98, 128.91, 128.50, 127.98, 126.69, 124.19, 79.05, 57.38.
3.1.16. 2-Fluoroethyl 6-(4-(pyridin-2-yl)phenyl)nicotinate (33)
6-(6-(Pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinic acid (32)
6-Cyanonicotinic acid (1.0 g, 6.75 mmol), 2-cyanopyridine (5.6 g, 45.01 mmol), and sulfur (0.43 g, 1.69 mmol) were suspended in EtOH (10 mL), followed by the addition of hydrazine hydrate (4.93 mL, 101.26 mmol). The reaction was heated to 90 °C for 2 h. The mixture was cooled to room temperature, and the formed precipitate was removed by filtration. Water (20 mL) and a solution of NaNO2 (9.31 g, 135.32 mmol) in 50 mL water were added, and the mixture was cautiously acidified to pH 2–3 by addition of AcOH. The mixture was extracted with CH2Cl2, and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated. The residue was purified by flash column chromatography (CH2Cl2/MeOH 95/5 + 0.1% AcOH) to afford 0.52 g (27%) of the desired compound as a pink solid. Rf = 0.21 (CH2Cl2/MeOH 95/5 + 0.1% AcOH); 1H NMR (400 MHz, DMSO) δ 9.37 (d, J = 2.1 Hz, 1H), 8.96 (q, J = 1.6 Hz, 1H), 8.71 (d, J = 8.1 Hz, 1H), 8.64 (d, J = 7.9 Hz, 1H), 8.58 (dd, J = 8.1, 2.1 Hz, 1H), 8.18 (td, J = 7.8, 1.8 Hz, 1H), 7.95–7.67 (m, 1H); 13C NMR (101 MHz, DMSO) δ 165.74, 163.21, 162.97, 153.13, 151.01, 150.70, 149.96, 138.66, 137.86, 128.69, 126.81, 124.53, 124.
2-Fluoroethyl 6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinate (33)
The compound was obtained from 6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinic acid (0.05 g, 0.18 mmol) as described for compound 3 to give 0.55 g (95%) of the desired product as a pink solid. Rf = 0.41 (heptane/EtOAc 20/80); 1H NMR (400 MHz, CDCl3) δ 9.56 (dd, J = 2.2, 0.9 Hz, 1H), 9.00 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H), 8.85 (dd, J = 8.2, 0.9 Hz, 1H), 8.77 (dt, J = 7.9, 1.1 Hz, 1H), 8.02 (td, J = 7.8, 1.8 Hz, 1H), 7.60 (ddd, J = 7.7, 4.7, 1.2 Hz, 1H), 4.88–4.79 (m, 1H), 4.72 (td, J = 5.9, 3.6 Hz, 2H), 4.64 (dd, J = 5.0, 3.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 164.34, 163.84, 163.50, 151.97, 149.86, 137.55, 127.81, 124.78, 123.95, 81.04 (d, J = 171.5 Hz), 64.62 (d, J = 20.3 Hz).
3.1.17. 2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinate (33a)
2-Hydroxyethyl 6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinate (34)
The compound was obtained from 32 (0.04 g, 0.14 mmol) as described for compound 11. The compound was used as a crude due to low solubility (0.035 g, 76%). Rf = 0.31 (CH2Cl2/MeOH 90/10); 1H NMR (400 MHz, CDCl3) δ 9.47 (dd, J = 2.1, 0.9 Hz, 1H), 9.01–8.90 (m, 1H), 8.86–8.74 (m, 1H), 8.70 (dd, J = 7.9, 1.1 Hz, 1H), 8.55 (dd, J = 8.2, 2.1 Hz, 1H), 7.96 (ddt, J = 10.4, 7.7, 2.2 Hz, 1H), 7.62–7.48 (m, 1H), 4.76–4.45 (m, 2H), 4.26–3.68 (m, 2H), 3.24 (s, 1H).
2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinate (33a)
The compound was obtained from 34 (0.035 g, 0.11 mmol) as reported for compound 3a. Purification by flash chromatography (CH2Cl2/MeOH 95/5) afforded 0.035 g (64%) of the desired compound as a red solid. Rf = 0.35 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, DMSO) δ 9.25 (d, J = 2.2 Hz, 1H), 8.76 (d, J = 8.4 Hz, 2H), 8.66 (d, J = 7.8 Hz, 1H), 8.50 (dd, J = 8.2, 2.2 Hz, 1H), 8.40 (d, J = 8.9 Hz, 2H), 8.24 (d, J = 8.9 Hz, 2H), 8.22–8.17 (m, 1H), 7.87–7.68 (m, 1H), 4.63 (s, 4H); 13C NMR (101 MHz, DMSO) δ 164.1, 163.71, 163.51, 154.88, 154.14, 151.23, 150.39, 141.06, 139.06, 138.39, 129.87, 127.37, 125.42, 125.08, 124.59, 123.78, 70.42, 63.28.
3.1.18. 5-((2-Fluoroethoxy)methyl)-2-(4-(pyridin-2-yl)phenyl)pyridine (38)
5-(Bromomethyl)picolinonitrile (35)
To a solution of 5-methylpicolinonitrile (7.00 g, 59.25 mmol) and N-bromo succinimide (13.71 g, 77.03 mmol) in CH3CN (100 mL) was added AIBN (3.89 g, 23.70 mmol). The resulting solution was refluxed for 12 h. The reaction was cooled down, and EtOAc (200 mL) was added. The organic layer was washed with water (2 × 100 mL) and brine (2 × 100 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (n-heptane/EtOAc 90/10) afforded 7.1 g (61%) of 35 as a white solid. Rf = 0.29 (n-heptane/EtOAc 80/20); 1H NMR (400 MHz, CDCl3) δ 8.72 (d, J = 2.2 Hz, 1H), 7.87 (dd, J = 8.1, 2.3 Hz, 1H), 7.69 (dd, J = 8.1, 0.9 Hz, 1H), 4.49 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 151.19, 137.50, 137.45, 133.40, 128.36, 116.84, 27.87.
5-((2-Hydroxyethoxy)methyl)picolinonitrile (36)
NaH (60% weight, 0.22 g, 5.58 mmol) was suspended in dry THF (10 mL), and ethylene glycol (2.83 mL, 50.75 mmol) was added dropwise under argon at 0 °C. The solution was left for 30 min at 0 °C before dropwise addition of 5-(bromomethyl)picolinonitrile (1.00 g, 5.87 mmol) in dry THF (10 mL) under argon at 0 °C. The reaction was stirred to room temperature for 10 min and refluxed for 3 h. The reaction was cooled to room temperature and quenched by adding EtOAc (40 mL), and the crude was washed with NH4Cl (sat, 50 mL × 2) and water (50 mL). The organic phase was dried over Na2SO4 and concentrated under reduced pressure. Purification by flash chromatography (DCM/MeOH 98/2) afforded 0.55 g (61%) of the desired product as a yellow oil. Rf = 0.31 (heptane/EtOAc 30/70); 1H NMR (400 MHz, CDCl3) δ 8.60 (d, J = 2.2 Hz, 1H), 7.82 (dd, J = 8.1, 2.2 Hz, 1H), 7.95–7.32 (m, 1H), 4.61 (s, 2H), 3.74 (dd, J = 5.4, 3.8 Hz, 2H), 3.61 (dd, J = 5.4, 3.8 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 149.94, 138.24, 135.89, 132.41, 128.30, 117.20, 72.47, 69.77, 61.51.
2-((6-(6-(Pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methoxy)ethan-1-ol (37)
Compound 36 (0.55 g, 3.08 mmol), 2-cyanopyridine (1.6 g, 15.43 mmol), and sulfur (0.2 g, 0.77 mmol) were suspended in EtOH (5 mL), followed by the addition of hydrazine hydrate (2.26 mL, 43.3 mmol). The reaction was heated to 90 °C for 2 h. The mixture was cooled to room temperature, and the formed precipitate was removed by filtration. Water (20 mL) and a solution of NaNO2 (4.26 g, 61.73 mmol) in 30 mL water were added, and the mixture was cautiously acidified to pH 2 by addition of AcOH. The mixture was extracted with CH2Cl2, and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated. The residue was purified by flash column chromatography (CH2Cl2/MeOH 95/5) to afford 0.4 g (42%) of the desired compound as a pink solid. Rf = 0.21 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, CDCl3) δ 9.03–8.94 (m, 1H), 8.90 (d, J = 2.1 Hz, 1H), 8.79–8.65 (m, 2H), 8.00 (ddd, J = 7.9, 5.5, 2.1 Hz, 3H), 7.57 (ddd, J = 7.6, 4.7, 1.2 Hz, 1H), 4.74 (s, 2H), 3.88–3.78 (m, 2H), 3.70 (dd, J = 5.3, 3.9 Hz, 2H), 2.76 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 163.79, 163.69, 150.99, 150.03, 149.30, 137.50, 137.21, 136.47, 126.59, 124.50, 124.25, 72.24, 70.26, 61.80.
3-(5-((2-Fluoroethoxy)methyl)pyridin-2-yl)-6-(pyridin-2-yl)-1,2,4,5-tetrazine (38)
The compound was obtained from 2-((6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methoxy)ethan-1-ol (0.1 g, 0.33 mmol) dissolved in 5 mL dry THF and PBSF (0.20 mg, 0.66 mmol), and DIPEA (0.34 mL, 1.93 mmol) was added. Et3N.3HF (0.11 mL, 0.66 mmol) was dissolved in 2 mL THF and added dropwise. The reaction was left at room temperature for 12 h. The reaction was diluted with 20 mL CH2Cl2 and washed with NH4Cl (sat). The aqueous phase was extracted with CH2Cl2 (2 × 10 mL), and the combined organic layers were dried over MgSO4 and concentrated under reduced pressure. Purification by flash chromatography (CH2Cl2/MeOH 95/5) afforded 0.075 g (75%) of the desired product as a pink solid. Rf = 0.45 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, CDCl3) δ 9.05–8.80 (m, 2H), 8.78–8.58 (m, 2H), 8.21–7.86 (m, 2H), 7.51 (ddd, J = 7.6, 4.7, 1.2 Hz, 1H), 4.71 (s, 2H), 4.65–4.58 (m, 1H), 4.57–4.42 (m, 1H), 3.97–3.78 (m, 1H), 3.77–3.68 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 163.82, 163.73, 150.98, 150.03, 149.97, 149.36, 137.50, 137.03, 136.43, 126.60, 124.48, 124.26, 82.98 (d, J = 169.5 Hz), 70.35, 70.00 (d, J = 19.6 Hz).
3.1.19. 2-((6-(4-(Pyridin-2-yl)phenyl)pyridin-3-yl)methoxy)ethyl 4-methylbenzene sulfonate (38a)
The compound was obtained from 37 ((0.05 g, 0.17 mmol) as reported for compound 3a. Purification by flash chromatography (CH2Cl2/MeOH 98/2) afforded 0.07 g (84%) of the desired compound as a red oil. Rf = 0.48 (CH2Cl2/MeOH 90/10); 1H NMR (600 MHz, CDCl3) δ 9.02 (dt, J = 4.5, 1.4 Hz, 1H), 8.86 (d, J = 2.1 Hz, 1H), 8.80–8.63 (m, 2H), 8.41–8.33 (m, 1H), 8.17–8.11 (m, 1H), 8.04 (td, J = 7.7, 1.8 Hz, 1H), 7.93 (dd, J = 8.0, 2.2 Hz, 1H), 7.61 (ddd, J = 7.6, 4.7, 1.1 Hz, 1H), 4.69 (s, 2H), 4.44–4.38 (m, 2H), 3.90–3.82 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 163.90, 163.68, 151.09, 150.76, 150.11, 150.00, 149.76, 141.81, 137.50, 136.40, 136.21, 129.25, 126.62, 124.58, 124.41, 124.22, 70.44, 70.15, 68.20.
3.1.20. 5-(2-Fluoroethoxy)-2-(4-(pyridin-2-yl)phenyl)pyridine (41)
5-(2-Fluoroethoxy)picolinonitrile (39)
The compound was obtained from 5-hydroxypicolinonitrile (0.5 g, 4.16 mmol) as described for compound 20 to give 0.56 g (81%) of the desired product as a pink solid. Rf = 0.34 (heptane/EtOAc 70/30); 1H NMR (400 MHz, CDCl3) δ 8.34 (d, J = 2.9 Hz, 1H), 7.59 (d, J = 8.6 Hz, 1H), 7.27–7.20 (m, 1H), 4.83–4.76 (m, 1H), 4.71–4.64 (m, 1H), 4.35–4.28 (m, 1H), 4.28–4.21 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 155.04, 138.50, 127.74, 123.96, 118.83, 115.55, 79.47 (d, J = 172.4 Hz), 66.05 (d, J = 20.4 Hz).
3-(5-(2-Fluoroethoxy)pyridin-2-yl)-6-(pyridin-2-yl)-1,2,4,5-tetrazine (41)
The compound was obtained from 39 (0.55 g, 3.31 mmol) as described for compound 37, followed by purification by preparative HPLC to give 0.04 g (4%) of the desired product as a pink solid. Rf = 0.32 (CH2Cl2/MeOH 97/3); 1H NMR (400 MHz, CDCl3) δ 8.94 (dt, J = 4.7, 1.4 Hz, 1H), 8.70 (d, J = 8.5 Hz, 2H), 8.65 (d, J = 2.9 Hz, 1H), 8.00 (td, J = 7.8, 1.7 Hz, 1H), 7.57 (ddd, J = 7.7, 4.8, 1.2 Hz, 1H), 7.46 (dd, J = 8.8, 2.9 Hz, 1H), 4.88–4.81 (m, 1H), 4.76–4.69 (m, 1H), 4.44–4.38 (m, 1H), 4.38–4.25 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 163.26, 163.21, 157.38, 150.46, 149.69, 142.33, 139.43, 138.19, 126.84, 125.92 124.58, 121.99, 81.39 (d, J = 172.7 Hz), 67.99 (d, J = 20.4 Hz); HPLC-MS [M+H]+ m/z calc. for [C14H12FN6O]+: 299.10; found: 299.12.
3.1.21. 2-((6-(4-(Pyridin-2-yl)phenyl)pyridin-3-yl)oxy)ethyl 4-nitrobenzenesulfonate (41a)
5-(2-Hydroxyethoxy)picolinonitrile (40)
The compound was obtained from 5-hydroxypicolinonitrile (1.4 g, 11.65 mmol) as described for compound 21. Purification by flash chromatography (n-heptane/EtOAc 40/60) afforded 1.45 g (76%) of the desired product as a white solid. Rf = 0.21 (heptane/EtOAc 40/60); 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 2.9 Hz, 1H), 7.59 (d, J = 8.6 Hz, 1H), 7.42–6.71 (m, 1H), 4.66–4.05 (m, 2H), 3.97 (dd, J = 5.1, 3.9 Hz, 2H), 2.27 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 157.27, 140.42, 129.65, 125.41, 120.58, 117.39, 70.21, 60.86.
2-((6-(6-(Pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)oxy)ethan-1-ol (42)
The compound was obtained from 40 (0.5 g, 3.04 mmol) as described for compound 37. Purification by flash chromatography (CH2Cl2/MeOH 97/3) afforded 0.27 g (30%) of the desired product as a pink solid. Rf = 0.25 (CH2Cl2/MeOH 95/5); 1H NMR (600 MHz, DMSO) δ 8.93 (ddd, J = 4.7, 1.8, 0.9 Hz, 1H), 8.65 (dd, J = 2.9, 0.6 Hz, 1H), 8.62–8.49 (m, 2H), 8.16 (td, J = 7.7, 1.8 Hz, 1H), 7.82–7.56 (m, 2H), 5.00 (s, 1H), 4.50–3.97 (m, 2H), 3.81 (td, J = 5.4, 4.3 Hz, 2H); 13C NMR (151 MHz, DMSO) δ 163.49, 163.30, 157.81, 151.05, 150.71, 142.37, 139.97, 138.25, 127.00, 126.05, 124.59, 121.95, 70.96, 59.88.
2-((6-(4-(Pyridin-2-yl)phenyl)pyridin-3-yl)oxy)ethyl 4-nitrobenzenesulfonate (41a)
The compound was obtained from 42 (0.05 g, 0.17 mmol) as reported for compound 3a. Purification by flash chromatography (CH2Cl2/MeOH 98/2) afforded 0.03 g (37%) of the desired compound as a red solid (mixture of conformers 70/30). Rf = 0.61 (CH2Cl2/MeOH 90/10); 1H NMR (600 MHz, DMSO) δ 8.94 (ddd, J = 4.7, 1.8, 0.9 Hz, 1H), 8.70 (d, J = 2.9 Hz, 0.3H), 8.65 (d, J = 8.5 Hz, 0.3H), 8.60 (d, J = 7.8 Hz, 1H), 8.56 (d, J = 8.8 Hz, 0.7H), 8.51 (d, J = 2.9 Hz, 0.7H), 8.47–8.43 (m, 1.4H), 8.27–8.22 (m, 1.4H), 8.21–8.18 (m, 0.6H), 8.16 (td, J = 7.8, 1.8 Hz, 1H), 7.90–7.81 (m, 0.6H), 7.78 (dd, J = 8.8, 2.9 Hz, 0.3H), 7.73 (ddd, J = 7.6, 4.7, 1.2 Hz, 1H), 7.64 (dd, J = 8.8, 3.0 Hz, 0.7H), 4.81–4.71 (m, 0.6H), 4.68–4.59 (m, 1.4H), 4.57–4.54 (m, 0.6H), 4.51–4.43 (m, 1.4H); 13C NMR (151 MHz, DMSO) δ 163.57, 163.53, 163.26, 163.21, 156.74, 156.60, 154.89, 151.16, 151.08, 150.66, 147.70, 143.22, 142.95, 140.95, 139.86, 139.77, 138.28, 129.93, 127.38, 127.08, 127.05, 126.08, 125.89, 125.43, 124.63, 123.78, 122.36, 122.12, 74.80, 70.58, 67.80, 66.45.
3.1.22. 2,2′-(((6-(6-(5-(2-Fluoroethoxy)pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methyl)azanediyl)diacetic Acid (45)
Di-tert-butyl 2,2′-(((6-cyanopyridin-3-yl)methyl)azanediyl)diacetate (43)
To a solution of 5-(bromomethyl)picolinonitrile (1.5 g, 7.61 mmol) in CH3CN (50 mL) was added K2CO3 (3.15 g, 22.84 mmol) and di-tert-butyl iminodiacetate (1.96 g, 7.99 mmol). The reaction mixture was stirred at room temperature overnight, and then the solvent was concentrated under reduced pressure. The resulting mixture was diluted with water (100 mL), extracted with EtOAc (3 × 40 mL), washed with brine (50 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (n-heptane/EtOAc 90/10) afforded 2.7 g (98%) of the desired compound as a white solid. Rf = 0.33 (n-heptane/EtOAc 80/20); 1H NMR (400 MHz, CDCl3) δ 8.61 (dd, J = 2.1, 0.9 Hz, 1H), 7.96 (dd, J = 8.0, 2.1 Hz, 1H), 7.60 (dd, J = 7.9, 0.9 Hz, 1H), 3.93 (s, 2H), 3.33 (s, 4H), 1.40 (s, 18H); 13C NMR (101 MHz, CDCl3) δ 170.09, 151.41, 138.96, 137.51, 132.77, 128.27, 117.35, 81.52, 55.41, 54.57, 28.16.
Di-tert-butyl 2,2′-(((6-(6-(5-(2-fluoroethoxy)pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl) methyl)-azanediyl)diacetate (44)
Compound 43 (0.2 g, 0.55 mmol), compound 39 (0.46 g, 2.77 mmol), and sulfur (0.36 g, 0.14 mmol) were suspended in EtOH (5 mL), followed by the addition of hydrazine hydrate (0.40 mL, 8.31 mmol). The reaction was heated to 90 °C for 2 h. The mixture was cooled to room temperature and the formed precipitate was removed by filtration. Water (20 mL) and a solution of NaNO2 (0.76 g, 11.07 mmol) in 10 mL water were added, and the mixture was cautiously acidified to pH 2–3 by addition of AcOH. The mixture was extracted with CH2Cl2, and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated. The residue was purified by flash column chromatography (CH2Cl2/MeOH 95/5) and crystallized from MeOH to afford 0.07 g (23%) of the desired compound as a pink solid. Rf = 0.36 (CH2Cl2/MeOH 95/5); 1H NMR (600 MHz, DMSO) δ 8.86 (d, J = 2.1 Hz, 1H), 8.68 (dd, J = 7.9, 3.1 Hz, 1H), 8.62 (d, J = 8.8 Hz, 1H), 8.58 (d, J = 8.0 Hz, 1H), 8.11 (dd, J = 8.1, 2.1 Hz, 1H), 7.76 (dd, J = 8.8, 3.0 Hz, 1H), 4.97–4.86 (m, 1H), 4.81 (dd, J = 4.8, 2.8 Hz, 1H), 4.54 (dd, J = 4.8, 2.7 Hz, 1H), 4.51–4.43 (m, 1H), 4.01 (s, 2H), 3.45 (s, 4H), 1.43 (s, 18H); 13C NMR (151 MHz, DMSO) δ 170.35, 163.47, 163.24, 157.21, 151.20, 149.63, 142.89, 139.86, 138.19, 138.11, 126.00, 124.30, 82.43 (d, J = 166.8 Hz), 80.97, 68.39 (d, J = 18.9 Hz), 55.57, 54.87, 28.29.
2,2′-(((6-(6-(5-(2-Fluoroethoxy)pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methyl)azane-diyl)diacetic acid (45)
To a solution of 44 (0.006 g, 0.83 mmol) in 5 mL of CH2Cl2 was added 2 mL of TFA. The mixture was stirred at room temperature for 4 h. The solvent was then removed under reduced pressure. Purification by preparative HPLC afforded 0.35 g (58%) of the desired compound (TFA salt) as a red solid. 1H NMR (600 MHz, DMSO) δ 8.89 (d, J = 2.1 Hz, 1H), 8.68 (d, J = 2.9 Hz, 1H), 8.62 (d, J = 8.8 Hz, 1H), 8.57 (dd, J = 8.0, 0.8 Hz, 1H), 8.14 (dd, J = 8.1, 2.1 Hz, 1H), 7.76 (dd, J = 8.8, 3.0 Hz, 1H), 4.91–4.86 (m, 1H), 4.83–4.79 (m, 1H), 4.57–4.52 (m, 1H), 4.52–4.47 (m, 1H), 4.06 (s, 2H), 3.53 (s, 4H); 13C NMR (151 MHz, DMSO) δ 172.62, 163.47, 163.23, 157.22, 151.33, 149.58, 142.89, 139.86, 138.28, 126.00, 124.27, 122.16, 82.43 (d, J = 166.8 Hz), 68.39 (d, J = 18.8 Hz), 55.00, 54.49; HPLC-MS [M+H]+ m/z calc. for [C19H19FN7O5]+: 444.14; found: 444.13.
3.1.23. Di-tert-butyl 2,2′-(((6-(6-(5-(2-(((4-nitrophenyl)sulfonyl)oxy)ethoxy)pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methyl)azanediyl)diacetate (45a)
Di-tert-butyl 2,2′-(((6-(6-(5-(2-hydroxyethoxy)pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methyl)azanediyl)diacetate (46)
Compound 43 (1.05 g, 2.92 mmol), compound 40 (0.12 g, 0.73 mmol), and sulfur (0.05 g, 0.18 mmol) were suspended in EtOH (3 mL), followed by the addition of hydrazine hydrate (0.53 mL, 10.96 mmol). The reaction was heated to 90 °C for 2 h. The mixture was cooled to room temperature and the formed precipitate was removed by filtration. Water (10 mL) and a solution of NaNO2 (1.0 g, 14.62 mmol) in 10 mL water were added, and the mixture was cautiously acidified to pH 2 by addition of AcOH. The mixture was extracted with CH2Cl2, and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated. The residue was purified by flash column chromatography (CH2Cl2/MeOH 95/5) to give 0.11 g (27%) of the desired compound as a pink solid. Rf = 0.33 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, MeOD) δ 8.88 (d, J = 2.0 Hz, 1H), 8.81–8.69 (m, 2H), 8.56 (d, J = 2.9 Hz, 1H), 8.22 (dd, J = 8.1, 2.1 Hz, 1H), 7.71 (dd, J = 8.9, 2.9 Hz, 1H), 4.45–4.25 (m, 2H), 4.10 (s, 2H), 4.04–3.93 (m, 2H), 3.50 (s, 4H), 3.33 (t, J = 1.6 Hz, 1H), 1.50 (s, 18H); 13C NMR (101 MHz, MeOD) δ 170.55, 163.15, 163.04, 158.24, 150.67, 148.89, 141.66, 139.29, 138.58, 138.49, 125.57, 123.75, 121.49, 81.08, 70.31, 59.99, 55.12, 54.74, 27.04.
Di-tert-butyl 2,2′-(((6-(6-(5-(2-(((4-nitrophenyl)sulfonyl)oxy)ethoxy)pyridin-2-yl)-1,2,4,5-tetrazine-3-yl)pyridin-3-yl)methyl)azanediyl)diacetate (45a)
The compound was obtained from 46 (0.05 g, 0.09 mmol) as reported for compound 3a. Purification by flash chromatography (heptane/EtOAc 40/60) afforded 0.045 g (67%) of the desired compound as a red solid. Rf = 0.26 (heptane/EtOAc 30/70); 1H NMR (600 MHz, CDCl3) δ 8.82 (d, J = 2.1 Hz, 1H), 8.70–8.59 (m, 2H), 8.48 (d, J = 2.9 Hz, 1H), 8.35 (d, J = 8.8 Hz, 2H), 8.15–7.98 (m, 3H), 7.32 (d, J = 2.9 Hz, 1H), 4.76–4.47 (m, 2H), 4.41–4.31 (m, 2H), 4.01 (s, 2H), 3.40 (s, 4H), 1.41 (s, 18H); 13C NMR (151 MHz, CDCl3) δ 169.22, 162.64, 162.15, 155.32, 150.32, 149.95, 148.26, 142.30, 140.49, 138.34, 137.23, 137.03, 128.33, 124.54, 123.55, 123.24, 120.47, 80.37, 67.51, 65.00, 54.31, 53.61, 27.18; HPLC-MS [M+H]+ m/z calc. for [C33H39N8O10S]+: 739.25; found: 739.26.
3.2. Radiochemistry
3.2.1. [18F]Fluoride Production and General Methods
The [18F]Fluoride was produced by a cyclotron CTI Siemens Eclipse, Rigshospitalet, Denmark, by irradiating [18O]H2O via a (p,n) reaction. Automated synthesis was performed on a Scanys synthesis module (Scansys Laboratorieteknik, Denmark), and analytical HPLC was performed on a Thermo Fisher UltiMate 3000 equipped with a C18 column (Luna 5 μm C18(2) 100 Å, 150 mm × 4.6 mm). Eluents: A, H2O with 0.1% TFA; B, MeCN with 0.1% TFA. Gradient: from 100% A to 100% B over 15 min, back to 100% A over 4 min, flow rate 1.5 mL/min. Detection by UV absorption was at λ = 254 nm on a UVD 170U detector, and radioactivity was analyzed with a flow-through GM-tube-based radiodetector (Scansys).
3.2.2. Radiolabeling
The aqueous [18F]fluoride solution received from the cyclotron was passed through a preconditioned anion exchange resin (Sep-Pak Light QMA cartridge). The QMA was preconditioned by flushing it with 10 mL 0.5 M K3PO4 and washing it with 10 mL H2O afterward. The [18F]F- was eluted from the QMA into a 4 mL v-shaped vial using 1 mL Bu4NOMs (20 µmol, 6,8 mg) dissolved in MeOH. The eluate was dried at 100 °C for 5 min under N2 flow. Compound 45a was dissolved in 167 µL DMSO and then diluted with 833 µL tBuOH (1:5 ratio). The solution was added to the dried fluoride solution an allowed to react for 5 min at 100 °C. The reaction was cooled to 50 °C with air before addition of 3 mL H2O. This mixture was applied to a Sep-Pak C18 Plus solid phase extraction (SPE) cartridge that was preconditioned by flushing it with 10 mL EtOH followed by 10 mL of H2O. The SPE was flushed with another 5 mL of H2O and dried with N2. The product was eluted from the SPE with 2 mL MeCN into a 7 mL v-shaped vial containing 600 µL TFA. This mixture was reacted for 10 min at 80 °C. The reaction was then concentrated under N2 flow for 20 min to reduce the solvent volume to <0.1 mL. To this crude product mixture, 2.5 mL of H2O was added, and this solution was purified by semipreparative HPLC (Luna 5 μm C18(2) 100 Å, 250 mm × 10 mm, isocratic, 70% EtOH in H2O with 0.1% TFA 3 mL/min (rt: 13 min)). The product was collected in a 20 mL vial and diluted with 100 mM sterile phosphate buffer to adjust the pH to 5–8. The max EtOH concentration was 5%, and the activity concentration was 30–80 MBq/mL.
3.2.3. Tetrazine Core Reactivity Test
The reaction between [18F]45 and TCO-PNB was performed by mixing the formulated [18F]45 (200 µL) with 5 µL of the commercially available TCO-PNB ester dissolved in DMF (5 mg/mL) in an analytical HPLC vial. The solution was gently shaken and left for 1 min before it was injected into the analytical HPLC instrument for analysis.
3.3. Blocking Assay and Ex Vivo Studies
3.3.1. Establishing Tumor Xenografts in Mice
All animal studies were approved by the Danish Animal Experiments Inspectorate, Ministry of Food, Agriculture and Fisheries of Denmark (license no. 2016-15-0201-00920).
The human colon cancer cell line LS174T (ATCC, Manassas, VA, USA) was cultured in minimum essential medium (MEM) supplemented with 10% fetal bovine serum, 1% L-glutamine, 1% sodium pyruvate, 1% nonessential amino acids, and 1% penicillin–streptomycin (all from Thermo Fisher Scientific, Waltham, MA, USA). The cells were trypsinized and harvested for inoculation when they were in their exponential growth phase.
Subcutaneous tumors were established in the flank of five-week-old female nude BALB/c mice (Janvier Labs, Le Genest-Saint-Isle, France) by inoculation of ~5 × 106 LS174T cells (in 100 μL sterile PBS).
The tumor volume was estimated from caliper measurements using the formula: volume = ½ (length × width2).
3.3.2. Blocking Experiments
The blocking experiments were performed as previously described [17,32]. Briefly, tumor-bearing animals were grouped based on their tumor volume (~100–300 mm3, n = 3/group) and administered 100 µg/100 µL of CC49-TCO per mouse (~7 TCO/mAb). The ability of nonradioactive Tzs to block the binding between [111In]47 and CC49-TCO was evaluated three days later. First, the animals were injected with the (nonradioactive) Tzs (39 nmol) that were chosen for in vivo evaluation. After a lag time of 1 h, [111In]47 (~5 MBq, 3.9 nmol) was administered, and after 22 h, the mice were euthanized. Tissues were resected and weighted, and the radioactivity was measured using a gamma counter (Wizard2, Perkin Elmer). Data were corrected for decay, tissue weight, and injected amount of radioactivity. The setup also included a control group of mice receiving the precursor of [111In]47 instead of a test compound 1 h prior to [111In]47 (positive control), as well as a group exclusively receiving [111In]47. The tumor uptake of the different evaluated Tzs was normalized to the tumor uptake of the latter to determine the blocking effect.
3.3.3. Pretargeted Imaging
LS174T xenografts were established in mice as previously described. When tumors reached a size of ~100 mm3, animals were divided into two groups based on their tumor volume (n = 5/group), and injected iv with 50 μg in 100 μL of either CC49-TCO or CC49. After 72 h the animals were administered intravenously with [18F]45 (4.74 ± 1.39 MBq in 100 μL of PBS) and scanned using PET/CT (Inveon, Siemens Medical Solutions, USA) 1 h later, using a PET acquisition time of 5 min, an energy window of 350–650 KeV, and a time resolution of 6 ns; followed by a continuous 360 projection/360° CT scan, acquired with an X-ray tube voltage of 65 kV, a tube anode current of 500 μA, and an exposure time per projection of 400 ms. Mice were anesthetized by breathing 3% sevoflurane (5% for induction) mixed in 35% O2 in N2, and during scans the body temperature of the mice was kept stable using a heating pad.
Signograms from PET scans were reconstructed using a three-dimensional maximum a posteriori algorithm with correction for scatter and attenuation. The mean percentage of injected dose per grams (%ID/mL) was determined by manually creating regions of interest (ROI) on coregistered PET/CT images (Inveon Research Workplace software, Siemens Medical Solutions, Malvern, PA, USA).
GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA) was used for statistical analysis, and an unpaired t-test with Welch’s correction was used to compare the tumor uptake in the two groups. Results were considered significant when p < 0.05.
4. Conclusions
In this study, we reported the development of the first bispyridyl Tz directly labeled with fluorine-18. The [18F]45 was obtained with sufficient yield, purity, and molar activity for in vivo evaluation. This imaging agent had comparable performances and target-to-background ratios compared to previously reported successful Tz tracers. Further evaluation studies are needed in order to optimize its potential for pretargeted imaging. The developed [18F]45 is of special interest in pretargeted imaging, as it may be used—as a bispyridyl-based pretargeted imaging agent—to quantify drug release based on ‘click-to-release’ approaches. Bispyridyls have recently been shown to near-quantitatively release drugs from iTCOs within <10 min [41]. This is so far the fastest release demonstrated. The ability to quantify in vivo drug release is essential to precisely fine-tune dosing—one of the most important factors influencing the effectiveness of a treatment.
Acknowledgments
The modified antibody used in this study was kindly provided by Tagworks Pharmaceuticals.
Abbreviations
| NIR | Near-infrared |
| TPSA | Topological polar surface area |
| TCO-PNP | Para-nitrophenol trans-cyclooctene |
| EOS | End of synthesis |
| Bu4NOMs | Tetrabutylammonium mesylate |
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph15020245/s1. The supplementary material is NMR spectra of all the compounds and radio-HPLC of the final compounds.
Author Contributions
R.G.-V. and J.T.J. contributed equally to the work. Organic synthesis was performed by U.M.B., and subsequent radiolabeling experiments were performed by R.G.-V. and K.E.B. In vivo studies and PET experiments were performed by J.T.J., L.H. and V.S. The study was conceptionally designed by U.M.B., A.K. and M.M.H. with the help of all authors. The manuscript was written by U.M.B. and M.M.H. with contributions from all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This project received funding from the European Union’s EU Framework Programme for Research and Innovation Horizon 2020 under grant agreement no. 668,532, and from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 813528. A.K. and M.M.H. received funding from the European Union’s EU Framework Programme for Research and Innovation Horizon 2020 (grant agreement no. 670261). V.S. was supported by the Lundbeck Foundation (R303-2018-3567) and by the BRIDGE Translational Excellence Programme at the Faculty of Health and Medical Sciences, University of Copenhagen, funded by the Novo Nordisk Foundation (grant agreement no. NNF18SA0034956). The Lundbeck Foundation, the Novo Nordisk Foundation, the Innovation Fund Denmark, and the Research Council for Independent Research (grant agreement no. 8022-00187B) are further acknowledged.
Institutional Review Board Statement
All animal experiments in this study were approved by national animal welfare committees in Denmark, and the experiments were performed in accordance with European guidelines. All animal experiments were performed under a protocol approved by the Animal Research Committee of the Danish Ministry of Environment and Food (license no. 2016-15-0201-00920) and the Animal Ethics Committee of the University of Copenhagen, and were in compliance with the guidelines in Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no competing financial interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kraeber-Bodéré F., Bodet-Milin C., Rousseau C., Eugène T., Pallardy A., Frampas E., Carlier T., Ferrer L., Gaschet J., Davodeau F., et al. Radioimmunoconjugates for the Treatment of Cancer. Semin. Oncol. 2014;41:613–622. doi: 10.1053/j.seminoncol.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Bourgeois M., Bailly C., Frindel M., Guérard F., Chérel M., Faivre-Chauvet A., Kraeber-Bodéré F., Bodet-Milin C. Radioimmunoconjugates for treating cancer: Recent advances and current opportunities. Expert Opin. Biol. Ther. 2017;17:813–819. doi: 10.1080/14712598.2017.1322577. [DOI] [PubMed] [Google Scholar]
- 3.Stéen E.J.L., Edem P., Norregaard K., Jørgensen J.T., Shalgunov V., Kjaer A., Herth M.M. Pretargeting in nuclear imaging and radionuclide therapy: Improving efficacy of theranostics and nanomedicines. Biomaterials. 2018;179:209–245. doi: 10.1016/j.biomaterials.2018.06.021. [DOI] [PubMed] [Google Scholar]
- 4.Nayak T., Brechbiel M.W. Radioimmunoimaging with Longer-Lived Positron-Emitting Radionuclides: Potentials and Challenges. Bioconjug. Chem. 2009;20:825–841. doi: 10.1021/bc800299f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garousi J., Orlova A., Frejd F.Y., Tolmachev V. Imaging using radiolabelled targeted proteins: Radioimmunodetection and beyond. EJNMMI Radiopharm. Chem. 2020;5:16. doi: 10.1186/s41181-020-00094-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossin R., Verkerk P.R., Bosch S.M.V.D., Vulders R.C.M., Verel I., Lub J., Robillard M.S. In Vivo Chemistry for Pretargeted Tumor Imaging in Live Mice. Angew. Chem. Int. Ed. 2010;49:3375–3378. doi: 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]
- 7.Zeglis B.M., Sevak K.K., Reiner T., Mohindra P., Carlin S., Zanzonico P., Weissleder R., Lewis J.S. A Pretargeted PET Imaging Strategy Based on Bioorthogonal Diels–Alder Click Chemistry. J. Nucl. Med. 2013;54:1389–1396. doi: 10.2967/jnumed.112.115840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adumeau P., Carnazza K.E., Brand C., Carlin S.D., Reiner T., Agnew B.J., Lewis J.S., Zeglis B.M. A Pretargeted Approach for the Multimodal PET/NIRF Imaging of Colorectal Cancer. Theranostics. 2016;6:2267–2277. doi: 10.7150/thno.16744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.García M.F., Zhang X., Shah M., Newton-Northup J., Cabral P., Cerecetto H., Quinn T. 99mTc-bioorthogonal click chemistry reagent for in vivo pretargeted imaging. Bioorg. Med. Chem. 2016;24:1209–1215. doi: 10.1016/j.bmc.2016.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyer J.-P., Houghton J.L., Kozlowski P., Abdel-Atti D., Reiner T., Pillarsetty N.V.K., Scholz W.W., Zeglis B.M., Lewis J.S. 18F-Based Pretargeted PET Imaging Based on Bioorthogonal Diels–Alder Click Chemistry. Bioconjug. Chem. 2016;27:298–301. doi: 10.1021/acs.bioconjchem.5b00504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer J.-P., Kozlowski P., Jackson J., Cunanan K.M., Adumeau P., Dilling T.R., Zeglis B.M., Lewis J.S. Exploring Structural Parameters for Pretargeting Radioligand Optimization. J. Med. Chem. 2017;60:8201–8217. doi: 10.1021/acs.jmedchem.7b01108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossin R., Läppchen T., Bosch S.M.V.D., Laforest R., Robillard M.S. Diels–Alder Reaction for Tumor Pretargeting: In Vivo Chemistry Can Boost Tumor Radiation Dose Compared with Directly Labeled Antibody. J. Nucl. Med. 2013;54:1989–1995. doi: 10.2967/jnumed.113.123745. [DOI] [PubMed] [Google Scholar]
- 13.Rossin R., Bosch S.M.V.D., Hoeve W.T., Carvelli M., Versteegen R.M., Lub J., Robillard M.S. Highly Reactive trans-Cyclooctene Tags with Improved Stability for Diels–Alder Chemistry in Living Systems. Bioconjug. Chem. 2013;24:1210–1217. doi: 10.1021/bc400153y. [DOI] [PubMed] [Google Scholar]
- 14.Rossin R., van Duijnhoven S.M.J., Läppchen T., Bosch S.M.V.D., Robillard M.S. Trans-Cyclooctene Tag with Improved Properties for Tumor Pretargeting with the Diels–Alder Reaction. Mol. Pharm. 2014;11:3090–3096. doi: 10.1021/mp500275a. [DOI] [PubMed] [Google Scholar]
- 15.van Duijnhoven S.M., Rossin R., Bosch S.M.V.D., Wheatcroft M.P., Hudson P.J., Robillard M.S. Diabody Pretargeting with Click Chemistry In Vivo. J. Nucl. Med. 2015;56:1422–1428. doi: 10.2967/jnumed.115.159145. [DOI] [PubMed] [Google Scholar]
- 16.Zeglis B.M., Brand C., Abdel-Atti D., Carnazza K.E., Cook B.E., Carlin S., Reiner T., Lewis J.S. Optimization of a Pretargeted Strategy for the PET Imaging of Colorectal Carcinoma via the Modulation of Radioligand Pharmacokinetics. Mol. Pharm. 2015;12:3575–3587. doi: 10.1021/acs.molpharmaceut.5b00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stéen E.J.L., Jørgensen J.T., Denk C., Battisti U.M., Nørregaard K., Edem P.E., Bratteby K., Shalgunov V., Wilkovitsch M., Svatunek D., et al. Lipophilicity and Click Reactivity Determine the Performance of Bioorthogonal Tetrazine Tools in Pretargeted In Vivo Chemistry. ACS Pharmacol. Transl. Sci. 2021;4:824–833. doi: 10.1021/acsptsci.1c00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stéen E.J.L., Jørgensen J.T., Johann K., Norregaard K., Sohr B., Svatunek D., Birke A., Shalgunov V., Edem P.E., Rossin R., et al. Trans-Cyclooctene-Functionalized PeptoBrushes with Improved Reaction Kinetics of the Tetrazine Ligation for Pretargeted Nuclear Imaging. ACS Nano. 2019;14:568–584. doi: 10.1021/acsnano.9b06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edem P.E., Sinnes J.-P., Pektor S., Bausbacher N., Rossin R., Yazdani A., Miederer M., Kjær A., Valliant J.F., Robillard M.S., et al. Evaluation of the inverse electron demand Diels-Alder reaction in rats using a scandium-44-labelled tetrazine for pretargeted PET imaging. EJNMMI Res. 2019;9:49. doi: 10.1186/s13550-019-0520-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herth M.M., Andersen V.L., Lehel S., Madsen J., Knudsen G.M., Kristensen J.L. Development of a 11C-labeled tetrazine for rapid tetrazine–trans-cyclooctene ligation. Chem. Commun. 2013;49:3805–3807. doi: 10.1039/c3cc41027g. [DOI] [PubMed] [Google Scholar]
- 21.Poulie C., Jørgensen J., Shalgunov V., Kougioumtzoglou G., Jeppesen T., Kjaer A., Herth M. Evaluation of [64Cu]Cu-NOTA-PEG7-H-Tz for Pretargeted Imaging in LS174T Xenografts—Comparison to [111In]In-DOTA-PEG11-BisPy-Tz. Molecules. 2021;26:544. doi: 10.3390/molecules26030544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stéen E.J.L., Jørgensen J.T., Petersen I.N., Norregaard K., Lehel S., Shalgunov V., Birke A., Edem P.E., ’Estrade E.T.L., Hansen H.D., et al. Improved radiosynthesis and preliminary in vivo evaluation of the 11C-labeled tetrazine [11C]AE-1 for pretargeted PET imaging. Bioorg. Med. Chem. Lett. 2019;29:986–990. doi: 10.1016/j.bmcl.2019.02.014. [DOI] [PubMed] [Google Scholar]
- 23. [(accessed on 2 December 2021)]. Available online: https://cen.acs.org/pharmaceuticals/Click-chemistry-sees-first-use/98/web/2020/10.
- 24.Li Z., Cai H., Hassink M., Blackman M.L., Brown R.C.D., Conti P.S., Fox J.M. Tetrazine–trans-cyclooctene ligation for the rapid construction of 18F labeled probes. Chem. Commun. 2010;46:8043–8045. doi: 10.1039/c0cc03078c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Šečkutė J., Devaraj N.K. Expanding room for tetrazine ligations in the in vivo chemistry toolbox. Curr. Opin. Chem. Biol. 2013;17:761–767. doi: 10.1016/j.cbpa.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knight J.C., Richter S., Wuest M., Way J.D., Wuest F. Synthesis and evaluation of an 18F-labelled norbornene derivative for copper-free click chemistry reactions. Org. Biomol. Chem. 2013;11:3817–3825. doi: 10.1039/c3ob40548f. [DOI] [PubMed] [Google Scholar]
- 27.Zhu J., Li S., Wängler C., Wängler B., Lennox R.B., Schirrmacher R. Synthesis of 3-chloro-6-((4-(di-tert-butyl[18F]fluorosilyl)-benzyl)oxy)-1,2,4,5-tetrazine ([18F]SiFA-OTz) for rapid tetrazine-based18F-radiolabeling. Chem. Commun. 2015;51:12415–12418. doi: 10.1039/C5CC03623B. [DOI] [PubMed] [Google Scholar]
- 28.Denk C., Svatunek D., Filip T., Wanek T., Lumpi D., Fröhlich J., Kuntner P.-D.D.C., Mikula H. Development of a18F-Labeled Tetrazine with Favorable Pharmacokinetics for Bioorthogonal PET Imaging. Angew. Chem. Int. Ed. 2014;53:9655–9659. doi: 10.1002/anie.201404277. [DOI] [PubMed] [Google Scholar]
- 29.Otaru S., Imlimthan S., Sarparanta M., Helariutta K., Wähälä K., Airaksinen A.J. Evaluation of Organo [18F]Fluorosilicon Tetrazine as a Prosthetic Group for the Synthesis of PET Radiotracers. Molecules. 2020;25:1208. doi: 10.3390/molecules25051208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Devaraj N.K., Thurber G., Keliher E.J., Marinelli B., Weissleder R. Reactive polymer enables efficient in vivo bioorthogonal chemistry. Proc. Natl. Acad. Sci. USA. 2012;109:4762–4767. doi: 10.1073/pnas.1113466109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keinänen O., Li X.-G., Chenna N.K., Lumen D., Ott J., Molthoff C.F.M., Sarparanta M., Helariutta K., Vuorinen T., Windhorst A.D., et al. A New Highly Reactive and Low Lipophilicity Fluorine-18 Labeled Tetrazine Derivative for Pretargeted PET Imaging. ACS Med. Chem. Lett. 2015;7:62–66. doi: 10.1021/acsmedchemlett.5b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.García-Vázquez R., Battisti U.M., Jørgensen J.T., Shalgunov V., Hvass L., Stares D.L., Petersen I.N., Crestey F.C., Löffler A., Svatunek D., et al. Direct Cu-mediated aromatic 18F-labeling of highly reactive tetrazines for pretargeted bioorthogonal PET imaging. Chem. Sci. 2021;12:11668–11675. doi: 10.1039/D1SC02789A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bratteby K., Shalgunov V., Battisti U.M., Petersen I.N., van den Broek S.L., Ohlsson T., Gillings N., Erlandsson M., Herth M.M. Insights into Elution of Anion Exchange Cartridges: Opening the Path toward Aliphatic 18F-Radiolabeling of Base-Sensitive Tracers. ACS Pharmacol. Transl. Sci. 2021;4:1556–1566. doi: 10.1021/acsptsci.1c00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Battisti U.M., Bratteby K., Jørgensen J.T., Hvass L., Shalgunov V., Mikula H., Kjær A., Herth M.M. Development of the First Aliphatic 18F-Labeled Tetrazine Suitable for Pretargeted PET Imaging—Expanding the Bioorthogonal Tool Box. J. Med. Chem. 2021;64:15297–15312. doi: 10.1021/acs.jmedchem.1c01326. [DOI] [PubMed] [Google Scholar]
- 35.Turlik A., Houk K.N., Svatunek D. Origin of Increased Reactivity in Rhenium-Mediated Cycloadditions of Tetrazines. J. Org. Chem. 2021;86:13129–13133. doi: 10.1021/acs.joc.1c01564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eising S., Engwerda A.H.J., Riedijk X., Bickelhaupt F.M., Bonger K.M. Highly Stable and Selective Tetrazines for the Coordination-Assisted Bioorthogonal Ligation with Vinylboronic Acids. Bioconjug. Chem. 2018;29:3054–3059. doi: 10.1021/acs.bioconjchem.8b00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darko A., Wallace S.C., Dmitrenko O.G., Machovina M.M., Mehl R.A., Chin J.W., Fox J.M. Conformationally strained trans-cyclooctene with improved stability and excellent reactivity in tetrazine ligation. Chem. Sci. 2014;5:3770–3776. doi: 10.1039/C4SC01348D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Svatunek D., Wilkovitsch M., Hartmann L., Houk K., Mikula H. Uncovering the key role of dis-tortion in tetrazine ligations guides the design of bioorthogonal tools with high reactivity and superior stability. ChemRxiv. 2021 doi: 10.33774/chemrxiv-2021-vnwzk. [DOI] [Google Scholar]
- 39.Carlson J., Meimetis L.G., Hilderbrand S.A., Weissleder R. BODIPY-Tetrazine Derivatives as Superbright Bioorthogonal Turn-on Probes. Angew. Chem. Int. Ed. 2013;52:6917–6920. doi: 10.1002/anie.201301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loredo A., Tang J., Wang L., Wu K.-L., Peng Y., Xiao H. Tetrazine as a general phototrigger to turn on fluorophores. Chem. Sci. 2020;11:4410–4415. doi: 10.1039/D0SC01009J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robillard Marc S., Mikula H., Ten Hoeve W., Rossin R. Compounds for Fast and Efficient Click Release. WO 2020/256546 A1. U.S. Patent. 2020 June 17;
- 42.Qu Y., Sauvage F.-X., Clavier G., Miomandre F., Audebert P. Metal-Free Synthetic Approach to 3-Monosubstituted Unsymmetrical 1,2,4,5-Tetrazines Useful for Bioorthogonal Reactions. Angew. Chem. Int. Ed. 2018;57:12057–12061. doi: 10.1002/anie.201804878. [DOI] [PubMed] [Google Scholar]
- 43.Herth M.M., Ametamey S., Antuganov D., Bauman A., Berndt M., Brooks A.F., Bormans G., Choe Y.S., Gillings N., Häfeli U.O., et al. On the consensus nomenclature rules for radiopharmaceutical chemistry—Reconsideration of radiochemical conversion. Nucl. Med. Biol. 2021;93:19–21. doi: 10.1016/j.nucmedbio.2020.11.003. [DOI] [PubMed] [Google Scholar]
- 44.McNitt K.A., Parimal K., Share A.I., Fahrenbach A.C., Witlicki E.H., Pink M., Bediako K., Plaisier C.L., Le N., Heeringa L.P., et al. Reduction of a Redox-Active Ligand Drives Switching in a Cu(I) Pseudorotaxane by a Bimolecular Mechanism. J. Am. Chem. Soc. 2009;131:1305–1313. doi: 10.1021/ja8085593. [DOI] [PubMed] [Google Scholar]
- 45.Sano S., Nakao M., Tanaka K., Kitaike S. Synthesis of Fluorine-Containing Analogues of 1-Lysoglycerophospholipids via Horner–Wadsworth–Emmons Reaction. Synthesis. 2017;49:3654–3661. doi: 10.1055/s-0036-1588826. [DOI] [Google Scholar]
- 46.Jacobson O., Kiesewetter D.O., Chen X. Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjug. Chem. 2015;26:1–18. doi: 10.1021/bc500475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is contained within the article and Supplementary Materials.