

Abstract
The complex syndrome of heart failure (HF) is characterized by increased left ventricular pressures. Cardiomyocytes increase in size, cardiac fibroblasts transform and make extracellular matrix, and leukocytes infiltrate the cardiac tissue and alter cardiomyocyte and cardiac fibroblast function. Here we review recent advances in our understanding of the cellular composition of the heart during homeostasis and in response to cardiac pressure overload, with an emphasis on immune cell communication with cardiac fibroblasts and its consequences in cardiac remodeling.
Keywords: Inflammation, fibrosis, heart, leukocytes
Introduction
The physiologic function of the heart is to pump blood through the body providing the necessary nutrients and oxygen for continued function. Pressure overload caused by conditions such as aortic stenosis and hypertension leads to increased strain on cardiac resident cells, local inflammation, and compensatory responses that result in left ventricular derangements in structure, such as cardiac hypertrophy and ventricular dilation. Sustained pressure leads to adverse remodeling, marked at the tissue and cellular level by cardiomyocyte hypertrophy, various stages of cardiac fibrosis, and inflammation, hallmarks of the of the deadly syndrome of heart failure (HF) which affects over 24 million people worldwide [1]. Resident immune cells act as sentinels against possible infection and injury in a tissue such as the heart that largely lacks regenerative capacity. In addition, cardiac fibroblasts synthesize extracellular matrix (ECM) to provide important biochemical and structural support. These important functions of cardiac resident immune cells and fibroblasts are becoming appreciated to contribute to cardiac homeostasis. During HF, the number of immune cells is increased in the heart, and they acquire a more proinflammatory state; simultaneously, fibroblasts transform to myofibroblasts that make excess ECM resulting in pathological fibrosis. Preclinical models of cardiac remodeling serve as important tools for HF investigation and have shed new light into how cardiac homeostasis is disrupted in the heart under pressure. Cardiac infiltrated immune cells actively contribute to cardiac remodeling by communicating with cardiomyocytes, endothelial cells, and cardiac fibroblasts. Here we review recent advancements in knowledge of the immune system’s role in cardiac homeostasis, in acute and chronic inflammation, and in adverse fibrotic remodeling in response to cardiac pressure overload (Figure 1).
Figure 1. Immune Cells in Fibrotic Remodeling.
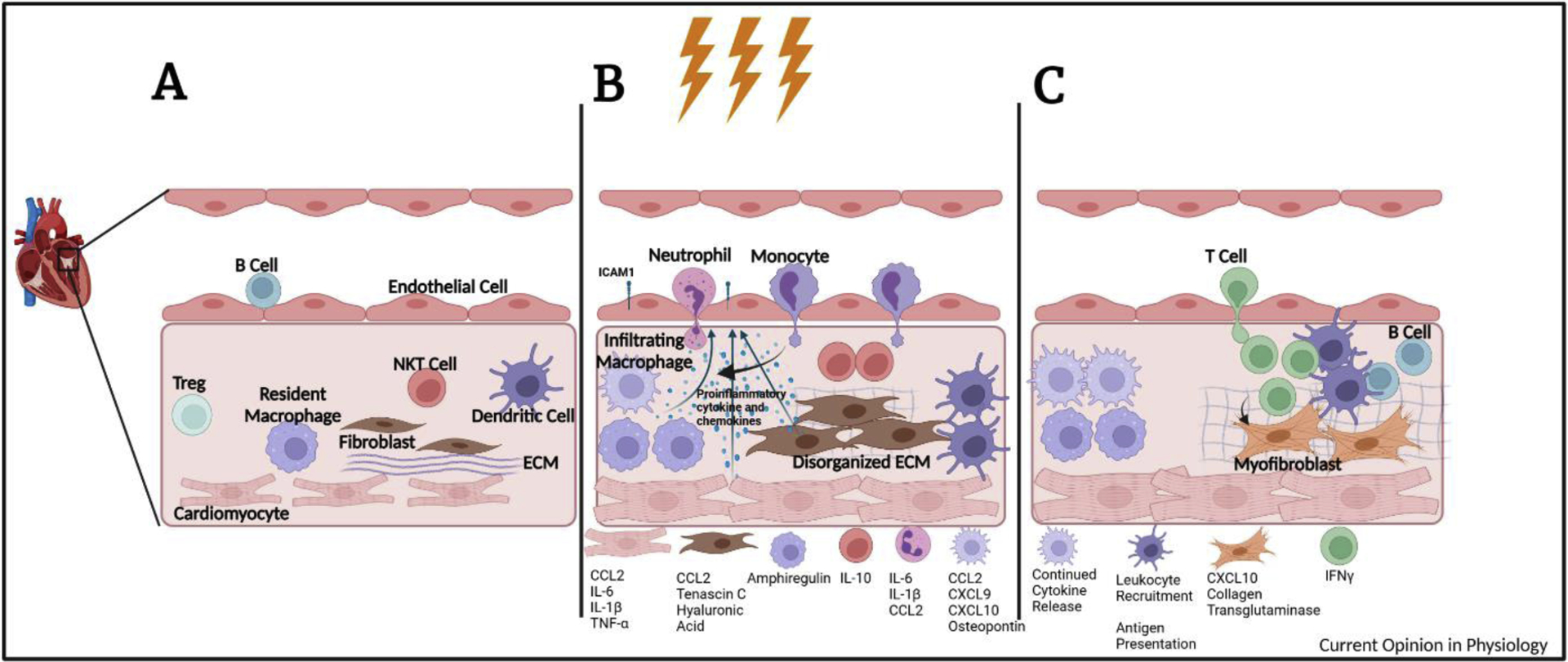
A) Cardiac Homeostasis. Immune cells, stromal cells, and cardiomyocytes are in close contact with each other and maintain cardiac homeostasis in several ways. Cardiac resident CCR2- macrophages participate in clearance of cell debris, lymphangiogenesis, and electrical conduction. A population of transcriptionally distinct B cells is associated with the cardiac microvasculature. And fibroblasts make ECM and sustain cardiomyocyte homeostatic function. B) Early inflammation in cardiac pressure overload. Cardiomyocytes are among the earliest cells sensing pressure and initiating inflammatory responses. Together with fibroblasts and resident and infiltrating macrophages produce proinflammatory chemokines and cytokines that recruit circulating immune cells. Resident macrophages proliferate and NKT cells increase playing important roles in compensatory cardiac remodeling. Neutrophils are the among the earliest infiltrating cells and precede the recruitment of CCR2+ monocytes, which participate in fibroblast activation and ECM deposition, promote cardiomyocyte hypertrophy, and secrete chemokines that recruit CXCR3+ T cells. DCs begin to accumulate during the first week after TAC. C) Sustained inflammation in cardiac pressure overload. Continued pressure overload leads to the recruitment of B and T lymphocytes. Infiltrating T cells are necessary for the progression of HF through the recognition of cardiac neoantigen presented by DC. Th1 cells adhere to cardiac fibroblasts and induce myofibroblast transformation. Fibrotic remodeling of the heart is promoted by the excessive ECM deposition, increased cross-linking of collagen, and accumulation of proteoglycans.
Immune Cells In Cardiac Homeostasis
Cardiomyocytes make up the largest volume of the murine cardiac tissue. Within the non-myocytes, endothelial cells make up 60% of cells, fibroblasts <20%, and leukocytes make up approximately 10% of cells as determined by flow cytometry [2]. Single cell sequencing of immune cells from both murine and deceased transplant donors without cardiac pathology show the healthy adult heart contains most major immune cell classes including macrophages, monocytes, neutrophils, mast cells, dendritic cells, T cells, T regulatory cells, and B cells [3, 4]. The distribution of immune cells is not uniform as samples from human atria and ventricles showed different proportions of immune cells with a greater number of myeloid and lymphoid cells in the atria [4].
Besides their role in protection from infectious pathogens, there is a growing appreciation for homeostatic roles of immune cells in the heart. Our greatest understanding comes from cardiac resident macrophages which are embryonically derived and can be distinguished from hematopoietic derived macrophages by the lack of expression of C-C Motif Chemokine Receptor 2(CCR2) [5, 6]. Resident cardiac macrophages are important for cardiac development, lymphangiogenesis, clearing local cell debris, and for proper electrical conduction [5, 7–10]. Similarly, B cells seem to be central to cardiac homeostasis, since B cell deficient mice show alterations in myocardial mass and ventricular contractility[11]. Recent single cell sequencing analysis of the human heart predicts extensive cell-cell interactions based on receptor- ligand circuits between immune cells, cardiomyocytes, and stromal cells, supporting cell-cell communication being central to cardiac homeostasis, but the physiologic functions of these interactions still need to be elucidated[4, 12]. Taken together analyzing RNA transcripts by bulk or single cells sequencing, and protein expression using cell specific markers, significant progress has been made in our understanding of the cellular composition of the heart. Sequencing studies have the advantage of being unbiased and of identifying low proportion cell subsets, yet protein validation of such low proportion cell populations as well as physiologic studies to confirm their predicted function still needs to be conducted.
Early Inflammation in Cardiac Pressure Overload
Most of the mechanistic insight on the spatial and temporal pathophysiological roles of immune cells and cardiac stromal cells comes from reproducible pre-clinical experimental models that result in cardiac pressure overload. Trans aortic constriction (TAC) is one of them, which mimics aortic stenosis by reducing the diameter of the aorta. Although the acute sudden left ventricular pressure induced by TAC does not replicate how human heart failure develops, it is well-established that with time, TAC induces hallmarks of HF that include cardiac hypertrophy, cardiac fibrosis and systolic and diastolic dysfunction progressively. Compensatory changes in response to sudden pressure transition into decompensation, and finally chronic HF. Recent single cell sequencing data have correlated transcriptional changes during the decompensation and chronic HF stages of TAC to bulk RNA sequencing data in patients with hypertrophic cardiomyopathy and HF, respectively, further supporting its use as a pre-clinical model system [13]. Systemic hypertension is modeled by chronic administration of Angiotensin-II (AngII), deoxycorticosterone acetate (DOCA), unilateral nephrectomy and high salt diet, which alters hemodynamic pressure and induce systemic and/or cardiac proinflammatory and ECM alterations.
Cardiomyocytes and Inflammation
Pressure overload induced by TAC or hypertension does not result in cardiomyocyte death early on. However, the pressure sensed by the heart induces the release of proinflammatory cytokines and initiates the recruitment of immune cells. Mechanical stretch or neurohormonal stimulation in the case of hypertension leads to increased intracellular Ca2+ and increased Ca/Calmodulin signaling [14]. Cardiomyocytes transform sensing of hemodynamic stress into inflammatory signals through the Ca2+/calmodulin dependent kinase protein kinase II δ(CaMKIIδ) leading to activation of the NOD-like receptor protein 3 (NLRP3) inflammasome. Indeed, cardiomyocyte specific deficiency of CaMKIIδ in mice results in reduced inflammatory cytokines (IL-1β, IL-6, TNF-α) and chemokines (CCL2, CCL3, CXCL1) as early as 3 days after TAC, and reduced cardiac macrophages 7 days after TAC. Thus cardiomyocytes are central players initiating inflammatory responses to pressure overload [15]. Additionally, sensing of danger associated molecular patterns contribute to early inflammation as knockdown of the cytosolic DNA sensor cGAS or deficiency of the DAMP receptor Toll Receptor 2 (TLR2) showed reduced cardiac hypertrophy and inflammation in TAC. Although DAMPS such as Il-β, ATP and HMGB-1 have been reported in TAC hearts, their cell specific actions through different receptors still need to be studied to understand the cell specific early inflammatory response to pressure overload [16, 17].
Cardiac Stromal Cells and Inflammation
Fibroblasts are important contributors to the proinflammatory environment in response to pressure overload. Single cell sequencing on sorted murine fibroblasts during the first three days after TAC highlighted a subset of proinflammatory fibroblasts that secrete CCL2 in a nuclear factor kappa B (NFκB) dependent manner and promoted the infiltration of Ly6Chi monocytes. Fibroblast deficiency of IκκB, an activator of NFkB, led to attenuated cardiac dysfunction in TAC [18]. Fibroblasts also participate in cardiac inflammation through surface receptors like fibroblast growth factor inducible 14, a highly inducible cytokine receptor, or production of matricellular proteins such as Tenascin-C that result in monocyte recruitment [19, 20]. Alterations in the intramyocardial vascular endothelium are also observed as early as 2 days post TAC, demonstrated by increased expression of intercellular adhesion molecule- 1 (ICAM1), promoting leukocyte infiltration [21]. Thus cardiac fibroblasts and endothelial cells actively contribute to inflammation.
Immune Cells and Inflammation
The onset of pressure overload leads to alterations in the immune cell composition of the heart [3]. Neutrophils are among the earliest cells to infiltrate the heart in response to pressure overload in a Wnt5a, a member of the Wnt family that activates noncanonical Wnt signaling pathways, dependent manner. Depletion of Neutrophils before TAC or myeloid specific deficient Wnt5a mice showed decreased cardiac dysfunction, diminished early immune cell recruitment, and decreased proinflammatory cytokines (IL-1β, IL-6) and chemokines (Cxcl1, Cxcl2, Cxcl5, CCL2) 3 days after TAC [22]. Additionally, local proliferation of cardiac resident macrophages as well as differentiation of infiltrated Ly6Clo and Ly6Chi monocytes into macrophages occurs within the first week after TAC [23–26]. Cardiac resident macrophages are required for survival to pressure overload, as their deletion drastically reduces survival during the first week after TAC, and myeloid specific Kruppel-like factor 4 deletion, a transcription factor that promotes cardiac macrophage proliferation, also worsened ventricular function post TAC [25]. This protection is in part due to resident Ly6Clo macrophage secretion of amphiregulin, an epidermal growth factor receptor (EGFR) ligand, that leads to compensatory hypertrophy and supports normal cardiomyocyte gap junction communication [9, 27]. Resident cardiac macrophages expressing lymphatic vessel endothelial hyaluronic acid receptor-1 (LYVE-1) are a recently discovered population that associate with lymphatic vessels and express lymphangiogenic factors. The severity of cardiac dysfunction was correlated with greater reductions in this population in response to pressure overload, supporting an additional cardioprotective mechanism [10].
Antagonism of CCR2 in the onset of TAC revealed that infiltrating CCR2+ macrophages derived from monocytes contribute to early proinflammatory chemokine release and expansion of CD4 T cells in the mediastinal lymph nodes [24, 28]. MicroRNA-21, the most highly expressed microRNA in cardiac macrophages, is increased after TAC, promotes a proinflammatory transcriptome, and is predicted by ligand receptor pairing analysis to increase crosstalk between macrophages and fibroblasts [29]. A more recent study analyzing single cell data at several points after TAC also supports increased inflammatory macrophages 2–5 weeks after TAC to be key contributors to cardiomyocyte pathology in response to pressure overload [13]. One example of this cross talk can be seen in a hypertensive model where macrophage IL-10 production induced secretion of osteopontin which promotes the transformation of myofibroblasts [30]. The transcription factor Megakaryocytic Leukemia 1 in macrophages also promotes pathological hypertrophy in cardiomyocytes possibly though exosomal release of miR-155 [31]. Macrophages also show important roles in the recruitment of immune cells through the secretion of CXCL9 and CXCL10 1 week after TAC which recruit CXCR3+ T cells to the heart [28].
In addition to cells of myeloid origin, thymic derived Natural Killer T (NKT), an innate type T lymphocyte at the interphase of innate and adaptive immune responses, have also recently been implicated in the inflammatory response to cardiac pressure overload. While their presence is increased in the heart in TAC, pharmacological or genetic deletion leads to worsened cardiac remodeling potentially through reduced cardiac levels of IL-10 [32]. Moreover, NKT cell activation with the CD1d ligand a-galactosylceramide resulted in attenuated cardiac fibrosis and hypertrophy in response to AngII infusion [33]. Thus, NKT cells seem to protect the heart from adverse cardiac remodeling induced by pressure overload.
Dendritic cells (DCs) also contribute to the inflammatory response to pressure overload. DCs begin to accumulate within the first week after TAC [26] and ablation of CD11c+ cells reduced cardiac hypertrophy, fibrosis, and leukocyte infiltration [34]. DCs also proved to be necessary for the induction of renal sodium transporters and the renin angiotensin system in hypertension [35], but dispensable for renal fibrosis and were not enriched in the kidneys of hypertensive mice [35, 36]. Since these conclusions were based on pan-CD11c+ cell depletion, it is possible that monocytes, which also express CD11c, may contribute to these phenotypes attributed to DCs.
In summary, pressure overload leads to the recruitment of neutrophils, monocytes, NKT cells, and DCs as well as the expansion of resident cardiac macrophages. Neutrophils and monocytes promote inflammation while NKT cells and resident macrophages play protective roles. In hypertension models, myeloid cells alter blood pressure and may have consequences in cardiac remodeling.
Sustained Inflammation in Cardiac Pressure Overload
Samples from HF patients also suggest a potential role of myeloid cells in cardiac remodeling. An increased neutrophil to lymphocyte ratio in patients newly diagnosed with hypertension was predictive of left ventricular hypertrophy [37]. Increasing numbers of CCR2+ macrophages are also associated with worsened ventricular function while patients with lower CCR2+ macrophages show better improvement following the implantation of left ventricular assistance devices [6]. In mice, however, depletion of mononuclear phagocytes starting two weeks after TAC, after the onset of left ventricular hypertrophy, did not alter pathologic cardiac remodeling [26], supporting that non-myeloid leukocytes, namely adaptive immune cells, are responsible for pathological cardiac remodeling.
In both hypertensive and TAC experimental models, deficiency in B cells resulted in decreased proinflammatory cytokines, IgG production, and adverse cardiac remodeling, and it was postulated that B cell production of cardiac specific autoantibodies contributes to cardiac remodeling [38, 39].
T cells from patients with nonischemic HF show increased adhesion to activated human primary endothelial cells in vitro, and myocardial tissue from HF patients show increased T cell infiltrates and CXCR3+ leukocytes, not seen in healthy donors by histology. Additionally, T cell deficient mice through deletion of the T cell receptor α, or mice with T cell costimulatory receptor blockade do not develop TAC induced cardiac fibrosis and dysfunction [40, 41]. TAC induces myeloid and cardiac fibroblast release of CXCL9 and CXCL10 that chemoattract CXCR3+ CD4 T cells to the heart in an LFA-1 integrin-ICAM-1 dependent manner. Indeed, CXCR3 deficient mice show reduced T cell infiltration and preserved cardiac function [28]. Once in the heart, T cell recognition of cardiac neoantigens induced by reactive oxygen species is required for sustaining T cell activation and systolic dysfunction. The presence of such neoantigens in the mouse and human failing hearts, and their induction of T cell proliferation further support a role for T cell immune response in the heart [42]. Moreover, deficiency in Tbet, the transcription factor necessary for Th1 cell development, alleviated cardiac fibrosis and hypertrophy in a model of abdominal aortic constriction in rats, confirming that Th1 cells are important in the development of pathologic remodeling [43]. In line with these findings, Th1 cells promote fibrosis through directly adhering to CFBs and secreting IFNγ which induces CFB production of TGFβ and myofibroblast transformation [44]. Advanced age, another risk factor for HF, may also facilitate T cell pathology in pressure overload as 12-month-old mice show an expansion in effector and IFNγ+ T cells within mLNs. These mLN T cells also demonstrated greater cardiotropism compared to cells from juvenile mice when adoptively transferred into T cell deficient mice [45]. The expansion of Th1 cells in aging mice may be due to disrupted mitochondrial function as T cell specific deficiency in mitochondrial transcription factor A, which stabilizes mitochondrial DNA, led to skewing towards Th1 cells [46]. CD8 T cells can also be activated in response to pressure overload induced by TAC, though they do not appear to play a role in the progression of HF [47]. While recent work has reviewed T cell contributions to the initiation of hypertension and vascular inflammation in hypertension, more work is needed to better define their role in hypertensive cardiac remodeling [48]. In all these studies, the read outs for cardiac fibrosis focus on collagen deposition as the dominant component of the ECM, and more studies are needed to identify whether other ECM components, in addition to collagen, are also modulated by immune cells.
T regulatory (Tregs) cells are characterized by the expression of the transcription factor FoxP3 and surface expression of the IL-2 receptor alpha chain CD25 and suppress effector T cell responses. Adoptive transfer of Tregs in mice with AngII resulted in reduced cardiac fibrosis and transformation of myofibroblasts [49] and endogenous expansion of Tregs through the induction of IL-2 leads to reduced LV remodeling induced by TAC and attenuates the progression of HF in mice with existing LV dysfunction, suggesting Tregs are protective in pressure overload induced HF [50].
Fibrosis and Extra Cellular Matrix Changes in Sustained Inflammation
Regardless of etiology, fibrosis is a hallmark of heart disease; and long-standing pressure overload induces interstitial and perivascular fibrosis through the deposition of excessive ECM proteins including collagens, fibronectin, hyaluronic acid, and proteoglycans. The extent of fibrosis is also associated with worse clinical outcomes [51]. The transformation of fibroblasts to myofibroblasts, the major contributors to fibrotic remodeling, can be triggered by neurohormonal activation, mechanosensitive pathways, and immune cells [52]. Inflammation and fibrosis are interrelated processes; fibroblasts make chemoattractants and ECM components such as hyaluronic acid that promote early inflammation and immune cell infiltration. Immune cells, in turn, directly or indirectly contribute to fibroblast activation [29, 30, 38, 44]. Inhibition of hyaluronic acid production through 4- methylumbelliferone (4-MU) in mice that underwent TAC showed reduced early inflammation with decreased levels of circulating monocytes and chemokine receptors (Ccr2, Ccr5, Cx3cr1) three days later. This was accompanied by reduced levels of hypertrophy and better systolic function as well as decreased fibrosis 7 weeks after TAC [53].
Recent studies using activated fibroblast specific deletions of Smad3, an important mediator in TGFβ signaling, showed that early fibroblast function is important in maintaining ECM structure and preventing early systolic dysfunction while continued activation is responsible for accumulation of fibrosis [54, 55]. In the presence of continued pressure overload, extensive collagen crosslinking by enzymes such as transglutaminase (tGT) promotes fibrosis and increased cross-linked collagen was associated with greater incidence of hospitalization for HF in hypertensive patients [56]. Inhibition of tGT prevented TAC induced fibrosis and diastolic dysfunction [57]. Continued pressure overload also leads to the accumulation of chondroitin sulfate proteoglycans that localize to fibrotic areas in both patients with HF and in rats that received TAC. Moreover, treatment with an enzyme that breaks down chondroitin sulfate resulted in improved ejection fraction 8 weeks after TAC [58]. The mechanisms of how immune cells contribute to these alterations in the ECM requires further investigation and analysis of predicted cell interactions may prove useful.
Concluding remarks
The work reviewed here highlights novel findings and mechanistic insights in nonischemic heart failure gained using pre-clinical models and novel sequencing technologies in the mouse and human hearts. Small molecule inhibitors to costimulatory pathways were effective in murine TAC models to reduce cardiac dysfunction through inhibiting immune cell activation [41, 59]. Recent advances in cancer research have also inspired a potential use for chimeric antigen receptor (CAR) T cells in HF as CD8 T cells engineered to recognize a protein on activated fibroblasts showed reduction in fibrosis and reduced loss in fractional shortening in a model of hypertension[60] However, to date, no anti-inflammatory or anti-fibrotic approaches have proven effective in clinical trials. How can these be leveraged to treat human heart failure patients? While in experimental models there is a clear characterization of each inflammatory and fibrotic step in response to cardiac pressure overload that can be timely prevented or treated, patients diagnosed with HF already present with hallmarks of inflammation and fibrosis. Moreover, most experimental models do not include aged animals, and thus exclude the inflammatory component of aging, prevalent in the HF population. Despite these challenges, the future is bright with new advances in cardio-immunology and our understanding of cardiac fibrosis. The development of transgenic animals with specific molecular immunological and fibrotic pathways altered, and the new single cell technologies in mouse and human hearts provide a wealth of information, including receptor-ligand pairing analysis that suggest active interplay between different cells in the heart. Once these predicted interactions are functionally validated, the field will be closer to develop new targeted immunomodulatory therapies with anti-fibrotic effects that are safe in patients with HF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng S, Delling FN, Elkind MSV, Evenson KR, Ferguson JF, Gupta DK, Khan SS, Kissela BM, Knutson KL, Lee CD, Lewis TT, Liu J, Loop MS, Lutsey PL, Ma J, Mackey J, Martin SS, Matchar DB, Mussolino ME, Navaneethan SD, Perak AM, Roth GA, Samad Z, Satou GM, Schroeder EB, Shah SH, Shay CM, Stokes A, Vanwagner LB, Wang N-Y, Tsao CW, Heart Disease and Stroke Statistics—2021 Update, Circulation 143(8) (2021). [DOI] [PubMed] [Google Scholar]
- [2].Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD, Revisiting Cardiac Cellular Composition, Circulation Research 118(3) (2016) 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Martini E, Kunderfranco P, Peano C, Carullo P, Cremonesi M, Schorn T, Carriero R, Termanini A, Colombo FS, Jachetti E, Panico C, Faggian G, Fumero A, Torracca L, Molgora M, Cibella J, Pagiatakis C, Brummelman J, Alvisi G, Mazza EMC, Colombo MP, Lugli E, Condorelli G, Kallikourdis M, Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation, Circulation 140(25) (2019) 2089–2107. [DOI] [PubMed] [Google Scholar]
- [4].Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, Nadelmann ER, Roberts K, Tuck L, Fasouli ES, Delaughter DM, McDonough B, Wakimoto H, Gorham JM, Samari S, Mahbubani KT, Saeb-Parsy K, Patone G, Boyle JJ, Zhang H, Zhang H, Viveiros A, Oudit GY, Bayraktar OA, Seidman JG, Seidman CE, Noseda M, Hubner N, Teichmann SA, Cells of the adult human heart, Nature 588(7838) (2020) 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wülfers EM, Seemann G, Courties G, Iwamoto Y, Sun Y, Savol AJ, Sager HB, Lavine KJ, Fishbein GA, Capen DE, Da Silva N, Miquerol L, Wakimoto H, Seidman CE, Seidman JG, Sadreyev RI, Naxerova K, Mitchell RN, Brown D, Libby P, Weissleder R, Swirski FK, Kohl P, Vinegoni C, Milan DJ, Ellinor PT, Nahrendorf M, Macrophages Facilitate Electrical Conduction in the Heart, Cell 169(3) (2017) 510–522.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, Epelman S, Kreisel D, Liu Y, Itoh A, Shankar TS, Selzman CH, Drakos SG, Lavine KJ, The human heart contains distinct macrophage subsets with divergent origins and functions, Nat Med 24(8) (2018) 1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Swirski FK, Nahrendorf M, Cardioimmunology: the immune system in cardiac homeostasis and disease, Nat Rev Immunol 18(12) (2018) 733–744. [DOI] [PubMed] [Google Scholar]
- [8].Sun K, Li YY, Jin J, A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair, Signal Transduct Target Ther 6(1) (2021) 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sugita J, Fujiu K, Nakayama Y, Matsubara T, Matsuda J, Oshima T, Liu Y, Maru Y, Hasumi E, Kojima T, Seno H, Asano K, Ishijima A, Tomii N, Yamazaki M, Kudo F, Sakuma I, Nagai R, Manabe I, Komuro I, Cardiac macrophages prevent sudden death during heart stress, Nat Commun 12(1) (2021) 1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bizou M, Itier R, Majdoubi M, Abbadi D, Pichery E, Dutaur M, Marsal D, Calise D, Garmy-Susini B, Douin-Echinard V, Roncalli J, Parini A, Pizzinat N, Cardiac macrophage subsets differentially regulate lymphatic network remodeling during pressure overload, Sci Rep 11(1) (2021) 16801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Adamo L, Rocha-Resende C, Lin CY, Evans S, Williams J, Dun H, Li W, Mpoy C, Andhey PS, Rogers BE, Lavine K, Kreisel D, Artyomov M, Randolph GJ, Mann DL, Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart, JCI Insight 5(3) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang L, Yu P, Zhou B, Song J, Li Z, Zhang M, Guo G, Wang Y, Chen X, Han L, Hu S, Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function, Nature Cell Biology 22(1) (2020) 108–119. [DOI] [PubMed] [Google Scholar]
- [13].Ren Z, Yu P, Li D, Li Z, Liao Y, Wang Y, Zhou B, Wang L, Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy, Circulation 141(21) (2020) 1704–1719. [DOI] [PubMed] [Google Scholar]
- [14].Saucerman JJ, Tan PM, Buchholz KS, McCulloch AD, Omens JH, Mechanical regulation of gene expression in cardiac myocytes and fibroblasts, Nature Reviews Cardiology 16(6) (2019) 361–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, Brown JH, Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca(2+)/Calmodulin-Dependent Protein Kinase II delta Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling, Circulation 138(22) (2018) 2530–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hu D, Cui Y-X, Wu M-Y, Li L, Su L-N, Lian Z, Chen H, Cytosolic DNA sensor cGAS plays an essential pathogenetic role in pressure overload-induced heart failure, American Journal of Physiology-Heart and Circulatory Physiology 318(6) (2020) H1525–H1537. [DOI] [PubMed] [Google Scholar]
- [17].Higashikuni Y, Tanaka K, Kato M, Nureki O, Hirata Y, Nagai R, Komuro I, Sata M, Toll-Like Receptor-2 Mediates Adaptive Cardiac Hypertrophy in Response to Pressure Overload Through Interleukin-1β Upregulation via Nuclear Factor κB Activation, Journal of the American Heart Association 2(6) (2013) e000267–e000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Abe H, Tanada Y, Omiya S, Podaru M-N, Murakawa T, Ito J, Shah AM, Conway SJ, Ono M, Otsu K, NF-kB activation in cardiac fibroblasts results in the recruitment of inflammatory Ly6Chi monocytes in pressure-overloaded hearts, Science Signaling 14(704) (2021) eabe4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Unudurthi SD, Nassal DM, Patel NJ, Thomas E, Yu J, Pierson CG, Bansal SS, Mohler PJ, Hund TJ, Fibroblast growth factor-inducible 14 mediates macrophage infiltration in heart to promote pressure overload-induced cardiac dysfunction, Life Sci 247 (2020) 117440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Abbadi D, Laroumanie F, Bizou M, Pozzo J, Daviaud D, Delage C, Calise D, Gaits-Iacovoni F, Dutaur M, Tortosa F, Renaud-Gabardos E, Douin-Echinard V, Prats AC, Roncalli J, Parini A, Pizzinat N, Local production of tenascin-C acts as a trigger for monocyte/macrophage recruitment that provokes cardiac dysfunction, Cardiovasc Res 114(1) (2018) 123–137. [DOI] [PubMed] [Google Scholar]
- [21].Salvador AM, Nevers T, Velazquez F, Aronovitz M, Wang B, Abadia Molina A, Jaffe IZ, Karas RH, Blanton RM, Alcaide P, Intercellular Adhesion Molecule 1 Regulates Left Ventricular Leukocyte Infiltration, Cardiac Remodeling, and Function in Pressure Overload-Induced Heart Failure, J Am Heart Assoc 5(3) (2016) e003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang Y, Sano S, Oshima K, Sano M, Watanabe Y, Katanasaka Y, Yura Y, Jung C, Anzai A, Swirski FK, Gokce N, Walsh K, Wnt5a-Mediated Neutrophil Recruitment Has an Obligatory Role in Pressure Overload-Induced Cardiac Dysfunction, Circulation 140(6) (2019) 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu X, Shi GP, Guo J, Innate Immune Cells in Pressure Overload-Induced Cardiac Hypertrophy and Remodeling, Front Cell Dev Biol 9 (2021) 659666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, Prabhu SD, CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload, JACC Basic Transl Sci 3(2) (2018) 230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liao X, Shen Y, Zhang R, Sugi K, Vasudevan NT, Alaiti MA, Sweet DR, Zhou L, Qing Y, Gerson SL, Fu C, Wynshaw-Boris A, Hu R, Schwartz MA, Fujioka H, Richardson B, Cameron MJ, Hayashi H, Stamler JS, Jain MK, Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy, Proc Natl Acad Sci U S A 115(20) (2018) E4661–E4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Patel B, Ismahil MA, Hamid T, Bansal SS, Prabhu SD, Mononuclear Phagocytes Are Dispensable for Cardiac Remodeling in Established Pressure-Overload Heart Failure, PLoS One 12(1) (2017) e0170781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fujiu K, Shibata M, Nakayama Y, Ogata F, Matsumoto S, Noshita K, Iwami S, Nakae S, Komuro I, Nagai R, Manabe I, A heart–brain–kidney network controls adaptation to cardiac stress through tissue macrophage activation, Nature Medicine 23(5) (2017) 611–622. [DOI] [PubMed] [Google Scholar]
- [28].Ngwenyama N, Salvador AM, Velazquez F, Nevers T, Levy A, Aronovitz M, Luster AD, Huggins GS, Alcaide P, CXCR3 regulates CD4+ T cell cardiotropism in pressure overload-induced cardiac dysfunction, JCI Insight 4(7) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ramanujam D, Schon AP, Beck C, Vaccarello P, Felician G, Dueck A, Esfandyari D, Meister G, Meitinger T, Schulz C, Engelhardt S, MicroRNA-21-Dependent Macrophage-to-Fibroblast Signaling Determines the Cardiac Response to Pressure Overload, Circulation 143(15) (2021) 1513–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, Osborne MT, Hung J, Vinegoni C, Naxerova K, Sosnovik DE, Zile MR, Bradshaw AD, Liao R, Tawakol A, Weissleder R, Rosenzweig A, Swirski FK, Sam F, Nahrendorf M, Cardiac macrophages promote diastolic dysfunction, J Exp Med 215(2) (2018) 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu L, Zhao Q, Lin L, Yang G, Yu L, Zhuo L, Yang Y, Xu Y, Myeloid MKL1 Disseminates Cues to Promote Cardiac Hypertrophy in Mice, Front Cell Dev Biol 9 (2021) 583492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Takahashi M, Kinugawa S, Takada S, Kakutani N, Furihata T, Sobirin MA, Fukushima A, Obata Y, Saito A, Ishimori N, Iwabuchi K, Tsutsui H, The disruption of invariant natural killer T cells exacerbates cardiac hypertrophy and failure caused by pressure overload in mice, Exp Physiol 105(3) (2020) 489–501. [DOI] [PubMed] [Google Scholar]
- [33].Wang HX, Li WJ, Hou CL, Lai S, Zhang YL, Tian C, Yang H, Du J, Li HH, CD1d-dependent natural killer T cells attenuate angiotensin II-induced cardiac remodelling via IL-10 signalling in mice, Cardiovasc Res 115(1) (2019) 83–93. [DOI] [PubMed] [Google Scholar]
- [34].Wang H, Kwak D, Fassett J, Liu X, Yao W, Weng X, Xu X, Xu Y, Bache RJ, Mueller DL, Chen Y, Role of bone marrow-derived CD11c(+) dendritic cells in systolic overload-induced left ventricular inflammation, fibrosis and hypertrophy, Basic Res Cardiol 112(3) (2017) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hevia D, Araos P, Prado C, Fuentes Luppichini E, Rojas M, Alzamora R, Cifuentes-Araneda F, Gonzalez AA, Amador CA, Pacheco R, Michea L, Myeloid CD11c(+) Antigen-Presenting Cells Ablation Prevents Hypertension in Response to Angiotensin II Plus High-Salt Diet, Hypertension 71(4) (2018) 709–718. [DOI] [PubMed] [Google Scholar]
- [36].Araos P, Prado C, Lozano M, Figueroa S, Espinoza A, Berger T, Mak TW, Jaisser F, Pacheco R, Michea L, Amador CA, Dendritic cells are crucial for cardiovascular remodeling and modulate neutrophil gelatinase-associated lipocalin expression upon mineralocorticoid receptor activation, J Hypertens 37(7) (2019) 1482–1492. [DOI] [PubMed] [Google Scholar]
- [37].Abdulmecit A, Ramazan A, Ertuğrul K, Hakan K, Neutrophil to Lymphocyte Ratio as a Predictor of Left Ventricular Hypertrophy in Patients with Newly Diagnosed Hypertension, Journal of Hypertension and Management 5(2) (2019). [Google Scholar]
- [38].Cordero-Reyes AM, Youker KA, Trevino AR, Celis R, Hamilton DJ, Flores-Arredondo JH, Orrego CM, Bhimaraj A, Estep JD, Torre-Amione G, Full Expression of Cardiomyopathy Is Partly Dependent on B-Cells: A Pathway That Involves Cytokine Activation, Immunoglobulin Deposition, and Activation of Apoptosis, Journal of the American Heart Association 5(1) (2016) e002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ma X-L, Lin Q-Y, Wang L, Xie X, Zhang Y-L, Li H-H, Rituximab prevents and reverses cardiac remodeling by depressing B cell function in mice, Biomedicine & Pharmacotherapy 114 (2019) 108804. [DOI] [PubMed] [Google Scholar]
- [40].Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velazquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM, Alcaide P, Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure, Circ Heart Fail 8(4) (2015) 776–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kallikourdis M, Martini E, Carullo P, Sardi C, Roselli G, Greco CM, Vignali D, Riva F, Ormbostad Berre AM, Stolen TO, Fumero A, Faggian G, Di Pasquale E, Elia L, Rumio C, Catalucci D, Papait R, Condorelli G, T cell costimulation blockade blunts pressure overload-induced heart failure, Nat Commun 8 (2017) 14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ngwenyama N, Kirabo A, Aronovitz M, Velazquez F, Carrillo-Salinas F, Salvador AM, Nevers T, Amarnath V, Tai A, Blanton RM, Harrison DG, Alcaide P, Isolevuglandin-Modified Cardiac Proteins Drive CD4+ T-Cell Activation in the Heart and Promote Cardiac Dysfunction, Circulation 143(12) (2021) 1242–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ma ZG, Dai J, Yuan YP, Bian ZY, Xu SC, Jin YG, Zhang X, Tang QZ, T-bet deficiency attenuates cardiac remodelling in rats, Basic Res Cardiol 113(3) (2018) 19. [DOI] [PubMed] [Google Scholar]
- [44].Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, Blanton RM, Alcaide P, Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure, J Exp Med 214(11) (2017) 3311–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ramos GC, Van Den Berg A, Nunes-Silva V, Weirather J, Peters L, Burkard M, Friedrich M, Pinnecker J, Abeßer M, Heinze KG, Schuh K, Beyersdorf N, Kerkau T, Demengeot J, Frantz S, Hofmann U, Myocardial aging as a T-cell–mediated phenomenon, Proceedings of the National Academy of Sciences 114(12) (2017) E2420–E2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Desdin-Mico G, Soto-Heredero G, Aranda JF, Oller J, Carrasco E, Gabande-Rodriguez E, Blanco EM, Alfranca A, Cusso L, Desco M, Ibanez B, Gortazar AR, Fernandez-Marcos P, Navarro MN, Hernaez B, Alcami A, Baixauli F, Mittelbrunn M, T cells with dysfunctional mitochondria induce multimorbidity and premature senescence, Science 368(6497) (2020) 1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Groschel C, Sasse A, Monecke S, Rohrborn C, Elsner L, Didie M, Reupke V, Bunt G, Lichtman AH, Toischer K, Zimmermann WH, Hasenfuss G, Dressel R, CD8(+)-T Cells With Specificity for a Model Antigen in Cardiomyocytes Can Become Activated After Transverse Aortic Constriction but Do Not Accelerate Progression to Heart Failure, Front Immunol 9 (2018) 2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Drummond GR, Vinh A, Guzik TJ, Sobey CG, Immune mechanisms of hypertension, Nat Rev Immunol 19(8) (2019) 517–532. [DOI] [PubMed] [Google Scholar]
- [49].Mian MOR, Barhoumi T, Briet M, Paradis P, Schiffrin EL, Deficiency of T-regulatory cells exaggerates angiotensin II-induced microvascular injury by enhancing immune responses, Journal of Hypertension 34(1) (2016) 97–108. [DOI] [PubMed] [Google Scholar]
- [50].Wang H, Hou L, Kwak D, Fassett J, Xu X, Chen A, Chen W, Blazar BR, Xu Y, Hall JL, Ge J-B, Bache RJ, Chen Y, Increasing Regulatory T Cells With Interleukin-2 and Interleukin-2 Antibody Complexes Attenuates Lung Inflammation and Heart Failure Progression, Hypertension 68(1) (2016) 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schelbert EB, Piehler KM, Zareba KM, Moon JC, Ugander M, Messroghli DR, Valeti US, Chang CCH, Shroff SG, Diez J, Miller CA, Schmitt M, Kellman P, Butler J, Gheorghiade M, Wong TC, Myocardial Fibrosis Quantified by Extracellular Volume Is Associated With Subsequent Hospitalization for Heart Failure, Death, or Both Across the Spectrum of Ejection Fraction and Heart Failure Stage, Journal of the American Heart Association 4(12) (2015) e002613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Frangogiannis NG, Cardiac fibrosis, Cardiovascular Research 117(6) (2021) 1450–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hackert K, Homann S, Mir S, Beran A, Gorreßen S, Funk F, Fischer JW, Grandoch M, Schmitt JP, 4-Methylumbelliferone Attenuates Macrophage Invasion and Myocardial Remodeling in Pressure-Overloaded Mouse Hearts, Hypertension 77(6) (2021) 1918–1927. [DOI] [PubMed] [Google Scholar]
- [54].Russo I, Cavalera M, Huang S, Su Y, Hanna A, Chen B, Shinde AV, Conway SJ, Graff J, Frangogiannis NG, Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program, Circulation Research 124(8) (2019) 1214–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J, Molkentin JD, Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis, J Clin Invest 127(10) (2017) 3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lopez B, Ravassa S, Gonzalez A, Zubillaga E, Bonavila C, Berges M, Echegaray K, Beaumont J, Moreno MU, San Jose G, Larman M, Querejeta R, Diez J, Myocardial Collagen Cross-Linking Is Associated With Heart Failure Hospitalization in Patients With Hypertensive Heart Failure, Journal of the American College of Cardiology 67(3) (2016) 251–260. [DOI] [PubMed] [Google Scholar]
- [57].Shinde AV, Su Y, Palanski BA, Fujikura K, Garcia MJ, Frangogiannis NG, Pharmacologic inhibition of the enzymatic effects of tissue transglutaminase reduces cardiac fibrosis and attenuates cardiomyocyte hypertrophy following pressure overload, Journal of Molecular and Cellular Cardiology 117 (2018) 36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhao R-R, Ackers-Johnson M, Stenzig J, Chen C, Ding T, Zhou Y, Wang P, Ng SL, Li PY, Teo G, Rudd PM, Fawcett JW, Foo RSY, Targeting Chondroitin Sulfate Glycosaminoglycans to Treat Cardiac Fibrosis in Pathological Remodeling, Circulation 137(23) (2018) 2497–2513. [DOI] [PubMed] [Google Scholar]
- [59].Bosch L, de Haan J, Seijkens T, van Tiel C, Brans M, Pasterkamp G, Lutgens E, de Jager S, Small molecule-mediated inhibition of CD40-TRAF6 reduces adverse cardiac remodelling in pressure overload induced heart failure, Int J Cardiol 279 (2019) 141–144. [DOI] [PubMed] [Google Scholar]
- [60].Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, Wang T, Bedi K, Morley MP, Linares Saldana RA, Bolar NA, McDaid K, Assenmacher CA, Smith CL, Wirth D, June CH, Margulies KB, Jain R, Pure E, Albelda SM, Epstein JA, Targeting cardiac fibrosis with engineered T cells, Nature 573(7774) (2019) 430–433. [DOI] [PMC free article] [PubMed] [Google Scholar]