

Abstract
The recent study investigated the in vitro anti-diabetic impact of the crude extract (MeOH) and subfractions ethyl acetate (EtOAc); chloroform; n-butanol; n-hexane; and aqueous fraction of S. edelbergii and processed the active EtOAc fraction for the identification of chemical constituents for the first time via ESI-LC-MS analysis through positive ionization mode (PIM) and negative ionization mode (NIM); the identified compounds were further validated through computational analysis via standard approaches. The crude extract and subfractions presented appreciable activity against the α-glucosidase inhibitory assay. However, the EtOAc fraction with IC50 = 0.14 ± 0.06 µg/mL revealed the maximum potential among the fractions used, followed by the MeOH and n-hexane extract with IC50 = 1.47 ± 0.14 and 2.18 ± 0.30 µg/mL, respectively. Moreover, the acarbose showed an IC50 = 377.26 ± 1.20 µg/ mL whereas the least inhibition was observed for the chloroform fraction, with an IC50 = 23.97 ± 0.14 µg/mL. Due to the significance of the EtOAc fraction, when profiled for its chemical constituents, it presented 16 compounds among which the flavonoid class was dominant, and offered eight compounds, of which six were identified in NIM, and two compounds in PIM. Moreover, five terpenoids were identified—three and two in NIM and PIM, respectively—as well as two alkaloids, both of which were detected in PIM. The EtOAc fraction also contained one phenol that was noticed in PIM. The detected flavonoids, terpenoids, alkaloids, and phenols are well-known for their diverse biomedical applications. The potent EtOAc fraction was submitted to computational analysis for further validation of α-glucosidase significance to profile the responsible compounds. The pharmacokinetic estimations and protein-ligand molecular docking results with the support of molecular dynamic simulation trajectories at 100 ns suggested that two bioactive compounds—dihydrocatalpol and leucosceptoside A—from the EtOAc fraction presented excellent drug-like properties and stable conformations; hence, these bioactive compounds could be potential inhibitors of alpha-glucosidase enzyme based on intermolecular interactions with significant residues, docking score, and binding free energy estimation. The stated findings reflect that S. edelbergii is a rich source of bioactive compounds offering potential cures for diabetes mellitus; in particular, dihydrocatalpol and leucosceptoside A could be excellent therapeutic options for the progress of novel drugs to overcome diabetes mellitus.
Keywords: S. edelbergii Rech. f, in-vitro diabetic, ESI-LC-MS, computational analysis, intermolecular interactions, α-glucosidase
1. Introduction
The genus Scutellaria L. (Lamiaceae) includes over 350 species and is extensively part of the conventional traditions for ailment purposes and is distributed mostly over mountain trails in the moderate hot and humid regions of America, Europe, and East Asia [1]. In Pakistan, the species of Scutellaria are found in the northern areas of Swat and Chitral. The genus Scutellaria has diverse biomedical applications due to its bioactive constituents, the screening of which commenced in 1889, and scutellarin (a flavone) from Scutellaria altissima was isolated for the first time in 1910 by Goldschmiedt and Lerner [2].
Around 295 chemical constituents in around 35 species of the genus Scutellaria have been isolated to date and have a significant role in the production of novel drugs. Dominant among them are flavonoids, terpenoids, alkaloids, steroids, and glycoside [1]. Due to its abundance of flavonoids, including baicalin, baicalein, and wogonin, and terpenoids comprising jodrellin A, jodrellin B, scutalbin A, and scutecyprol B, Scutellaria shows substantial value as a source of remedies such as antimicrobial, anti-inflammatory, and analgesic agents for various recent health complications [3]. Some species were also reported as prospective sources of agents that resist microbes, scavenge free radicals, inhibit inflammation, and relieve pain [3], and have been reported locally as well for their anti-diabetic significance [4].
Diabetes mellitus (DM) is a challenging disease related to insulin secretion, which is responsible for the balance of glucose in the blood and mainly regulated by flavonoids and terpenoids with the addition of alkaloids and steroids, which normalize active size responsible for insulin production and help cope with DM [5]. One of the most severe consequences of DM is hyperglycemia, which leads to long-term health complications, including cardiovascular diseases, metastatic cancer retinopathy, and nephropathy [1]. The prevalence rate varies over the globe; in Pakistan, the prevalence rate of diabetes type II (T2DM) is about 11.77 %, reflecting a high disease burden and increasing prevalence rate [2]. According to global estimates, it is predicted that some factors such as population growth, aging, urbanization, and lifestyle will lead to an increase in the global prevalence of DM, to 54% by the year 2030 [3]. α-glucosidase has become a valid target for drug design and problems related to hyperglycemia. Therefore, α-glucosidase inhibitors (AGIs) have displayed noteworthy consideration for clinical use in the treatment of hyperglycemia and, as well, use as antiviral agents [4]. Overcoming hyperglycemia by lowering the digestion and absorbance of carbohydrates is a key therapeutic strategy insight into the significant function of α-glucosidase. Hence, it is required to introduce new AGIs with relatively few side effects compared to the available clinical AGIs such as acarbose, voglibose, and miglitol [5,6].
As mentioned, some species of Scutellaria are used by local practitioners for blood cleansing and the treatment of inflammation, pain, and DM [3]. Thus, this study aimed to screen their antidiabetic potential, profile the active extracts via liquid chromatography-mass spectrometry to identify promising compounds for the treatment of various health complications, validate scientifically their local antidiabetic potential through in vitro antidiabetic assays, submit the most effective fractions to computational analysis for further validation of promising compounds, and, through isolation of the constituents considered useful for the treatment of the above-mentioned disorders, provide isolation experts and pharmacists with bioactive compounds for antidiabetic therapy.
2. Result and Discussion
The prepotency of medicinal plants is ratified through their bioactive constituents, which directly or indirectly depend upon edaphic and climatic characteristics, the quality and availability of water, and numerous other factors. In this ongoing study, S. edelbergii in crude extract and subfractions was screened for its in vitro antidiabetic capacity, and the most active fraction was submitted for qualitative assessment of its phytoconstituents through ESI-LC-MS analysis to highlight the active compounds, which were further subjected to computational screening to authenticate their anti-diabetic significance for scientific validation and contribute to the literature on S. edelbergii and its constituents as therapeutic agents. The selected genus had been reported earlier in the literature as a rich source of flavonoids, terpenoids, alkaloids, glycosides, and tannins [2]. The flavonoids, terpenoids, alkaloids, steroids, tannins, and phenols are well-known for their diverse biomedical applications [6,7].
2.1. Antidiabetic Significance
To substantiate the local applications and implications of S. edelbergii, the selected plant in the form of crude extract and subfractions was tested in vitro for anti-diabetic ability against yeast α-glucosidase enzymes (Figure 1). As we know, diverse medicinal plants and their yields have multiple health benefits [3]. S. edelbergii provided insight into their key role in the treatment of DM. The crude extract and subtractions showed potent α-glucosidase inhibitory potential with an IC50 in the range of 0.14–23 µg/mL, reflecting a therapeutic approach for the development of new drug candidates and molecules for the treatment of T2DM. The ethyl acetate fraction, followed by crude methanolic, n-hexane and n-butanol, presented appreciable antidiabetic activity with IC50 values of 0.14 ± 0.06 µg/mL, 1.47 ± 0.14 µg/mL, 2.18 ± 0.30 µg/mL, and 6.21 ± 0.10 µg/mL, respectively, in comparison with the standard acarbose with IC50 = 377.26 ± 1.20 µg/mL, indicating the main anti-diabetic molecules, while the least potential was observed in the aqueous and chloroform fractions with IC50 = 9.26 ± 0.16 µg/mL and 23.97 ± 0.14 µg/mL, respectively. Thus, screening showed that our data matched with the findings reported by [8] for S. baicalensis antidiabetic assay, and this potential was due to the presence of the active constituents described by [9].
Figure 1.

In vitro antidiabetic significance of S. edelbergii extract, (A) SEEA = S. edelbergii ethyl acetate extract, (B) SECR = S. edelbergii methanolic (crude) extract, (C) SECH = S. edelbergii chloroform extract, (D) SENB = S. edelbergii n-butanol extract, (E) SENH = S. edelbergii n-hexane extract, (F) SEAQ = S. edelbergii aqueous extract, (G) standard = acarbose.
ESI-LC-MS Analysis
Based on its significant in vitro antidiabetic potential, the EtOAc fraction was explored for the qualitative detection of the responsible bioactive compounds through ESI-LC-MS profiling using both positive ionization mode (PIM) and negative ionization mode (NIM). The EtOAc fraction presented 16 bioactive compounds corresponding to four major groups: alkaloids, flavonoids, terpenoids, and phenol (Table 1). The ESI-LC-MS full scan chromatograms in PIM and NIM, as well the chromatograms and structures of the identified compounds, are shown in Figures S1 and S2, respectively.
Table 1.
Identification of chemical constituents in the EtOAc fraction of S. edelbergii via ESI-LC-MS.
| No. | RT (Min) |
MW IM |
Fragmentation | Tentative Identification | Reference Species |
Class |
|---|---|---|---|---|---|---|
| 1 | 4.87 | [249]+ | 240.92, 234.17 | Isomatrine | S. flavescens [22] | Alkaloid |
| 2 | 5.15 | [255]− | 245.25, 237.17 | Pinocembrin | S. altissima [10] | Flavonoid |
| 3 | 5.35 | [285]− | 270.08, 267.08 | 5,7,2,6-Tetrahydroxy-flavone | S. baicalensis [11] | Flavonoid |
| 4 | 5.50 | [301]− | 286.08, 284.0 | Scuteamoenin | S. amoena [12] | Flavonoid |
| 5 | 5.74 | [365]+ | 350.08, 347.25 | Dihydrocatalpol | S. albida [18] | Terpenoid |
| 6 | 6.08 | [359]− | 344.08, 315.17 | 5,2,5-Trihydroxy-6,7,8-trimethoxyflavone | S. baicalensis [11] | Flavonoid |
| 7 | 6.43 | [445]+ | 430.17, 427.25 | Aurantiamide acetate | S. rivularis [23] | Alkaloid |
| 8 | 6.53 | [475]+ | 460.25, 457.17 | OroxylinA-7-O-d-glucuronopyranosidemethylester | S. amoena [14] | Flavonoid |
| 9 | 6.97 | [507]+ | 489.17, 475.25 | Scutalpin C | S. alpina [19] | Terpenoid |
| 10 | 7.04 | [447]− | 429.17, 403.17 | Luteloin-7-O-d-glucuronopyranoside | S. prostrata [13] | Flavonoid |
| 11 | 7.17 | [451]− | 433.25, 407.25 | Scutellone F | S. rivularis [17] | Terpenoid |
| 12 | 7.31 | [469]− | 451.17, 425.17 | Scutellone D | S. rivularis [17] | Terpenoid |
| 13 | 7.43 | [595]+ | 565.25, 523.42 | Scutellarein-7-O-neohesperidoside | S. multicaulis [15] | Flavonoid |
| 14 | 7.72 | [639]+ | 621.42, 567.42 | Leucosceptoside A | S. baicalensis [26] | Phenol |
| 15 | 8.02 | [581]− | 545.08, 519.25 | Lupulin A | S. linearis [18] | Terpenoid |
| 16 | 8.24 | [609]− | 577.33, 565.08 |
Hesperetin-7-O-neohesperidoside | S. multicaulis [15] | Flavonoid |
IM = ionization mode, MW = Molecular weight, Rt = Retention time.
Among the tentatively identified compounds from the EtOAc fraction, the flavonoid was the dominant group, containing eight chemical constituents, six of which 2, 3, 4, 6, 10, and 16 were detected in NIM, while two 8 and 13 were observed in PIM. All the flavonoids were previously reported from S. altissima [10], S. baicalensis [11] S. amoena [12], S. prostrata [13], S. amoena [14], and S.multicaulis [15], which have significant antimicrobial, anti-inflammatory, antioxidant and anticancer potential [6] and [16]. These mentioned compounds are reported for the first time from S. edelbergii; the EtOAc fraction showed significant ability against various in vitro and in vivo activities, as described by Shah et al. [3]. Moreover, the selected fraction offered five terpenoids, among which 11, 12, and 15 were tentatively identified in NIM and two—compounds 5 and 9—were observed in PIM. These compounds from S. edelbergii are also explained for the first time, reflecting the literature on S. rivularis [17], S. linearis [18], S. albida [18], and S. alpina [19]. The terpenoids were found to have effective anti-cancer, bacteriostasis, anti-virus, and anti-inflammatory properties [20,21]. Two alkaloids—1 and 7—were detected in PIM earlier in S. flavescens [22] and S. rivularis [23] and showed key anti-tumor, anti-stress, and anti-hypoglycemia properties [24]. One phenol (14) was also noted in the PIM for its ability to scavenge free radicals and act as an antioxidant and exhibited resistance to human pathogenic bacteria and the ability to serve as an antibacterial agent [25]; it was isolated for the first time from S.edelbergii and had been mentioned in the literature on S. baicalensis [26].
2.2. Computational Analysis
2.2.1. Database of the Compounds Identified in EtOAc Extract
The compounds identified from the EtOAc fraction of S.edelbergii via ESI-LC-MS analysis were used to generate a database with MOE software [27]. Among the tested crude extract and subfractions, the EtOAc fraction presented the maximum potential in vitro as an antidiabetic agent, and on that basis was selected as a significant candidate for further validation using a computational approach; inhibitory constant values reported in studies are included in Table 1. Sixteen compounds were identified from the EtOAc fraction, the chemical structure of which is presented in Figure 2. Each compound was studied for multiple therapeutic options and manages blood glucose levels. Therefore, in this study, computational techniques were used to screen α-glucosidase drugs by computer-aided drug design (CADD) applications, which showed that the drugs could bind significantly within the substrate-binding pocket.
Figure 2.

Compounds (1–16) identified in the EtOAc fraction of S. edelbergii.
2.2.2. Protein-Ligand Interaction Analysis
The structural information on the α-glucosidase enzyme (PDB ID: 5NN5) was used. The prepared enzyme/protein structure without any bounded ligand and considerable active sites for molecular docking and interaction analysis was identified from literature [28], as shown in Figure 3. A database of 16 bioactive compounds was identified through ESI-LC-MS and subjected to MOE with mdb extension, and protein-ligand docking simulations in the active site of the α-glucosidase enzyme were performed using the Dock module of MOE software [29] (Figure 3). Phytochemicals with the best binding poses and molecular interactions with the significant amino acids elucidated the mechanism of inhibition of α-glucosidase enzyme to manage DM disease. Through evaluation of selected active binding sites, the following residues (TRP376, ASP404, LUE405, ILE441, TRP481, ASP518, MET519, ARG600, TRP613, ASP616, PHE649, and HIS674) were highlighted for significant activity. Previously reported domains and active binding sites of the α-glucosidase enzyme are highlighted in Figure 3 in different colors. The small substrate-binding regions of amino acids located near the C-terminal of beta-strands of the catalytic domain presented as a loop from the region of the N-terminal beta-sheet domain (green and yellow colored representation). Table 2 summarizes the findings of molecular docking analyses of the 16 selected bioactive compounds. The selected compounds demonstrated the best binding poses with a very good dock score within the range of −8.2278 to −5.6735 kcal/mol. All compounds demonstrated a significant dock score along with the number of hydrogen bonds (HBs) within the range of 4.5 Å.
Figure 3.
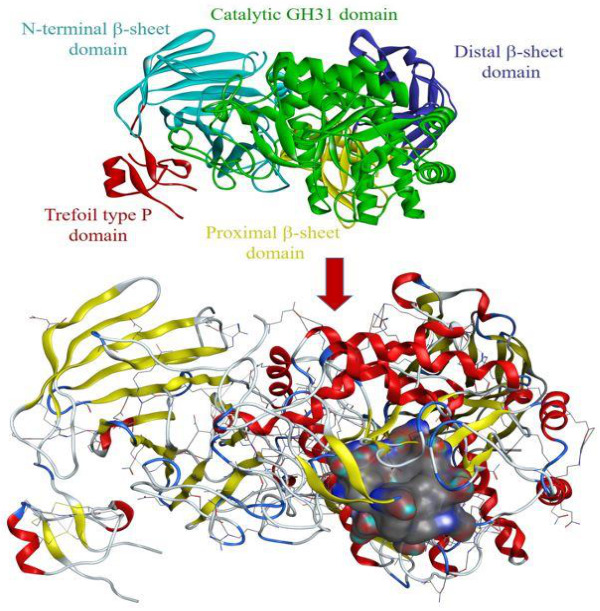
Three-dimensional representation of protein alpha-glucosidase (PDB ID: 5NN5) demonstrating identified functional domains with substrate-binding active sites (structure in surface representation) by Molecular Operating Environment (MOE) software.
Table 2.
Summary of results of molecular docking analysis of 16 selected compounds.
| No. | Dock Score | RMSD | Interacting Residues in the Binding Pocket | ||||
|---|---|---|---|---|---|---|---|
| Ligand | Receptor | Interaction | Distance | E (kcal/mol) | |||
| 1 | −5.7671 | 0.81 | - | - | - | - | - |
| 2 | −5.6933 | 0.97 | C6 O12 |
SD:MET519(A) OD1:ASP404(A) |
H-donor H-donor |
3.76 3.06 |
−0.5 −3.0 |
| 3 | −5.6735 | 2.40 | O20 | OD1:ASP518(A) | H-donor | 2.93 | −3.2 |
| 4 | −6.1364 | 1.22 | O20 | OD1:ASP518(A) | H-donor | 2.87 | −2.1 |
| 5 | −6.6079 | 1.09 | O14 O20 O21 O22 O28 O28 |
OD2:ASP616(A) OD1:ASP518(A) OD1:ASP404(A) OD1:ASP404(A) OD1:ASP282(A) N:ALA284(A) |
H-donor H-donor H-donor H-donor H-donor H-acceptor |
3.15 2.99 3.03 3.00 2.93 3.12 |
−0.5 −1.1 −0.9 −3.0 −0.9 −0.7 |
| 6 | −6.523 | 1.64 | - | - | - | - | - |
| 7 | −7.1585 | 1.63 | O27 | N:ALA284(A) | H-acceptor | 3.17 | −0.8 |
| 8 | −7.1405 | 1.09 | C17 O19 |
OD1:ASP282(A) N:ALA284(A) |
H-donor H-acceptor |
3.28 3.04 |
−0.7 −1.6 |
| 9 | −7.0130 | 1.95 | - | - | - | - | - |
| 10 | −6.9602 | 1.15 | O20 O30 O19 |
O:ASN524(A) OD2:ASP616(A) N:ALA284(A) |
H-donor H-donor H-acceptor |
3.00 2.97 3.23 |
−2.7 −2.3 −1.0 |
| 11 | −6.8364 | 1.60 | O20 | OD1:ASP282(A) | H-donor | 3.02 | −1.9 |
| 12 | −6.4003 | 1.95 | O33 | NH1:ARG600(A) | H-acceptor | 3.16 | −1.4 |
| 13 | −8.2278 | 2.17 | O38 | OD2:ASP282(A) | H-donor | 3.35 | −0.5 |
| 14 | −7.5862 | 2.08 | O29 O45 C28 |
OD1:ASP518(A) OD1:ASP282(A) 6ring:TRP376(A) |
H-donor H-donor H-pi |
3.32 2.78 4.54 |
−0.5 −3.4 −0.6 |
| 15 | −77456 | 2.33 | - | - | - | - | - |
| 16 | −8.169 | 2.96 | O35 | OD1:ASP518(A) | H-donor | 2.17 | −1.2 |
Compound 5 (dihydrocatalpol) and compound 14 (leucosceptoside A) presented very good docking findings in terms of binding energy and interactions with key amino acid residues. The threshold value for the acceptable docking score was −6.0 Kcal/mol. Thirteen compounds presented the best pose with a significant docking score above −6.0 kcal/mol. Four compounds (1, 6, 9, and 15) did not present interactions with the active site residues of the α-glucosidase protein. However, compound 5 showed the best-bounded conformation at energy value of −6.6079 kcal/mol, and a maximum of six hydrogen bonds were generated with the ASP282(A), ALA284(A), ASP404(A), ASP518(A), and ASP616(A) active residues of target protein (Figure 4). Compound 14 presented more significant binding energy at −7.5862 kcal/mol and generated two hydrogen bonds with ASP282(A) and ASP518(A) residues in the vicinity of the active bonding cleft of the target protein, while one Pi interaction also assisted by TRP376(A) residues, yielding a stable conformation in the docked complex (Figure 5).
Figure 4.

Schematic representation of dihydrocatalpol in the active site of protein alpha-glucosidase (PDB ID 5NN5). The active binding site shows hydrogen bonding capacity as donor atoms (purple) and acceptor atoms (green) (a); a two-dimensional plot showing hydrogen bonding interactions and other important hydrophobic interacting residues of the target protein (b).
Figure 5.

Schematic representation of leucosceptoside A in the active site of protein alpha-glucosidase (PDB ID 5NN5). The active binding site shows hydrogen bonding capacity as donor atoms (purple) and acceptor atoms (green) (a); a two-dimensional plot showing hydrogen bonding interactions and other important hydrophobic interacting residues of the target protein (b).
2.2.3. Molecular Dynamics (MD) Simulations
To validate the structural behavior of selected bioactive compounds within the substrate-binding active cavity of α-glucosidase, MD simulations were performed at 100 ns. α-glucosidase enzyme stability is the description of all the total forces to determine whether the protein will remain in folded state or assume non-native congregating structures. Superimposition of α-glucosidase protein and dihydrocatalpol inhibitor complex at 0 ns and 100 ns is illustrated in Figure 6. Therefore, comparing the protein-ligand complex conformations at a different level of stimulation until 100 ns provided highly useful structural insights that help to understand the possible changes in the ligand pose generated as a result. Hence, an RMSD of 1.08 Å was noted during complex comparison, protein at 0 ns (coral color) and ligand (gold color), while at 100 ns the protein (sky blue color) and ligand (cyan color) showed the stable conformation during simulation; the complex structural comparison indicates minor divergence in the conformation due to little movement in ligand pose from the docked pose, and the correlation of interactions proved that the simulation results confirmed the stability of α-glucosidase protein and dihydrocatalpol inhibitor complex. The superimposition of α-glucosidase and leucosceptoside A inhibitor complex at 0 ns and 100 ns is illustrated in Figure 7. During the critical investigation of structural changes, the RMSD value was calculated as 1.17 Å in the superimposed complex. In graphical representation, the protein at 0 ns is in violet color and ligand is in gold, while protein at 100 ns is lime green in color and ligand is in cyan.
Figure 6.
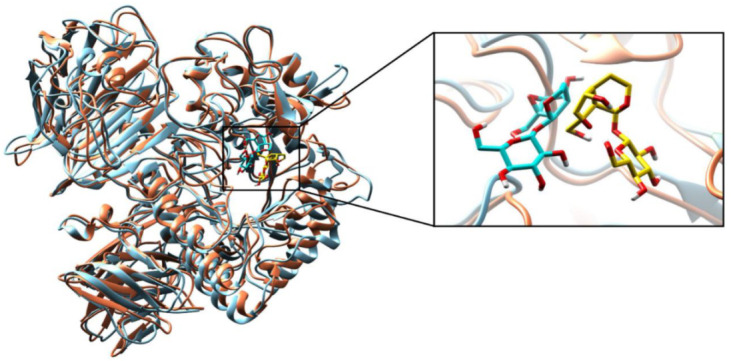
Superimposition of protein alpha-glucosidase complex with dihydrocatalpol at 0 ns and 100 ns. The calculated RMSD value is 1.08 Å; protein at 0 ns is presented in coral and ligand in gold color, while protein at 100 ns is presented in sky blue and ligand in cyan color. Critical structural changes are shown in zoomed view.
Figure 7.
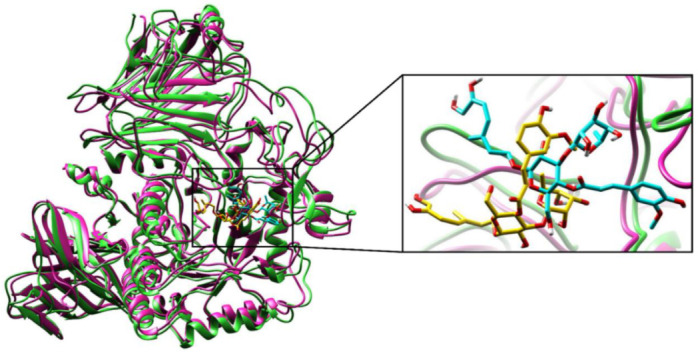
Superimposition of protein alpha-glucosidase complex with leucosceptoside A at 0 ns and 100 ns. The calculated RMSD value is 1.17 Å; protein at 0 ns is presented in violet and ligand in gold color, while protein at 100 ns is presented in lime green color and ligand in cyan color. Critical structural changes are shown in zoomed view.
During the simulation, 2D plots were generated to explain the fluctuating behavior of the docked complex at different time frames during MD simulation productions. These plots were important for statistical analysis of the MD simulations; hence, it could be significant to decode the backbone stability and flexibility of the residues during the different time frames of MD simulations [27]. RMSD was calculated as the small atoms convergence from a reference state (Figure 8A), while the RMSF of a protein explained the protein’s dynamic nature that contributed to the system’s overall versatility and residual mobility from its mean position (Figure 8B). The average RMSD value was noted as 0.904 Å, and the average RMSF value was noted as 2.019 Å for the dihydrocatalpol–α-glucosidase complex. However, it is noted that the average RMSD value was 1.863 Å and the average RMSF value was 0.847 Å for leucosceptoside A–α-glucosidase complex; it was observed that key residues were involved in the binding interactions at 0 ns and 100 ns, showing maximum correlation with the docking results. Additionally, minor fluctuations of the α-glucosidase macromolecule residues were observed from the initial state during MD simulation runs during the 0–100 ns simulation period.
Figure 8.

Root mean square deviation (RMSD) of protein and α-glucosidase inhibitor complexes (A) in Angstrom (Å) units is plotted on Y-axis, while the variation of bonded conformation through time in nanoseconds (ns) is plotted on the X-axis. Root mean square fluctuation (RMSF) of protein α-glucosidase (B) in Angstrom (Å) units is plotted on the Y-axis while the X-axis plots the index of the residue during from 0 ns to 100 ns. The radius of gyration (Rg) of protein and alpha-glucosidase inhibitor complexes (C) is plotted on the Y-axis in Angstrom (Å) units, while the X-axis plots the variation of bonded conformations throughout the simulations. A beta factor of protein alpha-glucosidase (D) is plotted on the Y-axis in Angstrom (Å) units, while the X-axis plots the index of the residue from 0 ns to 100 ns.
The radius of gyration (Rg) was evaluated to confirm the compactness and equilibrium conformation of the system; it is expected that the high and low values of the radius of gyration describe the magnitude of system compactness and a less-tight packing scheme. The estimated average Rg of the system was 28.616 Å with a maximum value of 28.741 Å, reflecting the highest peak of trajectories and compact nature of the system for dihydrocatalpol–α-glucosidase complex; and an average value of 28.644 Å with the maximum value of 28.703 Å, reflecting the highest peak of trajectories for leucosceptoside A–α-glucosidase complex (Figure 8C). The thermal residual deviation was calculated afterward via beta-factor (BF) (Figure 8D) and seemed effectively correlated with RMSF, thus validating the stability of the system. BF and RMSF are complements of each other in terms of overall system stability and residual flexibility. The average BF of the system analyzed for dihydrocatalpol and the enzyme complex was 28.839 Å, and for leucosceptoside A and enzyme complex, it was 24.7687 Å.
Solvent accessible surface area (SASA) is a very expedient examination in which changes in the accessibility of the α-glucosidase to solvent were determined. The stability of each complex was perceived during the simulation, and the average value of SASA was calculated at 151.65 nm2 for dihydrocatalpol and 150.41 nm2 for leucosceptoside A (Figure 9A).
Figure 9.

Solvent-accessible surface area (SASA) of α-glucosidase protein (A). Y-axis plots the thermodynamic calculations of changes in alpha-glucosidase complex surface area in nanometers squared (nm2) (Å) while X-axis plots residue stability time from 0 ns to 100 ns. The number of hydrogen bonds of alpha-glucosidase–dihydrocatalpol complex from 0 ns to 100 ns (B) and leucosceptoside A (C). Y-axis plots the calculated number of hydrogen bonds while X-axis plots variation in time during molecular dynamic simulations.
The frequency of the hydrogen bonds (HBs) plays an important role in overall complex stability; an increase in HBs boosts the binding affinity of the biomolecule for the protein binding site and thus strengthens the complex stability. These hydrogen bonds were obtained by VMD hydrogen bond plugin. During MD analysis from 0–100 ns, the estimated maximum number of HBs between α-glucosidase residues and dihydrocatalpol atom was 2, and the minimum number of HBs was 1 (Figure 9B), while the estimated maximum number of HBs between α-glucosidase residues and leucosceptoside A atom was 2 and the minimum number of HBs was 1 ns (Figure 9C).
2.2.4. Binding Free Energy Calculations (MM-GBSA/ MM-PBSA)
In molecular mechanics (MM), PBSA and GBSA energy model generation are the acceptable and necessary techniques used in the biomolecular investigation of binding free energy calculations of selected protein-ligand complexes and support the analysis of protein folding and stability in drug design and discovery [27]. The MM energy of the selected complex (ΔTOTAL) followed by generalized Born surface area (MM/GBSA) outline the well-organized estimations without any loss in terms of accuracy, and Poisson Boltzmann (MM/PBSA) benchmark parameters could be significant to expose the promising macromolecule α-glucosidase-dihydrocatalpol and -leucosceptoside A ligand complexes in pure water.
The total energy estimated for the MM/GBSA and MM/PBSA models of the dihydrocatalpol complex were −26.2923 and −10.5697 kcal/mol, respectively, while those for the leucosceptoside A complex were −18.3588 and −15.3721 kcal/mol, respectively. For molecular mechanics energy parameters, a strong influence was observed from the gas-phase energy (ΔG gas) when assessed against it, extremely minor contributions from the solvation energy (ΔG solv).
In the MM/GBSA model of the dihydrocatalpol complex, the ΔG gas energy was estimated at −78.0531 kcal/mol; however, in the MM/PBSA model, the same energy parameters estimation was recorded at −78.0531 kcal/mol. In the MM/GBSA model of the leucosceptoside A complex, the ΔG gas energy was estimated at −41.0979 kcal/mol, while in the MM/PBSA model, the ΔG gas energy estimate was the same, at −41.0979 kcal/mol. For the dihydrocatalpol complex, the ΔG solv energy for the MM/GBSA model was projected at 51.7608 kcal/mol, though in the case of MM/PBSA, it was observed to be 67.4834 kcal/mol. For leucosceptoside A complex, the ΔG solv energy in the case of the MM/GBSA model was 22.7391 kcal/mol and for the MM/PBSA model, 25.7258 kcal/mol. For the dihydrocatalpol complex, the electrostatic forces play a role in the stability of the complex. The system estimated by MM forces field in PBSA and GBSA models were in the range of total premeditated energy calculated at −44.2017 kcal/mol for dihydrocatalpol complex and -14.9926 kcal/mol for leucosceptoside A complex. Similarly, the van der Waals forces were also estimated from MM associated with the system stability as −33.8515 kcal/mol for dihydrocatalpol complex and −26.1053 kcal/mol for leucosceptoside A complex. For the dihydrocatalpol complex, an electrostatic energy impact (EGB and EPB) to the ΔG solv was observed, the prime constraint leading towards the out of the range calculations in MM/GBSA solv energy. The surface area energy highlighted as ESURF computed in the MM/GBSA model was −4.6737 kcal/mol. In the MM/PBSA model, the two important terms ENPOLAR and EDISPER, the repulsive and attractive free energies, were presented as −3.9887 and zero kcal/mol. For leucosceptoside A complex, the electrostatic energy impact, EGB and EPB, to the ΔG solv was counted as a non-favorable result in MM/GBSA solv energy. Total ESURF in the MM/GBSA was calculated at −2.9626 kcal/mol, whereas in the MM/PBSA model, ENPOLAR and EDISPER were −2.4337 and zero kcal/mol, correspondingly. The individual total bounded conformations presenting the free energy for the selected α-glucosidase macromolecule as receptor and selected phytochemical complex as ligand (5 and 14) are explained in Table 3.
Table 3.
Binding free energies of the α-glucosidase protein and selected potential bioactive compound complexes.
| C. No | MM/GBSA Model | MM/PBSA Model | ||||||
|---|---|---|---|---|---|---|---|---|
| 5 | VDWAALS | −33.8515 | 2.3527 | 0.2353 | VDWAALS | −33.8515 | 2.3527 | 0.2353 |
| EEL | −44.2017 | 85.0494 | 0.5049 | EEL | −44.2017 | 5.0494 | 0.5049 | |
| EGB | −56.4345 | 4.7492 | 0.4749 | EPB | 71.4721 | 6.3198 | 0.6321 | |
| ESURF | −4.6737 | 0.3065 | 0.0306 | ENPOLAR | −3.9887 | 0.1867 | 0.0187 | |
| EDISPER | 0 | 0 | 0 | |||||
| ΔG gas | −78.0531 | 5.3641 | 0.5364 | ΔG gas | −78.0531 | 5.3641 | 0.5364 | |
| ΔG solv | 51.7608 | 4.6677 | 0.4668 | ΔG solv | 67.4834 | 6.2590 | 0.6259 | |
| ΔTOTAL | −26.2923 | 2.3706 | 0.2371 | ΔTOTAL | −10.5697 | 4.2572 | 0.4257 | |
| 14 | VDWAALS | −26.1053 | 1.7151 | 0.1715 | VDWAALS | −26.1053 | 1.7151 | 0.1715 |
| EEL | −14.9926 | 4.3080 | 0.4308 | EEL | −14.9926 | 4.3080 | 0.4308 | |
| EGB | 25.7016 | 4.7242 | 0.4724 | EPB | 28.1595 | 5.1235 | 0.5123 | |
| ESURF | −2.9626 | 0.1831 | 0.0183 | ENPOLAR | −2.4337 | 0.0970 | 0.0097 | |
| EDISPER | 0 | 0 | 0 | |||||
| ΔG gas | −41.0979 | 5.3198 | 0.5320 | ΔG gas | −41.0979 | 5.3198 | 0.5320 | |
| ΔG solv | 22.7391 | 4.6019 | 0.4602 | ΔG solv | 25.7258 | 5.0694 | 0.5069 | |
| ΔTOTAL | −18.3588 | 1.6221 | 0.1622 | ΔTOTAL | −15.3721 | 2.0689 | 0.2069 | |
Where 5 = dihydrocatalpol, 14 = leucosceptoside A.
2.2.5. Pharmacokinetic /ADMET Profile Estimation
Several studies have proved the importance of pharmacokinetic/ADMET profile estimation for the screening of databases to identify potential drug-like and lead-like compounds that could be better tolerated in the design and development of new drugs [29]. ADMET properties were calculated by Swiss ADME [27] and Data warrior tools [29]. Physicochemical properties of the two selected compounds, such as partition coefficient/Lipophilic parameters (log P values), hydrogen bond acceptor (HBA), hydrogen bond donor (HDB), total polar surface area (TPSA), molar refractivity (MR), and rotatable bond (RB) were calculated for each compound. As initial phase drug discovery protocols promote the calculation of drug-likeness, water solubility, pharmacokinetics, and toxicity estimations, these parameters are presented along with the medicinal chemistry perspective for the 10 top-scored compounds in Table 4.
Table 4.
Summary of drug-likeness and in silico pharmacokinetic properties of the two selected α-glucosidase inhibitors calculated by SwissADME server and Data-Warrior software.
| Chemical Parameters | Dihydrocatalpol | Leucosceptoside A |
|---|---|---|
| Physicochemical Properties | ||
| Molecular weight (MW) (g/mol) | 364.35 | 638.61 |
| Rotatable bonds | 4 | 12 |
| Hydrogen bond acceptors (HBA) | 10 | 15 |
| Hydrogen bond donors (HBD) | 6 | 8 |
| Molar Refractivity (MR) | 77.11 | 152.89 |
| Total polar surface area (TPSA) (Å) | 161.60 | 234.29 |
| Bioavailability Score | 0.55 | 0.17 |
| Lipophilicity | ||
| Log Po/w (iLOGP) | 1.38 | 2.78 |
| LogPo/w (XLOGP3) | −3.34 | −0.18 |
| LogPo/w (WLOGP) | −3.71 | −0.82 |
| Lpg Po/w (MLOGP) | −2.81 | −2.18 |
| LogPo/w (SILICOS-IT) | −2.50 | −0.58 |
| Consensus Log Po/w | −2.20 | −0.20 |
| Water Solubility | ||
| Class | Highly soluble | Soluble |
| Pharmacokinetics | ||
| GI absorption | Low | Low |
| BBB permeant | No | No |
| P-gp substrate | Yes | Yes |
| CYP1A2 inhibitor | No | No |
| CYP2C19 Inhibitor | No | No |
| CYP2C9 inhibitor | No | No |
| CYP2D6 inhibitor | No | No |
| CYP3A4 inhibitor | No | No |
| Log Kp (skin permeation) (cm/s) | −10.89 | −10.32 |
| Toxicity estimation | ||
| Mutagenicity | Toxic effects | No toxic effects |
| Tumorigenicity | No toxic effects | No toxic effects |
| Reproductive effects | No toxic effects | No toxic effects |
| Irritant effects | No toxic effects | No toxic effects |
| Medicinal chemistry-related properties | ||
| PAINS | No alert | I alert Catechol_A |
| Brenk | 1 alert Three_membered_ heterocycle |
2 alert Catechol, Micheal_acceptor_1 |
| Synthetic accessibility | 5.39 | 6.47 |
Due to the large chemical structure of natural compounds in the selected dataset, physicochemical properties described by Ismail et al. [27] and Shaker et al. [29] theory of drug-likeness violate one or more parameters shown in Table 4. Compound 4 (dihydrocatalpol) has good bioavailability compared to compound 7 (leucosceptoside A). Toxicity estimates for both compounds presented significant results with minor violations. While dihydrocatalpol presented the expected mutagenic effect, leucosceptoside A presented no predicted toxicity in any of the four major parameters consisting of mutagenicity, tumorigenicity, reproductive effects and irritant effects. The lipophilic profile and water solubility of both the dihydrocatalpol and leucosceptoside A compounds were quite good values. Both phytochemicals presented structural alerts in the medicinal chemistry perspective evaluation, which are important preliminary predictions. Pharmacokinetic properties of both selected compounds varied in other drug-like parameters because most plant-derived compounds do not fulfill the Lipinski rule of drug-likeness due to their large chemical structures [29].
3. Material and Methods
The pipeline used for the identification and screening of small drug-like potential α -glucosidase inhibitors of DM explaining the significance of in vitro and computational approaches is shown in Figure 10. Phytocompounds were isolated and subjected to computational investigation to define the mechanism of α -glucosidase inhibition by molecular docking and dynamic simulations.
Figure 10.

Pipeline for identification and screening of small drug-like potential alpha-glucosidase inhibitors for diabetes mellitus.
3.1. Apparatus Used
LC-MS/MS (LTQ XL, Thermo Electron Corporation, Waltham, MA, USA) and Xcalibur 2.2 software (Thermo Fisher Scientific, Waltham, MA, USA) were purchased from Fisher Scientific (Illkirch, France). The solvents used for liquid chromatography were LC-MS grade acetonitrile (Fisher Scientific). Deionized water was purified by a Milli-Q (Millipore, Bedford, MA, USA) water purification system.
3.2. Plant Collection and Identification
S. edelbergii was harvested in the flowering season (April–June 2019) from mountain trails (1660–2200 m) in Kalam, District Swat, Khyber Pakhtunkhwa, Pakistan, and identified by the plant taxonomist Prof. Mehboob Rahman, Matta College Swat, KPK, Pakistan. The plant specimens were deposited at the herbarium (AWKUM/Herb/ 2234), Department of Botany, Abdul Wali Khan University, Mardan, Pakistan [3]. S. edelbergii is a perennial herb, propagating through its woody rootstock, with slender stems, procumbent or weakly ascending, round-quadrangular, leaves much-branched, with ovate or acute margins. The flowers are subtended, small calyx with purple scutellum, enlarged during fruit. The petals are yellow or sometimes blue-violet, lower lip darker, spreading erect or erect, densely glandular pilose. Flowering is produced in April to July, found between 1660–2200 m.
3.3. Extraction and Fractionation
The S. edelbergii specimens were cleaned via tap water to eliminate the unwanted particles and dehydrated at room temperature under shade for 3 weeks. The dried samples were mashed into fine powder with an electric blender. The obtained plant powder was weighed (2 kg) and placed in a refrigerator at 4 °C. The plant powder was subjected to extraction and fractionation using a systematic technique. Two kg of the plant powder was soaked in a glass container (6 L) with commercial-grade methanol, followed by shaking, and then filtered through a muslin cloth after 21 days. The resulting filtrates underwent solvent evaporation via Rota-vapor under controlled temperature (40 °C). The attained semi-solid paste was placed in the open air to dry, yielding a 600-g crude extract that was then kept in a sealed container for further use. To prepare different fractions, 500 g of crude extract was immersed in 1 L distilled water and then subjected to solvent-solvent extraction in a separating funnel from less to more polar solvents (n-hexane < chloroform < EtOAc < n-BuOH). The resulting extracts were vaporized through a rotary evaporator at 40 °C, yielding 21, 19, 20, 18, and 35 g of n-hexane, chloroform, EtOAc, n-BuOH, and aqueous fractions, respectively [3].
3.4. α-Glucosidase Inhibitory Assay
The α-glucosidase assay was performed using 50 mM phosphate buffer (pH 6.8). All samples were dissolved in DMSO. An enzyme (2 U/2 mL) 20 µL/well and test samples (0.5 mg/mL) 20 µL/well and 135 µL/well phosphate buffer were loaded into a 96-well plate, followed by 15 min of incubation at 37 °C. Substrate p-nitrophenyl-α-D-glucopyranoside (0.7 mM) 25 µL/well was added to the 96-well plate after 15 min of incubation, and readings were measured at 400 nm for 30 min. DMSO 7% was used as a positive control. Percent inhibition was calculated by using the following formula [30].
OD is the optical density of the tested samples.
3.5. LC-MS/MS Analysis
The EtOAc fraction of S.edelbergii displayed significant in vitro antidiabetic potential, based on qualitative detection of the chemical constituents in the selected fraction using linear ion trap mass spectrometer coupled with electrospray ionization source using positive and negative ionization mode via standard approach [31]. The (identification) detection was achieved via direct injection mode (DIM) using electron spray ionization (ESI). The capillary voltage was 3.3 kV, with a temperature of 280 °C, whereas the sample flow rate was set up at 10.5 μL/min. The optimized mass range was from 50–2000 m/z. The collision-induced dissociation (CID) energy ranged from 10–30, and during MS/MS was maintained in the range of 10–45, depending upon the nature of the parent molecular ion. As a mobile phase, the ratio of methanol and acetonitrile was 80:20 (v/v). The MS parameters for each compound were adjusted to verify the most satisfactory ionization and ion transfer conditions. The best possible signals of both the precursor and fragment ions were attained by infusing the analytes and manually adjusting the parameters. The source parameters were identical for all analytes.
3.6. Computational Analysis
3.6.1. Construction of Chemical Database for In Silico Screening
Recent literature highlights the importance of identifying promising drugs for DM because previously reported synthetic drugs have certain unhealthy side effects [28]. Virtual screening techniques are highly recommended to find potential drugs for fatal and infectious diseases [29]. An integrated CADD scheme was employed using 16 bioactive compound databases to identify potential drugs to combat DM. The structure of each compound was drawn using ChemDraw software [32], and information on each structure was crosschecked from the PubChem database [33] to reduce the chance of ambiguity and saved in SDF format for subsequent investigations.
3.6.2. Selection of Target Protein
The selection of an appropriate protein structure to initiate the drug design pipeline defines the important parameters necessary to clarify the action of bound ligands [34], which could selectively inhibit the activity of the α-glucosidase enzyme and hence improve insulin expression in DM patients [35]. Therefore, α-glucosidase protein (PDB ID:5NN5) was selected to perform a molecular docking experiment in this study to evaluate the protein–ligand binding interactions [36].
3.6.3. Protein-Ligand Docking and Interactions Analysis
Molecular docking is an important CADD application for the identification of protein-ligand interactions to understand the molecular mechanism of small drug-like entities in cellular pathways [28]. ESI-LC-MS identified 16 compounds that were used for molecular docking, and the 3D structure of the α-glucosidase protein (PBD ID: 5NN5) in pdb format was imported to the MOE software [37]. Heteroatoms, 3D protonation, and water molecules along with the default ligand attached to the target protein were removed to prepare the protein for the docking procedure shown in Figure 3. An active site was identified in the selected protein (5NN5) based on previous literature [36], and structural optimization was performed using steps such as the addition of hydrogen atoms and energy minimization with the Amber14 force field method applied with chiral constraints and geometrical parameters. By using the surfaces and maps panel module, transparency of the front and back surfaces was adjusted, resulting in information on significant residues in the selected substrate binding site of 5NN5 protein in the native conformation [36]. MOE software created a database of 16 compounds identified from experimental studies to perform molecular docking simulations and saved it with the mdb extension for further analysis. Top-ranked poses were subjected to refinement and calculation of binding free energies (ΔG), which were evaluated by the scoring function GBVI/WSA dg [38]. A reliable scoring scheme that resulted in the docking score of the correct binding poses was established by the number of molecular interactions (hydrogen, Pi, and hydrophobic interactions) [39]. The MOE database of the docked complex was carefully visualized to understand the mode of binding interactions of α-glucosidase inhibitors bound in the selected pocket of the target protein.
3.6.4. Molecular Dynamics Simulations
The two selected lead compound complexes were used for further MD simulation analysis at 100 ns to understand the behaviors of the docked complex and conformational stability. MD simulation steps were (1) preparation (2) pre-processing and (3) simulation run [40]. Amber 20 software was used for the MD simulations. Initial system preparation was performed with the antechamber module [41], followed by complex library adjustments, and the parameters were fixed for α-glucosidase protein and selected ligands. In the solvation system, the complex was solvated (12 Å) with the Leap module of the AMBER 20 software. Macromolecule binding was resolved by ff14SB force field [42] and charge neutralization, Na+ ions were also processed [43].
Pre-processing fixed the selected complex energy calculations and completed the set of hydrogen bonds (HBs), which were intended for 500 steps, minimization of solvation system energy for 1000 steps, limited to 200 kcal/mol-Å2 on the residual system; minimization of the complete set of system atoms was performed once more for 1000 steps with pragmatic restraint of 5 kcal/mol-Å2 applied to system carbon α atoms and 300 steps of minimization on system non-heavy atoms with the restraint of 100 kcal/mol-Å2 on other system modules. The protein-ligand complexes were heated progressively to 300 K by NVT ensemble and preserved by the Langevin dynamics [44] and SHAKE algorithm [45] to confine the HBs. The equilibration stage of the protein-ligand complexes was recovered at 100 ps. The pressure was applied in the MD simulation system using NPT ensemble and restraining the system to Cα atoms of 5 kcal/mol-Å2. The MD simulation plots of 0 ns to 100ns were generated using a time scale of 2 fs.
Before and after binding, significant interaction estimations were performed by maintaining a cut-off range of 8.0 Å. The CPPTRAJ module was applied for statistical estimations to ensure structural stability in the best conformation [46]. The MD simulation plots were generated by Visual Molecular Dynamics (VMD) software [47].
3.6.5. Binding Free Energy (BFE) Estimation
Amber 20 was used for the estimation of binding interactions and solvation free energies for the α-glucosidase protein target. Docked protein-ligand complex was subjected to MM-PBSA calculation for absolute binding free energy estimation, which was the sum of gas-phase and solvation free energies during MD simulation for 100 ns [48]; corresponding MM-GBSA calculations were performed to identify differences between the bound and unbound presentations of solvated conformations of the selected potential target [47]. Mathematically, the BFE can be analyzed by the following Equation (1).
| (1) |
For all three conditions of the MM system, the solvation energy for the transfer of molecules from the gas phase to solvent was assessed by resolving any one Poisson-Boltzmann (PB) or generalized Born (GB) equation. Therefore, it contributed to the electrostatic role of the solvation phase. Similarly, it allowed the calculation of empirical terms for hydrophobic forces as presented in Equation (2).
| (2) |
The estimation of the average interaction energy between the ligand and protein yields ΔG vacuum (Equation (3)).
| (3) |
3.6.6. Pharmacokinetic /ADMET Profile Estimation
Based on docking results, the best bioactive compounds were used for the calculation of the ADMET (absorption, distribution, metabolism, excretion, and toxicity) profile, which is an essential criterion for the screening of drug-like chemical compounds [49,50]. For ADMET profile estimation of selected phytochemicals, Swiss ADME [34], Data Warrior [51], and computational tools [52] were used.
3.7. Data Analysis
The compounds detected through ESI-LC-MS screening were identified using online database software [47,48] with already-reported literature on the genus Scutellaria cited against each compound; further, Xcalibur 2.0.7 (software) was applied for data analysis and acquisition.
4. Conclusions
Medicinal plants have provided numerous therapies due to the presence of bioactive compounds. The current study highlighted the importance of S. edelbergii crude extract and subfractions against the antidiabetic assay, demonstrating significant potential in general, whereas the EtOAc fraction was the most effective. The active fraction was profiled for the identification of biopotent compounds, yielding 16 bioactive constituents. Their significance was further validated through computational analysis. Molecular docking investigations of the 16 compounds led to the identification of two potential hits (dihydrocatalpol and leucosceptoside A). Dihydrocatalpol, a very important anti-inflammatory medicinal agent found in several traditional medicines, presented the best results in terms of energy value (−6.6079 kcal/mol), and generated six hydrogen bonds with the ASP282 (A), ALA284 (A), ASP404 (A), ASP518 (A), and ASP616 (A) residues of the target protein. While leucosceptoside A has benefits in traditional medicine for hyperglycemia and chronic diseases, it presented more significant binding energy (−7.5862 kcal/mol) and generated two hydrogen bonds with ASP282 (A) and ASP518 (A) residues in the vicinity of the active bonding cleft of the target protein, as well as one Pi interaction assisted by TRP376(A) residues, achieving a stable conformation in docked complex with a mechanism of α-glucosidase inhibition that assists the improvement of insulin secretion and manages blood glucose in diabetic patients. Furthermore, MD simulations allowed observation of the stable conformational parameters presenting estimated binding free energies and important trajectories explaining major features of the bounded complex. Very minor fluctuations were observed in terms of RMSD and RMSF values that correlated with the results of molecular docking. Moreover, an additional parameter was applied for ADMET estimations that offered insight into the drug-like characteristics of the two selected potential hits. Hence, these results are highly recommended for further clinical investigations and could facilitate the drug development phase for therapeutic agents against diabetes mellitus.
Acknowledgments
The authors are thankful to the University of Nizwa, Oman by providing PhD exchange programme to the PhD scholars.
Supplementary Materials
The related data are available online as Figure S1: Full-scan ESI-LCMS chromatogram of the EtOAc fraction of S.edelbergii. (A) Negative ionization mode and (B) positive ionization mode, Figure S2: Chromatograms of ESI-LCMS identified compounds in the EtOAc fraction of S.edelbergii.
Author Contributions
M.S., H.R., N.U.R. and W.M. designed the project and wrote the original manuscript. N.U.R. and O.U. performed the ESI-LC-MS analysis and interprets the data. S.B. performed the computational studies as well as helped in data interpretation. While A.K. and S.U. conducted the in vitro antidiabetic assay. W.M., N.U.R. and A.A.-H. supervised the project and assisted in reviewing and editing the manuscript. All authors have read and agreed to publish this version of the manuscript.
Funding
This research was funded by The Oman Research Council (TRC) through the funded project (BFP/RGP/CBS/21/002). The APC was funded by the same project.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are available to researchers upon request.
Conflicts of Interest
The authors declare that they have no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bruno M., Piozzi F., Rosselli S. Natural and hemisynthetic neoclerodane diterpenoids from Scutellaria and their antifeedant activity. Nat. Prod. Rep. 2002;19:357–378. doi: 10.1039/b111150g. [DOI] [PubMed] [Google Scholar]
- 2.Shang X., He X., He X., Li M., Zhang R., Fan P., Zhang Q., Jia Z. The genus Scutellaria an ethnopharmacological and phytochemical review. J. Ethnopharmacol. 2010;128:279–313. doi: 10.1016/j.jep.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Shah M., Murad W., Ur Rehman N., Halim S.A., Ahmed M., Rehman H., Zahoor M., Mubin S., Khan A., Nassan M.A. Biomedical applications of Scutellaria edelbergii Rech. f.: In vitro and in vivo approach. Molecules. 2021;26:3740. doi: 10.3390/molecules26123740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majid A., Ahmad H., Saqib Z., Ali H. Potential distribution of endemic Scutellaria chamaedrifolia; Geographic Information System and statistical model approach. Pak. J. Bot. 2015;47:51–56. [Google Scholar]
- 5.Asgarpanah J., Kazemivash N. Phytochemistry, pharmacology and medicinal properties of Carthamus tinctorius L. Chin. J. Integr. Med. 2013;19:153–159. doi: 10.1007/s11655-013-1354-5. [DOI] [PubMed] [Google Scholar]
- 6.Rasul A., Millimouno F.M., Ali Eltayb W., Ali M., Li J., Li X. Pinocembrin: A novel natural compound with versatile pharmacological and biological activities. BioMed Res. Int. 2013;2013:379850. doi: 10.1155/2013/379850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krishna P.M., KNV R., Banji D. A review on phytochemical, ethnomedical and pharmacological studies on genus Sophora, Fabaceae. Rev. Bras. Farmacogn. 2012;22:1145–1154. doi: 10.1590/S0102-695X2012005000043. [DOI] [Google Scholar]
- 8.Waisundara V.Y., Hsu A., Huang D., Tan B.K.-H. Scutellaria baicalensis enhances the anti-diabetic activity of metformin in streptozotocin-induced diabetic Wistar rats. Am. J. Chinese Med. 2008;36:517–540. doi: 10.1142/S0192415X08005953. [DOI] [PubMed] [Google Scholar]
- 9.Salehi B., Ata A., V Anil Kumar N., Sharopov F., Ramírez-Alarcón K., Ruiz-Ortega A., Abdulmajid Ayatollahi S., Valere Tsouh Fokou P., Kobarfard F., Amiruddin Zakaria Z. Antidiabetic potential of medicinal plants and their active components. Biomolecules. 2019;9:551. doi: 10.3390/biom9100551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomimori T., Miyaichi Y., Imoto Y., Kizu H., Namba T. Studies on the nepese crude drugs. VI.: On the flavonoid constituents of the root of Scutellaria discolor Colebr.(2) Chem. Pharm. Bull. 1986;34:406–408. doi: 10.1248/cpb.34.406. [DOI] [Google Scholar]
- 11.De Smet P. Adverse Effects of Herbal Drugs 2. Springer; Berlin/Heidelberg, Germany: 1993. Scutellaria Species; pp. 289–296. [Google Scholar]
- 12.Hu B., Liu Y. Studies on the structures of new flavonoids from the root of Scutellaria amoena. Acta pharm. Sin. 1989;24:200–206. [PubMed] [Google Scholar]
- 13.Kikuchi Y., Miyaichi Y., Yamaguchi Y., Kizu H., Tomimori T. Studies on the nepalese crude drugs. XII. On the phenolic compounds from the root of Scutellaria prostrata Jacq. ex Benth. Chem. Pharm. Bull. 1991;39:1047–1050. doi: 10.1248/cpb.39.1047. [DOI] [Google Scholar]
- 14.Zhou Z.-H., Zhang Y.-J., Yang C.-R. New flavonoid glycosides from Scutellaria amoena. Plant Sci. J. 1999;6:305–310. [Google Scholar]
- 15.Dehkordi F.J., Kharazian N., Lorigooini Z. Characterization of flavonoid components in Scutellaria L. species (Lamiaceae) using finger-printing analysis. Acta Biol. Crac. Ser. Bot. 2020;62:79–96. [Google Scholar]
- 16.Rashid M., Fareed M., Rashid H., Aziz H., Ehsan N., Khalid S., Ghaffar I., Ali R., Gul A., Hakeem K.R. Flavonoids, and their biological secrets. Plant and Hum. Health. 2019;2:579–605. [Google Scholar]
- 17.Lin Y.-L., Kuo Y.-H., Cheng M.-C., Wang Y. Structures of scutellones D and E determined from X-ray diffraction, spectral and chemical evidence. Neoclerodane-type diterpenoids from Scutellaria rivularis WALL. Chem. Pharm. Bull. 1988;36:2642–2646. doi: 10.1248/cpb.36.2642. [DOI] [Google Scholar]
- 18.Hussain H., Ahmad V.U., Anwar S., Miana G.A., Krohn K. Chemical constituents of Scutellaria linearis. Biochem. Syst. Ecol. 2008;5:490–492. doi: 10.1016/j.bse.2007.12.008. [DOI] [Google Scholar]
- 19.Muñoz D.M., Maria C., Rodríguez B., Simmonds M.S., Blaney W.M. Neo-clerodane insect antifeedants from Scutellaria alpina subsp. javalambrensis. Phytochemistry. 1997;44:593–597. doi: 10.1016/S0031-9422(96)00608-5. [DOI] [Google Scholar]
- 20.Chen Q., Rahman K., Wang S.-J., Zhou S., Zhang H. Scutellaria barbata: A review on chemical constituents, pharmacological activities, and clinical applications. Curr. Pharm. Des. 2020;26:160–175. doi: 10.2174/1381612825666191216124310. [DOI] [PubMed] [Google Scholar]
- 21.Denaro M., Smeriglio A., Trombetta D. Antioxidant and anti-inflammatory activity of citrus flavanones mix and its stability after in vitro simulated digestion. Antioxidant. 2021;10:140. doi: 10.3390/antiox10020140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma S.-C., Du J., But P.P.-H., Deng X.-L., Zhang Y.-W., Ooi V.E.-C., Xu H.-X., Lee S.H.-S., Lee S.F. Antiviral Chinese medicinal herbs against respiratory syncytial virus. J. Ethnopharmacol. 2002;79:205–211. doi: 10.1016/S0378-8741(01)00389-0. [DOI] [PubMed] [Google Scholar]
- 23.Lin Y.-L. Aurantiamide from the aerial parts of Scutellaria rivularis. Planta Med. 1987;53:507–508. doi: 10.1055/s-2006-962795. [DOI] [PubMed] [Google Scholar]
- 24.Lim D.W., Kim Y.T. Dried root of Rehmannia glutinosa prevents bone loss in ovariectomized rats. Molecules. 2013;18:5804–5813. doi: 10.3390/molecules18055804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brito A., Ramirez J.E., Areche C., Sepúlveda B., Simirgiotis M.J. HPLC-UV-MS profiles of phenolic compounds and antioxidant activity of fruits from three citrus species consumed in Northern Chile. Molecules. 2014;19:17400–17421. doi: 10.3390/molecules191117400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Y., Hirotani M., Yoshikawa T., Furuya T. Flavonoids and phenylethanoids from hairy root cultures of Scutellaria baicalensis. Phytochemistry. 1997;44:83–87. doi: 10.1016/S0031-9422(96)00443-8. [DOI] [Google Scholar]
- 27.Ismail S., Ahmad S., Azam S.S. Immunoinformatics characterization of SARS-CoV-2 spike glycoprotein for prioritization of epitope based multivalent peptide vaccine. J. Mol. Liq. 2020;314:113612. doi: 10.1016/j.molliq.2020.113612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bibi S., Sakata K. Current status of computer-aided drug design for type 2 diabetes. Curr. Comp. Aided Drug Des. 2016;12:167–177. doi: 10.2174/1573409912666160426120709. [DOI] [PubMed] [Google Scholar]
- 29.Shaker B., Ahmad S., Thai T.D., Eyun S.-i., Na D. Rational drug design for Pseudomonas aeruginosa PqsA enzyme: An in silico guided study to block biofilm formation. Front. Mol. Biosci. 2020;7:284. doi: 10.3389/fmolb.2020.577316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kausar N., Ullah S., Khan M.A., Zafar H., Choudhary M.I., Yousuf S. Celebrex derivatives: Synthesis, α-glucosidase inhibition, crystal structures and molecular docking studies. Bioorg. Chem. 2021;106:104499. doi: 10.1016/j.bioorg.2020.104499. [DOI] [PubMed] [Google Scholar]
- 31.Khan I., Rahman H., Abd El-Salam N.M., Tawab A., Hussain A., Khan T.A., Khan U.A., Qasim M., Adnan M., Azizullah A. Punica granatum peel extracts: HPLC fractionation and LC-MS analysis to quest compounds having activity against multidrug resistant bacteria. BMC Complement. Altern. Med. 2017;17:1–6. doi: 10.1186/s12906-017-1766-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Sousa Luisa J.A., Barrosb R.P.C., de Sousab N.F., Muratovc E., Scottib L., Scottib M.T. Virtual screening of natural products database. Mini Rev. Med. Chem. 2021;21:2657–2730. doi: 10.2174/1389557520666200730161549. [DOI] [PubMed] [Google Scholar]
- 33.Kim S., Thiessen P.A., Bolton E.E., Chen J., Fu G., Gindulyte A., Han L., He J., He S., Shoemaker B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016;44:1202–1213. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daina A., Michielin O., Zoete V. iLOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014;54:3284–3301. doi: 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- 35.Dirir A.M., Daou M., Yousef A.F., Yousef L.F. A review of alpha-glucosidase inhibitors from plants as potential candidates for the treatment of type-2 diabetes. Phytochem. Rev. 2021:1–31. doi: 10.1007/s11101-021-09773-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roig-Zamboni V., Cobucci-Ponzano B., Iacono R., Ferrara M.C., Germany S., Bourne Y., Parenti G., Moracci M., Sulzenbacher G. Structure of human lysosomal acid α-glucosidase–a guide for the treatment of Pompe disease. Nat. Comm. 2017;8:1–10. doi: 10.1038/s41467-017-01263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vilar S., Cozza G., Moro S. Medicinal chemistry, and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008;8:1555–1572. doi: 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- 38.Yousafi Q., Batool J., Khan M.S., Perveen T., Sajid M.W., Hussain A., Mehmood A., Saleem S. In Silico Evaluation of Food Derived Bioactive Peptides as Inhibitors of Angiotensin Converting Enzyme (ACE) Int. J. Pept. Res. Ther. 2021;27:341–349. doi: 10.1007/s10989-020-10090-y. [DOI] [Google Scholar]
- 39.Aldeghi M., Heifetz A., Bodkin M.J., Knapp S., Biggin P.C. Accurate calculation of the absolute free energy of binding for drug molecules. Chem. Sci. 2016;7:207–218. doi: 10.1039/C5SC02678D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmad S., Ranaghan K.E., Azam S. Combating tigecycline resistant Acinetobacter baumannii: A leap forward towards multi-epitope-based vaccine discovery. Eur. J. Pharm. Sci. 2019;132:1–17. doi: 10.1016/j.ejps.2019.02.023. [DOI] [PubMed] [Google Scholar]
- 41.Lee T.-S., Allen B.K., Giese T.J., Guo Z., Li P., Lin C., McGee Jr T.D., Pearlman D.A., Radak B.K., Tao Y. Alchemical binding free energy calculations in AMBER20: Advances and best practices for drug discovery. J. Chem. Inf. Model. 2020;60:5595–5623. doi: 10.1021/acs.jcim.0c00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergonzo C., Cheatham III T.E. Improved force field parameters lead to a better description of RNA structure. J. Chem. Theory Comput. 2015;11:3969–3972. doi: 10.1021/acs.jctc.5b00444. [DOI] [PubMed] [Google Scholar]
- 43.Hyun T.K., Eom S.H., Kim J.-S. Molecular docking studies for discovery of plant-derived a-glucosidase inhibitors. Plant Omics. 2014;7:166–170. [Google Scholar]
- 44.Izaguirre J.A., Catarello D.P., Wozniak J.M., Skeel R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001;114:2090–2098. doi: 10.1063/1.1332996. [DOI] [Google Scholar]
- 45.Kräutler V., Van Gunsteren W.F., Hünenberger P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001;22:501–508. doi: 10.1002/1096-987X(20010415)22:5<501::AID-JCC1021>3.0.CO;2-V. [DOI] [Google Scholar]
- 46.Roe D.R., Cheatham III T.E. Ptraj and Cpptraj: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 47.Humphrey W., Dalke A., Schulten K. VMD: Visual molecular dynamics. J. Mol. Grap. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 48.Miller III B.R., McGee Jr T.D., Swails J.M., Homeyer N., Gohlke H., Roitberg A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 49.Khan M.S., Mehmood B., Yousafi Q., Bibi S., Fazal S., Saleem S., Sajid M.W., Ihsan A., Azhar M., Kamal M.A. Molecular Docking Studies Reveal Rhein from rhubarb (Rheum rhabarbarum) as a Putative Inhibitor of ATP-binding Cassette Super-family G member 2. Med. Chem. 2021;17:273–288. doi: 10.2174/1573406416666191219143232. [DOI] [PubMed] [Google Scholar]
- 50.Avdović E.H., Petrović I.P., Stevanović M.J., Saso L., Dimitrić Marković J.M., Filipović N.D., Živić M.Ž., Cvetić Antić T.N., Žižić M.V., Todorović N.V. Synthesis and Biological Screening of New 4-Hydroxycoumarin Derivatives and Their Palladium (II) Complexes. Oxid. Med. Cell. Longev. 2021;2021:8849568. doi: 10.1155/2021/8849568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sander T., Freyss J., von Korff M., Rufener C. Data Warrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 52.Avdović E.H., Milanović Ž.B., Molčanov K., Roca S., Vikić-Topić D., Mrkalić E.M., Jelić R.M., Marković Z.S. Synthesis, characterization and investigating the binding mechanism of novel coumarin derivatives with human serum albumin: Spectroscopic and computational approach. J. Mol. Struct. 2022;1254:132366. doi: 10.1016/j.molstruc.2022.132366. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data are available to researchers upon request.