

Abstract
Antibiotic resistance is a daunting challenge in modern medicine, and novel approaches that minimize the emergence of resistant pathogens are desperately needed. Antimicrobial peptides are newer therapeutics that attempt to do this; however, they fall short because of low to moderate antimicrobial activity, low protease stability, susceptibility to resistance development, and high cost of production. The recently developed random peptide mixtures (RPMs) are promising alternatives. RPMs are synthesized by incorporating a defined proportion of two amino acids at each coupling step rather than just one, making them highly variable but still defined in their overall composition, chain length, and stereochemistry. Because RPMs have extreme diversity, it is unlikely that bacteria would be capable of rapidly evolving resistance. However, their efficacy against pathogens in animal models of human infectious diseases remained uncharacterized. Here, we demonstrated that RPMs have strong safety and pharmacokinetic profiles. RPMs rapidly killed both Pseudomonas aeruginosa and Staphylococcus aureus efficiently and disrupted preformed biofilms by both pathogens. Importantly, RPMs were efficacious against both pathogens in mouse models of bacteremia and acute pneumonia. Our results demonstrate that RPMs are potent broad-spectrum therapeutics against antibiotic-resistant pathogens.
Keywords: Random peptide mixtures, antibiotic resistance, Pseudomonas aeruginosa, Staphylococcus aureus, bacteremia, acute pneumonia
Graphical Abstract
Antibiotic resistance is a pervasive problem in modern medicine, with more than 2.8 million antibiotic-resistant infections in the United States each year, causing upward of 35,000 mortalities.1 Broad reliance on antibiotics for treatment of bacterial infections and poor antibiotic stewardship further accelerate the crisis, and the establishment of distinct groups of drug-resistant bacteria has taken hold globally. These groups are multidrug-resistant (MDR), extensively drug-resistant (XDR), and pan-drug-resistant (PDR) bacteria,2 and all present overwhelming challenges to health care systems worldwide. Rampant antibiotic resistance exists within the ESKAPE pathogens, Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species, adding a significant risk factor to the frequent nosocomial infections caused by these bacteria.3–5
With the spread of antibiotic resistance in bacteria only hastening, it is vital that effective, non-antibiotic options be developed. One such type of agent is antimicrobial peptides (AMPs) produced by the innate immune system of many organisms.6–8 These peptides have broad-spectrum antimicrobial activities,9,10 with the majority of them functioning through disruption of bacterial membranes.8,11–14 Naturally derived AMPs and their synthetic analogs were recently demonstrated to be efficacious in multiple mouse models of infection.15–17 However, AMPs are not without their own shortcomings. AMPs exhibit high cell toxicity, weaker antimicrobial activity (as compared to traditional antibiotics), low protease stability, and high cost of production, and still, they are unable to evade the development of resistance against them by bacteria.18–20 Just as is the case with antibiotics, growing use of AMPs has prompted reports of bacterial strains resistant to them.21 Additionally, some bacteria have inherent strategies that enable them to be resistant to AMPs.22 Gram-positive bacteria, for example, express the protein Dlt that inserts d-alanine into cell wall teichoic acid,23 and Gram-negative bacteria incorporate 4-aminoarabinose into lipid A, reducing the charge of lipopolysaccharides (LPS).24 Both changes diminish the affinity of AMPs for the bacterial cell membrane.
Interestingly, a recent study had shown that S. aureus evolved resistance to AMPs at a slower rate than to antibiotics.25 These authors also showed that this resistance evolved even slower to a cocktail of two AMPs, suggesting that combination therapy with multiple AMPs would impede the development of resistance in bacteria. To capitalize on this concept, Hayouka and co-workers have developed a novel approach to synthesize ultradiverse, random antimicrobial peptide mixtures (RPMs). In contrast to the traditional method of solid-phase peptide synthesis, RPMs were synthesized by incorporating a defined proportion of two amino acids at each coupling step, rather than just one, making them highly variable but still defined in their overall composition, chain length, and stereochemistry (Figure 1).26 RPMs are effective antimicrobials in vitro in prevention of biofilm formation and eradication of mature biofilms by methicillin-resistant S. aureus (MRSA) and for reduction of disease severity in plant bacterial infectious disease models and inhibition of bacterial growth in dairy milk.26–29 However, the safety and efficacy of RPMs have not been analyzed in preclinical mouse models of human infectious diseases. In this study, we examine the safety of RPMs in vitro and in mice. Additionally, we examine the efficacy of RPMs in mouse models of human infectious diseases, bacteremia and acute pneumonia, caused by MRSA and P. aeruginosa.
Figure 1.
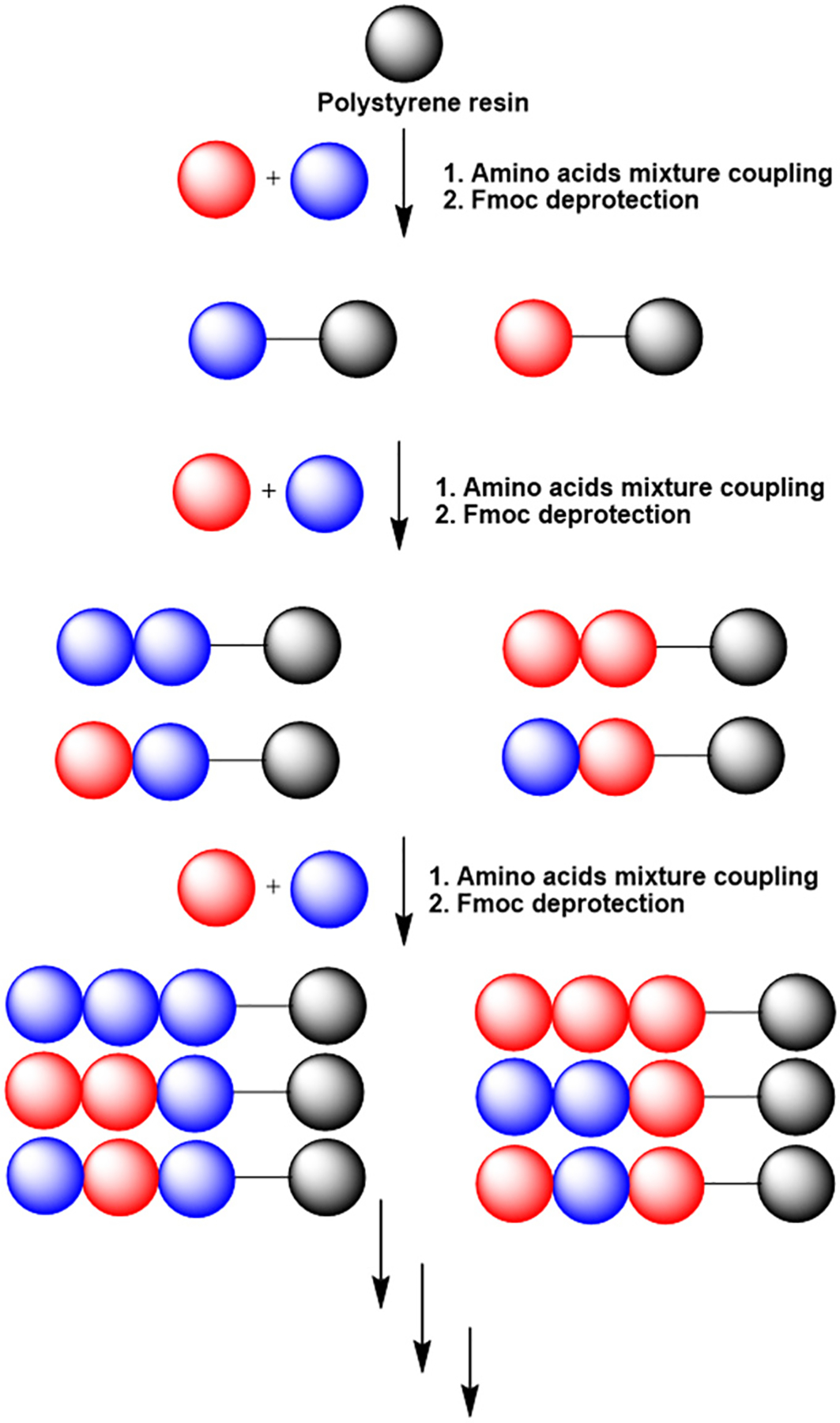
Illustration of the synthesis of random peptide mixtures (RPMs). Standard Fmoc-SPPS is used, with the addition of a mixture of two protected amino acids, rather than single protected amino acids, in each coupling step. In this process, each resin bead bears several different sequences. The results of three coupling steps are illustrated before the cleavage of the peptides from the solid support.
RESULTS AND DISCUSSION
Random Peptide Mixtures Rapidly and Effectively Kill P. aeruginosa and MRSA In Vitro.
In this study, we examined the safety and efficacy of four RPMs as novel antimicrobial agents: FK20 (l-phenylalanine–l-lysine, 20-mer), FdK (l-phenylalanine–d-Lysine, 20-mer), LK20 (l-leucine–l-lysine, 20-mer), and dFdK (d-phenylalanine–d-lysine, 20-mer). We tested these RPMs for their ability to kill P. aeruginosa and MRSA under in vitro conditions. Overnight cultures of MRSA strain USA300 LAC and P. aeruginosa strain PAO1 were serially diluted to ~108 colony forming units (CFUs) and incubated with 4, 20, and 100 μg mL−1 of RPMs for 10 min, as we have previously published.29 At 20 and 100 μg mL−1, all RPMs killed USA300 LAC and PAO1 at significantly higher levels than the saline control (Figure 2A,B). The most effective RPMs were FK20, FdK, and dFdK, which at 100 μg mL−1 were able to achieve up to 4.5 log decrease in CFUs of USA300 LAC (Figure 2A) and up to 7 log decrease in CFUs of PAO1 (Figure 2B). For comparison, in recent studies, conventional antibiotics have been able to reduce bacterial counts by 5 log for MRSA30 (vancomycin) and 2 log for P. aeruginosa (tobramycin).31
Figure 2.
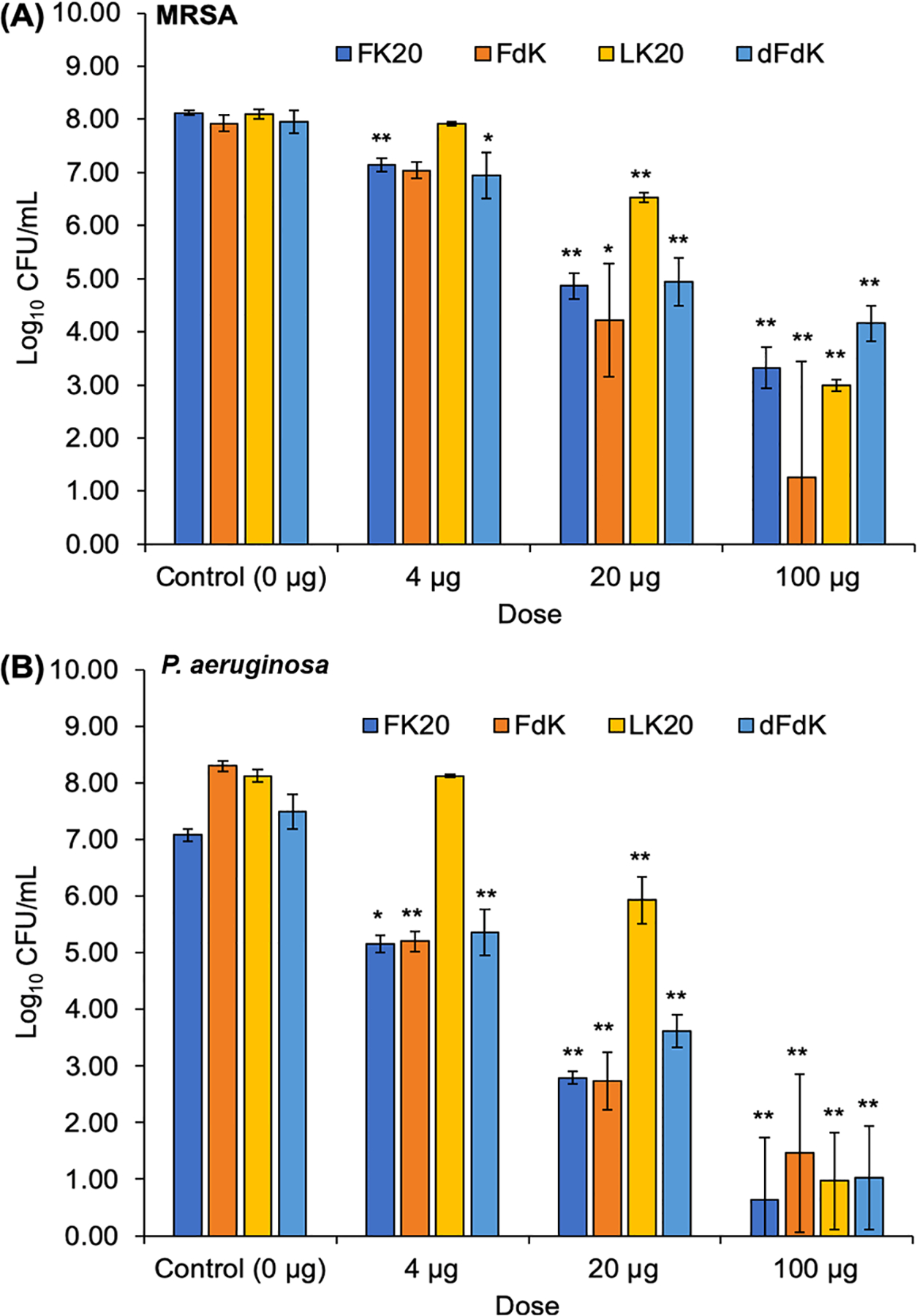
In vitro killing of MRSA strain USA300 LAC (A) and P. aeruginosa strain PA01 (B). Bacteria were cultured in lysogeny broth (LB) at 37 °C overnight and then washed with PBS. Bacteria were then incubated with RPMs for 10 min at 37 °C, serially diluted with PBS, and plated on LB (MRSA) or Pseudomonas isolation agar (PA01). Colonies were counted after overnight incubation at 37 °C, and data were log-transformed. Error bars represent SD for n = 3 from a typical experiment performed independently three times. Significance was determined with ANOVA and Tukey HSD. *p < 0.05; **p < 0.01 significance compared to control; data without symbols are not significantly different.
Random peptide mixtures disrupt preformed biofilms by both P. aeruginosa and MRSA.
For the subsequent experiments, we chose to focus our analysis on FK20 and FdK, because these RPMs had the best efficacy in the initial P. aeruginosa and MRSA killing assays. Because biofilms are recalcitrant to eradication by antimicrobials, we examined if RPMs would be effective in this setting. Using a protocol described by O’Toole,32 biofilms were grown overnight (12 h) and then incubated with RPMs FK20 and FdK for another night, after which biofilm levels were quantified. Tobramycin and daptomycin, antibiotics used to treat P. aeruginosa and S. aureus infections respectively, were included as positive controls. For both RPMs tested, biofilm levels decreased with increasing concentrations of peptide. FK20 was more efficacious in eradicating biofilms than FdK at 4 μg mL−1 level (Figure 3A), but both peptides were equally effective against P. aeruginosa strain PAO1 at all concentrations tested (Figures 3A and 3B). At 100 μg mL−1, both peptides were able to eradicate biofilms comparable to the levels by antibiotics daptomycin and tobramycin (Figure 3). These results show that RPMs are effective at eradicating biofilms.
Figure 3.
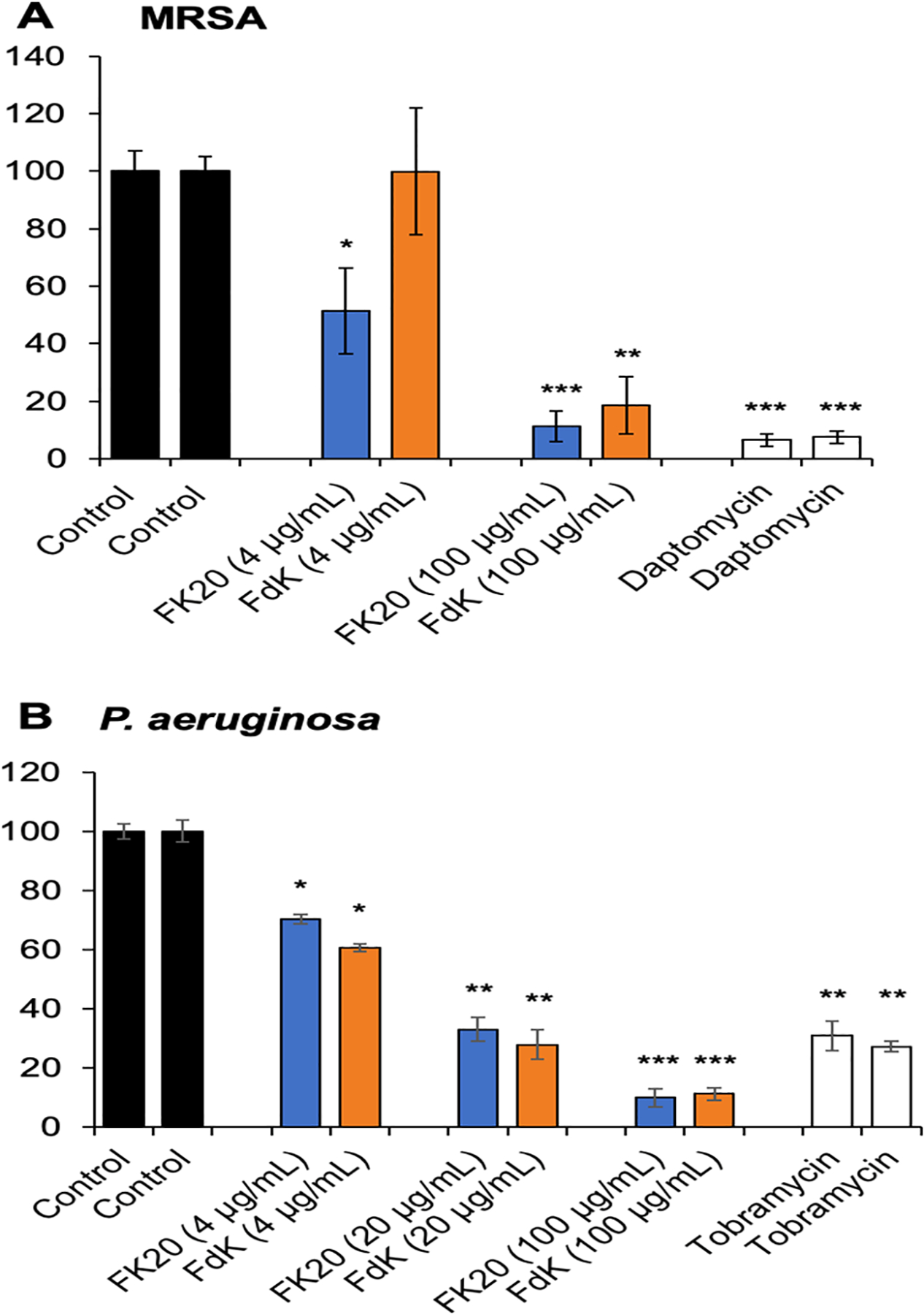
Biofilm eradication by RPMs FK20 and FdK. MRSA strain USA300 LAC (A) and P. aeruginosa strain PA01 (B) were cultured at 37 °C overnight in LB, then diluted 1:100 in M63 minimal media and aliquoted into a round-bottom 96-well plate. The plate was covered and incubated at 37 °C overnight. The liquid was shaken out from the plate, and it was rinsed with sterile H2O. Fresh M63 media with RPMs, antibiotics (daptomycin 10 μg mL−1, tobramycin 10 μg mL−1), or PBS added was pipetted into the same wells. The plate was covered and again incubated at 37 °C overnight. The plate was dumped and rinsed again, and 125 μL of 0.1% crystal violet was added to each well. Wells were incubated at room temperature for 15 min, then dumped and rinsed 3 times with distilled H2O. The plate was left to dry overnight at room temperature, uncovered. Acetic acid (125 μL of 30%) was added to each well, and the liquid from each of the wells was transferred to a new, flat-bottom 96-well dish. The plate was read at 550 nm, and absorbance was recorded. Error bars represent SD for n = 3 of a typical experiment performed independently three times. Significance was determined with ANOVA and Tukey HSD. *p < 0.05, **p < 0.01, and ***p < 0.001 significance compared to control; data without symbols are not significantly different.
Random Peptide Mixtures Are Not Cytotoxic to Cultured Human Cells and Do Not Cause Deleterious Toxicity in Mice.
Before examining the efficacy of FK20 and FdK in animal models of human infectious diseases, we examined the cytotoxicity and maximum tolerated dose (MTD) for both peptides. To assess the safety of both RPMs in vitro, we conducted cytotoxicity assays on the airway bronchial epithelial 16HBE cells. Cells were incubated with 2 different concentrations of each peptide for 24 h and viability was measured by using the WST-1 reagent. For both RPMs tested, the cell viability was not altered compared to control 16HBE cells treated with sterile PBS; the only significantly different treatment with FK20 resulted in slightly better viability in 16HBE cells than control (Figure 4A). These results indicate that RPMs are not cytotoxic.
Figure 4.
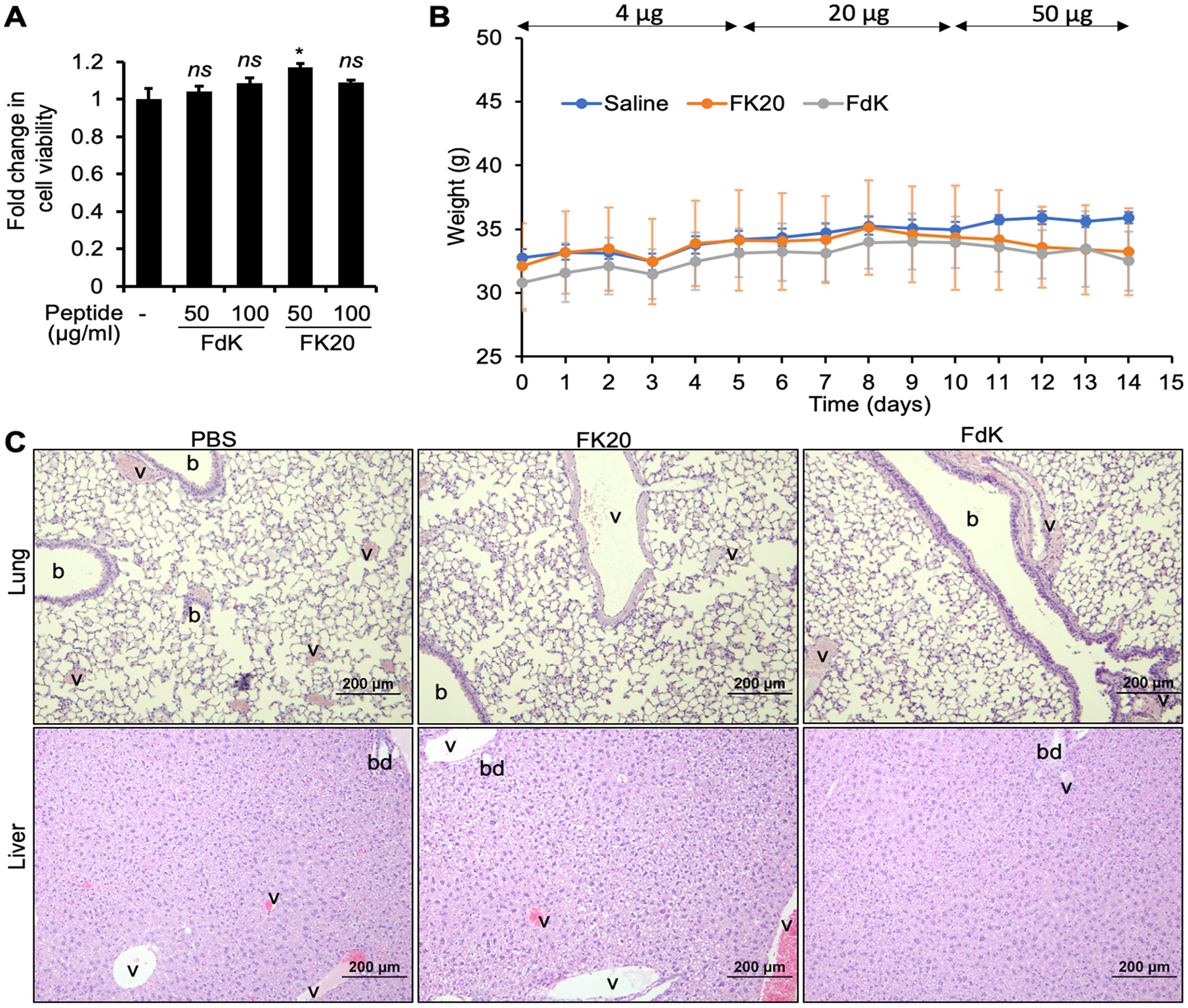
In vitro cytotoxicity and in vivo toxicity of RPMs. (A) Bronchial epithelial 16HBE cells seeded in a 96-well cell culture plate were exposed to 50 or 100 μg mL−1 of each RPM for 24 h. Cytotoxicity was determined by incubating with the WST-1 Cell Proliferation and Cytotoxicity Assay Kit for 2 h and measuring the absorbance at 440 nm. Error bars represent SEM for n = 3. Significance was determined with Student’s t test. *p < 0.05 significance compared to control; ns, not statistically significant. (B) Mouse weight change over a 14-day period of increasing doses with RPMs. Six-week old CD-1 mice (n = 5, males) were inoculated intranasally once daily with either FK20 or FdK at a concentration of 4 μg mL−1 in the first 5 days, 20 μg mL−1 in the next 5 days, and 100 μg mL−1 in the last 4 days. Mouse cohort instilled with an equal volume of daily sterile PBS (50 μL) was used as control. Error bars represent SD for n = 5. Significance was determined with ANOVA. Data across treatments are not significantly different from control. (C) Lung and liver histopathology after FK20 and FdK challenge. Mice were euthanized on day 15, and tissues were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin: b, bronchioles; v, blood vessels; bd, biliary duct.
To effectively determine the safety of RPMs in vivo, six-week old CD-1 mice (cohorts of 5) were intranasally inoculated daily with increasing concentrations of FK20 and FdK (Figure 4B). Mouse cohort challenged with sterile PBS was used as control. Mouse weight was determined daily. On day 15, the mice were euthanized, and analyses of blood chemistry and lung histopathology were performed at the University of Illinois College of Veterinary Medicine Veterinary Diagnostic Laboratory. There was no significant change in the mean weights of the mice in any group throughout the 14-day exposure (Figure 4B). Additionally, there was no distinct abnormality observed in the histopathology of lungs among the three cohorts (Figure 4C).
There were no significant differences between mouse cohorts treated with FK20 and FdK versus the PBS in profiles of both blood chemistry (Table 1) and hematology (Table 2), with the exception of low platelet count across all mouse cohorts, most likely caused by common clotting events during the blood collection process (Table 2). Another potential exception was potential hyperbilirubinemia in both FK20 and FdK cohorts. Major causes of hyperbilirubinemia include hepatic dysfunction and hemolysis (either pathologic or artificial). However, the normal ALT value and icteric indicator suggested that there was no underlying hepatic dysfunction (Table 1). The lack of hepatic toxicity was further supported by histopathology analysis of livers, which showed only mild to moderate degrees of physiological glycogen infiltration in the periportal and intermediate zones in all three cohorts, with no overt evidence of hepatocellular necrosis or associated inflammatory response (Figure 4C). Collectively, these results indicate that RPMs are not toxic and will be safe for in vivo applications against bacterial infections.
Table 1.
Blood Chemistry of CD-1 Mice after Exposure to FK20 and FdKa
| analytes | saline | FK20 | FdK | reference range |
|---|---|---|---|---|
| creatinine (mg/dl) | 0.20a ± 0 | 0.12b ± 0.04 | 0.13b ± 0.05 | 0.1–0.4 |
| BUN (mg/dL) | 23.60a ± 0.55 | 20.40b ± 2.07 | 17.40c ± 1.95 | 16–29 |
| total protein (g/dL) | 5.26 ± 0.17 | 5.42 ± 0.43 | 5.48 ± 0.23 | 5.0–6.3 |
| albumin (g/dL) | 2.74a ± 0.13 | 3.02ab ± 0.26 | 3.08b ± 0.13 | 3.0–4.1 |
| globulin (g/dL) | 2.52 ± 0.08 | 2.40 ± 0.20 | 2.40 ± 0.12 | 1.8–2.3 |
| albumin/globulin ratio | 1.10a ± 0.07 | 1.24b ± 0.09 | 1.28b ± 0.04 | |
| calcium (mg/dL) | 9.96 ± 0.18 | 9.52 ± 0.47 | 9.44 ± 0.38 | 9.8–10.8 |
| phosphorus (mg/dL) | 8.62a ± 1.14 | 6.14b ± 1.05 | 6.40b ± 1.53 | 6.1–13.1 |
| sodium (mmol/L) | 152.40a ± 1.14 | 149.00b ± 2.35 | 148.60b ± 1.67 | 153–159 |
| potassium (mmol/L) | 5.06 ± 0.32 | 6.10 ± 1.55 | 6.82 ± 1.12 | 8.3–12.7 |
| sodium/potassium ratio | 30.20 ± 1.79 | 25.60 ± 6.58 | 22.40 ± 4.04 | |
| chloride (mmol/L) | 107.40 ± 1.52 | 107.20 ± 1.79 | 106.40 ± 2.3 | 106.1–113.9 |
| glucose (mg/dL) | 114.80 ± 28.17 | 118.80 ± 44.87 | 119.60 ± 33.23 | 169–298 |
| alkaline phos total (U/L) | 76.80 ± 22.08 | 52.40 ± 30.48 | 34.80 ± 23.25 | 34–106 |
| ALT [SGPT] (U/L) | 32.40 ± 6.5 | 30.20 ± 17.7 | 35.60 ± 4.62 | 25–76 |
| GGT (U/L) | 0.00 ± 0 | 0.00 ± 0 | 0.00 ± 0 | 36–89 |
| total bilirubin (mg/dL) | 0.28 ± 0.08 | 0.52 ± 0.35 | 0.62 ± 0.20 | 0.16–0.31 |
| cholesterol total (mg/dL) | 190.40 ± 25.98 | 188.60 ± 33.75 | 180.60 ± 21.26 | 111–196 |
| triglycerides (mg/dL) | 190.60 ± 94.61 | 173.80 ± 62.75 | 190.25 ± 56.63 | 128–288 |
| bicarbonate [TCO2] (mmol/L) | 9.60 ± 2.51 | 10.40 ± 1.95 | 8.40 ± 3.36 | b |
| anion gap | 40.60 ± 2.51 | 37.40 ± 0.89 | 40.60 ± 3.05 | b |
| lipemic indicator | 0.00 ± 0 | 0.00 ± 0 | 0.00 ± 0 | b |
| icteric indicator | 0.00 ± 0 | 0.00 ± 0 | 0.00 ± 0 | b |
| hemolytic indicator | 3.40 ± 0.55 | 3.60 ± 0.55 | 4.00 ± 0 | b |
Letters a–c online in rows are to denote statistical significance: mean values in the same row with at least one letter in common are not significantly different. If no letters are present in a row, none of the mean values in that row are significantly different. Significance was determined in R with ANOVA and Tukey mean separation; p < 0.05 for significance.
Not available.
Table 2.
Complete Blood Count with Differential of CD-1 Mice after Exposure to FK20 and FdKa
| analytes | saline | FK20 | FdK | reference range |
|---|---|---|---|---|
| red blood cells (×106/μL) | 9.10 ± 0.57 | 8.85 ± 0.28 | 9.04 ± 0.16 | 8.49–10.43 |
| hemoglobin (g/dL) | 14.52 ± 0.57 | 14.16 ± 0.59 | 14.22 ± 0.13 | 13.8–16.8 |
| hematocrit % | 42.96a ± 1.22 | 40.72b ± 1.11 | 41.38ab ± 0.23 | 47.0–39.3 |
| mean cell volume (fL) | 47.30 ± 1.94 | 46.00 ± 1.3 | 45.76 ± 0.69 | 42.3–48.0 |
| MCH (pg) | 15.98 ± 0.64 | 15.98 ± 0.85 | 15.72 ± 0.38 | 14.7–17.5 |
| MCHC (g/dL) | 33.78 ± 0.4 | 34.76 ± 0.92 | 34.36 ± 0.29 | 34.1–36.7 |
| platelet estimate (×103/μL) | 232.40 ± 220.15 | 256.00 ± 55.71 | 144.00 ± 66.75 | 784–1812 |
| WBC count (×103/μL) | 6.31a ± 1.19 | 8.29b ± 0.86 | 7.89ab ± 1.29 | 7.06–17.29 |
| Neu % | 17.30 ± 8.86 | 16.56 ± 8.13 | 18.86 ± 10.61 | 8.74–55.68 |
| Lymph % | 80.12 ± 9.73 | 72.32 ± 10.37 | 75.30 ± 11.83 | 37.50–85.01 |
| Mono % | 1.06a ± 0.13 | 2.82b ± 1.29 | 1.64ab ± 1.13 | 2.84–13.09 |
| Eos % | 1.44 ± 1.1 | 7.08 ± 5.31 | 4.12 ± 1.43 | 0.30–5.20 |
| Baso % | 0.08 ± 0.13 | 0.02 ± 0.04 | 0.08 ± 0.11 | 0.00–0.17 |
| A Neu (×103/μL) | 1.13 ± 0.7 | 1.39 ± 0.74 | 1.41 ± 0.55 | 0.98–6.06 |
| A Lymph (×103/μL) | 5.01 ± 0.89 | 5.97 ± 0.92 | 6.02 ± 1.59 | 5.01–11.60 |
| A Mono (×103/μL) | 0.07a ± 0.02 | 0.24b ± 0.13 | 0.14ab ± 0.1 | 0.09–0.63 |
| A Eos (×103/μL) | 0.10a ± 0.08 | 0.69b ± 0.33 | 0.31a ± 0.07 | 0–0.75 |
| A Baso (×103/μL) | 0.01 ± 0.01 | 0.00 ± 0 | 0.01 ± 0.01 | 0–0.09 |
Letters a and b online in rows are to denote statistical significance: mean values with at least one letter in common are not significantly different. If no letters are present in a row, none of the mean values in that row are significantly different. Significance was determined in R with ANOVA and Tukey mean separation; p < 0.05 for significance.
Random Peptide Mixtures Are Efficacious in Attenuating P. aeruginosa and MRSA Burden in Mouse Models of Acute Pneumonia and Bacteremia.
As shown above, both in vitro and in vivo studies demonstrate the effectiveness and safety of the RPMs. To further corroborate our findings, next we examined the capability of FK20 and FdK to reduce bacterial burden in mouse models of human infectious diseases by P. aeruginosa and MRSA. P. aeruginosa-mediated pneumonia and sepsis, particularly of ventilated patients in intensive care units, are associated with high morbidity and mortality33,34 Additionally, P. aeruginosa is one of the dominant pathogens in patients with chronic lung diseases, including cystic fibrosis, chronic obstructive pulmonary disease, primary ciliary dyskinesis, and bronchiectasis35,36 P. aeruginosa is also the dominant pathogen causing chronic suppurative otitis media,37 severe ocular infection,38,39 and burn sepsis.40 S. aureus, including MRSA strains, are opportunistic asymptomatic colonizers but can cause serious infection in immunocompromised individuals. S. aureus is one of the five most common causes of nosocomial infections, ranging from minor skin infections and abscess formation41 to life-threatening diseases such as acute pneumonia, chronic pneumonia (e.g., in cystic fibrosis), endocarditis, toxic shock syndrome, bacteremia, and sepsis,33,34 as well as periprosthetic infections and osteomyelitis, due to its ability to form biofilms on implant surfaces.42–44
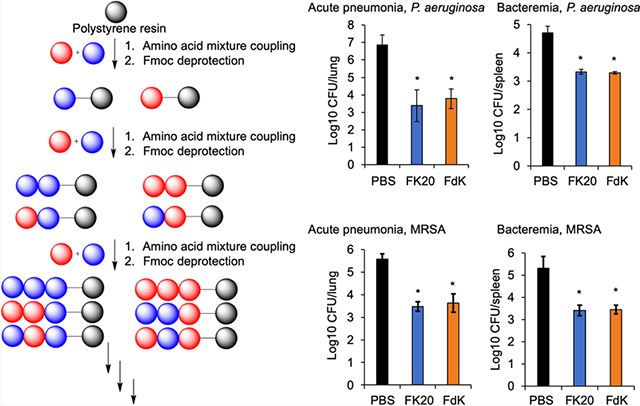
First, we tracked the stability and spread of the intramuscularly administered FK20 conjugated to the fluorescence dye Cy7.5 (FK20–Cy7.5) spatiotemporally. As shown in Figure 5A, FK20–Cy7.5 remained stable and continued to spread systemically over 24 h after injection. Some of the FK20–Cy7.5 remained at the site of injection at 24 h, suggesting that systemic spread is steady and prolonged. We examined the efficacy of FK20 and FdK against P. aeruginosa strain PAO1 and MRSA strain USA300 LAC in mouse models of acute pneumonia and bacteremia. In the acute pneumonia infection, FK20 and FdK at 10 mg/kg (given in 2 doses of 5 mg/kg) reduced lung burden of PAO1 by 3.49 and 3.09 log and that of USA300 LAC by 2.09 and 1.94 log, respectively (Figure 5B,C). In the bacteremia model, both FK20 and FdK attenuated the burden of PAO1 in spleens by 1.37 and 1.41 log and USA300 LAC burden in spleens by 1.90 and 1.86 log, respectively (Figure 5B,C). In mouse studies with conventional antibiotics, pneumonia models showed a 2.5- (P. aeruginosa, tobramycin)45 and 1.5-log (MRSA, vancomycin)46 decrease in CFUs, while sepsis models had a 3-log (P. aeruginosa, colistin, and MRSA, vancomycin)46,47 decrease in CFUs. One important note is that the therapeutic dose of FK20 and FdK (10 mg/kg) was ~5× higher than that of the FK20–Cy7.5 used for the live IVIS-based imaging. Collectively, these results indicate that RPMs are efficacious in reducing acute infection in mouse models of human infectious diseases mediated by these two ESKAPE pathogens.
Figure 5.
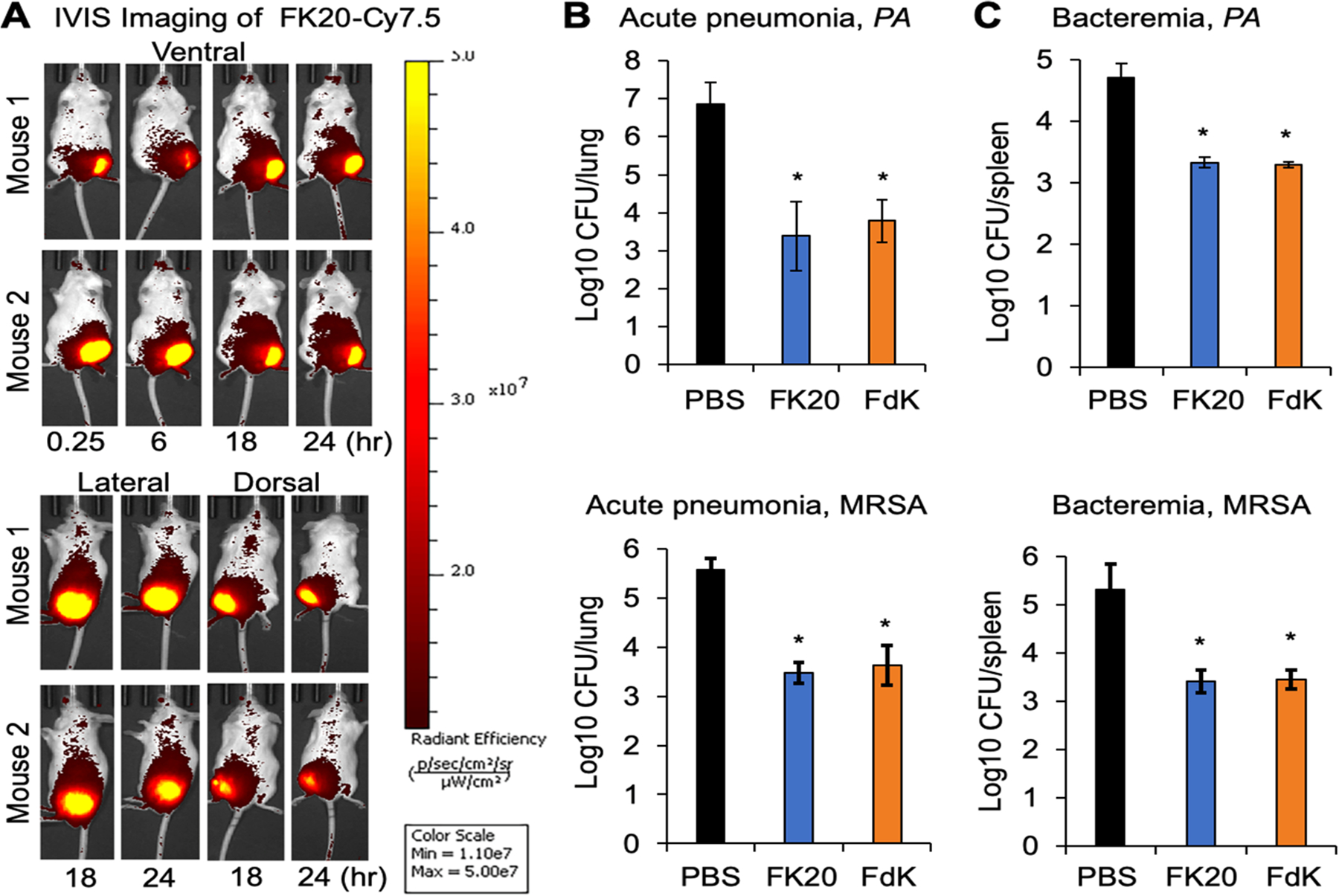
RPM efficacy in mouse models of pneumonia and bacteremia. (A) CD-1 mice (n = 2, males) were intramuscularly injected with FK20 conjugated to the fluorescence dye Cy7.5 (50 μg) in the left thigh and imaged in three positions over 24 h using an IVIS SpectrumCT imaging system. (B, C) CD-1 mice (7–9 week old, males and females) were inoculated with P. aeruginosa strain PAO1 or MRSA USA300 strain LAC. For the acute pneumonia model, mice were intranasally inoculated with 4.6 × 107 CFU of PAO1 for 24 h and 1.5 × 108 CFU of USA300 LAC for 48 h. For bacteremia, mice were intraperitoneally inoculated with 6.4 × 106 CFU of PAO1 and 7.6 × 106 CFU of USA300 LAC, and both models were followed for 24 h. Infected mice were treated intramuscularly twice daily with 5 mg/kg of FK20, FdK, or sterile PBS control. At the designated time, mouse lungs (acute pneumonia) or spleens (bacteremia) were harvested from euthanized mice, homogenized, and serially diluted and plated to determine bacterial burden. Error bars represent SD for n = 9 (pneumonia) or n = 6 (bacteremia). Significance was determined with the Student’s t test; *p < 0.05 significance compared to control.
In summary, we demonstrate that RPMs are potent broad-spectrum therapeutics against antibiotic-resistant pathogens. RPMs were not cytotoxic in vitro and did not exhibit abnormal levels of toxicity in preclinical mouse model. RPMs rapidly killed both P. aeruginosa and MRSA in vitro, eradicated preformed biofilms, and showed significant efficacy against both ESKAPE pathogens in mouse models of bacteremia and acute pneumonia. Future efforts will examine the efficacy of RPMs in additional models of human infectious diseases as well as synthesis of next-generation, more efficacious random peptide mixture drugs.
METHODS
Reagents.
All reagents were purchased from Millipore-Sigma-Aldrich (St. Louis, MO, USA), unless otherwise noted.
Random Peptide Mixtures.
RPMs were synthesized as previously described.26–29 Previous publications have confirmed the success of the synthesis using MALDI-TOF mass spectrometry and amino acid analysis to validate the chain length of the synthesized peptide and that the 1:1 molar ratio of the two amino acids (phenylalanine and l/d-lysine) in FK20 or FdK RPMs was preserved during the synthesis.48,49 The RPMs were dissolved in sterile deionized H2O to a concentration of 1 mg mL−1, distributed into small aliquots, and stored at −20 °C. When used in experiments, aliquots were first thawed at room temperature. Aliquots used in killing assays were always fresh to avoid loss of activity because of repeated freeze/thaw cycles.
Bacterial Strains, Media, and Growth Conditions.
The P. aeruginosa wild-type strain PAO1 was originally provided by Professor Michael Vasil (University of Colorado School of Medicine), as we have previously published.50–54 MRSA strain USA300 LAC was provided by Professor Jianjun Cheng of the University of Illinois at Urbana–Champaign. Bacteria were cultured in the lysogeny broth (LB) (Benton, Dickinson, and Company, Franklin Lakes, New Jersey, USA) for 16 h at 37 °C, resuspended in fresh LB with 20% sterile glycerol and frozen in aliquots at −80 °C. Before each assay, aliquots of the bacteria were cultured from frozen stocks in fresh LB to desirable growth phase and cell density. The optical density at 600 nm was determined using a spectrophotometer and correlated with numbers of viable bacteria (CFU) after serial dilution plating on agar plates.
In Vitro Bacterial Killing Assays.
Overnight cultures of P. aeruginosa PAO1 and MRSA USA300 LAC were washed 3× with sterile PBS, adjusted to 107–108 CFU mL−1 and added to 4 μg mL−1, 20 μg mL−1, and 100 μg mL−1 of RPMs or an equal volume of sterile PBS (negative control). Bacteria and RPMs were incubated at 37 °C for 10 min while rotating on a New Brunswick Scientific (Edison, NJ, USA) TC-7 rotary drum at a speed dial setting 2. Samples were then immediately chilled on ice, serially diluted 1:10 in ice-cold PBS, and then plated on LB agar for USA300 LAC or Pseudomonas isolation agar (agar 13.6 g L−1, magnesium chloride 1.4 g L−1, tryptone 20 g L−1, potassium sulfate 10 g L−1, glycerol 20 mL L−1, Irgasan 0.025 g L−1) for PAO1. Plates were incubated at 37 °C overnight, and then CFUs were enumerated. Bacterial killing experiments were performed in triplicate independently three times.
Cell Viability and Cytotoxicity Assays.
The 16HBE bronchial epithelial cells55 were a generous gift from Professor D. C. Gruenert (University of California, San Francisco, CA, USA). Approximately 20 000 cells were seeded into each well of a 96-well plate and cultured in MEM (Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA) supplemented with 10% FBS and penicillin/streptomycin solution (MilliporeSigma) at 37 °C in 5% CO2 until 70% confluency, as we have previously described.56,57 The cells were exposed to RPMs at a concentration of 50 or 100 μg mL−1 for 24 h. The cells were then incubated with WST-1 reagent (Roche Molecular Systems, Indianapolis, IN, USA) for 2 h using the protocol described by the manufacturer, measuring the absorbance at 440 nm with a SpectraMax Gemini EM Microplate Reader (San Jose, CA, USA). Treatments were performed in triplicate and independently three times.
Biofilm Disruption Assays.
P. aeruginosa strain PAO1 and MRSA strain USA300 LAC were cultured in LB overnight at 37 °C. The overnight culture was then diluted 1:100 in M63 minimal media,32 and 100 μL of the dilution was added to each well in a 96-well, round-bottom microtiter dish, and the dish was sealed with parafilm. The plate was incubated at 37 °C for 12 h. Unattached cells were then dumped out by turning over the dish and shaking out the liquid. The plate was submerged in deionized H2O, and water was shaken out. Fresh media with RPMs, PBS, tobramycin (10 μg mL−1), or daptomycin (10 μg mL−1) was added to the wells, and the plate was then incubated at 37 °C overnight. The culture supernatant in wells was dumped and rinsed again, then the contents were stained with 125 μL of a 0.1% solution of crystal violet. The plate was incubated at room temperature for 15 min, then rinsed with deionized water 3 times in the manner described previously and left to dry, uncovered and inverted, in a fume hood overnight. To quantify the biofilm, 125 μL of 30% acetic acid in water was added to each well, and the plate was incubated at room temperature for 15 min. The solution from each well was then transferred into wells in a new, flat-bottom 96-well plate. Absorbance was measured at a wavelength of 550 nm. Treatments were performed in triplicate independently three times.
Mouse Safety Studies, In Vivo Imaging, and Infection.
The mouse studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Illinois at Urbana–Champaign. Briefly, six-week old CD-1 mice (males and females) were purchased from Charles River (Wilmington, MA, USA), and acclimated for at least 1 week before use. All mice were housed in positively ventilated microisolator cages with automatic recirculating water located in a room with laminar, high efficiency particulate-filtered air. The animals received autoclaved food, water, and bedding.
For the toxicity studies, CD-1 mice (cohorts of 5) were dosed intranasally daily with FK20, FdK, or sterile saline control (in 50 μL). The concentration of RPMs was increased every 5 days, starting at 4 μg, then 20 μg, and finally 100 μg. The weights of each mouse were recorded each day, and averages were calculated. Mice were euthanized on the 15th day, and blood and major organs (e.g., lung, liver, etc.) were collected. Blood chemistry and complete blood count with differential analysis was performed at the University of Illinois at Urbana–Champaign Veterinary Diagnostic Laboratory. Lungs and livers were embedded in paraffin, sectioned, stained with hematoxylin and eosin, and imaged using an Olympus DP70 light microscope (Central Valley, PA, USA).
For the in vivo imaging, CD-1 mice (n = 2) were anesthetized with 3% isoflurane in an induction chamber. Mice were intramuscularly injected with FK20–Cy7.5 (50 μg) in the left thigh muscle, and imaged over 24 h using an IVIS SpectrumCT imaging system (PerkinElmer, Norwalk, CT, USA). Fluorescence images were acquired using the following settings: binning factor as 1, f number as 1, field of view as 25.4, and fluorescence exposure time for 60 s. Images were analyzed by Living Image Software (PerkinElmer, MA).
For the acute pneumonia model, CD-1 mice (cohorts of 9) were intranasally inoculated with 4.6 × 107 CFU of P. aeruginosa strain PAO1 for 24 h and 1.5 × 108 CFU of MRSA USA300 LAC for 48 h. For bacteremia, mice (cohorts of 6) were intraperitoneally inoculated with 6.4 × 106 CFU of PAO1 and 7.6 × 106 CFU of USA300 LAC, and both models were followed for 24 h. Infected mice were treated intramuscularly twice daily with 5 mg/kg (in 100 μL) FK20, FdK, or sterile PBS control, with the first treatment at 4 h postinfection. At designated time points, mouse lungs (acute pneumonia) or spleens (bacteremia) were harvested from euthanized mice, homogenized, serially diluted, and plated to determine bacterial burden.
Statistical Analysis.
Quantitative data were expressed as the mean ± standard deviation or standard error and were calculated using their respective functions. Statistical significance was determined by using ANOVA, Student’s t-test, and Tukey HSD.58 Statistical significance was expressed as p ≤ 0.05, p ≤ 0.01, p ≤ 0.001, or ns (not significant).
ACKNOWLEDGMENTS
We thank Professors Michael Vasil (University of Colorado Health Science Center) for the P. aeruginosa strain PAO1, Jianjun Cheng (University of Illinois at Urbana–Champaign) for MRSA strain USA300 LAC, and D.C. Gruenert (University of California, San Francisco, CA, USA) for the gift of 16HBE cells. This work was supported by the NIH (Grants HL090699, AI080710-06A1, and HL142626 to G.W.L.).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acsinfecdis.0c00871
The authors declare no competing financial interest.
Contributor Information
Richard C. Bennett, Department of Pathobiology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61802, United States
Myung Whan Oh, Department of Pathobiology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61802, United States.
Shanny Hsuan Kuo, Department of Pathobiology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61802, United States.
Yael Belo, Institute of Biochemistry, Food Science and Nutrition, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot 76100, Israel.
Bar Maron, Institute of Biochemistry, Food Science and Nutrition, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot 76100, Israel.
Einav Malach, Institute of Biochemistry, Food Science and Nutrition, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot 76100, Israel.
Jingjun Lin, Department of Pathobiology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61802, United States.
Zvi Hayouka, Institute of Biochemistry, Food Science and Nutrition, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot 76100, Israel;.
Gee W. Lau, Department of Pathobiology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61802, United States;.
REFERENCES
- (1).CDC/Centers for Disease Control and Prevention (2019) Antibiotic Resistance Threats in the United States, U.S. Department of Health and Human Services, CDC. [Google Scholar]
- (2).Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, and Monnet DL (2012) Multidrug-resistant, Extensively drug-resistant and Pandrug-resistant Bacteria: an International Expert Proposal for Interim Standard Definitions for Acquired Resistance. Clin. Microbiol. Infect 18, 268–281. [DOI] [PubMed] [Google Scholar]
- (3).Rice LB (2008) Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J. Infect. Dis 197, 1079–1081. [DOI] [PubMed] [Google Scholar]
- (4).WHO/World Health Organization (2017) Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics, WHO. [Google Scholar]
- (5).De Oliveira D, Forde BM, Kidd TJ, Harris P, Schembri MA, Beatson SA, Paterson DL, and Walker MJ (2020) Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev 33, e00181–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Diamond G, Beckloff N, Weinberg A, and Kisich KO (2009) The Roles of Antimicrobial Peptides in Innate Host Defense. Curr. Pharm. Des 15, 2377–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zasloff M (2002) Antimicrobial Peptides of Multicellular Organisms. Nature 415, 389–395. [DOI] [PubMed] [Google Scholar]
- (8).Rathinakumar R, Walkenhorst WF, and Wimley WC (2009) Broad-Spectrum Antimicrobial Peptides by Rational Combinatorial Design and High-Throughput Screening: The Importance of Interfacial Activity. J. Am. Chem. Soc 131, 7609–7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Aoki W, and Ueda M (2013) Characterization of Antimicrobial Peptides toward the Development of Novel Antibiotics. Pharmaceuticals 6, 1055–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lei J, Sun L, Huang S, Zhu C, Li P, He J, Mackey V, Coy DH, and He Q (2019) The Antimicrobial Peptides and their Potential Clinical Applications. Am. J. Transl. Res 11, 3919–3931. [PMC free article] [PubMed] [Google Scholar]
- (11).Oren Z, Ramesh J, Avrahami D, Suryaprakash N, Shai Y, and Jelinek R (2002) Structures and Mode of Membrane Interaction of a Short Alpha Helical Lytic Peptide and its Diastereomer Determined by NMR, FTIR, and Fluorescence Spectroscopy. Eur. J. Biochem 269, 3869–3880. [DOI] [PubMed] [Google Scholar]
- (12).Zasloff M (2007) Antimicrobial Peptides, Innate Immunity, and the Normally Sterile Urinary Tract. J. Am. Soc. Nephrol 18, 2810–2816. [DOI] [PubMed] [Google Scholar]
- (13).Zasloff M, Martin B, and Chen HC (1988) Antimicrobial Activity of Synthetic Magainin Peptides and Several Analogues. Proc. Natl. Acad. Sci. U. S. A 85, 910–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hayouka Z, Mortenson DE, Kreitler DF, Weisblum B, Forest KT, and Gellman SH (2013) Evidence for Phenylalanine Zipper-Mediated Dimerization in the X-ray Crystal Structure of a Magainin 2 Analogue. J. Am. Chem. Soc 135, 15738–15741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhou J, Liu Y, Shen T, Chen L, Zhang C, Cai K, Liao C, and Wang C (2019) Antimicrobial Activity of the Antibacterial Peptide PMAP-36 and its Analogues. Microb. Pathog 136, 103712. [DOI] [PubMed] [Google Scholar]
- (16).Gorr SU, Flory CM, and Schumacher RJ (2019) In vivo Activity and Low Toxicity of the Second-Generation Antimicrobial Peptide DGL13K. PLoS One 14, e0216669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mourtada R, Herce HD, Yin DJ, Moroco JA, Wales TE, Engen JR, and Walensky LD (2019) Design of Stapled Antimicrobial Peptides that are Stable, Nontoxic and Kill Antibiotic-Resistant Bacteria in Mice. Nat. Biotechnol 37, 1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fjell CD, Hiss JA, Hancock RE, and Schneider G (2012) Designing Antimicrobial Peptides: Form Follows Function. Nat. Rev. Drug Discovery 11, 37–51. [DOI] [PubMed] [Google Scholar]
- (19).Li J, Koh JJ, Liu S, Lakshminarayanan R, Verma CS, and Beuerman RW (2017) Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci 11, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Greber KE, and Dawgul M (2016) Antimicrobial Peptides Under Clinical Trials. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 17, 620–628. [DOI] [PubMed] [Google Scholar]
- (21).Perron GG, Zasloff M, and Bell G (2006) Experimental Evolution of Resistance to an Antimicrobial Peptide. Proc. R. Soc. London, Ser. B 273, 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Epand RM (2016) Host Defense Peptides and Their Potential as Therapeutic Agents. Anticancer Res. 36, 4375. [PubMed] [Google Scholar]
- (23).Neuhaus FC, and Baddiley J (2003) A Continuum of Anionic Charge: Structures and Functions of D-Alanyl-Teichoic Acids in Gram-Positive Bacteria. Microbiol. Mol. Biol. Rev 67, 686–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, and Miller SI (2000) Genetic and Functional Analysis of a PmrA-PmrB-Regulated Locus Necessary for Lipopolysaccharide Modification, Antimicrobial Peptide Resistance, and Oral Virulence of Salmonella enterica Serovar Typhimurium. Infect. Immun 68, 6139–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dobson AJ, Purves J, Kamysz W, and Rolff J (2013) Comparing Selection on S. aureus Between Antimicrobial Peptides and Common Antibiotics. PLoS One 8, e76521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Amso Z, and Hayouka Z (2019) Antimicrobial Random Peptide Cocktails: A New Approach to Fight Pathogenic Bacteria. Chem. Commun. (Cambridge, U. K.) 55, 2007–2014. [DOI] [PubMed] [Google Scholar]
- (27).Stern T, Zelinger E, and Hayouka Z (2016) Random Peptide Mixtures Inhibit and Eradicate Methicillin-Resistant Staphylococcus aureus Biofilms. Chem. Commun. (Cambridge, U. K.) 52, 7102–7105. [DOI] [PubMed] [Google Scholar]
- (28).Topman S, Tamir-Ariel D, Bochnic-Tamir H, Stern Bauer T, Shafir S, Burdman S, and Hayouka Z (2018) Random Peptide Mixtures as New Crop Protection Agents. Microb. Biotechnol 11, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hayouka Z, Bella A, Stern T, Ray S, Jiang H, Grovenor C, and Ryadnov MG (2017) Binary Encoding of Random Peptide Sequences for Selective and Differential Antimicrobial Mechanisms. Angew. Chem., Int. Ed 56, 8099–8103. [DOI] [PubMed] [Google Scholar]
- (30).Choi S, Moon SM, Park SJ, Lee SC, Jung KH, Sung HS, Kim MN, Jung J, Kim MJ, Kim SH, Lee SO, Choi SH, Jeong JY, Woo JH, Kim YS, and Chong YP (2020) Antagonistic Effect of Colistin on Vancomycin Activity against Methicillin-Resistant Staphylococcus aureus in In Vitro and In Vivo Studies. Antimicrob. Agents Chemother 64, e01925–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Meylan S, Porter C, Yang JH, Belenky P, Gutierrez A, Lobritz MA, Park J, Kim SH, Moskowitz SM, and Collins JJ (2017) Carbon Sources Tune Antibiotic Susceptibility in Pseudomonas aeruginosa via Tricarboxylic Acid Cycle Control. Cell Chem. Biol 24, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).O’Toole GA (2011) Microtiter Dish Biofilm Formation Assay. J. Vis. Exp 47, e2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Barbier F, Andremont A, Wolff M, and Bouadma L (2013) Hospital-Acquired Pneumonia and Ventilator-Associated Pneumonia: Recent Advances in Epidemiology and Management. Curr. Opin. Pulm. Med 19, 216–228. [DOI] [PubMed] [Google Scholar]
- (34).Jean SS, Chang YC, Lin WC, Lee WS, Hsueh PR, and Hsu CW (2020) Epidemiology, Treatment, and Prevention of Nosocomial Bacterial Pneumonia. J. Clin. Med 9, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Döring G, Parameswaran IG, and Murphy TF (2011) Differential Adaptation of Microbial Pathogens to Airways of Patients with Cystic Fibrosis and Chronic Obstructive Pulmonary Disease. FEMS Microbiol. Rev 35, 124–146. [DOI] [PubMed] [Google Scholar]
- (36).Riquelme SA, Ahn D, and Prince A (2018) Pseudomonas aeruginosa and Klebsiella pneumoniae Adaptation to Innate Immune Clearance Mechanisms in the Lung. J. Innate Immun 10, 442–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mittal R, Lisi CV, Gerring R, Mittal J, Mathee K, Narasimhan G, Azad RK, Yao Q, Grati M, Yan D, Eshraghi AA, Angeli SI, Telischi FF, and Liu XZ (2015) Current Concepts in the Pathogenesis and Treatment of Chronic Suppurative Otitis Media. J. Med. Microbiol 64, 1103–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Fleiszig S, Kroken AR, Nieto V, Grosser MR, Wan SJ, Metruccio M, and Evans DJ (2020) Contact Lens-Related Corneal Infection: Intrinsic Resistance and its Compromise. Prog. Retinal Eye Res 76, 100804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hilliam Y, Kaye S, and Winstanley C (2020) Pseudomonas aeruginosa and Microbial Keratitis. J. Med. Microbiol 69, 3–13. [DOI] [PubMed] [Google Scholar]
- (40).Shupp JW, Pavlovich AR, Jeng JC, Pezzullo JC, Oetgen WJ, Jaskille AD, Jordan MH, and Shoham S (2010) Epidemiology of Bloodstream Infections in Burn-Injured Patients: A Review of the National Burn Repository. Journal of Burn Care and Research: Official Publication of the American Burn Association 31, 521–528. [DOI] [PubMed] [Google Scholar]
- (41).Estrada S, Lodise TP, Tillotson GS, and Delaportas D (2020) The Real-World Economic and Clinical Management of Adult Patients with Skin and Soft Tissue Infections (SSTIs) with Oritavancin: Data From Two Multicenter Observational Cohort Studies. Drugs - Real World Outcomes 7, 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Arciola CR, An YH, Campoccia D, Donati ME, and Montanaro L (2005) Etiology of Implant Orthopedic Infections: A Survey on 1027 Clinical Isolates. Int. J. Artif. Organs 28, 1091–1100. [DOI] [PubMed] [Google Scholar]
- (43).Masters EA, Trombetta RP, de Mesy Bentley KL, Boyce BF, Gill AL, Gill SR, Nishitani K, Ishikawa M, Morita Y, Ito H, Bello-Irizarry SN, Ninomiya M, Brodell JD Jr., Lee CC, Hao SP, Oh I, Xie C, Awad HA, Daiss JL, Owen JR, Kates SL, Schwarz EM, and Muthukrishnan G (2019) Evolving Concepts in Bone Infection: Redefining “Biofilm”, “Acute vs. Chronic Osteomyelitis”, “the Immune Proteome” and “Local Antibiotic Therapy”. Bone Res. 7, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Urish KL, and Cassat JE (2020) Staphylococcus aureus Osteomyelitis: Bone, Bugs, and Surgery. Infect. Immun 88, e00932–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kirby BD, Al Ahmar R, Withers TR, Valentine ME, Valentovic M, Long TE, Gaskins JR, and Yu HD (2019) Efficacy of Aerosolized Rifaximin versus Tobramycin for Treatment of Pseudomonas aeruginosa Pneumonia in Mice. Antimicrob. Agents Chemother 63, e02341–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lakshmaiah Narayana J, Mishra B, Lushnikova T, Wu Q, Chhonker YS, Zhang Y, Zarena D, Salnikov ES, Dang X, Wang F, Murphy C, Foster KW, Gorantla S, Bechinger B, Murry DJ, and Wang G (2020) Two distinct amphipathic peptide antibiotics with systemic efficacy. Proc. Natl. Acad. Sci. U. S. A 117, 19446–19454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Cirioni O, Wu G, Li L, Orlando F, Silvestri C, Ghiselli R, Shen Z, Gabrielli E, Brescini L, Lezoche G, Provinciali M, Guerrieri M, and Giacometti A (2011) S-thanatin in vitro prevents colistin resistance and improves its efficacy in an animal model of Pseudomonas aeruginosa sepsis. Peptides 32, 697–701. [DOI] [PubMed] [Google Scholar]
- (48).Hayouka Z, Chakraborty S, Liu R, Boersma MD, Weisblum B, and Gellman SH (2013) Interplay among subunit identity, subunit proportion, chain length, and stereochemistry in the activity profile of sequence-random peptide mixtures. J. Am. Chem. Soc 135, 11748–11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Topman-Rakover S, Malach E, Burdman S, and Hayouka Z (2020) Antibacterial lipo-random peptide mixtures exhibit high selectivity and synergistic interactions. Chem. Commun. (Cambridge, U. K.) 56, 12053–12056. [DOI] [PubMed] [Google Scholar]
- (50).Holloway BW, Krishnapillai V, and Morgan AF (1979) Chromosomal genetics of Pseudomonas. Microbiol. Rev 43, 73–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Zhang S, Chen Y, Potvin E, Sanschagrin F, Levesque RC, McCormack FX, and Lau GW (2005) Comparative signature-tagged mutagenesis identifies Pseudomonas aeruginosa factors conferring resistance to pulmonary collectin SP-A. PLoS Pathog. 1, e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Kuang Z, Hao Y, Hwang S, Zhang S, Kim E, Akinbi HT, Schurr MJ, Irvin RT, Hassett DJ, and Lau GW (2011) The Pseudomonas aeruginosa flagellum confers resistance to pulmonary surfactant protein-A by impacting the production of exoproteases through quorum-sensing. Mol. Microbiol 79, 1220–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Tan RM, Kuang Z, Hao Y, and Lau GW (2014) Type IV pilus of Pseudomonas aeruginosa confers resistance to antimicrobial activities of the pulmonary surfactant protein-A. J. Innate Immun 6, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Tan RM, Kuang Z, Hao Y, Lee F, Lee T, Lee RJ, and Lau GW (2015) Type IV pilus glycosylation mediates resistance of Pseudomonas aeruginosa to opsonic activities of the pulmonary surfactant protein A. Infect. Immun 83, 1339–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, and Gruenert DC (1994) CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol 10, 38–47. [DOI] [PubMed] [Google Scholar]
- (56).Hao Y, Kuang Z, Walling BE, Bhatia S, Sivaguru M, Chen Y, Gaskins HR, and Lau GW (2012) Pseudomonas aeruginosa pyocyanin causes airway goblet cell hyperplasia and metaplasia and mucus hypersecretion by inactivating the transcriptional factor FoxA2. Cell. Microbiol 14, 401–415. [DOI] [PubMed] [Google Scholar]
- (57).Hao Y, Kuang Z, Xu Y, Walling BE, and Lau GW (2013) Pyocyanin-induced mucin production is associated with redox modification of FOXA2. Respir. Res 14 (1), 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Vasuvada N (2016) One-way ANOVA (ANalysis Of VAriance) with Post-Hoc Tukey HSD (Honestly Significant Difference) Test Calculator for Comparing Multiple Treatments. https://astatsa.com/OneWay_Anova_with_TukeyHSD/ (accessed October 29, 2020).