

Abstract
Changes in the levels and compositions of N-glycans released from serum and plasma glycoproteins have been assessed in many diseases across many large clinical sample cohorts. Assays used for N-glycan profiling in these fluids currently require multiple processing steps and have limited throughput, thus diminishing their potential for use as standard clinical diagnostic assays. A novel slide-based N-glycan profiling method was evaluated for sensitivity and reproducibility using a pooled serum standard. Serum was spotted on to an amine-reactive slide, delipidated and desalted with a series of washes, sprayed with peptide N-glycosidase F and matrix, and analyzed by MALDI-FTICR or MALDI-Q-TOF mass spectrometry. Routinely, over 75 N-glycan species can be detected from one microliter of serum in less than 6.5 h. Additionally, endoglycosidase F3 was applied to this workflow to identify core-fucosylated N-glycans and displayed the adaptability of this method for the determination of structural information. This method was applied to a small pooled serum set from either obese or nonobese patients that had breast cancer or a benign lesion. This study confirms the reproducibility, sensitivity, and adaptability of a novel method for N-glycan profiling of serum and plasma for potential application to clinical diagnostics.
Keywords: N-glycan, glycosylation, serum, plasma, imaging mass spectrometry, MALDI
Graphical Abstract
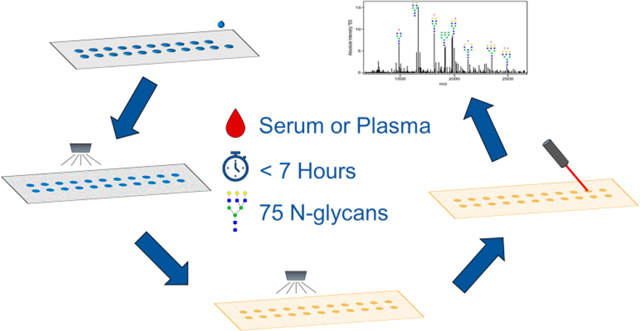
INTRODUCTION
The composition of serum and plasma collected during blood draws reflects dynamic decellularized biofluids composed of thousands of proteins and metabolites. A major component of the fluids is the multiple high-abundance glycoproteins produced by the liver and immunoglobulins secreted by B-cells. Extensive characterization of the proteomes of serum and plasma has indicated a 10-fold order of magnitude concentration difference from the most abundant proteins, i.e., albumin and immunoglobulins, to the lowest concentration ones.1,2 Current proteomic mass spectrometry studies have identified over 4300 individual proteins present in serum,3 yet by percentage of abundance, 22 proteins represent 99% of the content.1,2 Except for albumin, which is not a glycoprotein and accounts for approximately 50% of the total protein content, the majority of the 22 most abundant proteins and next 30 are glycoproteins secreted by the liver and B-cells. Interrogation of the glycan composition of these most abundant glycoproteins for potential clinical application to differentiate healthy from disease states has been ongoing for decades.4−6 In addition to advanced high-performance liquid chromatrography (HPLC)-based methods,7 in recent years, multiple high-resolution tandem liquid chromatography (LC) and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) options have been utilized for these types of analyses.8,9
Glycosylation is one of the most common types of posttranslational modifications that is estimated to occur on over 50% of proteins.10 The diversity of monosaccharides, intersaccharide bonding, and nontemplate derived branching structures creates a large and complex glycan repertoire called the glycome.11 Glycosylation is a highly regulated metabolic process shown to have essential roles in protein folding, molecular trafficking, signal transduction, and many other processes.12,13 The most common forms of glycosylation studied for serum or plasma glycoproteins are for those that are O-linked glycans, attached to proteins via threonine/serine residues, and N-linked glycans, attached to proteins via an asparagine residue. The majority of the most abundant serum and plasma glycoproteins have N-linked glycosylation. Investigating the changes in specific glycans or glycan structural motifs, such as branching or fucosylation, is a promising area for biomarker identification, and the minimally invasive and cost-effective properties of plasma and serum collection makes the total serum or plasma N-glycome an attractive area of focus.4,5 The large cohorts amenable for biomarker discovery and potential clinical applications require high-throughput methodologies that continue to be assessed and evolve.14
The currently used methods for high-throughput serum N-glycome analysis are hydrophilic interaction chromatography (ultra) high-performance liquid chromatography with fluorescence detection (HILIC-UHPLC-FLD),15 multiplexed capillary gel electrophoresis with laser-induced fluorescence detection (xCGE-LIF),16 and matrix-assisted laser desorption/ ionization time-of-flight mass spectrometry (MALDI-TOF-MS).17 While these methods have clear utility in understanding the role of serum N-glycans in a variety of diseases, there are elements of each that are incompatible with routine use in a clinical laboratory. Each of these methods requires labeling or derivatization of released glycans and further processing and purification steps. Not only do these steps involve specialized materials and expertise but also they require additional time, as analysis of 96 samples with these methods can take from 1 to 5 days.16−18
Our collective group has developed methods for analyzing released N-glycans from tissues, cells, and immuno-captured serum glycoproteins by MALDI imaging mass spectrometry (MALDI-IMS).19−24 The key to each of these methods is the spraying of a molecular coating of peptide N-glycosidase F (PNGase F) onto a slide surface containing the biological sample, followed by the detection of released N-glycans by MALDI-IMS. We hypothesized that we could adapt techniques from these workflows to create a rapid and sensitive serum N-glycan profiling method. The sensitivity and repeatability of this method were demonstrated using different control and diseased serum samples, as well as plasma. An additional enzyme, endoglycosidase F3 (Endo F3),25 was used to distinguish core fucose from outer arm fucose locations on the glycan structures. Finally, the utility of this method was demonstrated by analyzing a set of pooled serum samples from patients with either breast cancer or a benign lesion in their breast. The clinical applicability of this serum and plasma N-glycan profiling method improves the potential of glycans to be used as biomarkers. The speed and simplicity of the novel N-glycan profiling method described in this study are compatible with the requirements of clinical laboratories.
METHODS
Materials.
Hydrogel-coated slides (Nexterion Slide H) were obtained from Applied Microarrays (Tempe, AZ). The rotary tool was a Dremel 200 series. The well slide module (ProPlate Multi-Array Slide System, 64-well) was obtained from Grace Bio-Laboratories (Bend, OR). Sodium bicarbonate, trifluoroacetic acid, and α-cyano-4-hydroxycinnamic acid were obtained from Sigma-Aldrich (St. Louis, MO). HPLC grade water, acetonitrile, glacial acetic acid, and chloroform were obtained from Fisher Scientific (Hampton, NH). Ethanol was obtained from Decon Laboratories (King of Prussia, PA). Peptide-N-glycosidase F (PNGase F) PRIME was from N-Zyme Scientifics (Doylestown, PA). Endoglycosidase F3 (Endo F3) was cloned, expressed, and purified in-house as previously described.25
Samples.
A pooled human serum sample was used that represented over 360 healthy human donors.26 A control plasma-EDTA sample was obtained from a healthy donor. Serum from nonobese or obese patients with benign breast lesions or breast cancer was pooled from 5−10 patients, derived from a previously published study.27
Array Preparation.
Hydrogel-coated slides were used, and wells were created by attaching a 64 (4 × 16)-well module. The hydrogel-coated slides were equilibrated to room temperature in a moisture resistant pouch for 45 min. The slide was ground down with a rotary tool until it fit into a Bruker MTP Slide Adapter II. The well module was attached to the slide, the wells were outlined with a marker on the uncoated side of the slide, and then the well module was unattached.
Sample Capture and Washing.
A microliter of serum was diluted in 2 μL of sodium bicarbonate (100 mM, pH 8.0). After briefly mixing with the pipet, 1 μL was spotted within the outline of a well. Only wells from the two innermost rows were used. The pooled serum standard and plasma standard were spotted in quadruplicate, and the clinical serum samples were spotted in triplicate. Spots were left to immobilize to the slide at room temperature for 1 h in a humidity chamber made from a culture dish with a Wypall X 60 paper towel lining the bottom and two rolled KimWipes saturated with distilled water on opposite sides. For the immobilization, the slide was placed flat in the dish in between the two KimWipes. The slide was then dried in a desiccator for 15 min, and the well module was attached. Each well was filled with 50 μL of Carnoy’s solution (10% glacial acetic acid, 30% chloroform, and 60% 200 proof ethanol) without mixing or agitating for 3 min. The Carnoy’s solution was dumped out of the well, and the wash was repeated two more times. After the three Carnoy’s solution washes, each well was rinsed with 50 μL of double distilled water without mixing or agitating for 1 min. Following the removal of water, the slide with the well module attached was dried in a desiccator for 30 min.
N-Glycan Release and Matrix Application.
To release the N-glycans from the immobilized proteins, the well module was removed and a M5 TM-Sprayer (HTX Technologies) was used to spray a 0.1 mg/mL PNGase F PRIME solution in water with 15 passes at 25 μL/min, 1200 mm/min, 45 °C, 3 mm spacing between passes with 10 psi nitrogen gas. The slide was incubated for 2 h at 37 °C in a preheated humidity chamber. To apply the MALDI matrix α-cyano-4-hydroxycinnamic acid (CHCA, 7 mg/mL in 50% acetonitrile/0.1% trifluoracetic acid), the same automated sprayer was used with 10 passes at 100 μL/min, 1200 mm/min, 79 °C, 2.5 mm spacing between passes with 10 psi nitrogen gas. For Endo F3 treatment, a mix of 10% Endo F3 and 90% PNGase F Prime was sprayed at the same settings as those described for only PNGase F and all other steps remained the same.22,25
MALDI Imaging Mass Spectrometry.
A SolariX Legacy 7T Fourier-transform ion cyclotron resonance (FTICR) mass spectrometer (Bruker) equipped with a MALDI source was used to image the slides. Images were collected with a Smart-Beam II laser operating at 2000 Hz with a 25 μm laser spot size and a smartwalk pattern at a 250 μm raster with 200 laser shots per pixel. Samples were analyzed in positive ion, broadband mode using a 512k word time domain spanning a m/z range of 500−5000. The individual regions to be imaged were manually outlined to encompass each well with a sample, as well as one blank well that did not have any sample spotted in it. An on-slide resolving power of 48 000 at m/z = 1136 was calculated.
Data Analysis.
Data was imported at a 0.95 ICR reduction noise threshold and normalized to total ion current in to FlexImaging v5.0 (Bruker) for manual N-glycan peak selection on the basis of theoretical mass values. Data Analysis 5.0 (Bruker) was used for spectra recalibration with a linear function based on N-glycan theoretical masses. Spectra were imported into SCiLS Lab software 2017a (Bruker) for individual peak visualization and quantification. Each well was designated a unique region, and the area under monoisotopic peak values were exported from each well. Individual N-glycan area under monoisotopic peak values were divided by the sum of the area under monoisotopic peak values of all detected N-glycans for relative intensity values. Quantifications of N-glycan structural classes were calculated by summing the relative intensities of the individual N-glycans belonging to each class, as determined by the putative structures. Each slide contained nonsample wells processed in the same manner as wells with samples and can be used for background signal subtraction in subsequent data analysis steps.
MS/MS.
A timsTOF fleX mass spectrometer (Bruker) was used to acquire MS/MS data by collision-induced dissociation (CID) for N-glycans directly from the processed slides. A 2 Da window was used for MS/MS precursor selection. The collision energies were individually optimized for consistent and sensitive fragmentation and ranged from 100 to 140 eV. The number of laser shots summed were also optimized per N-glycan for fragmentation reporting. Detected glycan species were cross-referenced with existing in-house structural databases from previous MALDI-FTICR-MS studies.25,28,29
RESULTS
Serum N-Glycan Profiling Workflow.
This study was initiated to identify a serum and plasma glycan profiling strategy on the basis of direct spotting of sample on glass slides, spraying of PNGase F to release N-glycans, followed by detection using MALDI-IMS. The goal was to develop a protocol that was both rapid (relative to current methods used) and reproducible. Analysis workflows for processing tissues on slides for N-glycan MALDI-IMS analysis using sprayed PNGase F to release N-glycans were established and used in this study.22 It was the initial sample spotting and processing steps prior to PNGase F spraying that required extensive evaluation and optimization. Parameters like slide chemistry, washing and sample buffers, sample amount, and digestion conditions were assessed. Peak intensities and numbers of N-glycans detected by MALDI-FTICR-MS were the final determinants.
Initially, multiple slide chemistries were evaluated for serum spotting, including blank glass histology slides and different modifications like indium tin oxide (ITO), polylysine, and nitrocellulose. Because MALDI was being used for detection, a desalting component was needed. A denaturing component was also required in the sample preparation due to the high concentration of protein present in even 1 μL of human serum (∼75 μg) and the complexities of releasing N-glycans from native proteins. Each of the indicated slide chemistries had issues with sample loss during the washing and denaturing steps, leading to inconsistent or loss of N-glycan detection across replicates (data not shown). Attempts to fixate the samples to the slides using formalin cross-linking resulted in sample spot hardening and flaking off of the slide after washing. To address these limitations, amine-reactive slides were assessed, which provided covalent attachment of the serum proteins, facilitating efficient desalting and denaturation steps. These slides were used throughout the study, as summarized in the workflow schematic shown in Figure 1.
Figure 1.

Workflow for slide-based serum and plasma N-glycan profiling by MALDI IMS. (A) Sample preparation steps include diluting the serum or plasma 1:2 in sodium bicarbonate (100 mM, p.H. 8.0) and spotting 1 μL onto a hydrogel-coated slide. The sample spots are put in a humidity chamber for an hour to immobilize to the slide. Carnoy’s solution is used to delipidate the samples for sensitive N-glycan detection. (B) Sample spots are sprayed with PNGase F to enzymatically release N-glycans, and CHCA matrix is applied. (C) Sample spots are imaged in distinct regions by a MALDI-FTICR-MS, allowing for images and abundances of m/z peaks corresponding to N-glycans to be compared across samples.
The amine-reactive hydrogel slides used contain N-hydroxysuccinimide (NHS) esters free to covalently react with primary amines. Serum samples were mixed with 100 mM sodium bicarbonate at pH 8 to dilute the sample for more efficient capture on the hydrogel-coated slide. After 1 h incubation, the bound proteins on the slide can be washed with minimal loss of sample. Carnoy’s solution (10% glacial acetic acid, 30% chloroform, and 60% ethanol) was used to delipidate and denature the proteins, and water washes were done to remove residual Carnoy’s solution and salts. PNGase F was sprayed evenly over the slide, and after 2 h of incubation, a CHCA matrix coating was sprayed on the slide. The slide was analyzed using MALDI-FTICR MS, resulting in an overall mass spectrum that could be used to obtain heat map images correlating to the intensities of specific m/z peaks across the slide. The intensities of the detected N-glycans can be quantified for areas covering the wells of specific samples, allowing for comparisons of N-glycans between samples.
The current workflow described here (Figure 1) will analyze 28 spots per slide, and additional slides can be prepared together up to the point of the MALDI IMS with little additional time. The total time to prepare a slide for imaging is 6 h. Each spot can be imaged in 10 min with the given parameters. The entire workflow can be used to analyze a single spot in less than 6.5 h and 28 spots in less than 11 h. To define the breadth of N-glycans detected, a serum and plasma standard was spotted in quadruplicate, processed, and analyzed. Using this method, 75 N-glycans were detected in human serum (Supporting Information Table S1) with an average coefficient of variation (CV) of 6% for the 20 most abundant glycans. For sialylated N-glycans, predominant peaks detected in formalin-fixed paraffin embedded (FFPE) tissues have an extra Na ion per each sialic acid present.20 This serum and plasma protocol results in the detection of many single sodiated sialylated glycans, in addition to the extra Na adduct glycoforms. This is also seen in frozen tissue analyses of N-glycans21 and is likely related to the Carnoy’s and water washes not done with FFPE tissues. An equivalent number of N-glycans were detected in plasma, and the CV values for the top 20 most abundant N-glycans and structural classes were 9% and 5%, respectively. The average mass error for all detected glycans was 0.01 ± 0.01 Da. A representative spectrum obtained from the serum standard and plasma standard is shown in Figure 2.
Figure 2.

Representative spectra of a standard serum sample (top) and a standard plasma sample (bottom) analyzed with the N-glycan profiling method by MALDI-FTICR MS. A sample of detected N-glycans are annotated with putative structures. The experimental layout of the slide analyzed is displayed in the bottom right. The N-glycan compositions are represented by blue squares for N-acetylglucosamine, green circles for mannose, yellow circles for galactose, purple diamonds for sialic acid, and red triangles for fucose.
Repeatability Testing.
To evaluate the repeatability and day-to-day variation of this workflow, a human serum standard and human plasma standard were spotted and processed using the optimized workflow and the workflow was repeated on the next two successive days (days 1−3). Figure 3 displays the area of the 20 most abundant glycan compositions in serum using the area under the peak intensities compared to the total sum of peak intensity values for all of the detected glycan peaks. The most abundant glycan peaks included species across a broad m/z range (m/z = 1136−3071). For sialylated glycan species that had multiple sodiated adducts detected, only one species was included in Figure 3. These glycans showed little to moderate variability across the experiments, with the CV averaging to 11.8%, with some peaks having much lower variation. If the glycans were grouped based on structural features, this improved repeatability across the 3 days (CV = 5%). This type of grouping and improvement in variability has been previously reported for MALDI serum glycan analysis.14 The average relative intensity of the most abundant unique N-glycan peak (m/z = 1976.6666, Hex5HexNAc4NeuAc1 + 2Na) across all experiments was 10% (SD ± 1%). The results of the plasma standard showed larger variability within the repeatability parameters (16% and 8% CV for the 20 most abundant unique N-glycan peaks and N-glycan structural classes, respectively) (Supporting Information Figure S1). Further optimization studies for plasma and the different anticoagulants (EDTA, heparin, citrate) used clinically are still ongoing.
Figure 3.

Repeatability of the N-glycan profiling method. The same serum standard sample was analyzed by MALDI-FTICR MS on three successive days with a blank well that contained no serum on each run. (A) Graph shows the average relative intensities detected for the 20 most abundant N-glycans (normalized to the overall sum of N-glycan intensities) with putative structures. For determining the most abundant N-glycans, only N-glycans with no overlap with another N-glycan peak isotope were included and only one species was selected for sialylated N-glycans with multiple sodiated adducts. Structural classes were assigned to the N-glycans, and the sum of the N-glycans for the (B) high-abundance and (C) low-abundance classes are displayed. Error bars indicate the standard deviation of the four replicates.
PNGase F and Endo F3 Mixture Treatment for Fucose Isomers.
A mixture of Endo F3 and PNGase F was applied to the serum and plasma samples. Endo F3 is a glycosidase that cleaves N-glycans between the first two N-acetylglucosamines (GlcNAc) linked to asparagine, preferentially when the first GlcNAc is fucosylated. It can be used to differentiate N-glycans containing the core fucose versus glycans with fucose modifications on the antennae.25 Additionally, the m/z’s of Endo F3 cleaved N-glycans products can be calculated on the basis of the loss of 349 m.u. and confirmed by MS/MS. Use of the Endo F3/PNGase F mix allowed for comparison of the data already obtained for fucosylated N-glycans using PNGase F alone. This modification requires no increase in processing time and no changes to the MALDI IMS parameters.
As shown in Figure 4A, multiple fucosylated N-glycans from serum were decreased in abundance in the Endo F3/PNGase F treatment compared to that of the PNGase F only treatment. The Endo F3/PNGase F treatment did not affect the abundances of nonfucosylated glycans in serum and plasma (Figure 4B). While some N-glycans could be readily identified as core fucosylated using this method (Figure 4C,D), the identification of several other N-glycan fucosylation locations were obscured by several different factors. Various Endo F3 products have the m/z of a fucosylated N-glycan, making it difficult to attribute the signal intensity to either N-glycan. Other Endo F3 products that did not have an overlapping m/z with another N-glycan were not detected, but their parent N-glycan abundance did decrease. Collision-induced dissociation MS/MS was also used to ascertain structural information, with representative data examples provided in the Supporting Information Figure S2.
Figure 4.

Endo F3 modification to the workflow. (A) Representative spectrum for a serum standard with annotations for a set of putative N-glycan structures containing a fucose on the first core N-acetylglucosamine. The top half of the spectrum is PNGase F treated only. The bottom half of the spectrum is Endo F3 and PNGase F treated. (B−D) N-glycan profiles of a serum standard and a plasma standard were analyzed by MALDI-FTICR MS in quadruplicate by a treatment with PNGase F or a PNGase F and Endo F3 mix. The intensities of a noncore fucosylated N-glycan (m/z 2289.7326), core-fucosylated N-glycan (m/z 1809.6393), and Endo F3 product (m/z 1460.5020), respectively, are displayed for both treatments. The N-glycan compositions are represented by blue squares for N-acetylglucosamine, green circles for mannose, yellow circles for galactose, purple diamonds for sialic acid, and red triangles for fucose.
N-Glycan Profiling of Clinical Serum Sets.
The potential clinical application of this method was assessed by analyzing a small set of pooled serum samples from nonobese or obese patients with benign breast lesions or breast cancer.27 Most of the N-glycans that were detected in the pooled serum standard were also present in these samples, and there were N-glycans that were only found in the samples and not in the serum standard (marked with an asterisk in the Supporting Information Table S1). While major differences were not detected across groups for most of the detected N-glycans, some representative examples of differential detection per clinical group are shown in Figure 5. A higher abundance in a core fucosylated, nongalactosylated N-glycan at m/z = 1485.5337 (Hex3dHex1HexNAc4 + 1Na) was observed in the cancerous obese serum (Figure 5A). A monogalactosylated N-glycan m/z = 1501.5286 (Hex4HexNAc4 + 1Na) had a higher abundance in both nonobese samples (Figure 5B). The nonobese and obese benign samples also had higher amounts of the highly abundant fucosylated biantennary N-glycan m/z = 1809.6393 (Hex5dHex1HexNAc4 + 1Na) (Figure 5C). The benign obese serum had a lower abundance of the presumably bisected N-glycan m/z = 1866.6608 (Hex5HexNAc5 + 1Na) (Figure 5D). These N-glycans vary largely in their average relative intensities, with m/z = 1809.6393 accounting for 5.1% of the glycan signal and m/z = 1866.6608 accounting for 0.4%. The N-glycans detected in the clinical serum set had a similar level of variability as those found in the serum standards. The top 20 most abundant N-glycans averaged a CV of 8% (data not shown).
Figure 5.

Application of the workflow to a breast cancer clinical serum set. N-glycan profiles of pooled serum samples from obese and nonobese patients that had a benign lesion or breast cancer were analyzed in quadruplicate by MALDI-FTICR MS. The average relative intensities (normalized to the overall sum of N-glycan intensities) of (A) m/z 1485.5337, (B) m/z 1501.5286, (C) m/z 1809.6393, and (D) m/z 1866.6608 N-glycans are shown as a heat map image array with a color scale optimized for brightness and a bar chart with error bars displaying the standard deviation for the four replicates. The relative intensities of the blank wells were used for background signal subtraction. N-glycan compositions are represented by blue squares for N-acetylglucosamine, green circles for mannose, yellow circles for galactose, purple diamonds for sialic acid, and red triangles for fucose. BN = nonobese patient with a benign lesion, BO = obese patient with a benign lesion, CN = nonobese patient with breast cancer, CO = obese patient with breast cancer.
DISCUSSION
In this study, a comparatively rapid and reproducible slidebased method for N-glycan profiling analysis of serum and plasma N-glycan profiling was presented. The approach possesses the advantages of related glycan profiling protocols for tissues, cells, and antibody capture arrays that our collective group have previously reported,22−24 in that it requires minimal sample processing and no derivatization or purification of PNGase F released glycans. Using only 1−2 μL of serum or plasma, the sensitivity of this method allows for detection of a broad set of N-glycans, comparable to other N-glycan profiling methods, as well as single day analysis timelines. By repeating this workflow using the same samples on three successive days, the variation of glycan intensities dayto-day was shown to be consistent with the reported technical variation of MALDI MS applied to serum.14 As was also reported,14 the use of MALDI facilitates rapid analysis of structural class groupings of N-glycans, as shown in Figure 3. The current study was focused more on establishing the method; however, the use of a small cohort of clinical breast cancer samples was evaluated for proof-of-concept applications (Figure 5). Previous studies have examined the change of the total serum N-glycome in breast cancer patients and identified N-glycans and N-glycan structural motifs that are associated with the presence of cancer, metastasis, and cancer stages.30−35 Of the glycans detected in our pooled breast cancer samples, only a small set of glycans were found to have differing levels of abundance in one or more samples (Figure 5). While statistical analysis for clinical application is not possible with these samples, both the increase of the N-glycan m/z = 1485.5337 and decrease of the N-glycan m/z = 1809.6393 have been found in previous studies.30,33,36 Larger cohort studies are ongoing to better identify potential serum and plasma glycan biomarker candidates related to cancer and other diseases.
The amine-reactive hydrogels that were used offered several advantages compared to other slide chemistries tested. For example, the most direct approach would be to spot serum on a glass histology slide and apply PNGase F and matrix without any other processing steps. This was done in multiple iterations, and it resulted in no glycan signal being detected by MALDI-FTICR MS. Two factors are critical: First, the salts and ions present in serum and plasma lead to ion suppression. Second, the proteins are concentrated and nondenatured, limiting efficient access to PNGase F. The use of the aminereactive gel slide provided covalent attachment of the serum and plasma proteins, which allowed for denaturing and delipidating with Carnoy’s solution, followed by the removal of salts and ions with water washes. This covalent attachment also allows for any plasma formulation to be used. Plasma with EDTA and citrate anticoagulants has been successfully used with this workflow. Plasma with heparin anticoagulant has not yet been evaluated, but it should work like the other plasma samples or, due to its highly charged sulfation characteristics, be readily adaptable to additional wash step strategies. Similarly, the workflow could serve as a starting point for analysis of N-glycans in other types of nonblood derived biofluids. This report focused specifically on use of glass slides, but efforts are ongoing to adapt the workflow to standard MALDI steel plates. It should also be noted that only a CHCA matrix was used for analyses in positive ion mode. It is certainly feasible that other matrix compounds could be evaluated, as well as use of this method in negative ion mode.
One of the goals in the method development was to simplify the sample processing steps and thereby improve the timelines of analysis. There are multiple steps in the current protocol workflow that could be further adjusted to both improve reproducibility and decrease time of analysis. The slide-based approach for sample spotting and washing could easily be adapted to sample spotting platforms and automated liquid handling systems. This could potentially decrease sample spotting and processing time, increase numbers of samples analyzed per slide, and lower CV values. While this was not reported herein, the PNGase F PRIME eN-Zyme (N-Zyme Scientifics) is stable at temperatures up to 50 °C and results in shorter reaction times. This could decrease at least 60−90 min in the processing timeline and is under evaluation. Additionally, the MALDI-FTICR-MS analysis reported herein used a larger sample and slide area for analysis as part of the method optimization steps. As mentioned, automated sample spotting and robotic sample handling would integrate well at this step. Currently, the mass spectrometry detection aspects of the protocol are being assessed on the timsTOF fleX mass spectrometer (Bruker). Even using the current larger sample areas, this instrument will reduce the time for data acquisition to 30 min or less per slide. Overall, the time elapsed from sample preparation to data analysis is very quick compared to those of other clinical biofluid N-glycan profiling methods.14 Using the aforementioned strategies, we expect that the current workflow can be adapted to a 4−5 h analysis time frame from start to finish per slide.
The use of MALDI for this analysis comes with multiple limitations relative to other analytical techniques, as highlighted in the extensive multilaboratory evaluation of HPLC, capillary electrophoresis, and MALDI mass spectrometry evaluations of the same clinical serum cohort.14 In particular, MALDI MS is limited in determining the many structural isomers associated with fucosylation and sialylation, and stability of sialic acids on N-glycans is a well described issue. High-vacuum MALDI and localized heat of ionization contribute to sialic acid lability for N-glycans. The MALDIFTICR-MS used herein has a cooling gas in the source that mitigates some of this loss,19−21 and chemical amidation strategies have been reported that stabilize sialylated N-glycans and provide isomeric linkage information.14,37,38 Adapting these amidation reactions conditions to the slide-based workflow is currently ongoing. The application of a mixture of Endo F3 and PNGase F to serum and plasma (shown in Figure 4) represents an initial demonstration of feasibility for differentiating N-glycan fucose isomers within the protocol. Comparing data obtained using Endo F3 alone in comparison to the eN-Zyme mixture is in progress. Another modification to this workflow that can significantly increase the data acquired is utilizing an instrument like the timsTOF fleX (Bruker) that has ion mobility capabilities. Determining the collision cross section of the N-glycans is another dimension of data that could be used to confirm structure annotations and determine linkages, especially for distinguishing isomeric species.
CONCLUSIONS
Historically, the use of MALDI mass spectrometry for clinical diagnostic applications is frequently criticized for issues of reproducibility and laser performance over time. It should be noted that in the past decade use of MALDI instruments has completely transformed clinical microbiology laboratories and clinical diagnostic workflows.39−41 The key to those methods is the signature analyte barcodes used to identify individual bacterial genus species and other microorganisms. Especially for the most abundant N-glycan signatures detected in serum and plasma, there is potential to establish N-glycan barcode signatures reflective of healthy and disease states. Feasibility has already been established in principle for N-glycan analysis of immunoglobulin G in serum and plasma. We expect that the continued refinement of sample processing steps, incorporation of automation, and additional glycan characterization strategies could add more dimensions to the acquired data and allow for more thorough analysis with little additional time requirements. Overall, the simplification of the processing workflows and speed of detection present in our protocol is a step toward bringing routine N-glycan analysis closer to clinical diagnostic applications.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the following grants: U01 CA242096 (RRD, ASM, PMA); R01 CA212409 (RRD); Department of Defense PC160851 (RRD); U54 MD010706 (RRD, ASM); U01 CA226052 (ASM, RRD); R21 CA225474 (ASM); the NIH National Center for Advancing Translational Sciences (NCATS) through grant numbers TL1 TR001451 and UL1 TR001450 (APB).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.0c00213.
Table of peak list of N-glycans detected in serum and plasma and figures of repeatability of the N-glycan profiling method for plasma and MS/MS spectra (PDF)
The authors declare no competing financial interest.
Contributor Information
Calvin R. K. Blaschke, Department of Cell and Molecular Pharmacology, Medical University of South Carolina, Charleston 29425, South Carolina, United States.
Alyson P. Black, Department of Cell and Molecular Pharmacology, Medical University of South Carolina, Charleston 29425, South Carolina, United States.
Anand S. Mehta, Department of Cell and Molecular Pharmacology, Medical University of South Carolina, Charleston 29425, South Carolina, United States.
Peggi M. Angel, Department of Cell and Molecular Pharmacology, Medical University of South Carolina, Charleston 29425, South Carolina, United States.
Richard R. Drake, Department of Cell and Molecular Pharmacology, Medical University of South Carolina, Charleston 29425, South Carolina, United States
REFERENCES
- (1).Anderson NL; Anderson NG The Human Plasma Proteome: History, Character, and Diagnostic Prospects. Mol. Cell. Proteomics 2002, 1 (11), 845–867. [DOI] [PubMed] [Google Scholar]
- (2).Qian WJ; Kaleta DT; Petritis BO; Jiang H; Liu T; Zhang X; Mottaz HM; Varnum SM; Camp DG; Huang L; Fang X; Zhang WW; Smith RD Enhanced Detection of Low Abundance Human Plasma Proteins Using a Tandem IgY12-SuperMixImmunoaffinity Separation Strategy. Mol. Cell. Proteomics 2008, 7 (10), 1963–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dey KK; Wang H; Niu M; Bai B; Wang X; Li Y; Cho JH; Tan H; Mishra A; High AA; Chen PC; Wu Z; Beach TG; Peng J. Deep Undepleted Human Serum Proteome Profiling toward Biomarker Discovery for Alzheimer’s Disease. Clin. Proteomics 2019, 16 (1), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Adamczyk B; Tharmalingam T; Rudd PM Glycans as Cancer Biomarkers. Biochim. Biophys. Acta, Gen. Subj. 2012, 1820, 1347–1353. [DOI] [PubMed] [Google Scholar]
- (5).Kailemia MJ; Park D; Lebrilla CB Glycans and Glycoproteins as Specific Biomarkers for Cancer. Anal. Bioanal. Chem. 2017, 409 (2), 395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Dotz V; Wuhrer M N -glycome Signatures in Human Plasma: Associations with Physiology and Major Diseases. FEBS Lett. 2019, 593, 1–11. [DOI] [PubMed] [Google Scholar]
- (7).Adamczyk B; Stöckmann H; O’Flaherty R; Karlsson NG; Rudd PM High-Throughput Analysis of the Plasma N-Glycome by UHPLC; Humana Press: New York, NY, 2017; pp 97–108. [DOI] [PubMed] [Google Scholar]
- (8).Ruhaak LR; Xu G; Li Q; Goonatilleke E; Lebrilla CB Mass Spectrometry Approaches to Glycomic and Glycoproteomic Analyses. Chem. Rev. 2018, 118 (17), 7886–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Suttapitugsakul S; Sun F; Wu R. Recent Advances in Glycoproteomic Analysis by Mass Spectrometry. Anal. Chem. 2020, 92 (1), 267–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Apweiler R; Hermjakob H; Sharon N. On the Frequency of Protein Glycosylation, as Deduced from Analysis of the SWISS-PROT Database. Biochim. Biophys. Acta, Gen. Subj. 1999, 1473 (1), 4–8. [DOI] [PubMed] [Google Scholar]
- (11).Lauc G; Pezer M; Rudan I; Campbell H. Mechanisms of Disease: The Human N-Glycome. Biochim. Biophys. Acta, Gen. Subj. 2016, 1860 (8), 1574–1582. [DOI] [PubMed] [Google Scholar]
- (12).Moremen KW; Tiemeyer M; Nairn AV Vertebrate Protein Glycosylation: Diversity, Synthesis and Function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tanaka T; Yoneyama T; Noro D; Imanishi K; Kojima Y; Hatakeyama S; Tobisawa Y; Mori K; Yamamoto H; Imai A; Yoneyama T; Hashimoto Y; Koie T; Tanaka M; Nishimura SI; Kurauchi S; Takahashi I; Ohyama C. Aberrant N-Glycosylation Profile of Serum Immunoglobulins Is a Diagnostic Biomarker of Urothelial Carcinomas. Int. J. Mol. Sci. 2017, 18 (12), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Reiding KR; Bondt A; Hennig R; Gardner RA; O’Flaherty R; Trbojevic-´ Akmacič I; Shubhakar A; Wuhrer M;́ Lauc G; Rudd PM High-Throughput Serum N-Glycomics: Method Comparison and Application to Study Rheumatoid Arthritis and Pregnancy-Associated Changes. Mol. Cell. Proteomics 2019, 18, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).ckmann H; O’Flaherty R; Adamczyk B; Saldova R; Rudd PM Automated, High-Throughput Serum Glycoprofiling Platform. Integr. Biol. 2015, 7 (9), 1026–1032. [DOI] [PubMed] [Google Scholar]
- (16).Ruhaak LR; Hennig R; Huhn C; Borowiak M; Dolhain RJEM; Deelder AM; Rapp E; Wuhrer M. Optimized Workflow for Preparation of APTS-Labeled N-Glycans Allowing High Analysis of Human Plasma Glycomes Using 48-Channel Multiplexed CGE-LIF. J. Proteome Res. 2010, 9 (12), 6655–6664. [DOI] [PubMed] [Google Scholar]
- (17).Vreeker GCM; Nicolardi S; Bladergroen MR; van der Plas CJ; Mesker WE; Tollenaar RAEM; van der Burgt YEM; Wuhrer M. Automated Plasma Glycomics with Linkage-Specific Sialic Acid Esterification and Ultrahigh Resolution MS. Anal. Chem. 2018, 90, 11955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hanic M; Lauc G; Trbojević - Akmacič Í.N-Glycan Analysis by Ultra-Performance Liquid Chromatography and Capillary Gel Electrophoresis with Fluorescent Labeling. Curr. Protoc. Protein Sci 2019, 97 (1), No. e95. [DOI] [PubMed] [Google Scholar]
- (19).Powers TW; Neely BA; Shao Y; Tang H; Troyer DA; Mehta AS; Haab BB; Drake RR MALDI Imaging Mass Spectrometry Profiling of N-Glycans in Formalin-Fixed Paraffin Embedded Clinical Tissue Blocks and Tissue Microarrays. PLoS One 2014, 9 (9), e106255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Drake RR; Powers TW; Jones EE; Bruner E; Mehta AS; Angel PM MALDI Mass Spectrometry Imaging of N-Linked Glycans in Cancer Tissues. Adv. Cancer Res 2017, 134, 85–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Powers TW; Jones EE; Betesh LR; Romano PR; Gao P; Copland JA; Mehta AS; Drake RR Matrix Assisted Laser Desorption Ionization Imaging Mass Spectrometry Workflow for Spatial Profiling Analysis of N-Linked Glycan Expression in Tissues. Anal. Chem 2013, 85 (20), 9799–9806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Drake RR; Powers TW; Norris-Caneda K; Mehta AS; Angel PM In Situ Imaging of N-Glycans by MALDI Imaging Mass Spectrometry of Fresh or Formalin-Fixed Paraffin-Embedded Tissue. Curr. Protoc. Protein Sci 2018, 94 (1), e68. [DOI] [PubMed] [Google Scholar]
- (23).Black AP; Liang H; West CA; Wang M; Herrera HP; Haab BB; Angel PM; Drake RR; Mehta AS A Novel Mass Spectrometry Platform for Multiplexed N-Glycoprotein Biomarker Discovery from Patient Biofluids by Antibody Panel Based N-Glycan Imaging. Anal. Chem 2019, 91 (13), 8429–8435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Angel PM; Saunders J; Clift CL; White-Gilbertson S; Voelkel-Johnson C; Yeh E; Mehta A; Drake RR A Rapid Array-Based Approach to N -Glycan Profiling of Cultured Cells. J. Proteome Res 2019, 18 (10), 3630–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).West CA; Liang H; Drake RR; Mehta AS A New Enzymatic Approach to Distinguish Fucosylation Isomers of N-Linked Glycans in Tissues Using MALDI Imaging Mass Spectrometry. J. Proteome Res 2020, 19, 2989–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Semmes OJ; Feng Z; Adam B-L; Banez LL; Bigbee WL; Campos D; Cazares LH; Chan DW; Grizzle WE; Izbicka E; Kagan J; Malik G; McLerran D; Moul JW; Partin A; Prasanna P; Rosenzweig J; Sokoll LJ; Srivastava S; Srivastava S; Thompson I; Welsh MJ; White N; Winget M; Yasui Y; Zhang Z; Zhu L. Evaluation of Serum Protein Profiling by Surface-Enhanced Laser Desorption/Ionization Time-of-Flight Mass Spectrometry for the Detection of Prostate Cancer: I. Assessment of Platform Reproducibility. Clin. Chem 2005, 51 (1), 102–112. [DOI] [PubMed] [Google Scholar]
- (27).Schaub NP; Jones KJ; Nyalwidhe JO; Cazares LH; Karbassi ID; Semmes OJ; Feliberti EC; Perry RR; Drake RR Serum Proteomic Biomarker Discovery Reflective of Stage and Obesity in Breast Cancer Patients. J. Am. Coll. Surg 2009, 208 (5), 970–978. [DOI] [PubMed] [Google Scholar]
- (28).Scott DA; Casadonte R; Cardinali B; Spruill L; Mehta AS; Carli F; Simone N; Kriegsmann M; Del Mastro L; Kriegsmann J; Drake RR Increases in Tumor N-Glycan Polylactosamines Associated with Advanced HER2-Positive and Triple-Negative Breast Cancer Tissues. Proteomics: Clin. Appl 2019, 13 (1), 1800014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Drake RR; McDowell C; West C; David F; Powers TW; Nowling T; Bruner E; Mehta AS; Angel PM; Marlow LA; Tun HW; Copland JA Defining the Human Kidney N-Glycome in Normal and Cancer Tissues Using MALDI Imaging Mass Spectrometry. J. Mass Spectrom 2020, 55 (4), No. e4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Saldova R; Asadi Shehni A; Haakensen VD; Steinfeld I; Hilliard M; Kifer I; Helland A; Yakhini Z; Børresen-Dale A-L; Rudd PM Association of N-Glycosylation with Breast Carcinoma and Systemic Features Using High-Resolution Quantitative UPLC. J. Proteome Res 2014, 13 (5), 2314. [DOI] [PubMed] [Google Scholar]
- (31).Pierce A; Saldova R; Abd Hamid UM; Abrahams JL; McDermott EW; Evoy D; Duffy MJ; Rudd PM Levels of Specific Glycans Significantly Distinguish Lymph Node-Positive from Lymph Node-Negative Breast Cancer Patients. Glycobiology 2010, 20 (10), 1283–1288. [DOI] [PubMed] [Google Scholar]
- (32).Saldova R; Reuben JM; Abd Hamid UM; Rudd PM; Cristofanilli M. Levels of Specific Serum N-Glycans Identify Breast Cancer Patients with Higher Circulating Tumor Cell Counts. Ann. Oncol 2011, 22 (5), 1113–1119. [DOI] [PubMed] [Google Scholar]
- (33).Kyselova Z; Mechref Y; Kang P; Goetz JA; Dobrolecki LE; Sledge GW; Schnaper L; Hickey RJ; Malkas LH; Novotny MV Breast Cancer Diagnosis and Prognosis through Quantitative Measurements of Serum Glycan Profiles. Clin. Chem 2008, 54 (7), 1166–1175. [DOI] [PubMed] [Google Scholar]
- (34).Abd Hamid UM; Royle L; Saldova R; Radcliffe CM; Harvey DJ; Storr SJ; Pardo M; Antrobus R; Chapman CJ; Zitzmann N; Robertson JF; Dwek RA; Rudd PM A Strategy to Reveal Potential Glycan Markers from Serum Glycoproteins Associated with Breast Cancer Progression. Glycobiology 2008, 18 (12), 1105–1118. [DOI] [PubMed] [Google Scholar]
- (35).Ju L; Wang Y; Xie Q; Xu X; Li Y; Chen Z; Li Y. Elevated Level of Serum Glycoprotein Bifucosylation and Prognostic Value in Chinese Breast Cancer. Glycobiology 2016, 26, 460. [DOI] [PubMed] [Google Scholar]
- (36).Saldova R; Royle L; Radcliffe CM; Abd hamid UM; Evans R; Arnold JN; Banks RE; Hutson R; Harvey DJ; Antrobus R; Petrescu SM; Dwek RA; Rudd PM Ovarian Cancer Is Associated with Changes in Glycosylation in Both Acute-Phase Proteins and IgG. Glycobiology 2007, 17 (12), 1344–1356. [DOI] [PubMed] [Google Scholar]
- (37).Reiding KR; Blank D; Kuijper DM; Deelder AA; Wuhrer M. High-Throughput Profiling of Protein N-Glycosylation by MALDI-TOF-MS Employing Linkage-Specific Sialic Acid Esterification. Anal. Chem 2014, 86, 5784. [DOI] [PubMed] [Google Scholar]
- (38).Bladergroen MR; Reiding KR; Hipgrave Ederveen AL; Vreeker GCM; Clerc F; Holst S; Bondt A; Wuhrer M; Van Der Burgt YEM Automation of High-Throughput Mass Spectrometry-Based Plasma n-Glycome Analysis with Linkage- Sialic Acid Esterification. J. Proteome Res 2015, 14 (9), 4080–4086. [DOI] [PubMed] [Google Scholar]
- (39).Kostrzewa M. Application of the MALDI Biotyper to Clinical Microbiology: Progress and Potential. Expert Rev. Proteomics 2018, 15 (3), 193–202. [DOI] [PubMed] [Google Scholar]
- (40).Mellmann A; Bimet F; Bizet C; Borovskaya AD; Drake RR; Eigner U; Fahr AM; He Y; Ilina EN; Kostrzewa M; Maier T; Mancinelli L; Moussaoui W; Prevost G; Putignani L; Seachord CL; Tang YW; Harmsen D. High Interlaboratory Reproducibility of Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry-Based Species Identification of Nonfermenting Bacteria. J. Clin. Microbiol 2009, 47 (11), 3732–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Drake RR; Boggs SR; Drake SK Pathogen Identification Using Mass Spectrometry in the Clinical Microbiology Laboratory. J. Mass Spectrom 2011, 46 (12), 1223–1232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.