

Abstract
In this study, a series of naringenin-O-alkylamine derivatives were designed and obtained by introducing an alkylamine fragment into the naringenin skeleton. The in vitro biological activity results revealed that compounds 5f and 7k showed good antioxidant activity with ORAC values of 2.3eq and 1.2eq, respectively. Compounds 5f and 7k were reversible and excellent huAChE inhibitors with IC50 values of 0.91 μM and 0.57 μM, respectively. Moreover, compounds 5f and 7k could inhibit self-induced Aβ1–42 aggregation with 62.1% and 43.8% inhibition rate, respectively, and significantly inhibited huAChE-Aβ1–40 aggregation with 51.7% and 43.4% inhibition rate, respectively. In addition, compounds 5f and 7k were selective metal chelators and remarkably inhibited Cu2+-induced Aβ1–42 aggregation with 73.5% and 68.7% inhibition rates, respectively. Furthermore, compounds 5f and 7k could cross the blood-brain barrier in vitro and displayed good neuroprotective effects and anti-inflammatory properties. Further investigation showed that compound 5f did not show obvious hepatotoxicity and displayed a good hepatoprotective effect by its antioxidant activity. The in vivo study displayed that compound 5f significantly improved scopolamine-induced mice memory impairment. Therefore, compound 5f was a potential multifunctional candidate for the treatment of AD.
Keywords: Alzheimer’s disease, naringenin-O-alkylamine derivatives, multifunctional agents, scopolamine-induced mice memory impairment
1. Introduction
Alzheimer’s disease (AD), accounting for about 70% of all dementia cases, is a chronic, progressive neurodegenerative brain disease in elderly people. According to the World Alzheimer Report, there are more than 50 million people living with dementia worldwide and the figure of AD patients will triple by 20501. Accordingly, AD poses a great problem for global health. Despite many scientific efforts, the mechanism underlying the aetiology of AD is not exactly explained due to its complex and multifactorial characteristics. However, several factors including deficits of acetylcholine (ACh), amyloid-β (Aβ) oligomer deposits, elevated oxidative stress, dyshomeostasis of biometals and neuroinflammation, have been considered as crucial roles in AD onset and progression2.
The cholinergic hypothesis states that the degeneration of cholinergic neurons and the associated loss of cholinergic neurotransmission in the cerebral cortex are responsible for the deterioration of cognitive function observed in the brain of AD patients. There are two types of cholinesterases (ChEs) that can catalyse the hydrolysis of ACh, namely acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). AChE is mainly distributed in the nerve tissues like white matter and grey matter, while BuChE is widely spread in the plasma, liver and muscle tissues3,4. Therefore, selective inhibition of AChE would be beneficial to the treatment of AD, such as the approved anti-AD drugs, donepezil, galanthamine and tacrine3. In addition, evidence display that AChE can accelerate the aggregation of Aβ through peripheral anion site (PAS), producing stable AChE-Aβ complexes, which present more neurotoxicity than single Aβ peptide5. Moreover, Aβ is generated by the proteolytic processing of amyloid precursor protein (APP), including Aβ1–40 and Aβ1–42. Aβ1–42 is more likely to aggregate and form the Aβ oligomer, which triggers neuroinflammation, tau-related neurofibrillary tangles and neuronal degeneration, leading to AD6. Therefore, selective inhibition of AChE and the reduction of Aβ1–42 are promising therapeutic strategies for the treatment of AD.
Accumulation of evidence exhibits that neuroinflammation acts as a fundamental role in AD patients, which is related to microglia, astrocytes and inflammatory factors. Microglia activated by Aβ stimulation produce inflammatory factors and free radicals, such as tumour necrosis factor (TNF-α), interferon γ (INF-γ), ROS and so on7. These neurotoxic substances induce neuroinflammation and neurons damage, and increase Aβ deposition, leading to cognitive dysfunction and neuron loss. Therefore, the anti-inflammatory property might be a potent approach for treating AD.
Numerous studies show that oxidative damage is one of the earliest features of AD. The oxidative homeostasis imbalance in AD brain leads to the abnormal generation of reactive oxygen species (ROS)8. ROS can damage lipids, proteins and nucleic acids, and further accelerates the formation of amyloid and inflammation. Furthermore, the bio-metals (Cu2+, Zn2+, Al3+, Fe2+) have been verified to be related to AD, the concentration of bio-metals in the brain of AD patients is 3–7 times higher than healthy individuals. The metal dyshomeostasis may potentially cause neurotoxic effects by affecting Aβ aggregation and oxidative stress9. Thus, antioxidant activity or metal chelators could be beneficial to the treatment of AD.
Given that AD is a complex disease with multifactorial pathological nature, the multi-target-directed ligand strategy (MTDLs) to develop novel small molecules which can hit two or more AD-relevant complementary targets in the progression of AD, has drawn considerable attention for their advancement in the treatment of AD10–12. In particular, the multifunctional agents with AChE inhibitory potency present the ability to tacking the progression of AD while relieving symptoms13.
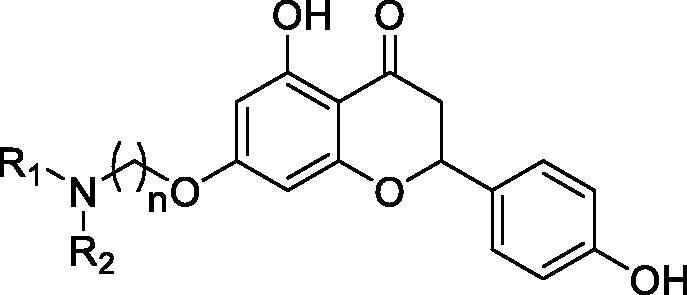
Naringenin, 5,7-dihydroxy-2–(4-hydroxyphenyl)-2,3-dihydrochromen-4-one, is a primary flavanone, present in orange, grape fruit, tangerine, raw lemon peels and raw lime peel. Naringenin possesses significant antioxidant and anti-inflammatory activities, which are beneficial to the treatment of AD14. Studies show that naringenin could improve cholinergic function in vivo due to its antioxidant property, and it attenuates Aβ-induced impairment of learning and memory15. However, the hydroxy of naringenin limited its clinical use as an anti-AD drug due to merely 15% oral bioavailability14. In order to improve bioavailability and develop naringenin as multifunctional agents anti-AD, naringenin carbamate derivatives have been designed and evaluated, however, these derivatives were selective BuChE inhibitors16. Donepezil is a selective AChE inhibitor approved by FDA, and 1-benzylpiperidine is the key pharmacophore, based on this, O-alkylamine fragments have been developed as effective AChE inhibition fragments, our group has developed natural flavonoids as multifunctional agents by introducing O-alkylamine fragments17–20. In our previous work, we had developed a series of apigenin/naringenin/genistein-O-alkylamine derivatives with the length of methylene was 4 based on MTDLs, and the results revealed that apigenin-O-alkylamine derivative candidate TM-4 was a promising multifunctional agent for treating AD20. In order to further develop naringenin skeleton as multifunctional agents, we plan to introduce different tertiary amino groups into the naringenin skeleton by changing the length of methylene to obtain novel naringenin-O-alkylamine derivatives (Figure 1), moreover, apigenin-O-alkylamine derivatives were also synthesised to explore the structure-activity-relationship of naringenin-O-alkylamine derivatives, hoping these derivatives possess multifunctional activity to treat AD, such as AChE inhibitory potency, antioxidant activity, anti-inflammatory property, metal chelation property and neuroprotective effect.
Figure 1.

The design strategy of naringenin-O-alkylamine derivatives.
Herein, a series of novel naringenin-O-alkylamine derivatives are rationally designed by MTDLs. These derivatives are synthesised and evaluated by AChE/BuChE inhibition, antioxidant property, inhibition of Aβ aggregation, metal chelation property, neuroprotective effect and anti-inflammatory effect. The leading compound was further investigated by hepatotoxicity and hepatoprotective activity in vitro, and scopolamine-induced mice memory impairment in vivo.
2. Results and discussion
2.1. Chemistry
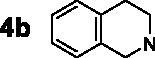
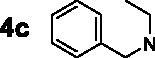
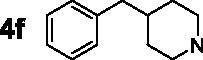
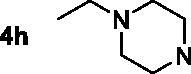
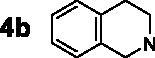
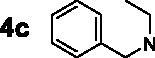
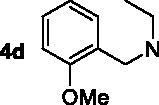
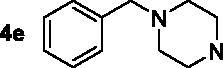
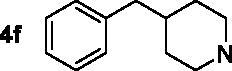
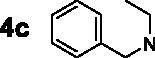
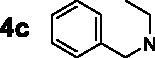
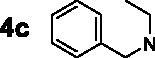
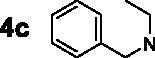
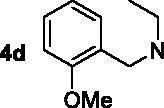
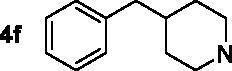
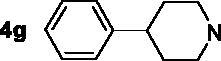
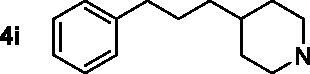
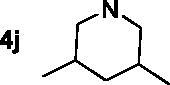
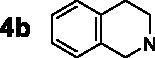
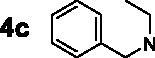
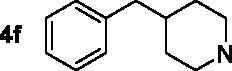
The target compounds 5a–5j, 7a–7l and 10a–10v were synthesised according to Scheme 1–3. Naringenin possessed three hydroxy groups at 4′-, 5-, 7-position, respectively. The 7-OH group showed more acidity than 4′-OH group, and the 5-OH group is the weaker nucleophilic of naringenin due to the formation of intramolecular hydrogen bond with a carbonyl group at 4 position21. Given the difference in reactivity of the OH group, it is easy to regioselective make substitutions at 7-OH or/and 4′-OH position. Synthesis of 7-O-modified naringenin derivatives was displayed in Scheme 1. The starting material naringenin 1 was treated with 1.2 equivalent dibromides (1,4-dibromobutane 2b or 1,5-dibromopentane 2c, 1,6-dibromohexane 2d, 1,10-dibromodecane 2e, and 1,12-dibromododecane, respectively) in the presence of 1.3 equivalent of K2CO3 at 65 °C for 8–12 h to get key intermediates 3a–3c. Subsequently, compounds 3a–3c were reacted with 2.0 equivalent of secondary amines 4a–4f in the presence of 1.3 equivalent of K2CO3 at 65 °C for 6–10 h to obtain 7-O-modified naringenin derivatives 5a–5j with good yields (Table 1).
Scheme 1.

Synthesis of 7-O-modified naringenin derivatives 5a–5j. Reagents and conditions: (i) Br(CH2)nBr (2b–2f), K2CO3, CH3CN, 65 °C, 8–12 h; (ii) R1R2NH (4a–4f), K2CO3, CH3CN, 65 °C 6–10 h.
Table 1.
The property and yield of 7-O-modified naringenin derivatives.
| ||||
|---|---|---|---|---|
| Compound | n | NR1R2 | Property | Yield (%) |
| 5a | 4 |
|
Light yellow oily matter | 76.2% |
| 5b | 4 |
|
Light yellow oily matter | 70.1% |
| 5c | 4 |
|
Light yellow oily matter | 73.9% |
| 5d | 4 |
|
Light yellow oily matter | 74.6% |
| 5e | 4 |
|
Light yellow oily matter | 70.3% |
| 5f | 5 |
|
Light yellow oily matter | 68.7% |
| 5g | 5 |
|
Light yellow oily matter | 60.7% |
| 5h | 6 |
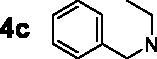
|
Light yellow oily matter | 62.4% |
| 5i | 10 |
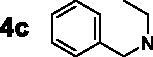
|
Light yellow oily matter | 52.6% |
| 5j | 12 |
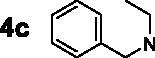
|
Light yellow oily matter | 51.7% |
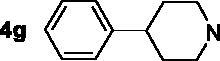
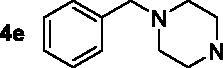
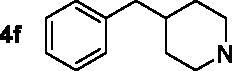
For the synthesis of 7,4′-O-modified naringenin derivatives 7a–7k (Scheme 2), compounds 6a–6c were the key intermediates, which were prepared by using dibromides 2a–2d in the presence of base following the described method. Thus, naringenin was alkylated with 3.0 equivalent of K2CO3 in CH3CN at 65 °C for 10–15 h followed by the addition of 5.5 equivalent dibromides (2a, 2b and 2d) to get intermediates 6a–6c, respectively. Then, the target compounds 7,4′-O-modified naringenin derivatives 7a–7k were obtained through reacting 6a–6c with 3.0 equivalent of secondary amines 4a–4h in the presence of 3.5 equivalent of K2CO3 at 65 °C for 8–12 h (Table 2).
Scheme 2.

Synthesis of 7,4’-O-modified naringenin derivatives 7a–7l. Reagents and conditions: (i) Br(CH2)nBr (2a, 2b and 2d), K2CO3, CH3CN, 65 °C, 10–15 h; (ii) R1R2NH (4a–4h), K2CO3, CH3CN, 65 °C 8–12 h.
Table 2.
The property and yield of 7,4’-O-modified naringenin derivatives 7a–7l.
| ||||
|---|---|---|---|---|
| Compound | n | NR1R2 | Property | Yield (%) |
| 7a | 3 |
|
Light yellow oily matter | 40.9% |
| 7b | 3 |
|
Light yellow oily matter | 57.2% |
| 7c | 3 |
|
Light yellow oily matter | 47.1% |
| 7d | 3 |
|
Light yellow oily matter | 56.8% |
| 7e | 3 |
|
Light yellow oily matter | 52.3% |
| 7f | 4 |
|
Light yellow oily matter | 61.3% |
| 7g | 4 |
|
Light yellow oily matter | 57.4% |
| 7h | 4 |
|
Light yellow oily matter | 43.9% |
| 7i | 4 |
|
Light yellow oily matter | 51.9% |
| 7j | 4 |
|
Light yellow oily matter | 51.7% |
| 7k | 5 |
|
Light yellow oily matter | 40.1% |
| 7l | 6 |
|
Light yellow oily matter | 46.7% |
In order to further explore the structure-activity-relationship of naringenin derivatives, we synthesised a series of 7,4′-O-modified apigenin derivatives 10a–10v in Scheme 3. The experiment procedure was similar to the synthesis of 7,4′-O-modified naringenin derivatives. The starting material apigenin 8 was treated with 5.5 equivalent dibromides 2a–2d in the presence of 3.0 equivalent of K2CO3 in CH3CN at 65 °C for 10–15 h to get compounds 9a–9d. Subsequently, compounds 9a–9d were treated with secondary amines 4a–4j (3.0 equivalent) in the presence of K2CO3 (3.5 equivalent) in CH3CN at 65 °C for 8–12 h (Table 3).
Scheme 3.

Synthesis of 7,4’-O-modified apigenin derivatives 10a–10v. Reagents and conditions: (i) Br(CH2)nBr (2a–2d), K2CO3, CH3CN, 65 °C, 10–15 h; (ii) R1R2NH (4a–4j), K2CO3, CH3CN, 65 °C 8–12 h.
Table 3.
The property and yield of 7,4’-O-modified apigenin derivatives 10a–10v.
| ||||
|---|---|---|---|---|
| Compound | n | NR1R2 | Property | Yield (%) |
| 10a | 3 |
|
Light yellow oily matter | 41.3% |
| 10b | 3 |
|
Light yellow oily matter | 56.8% |
| 10c | 3 |
|
Light yellow oily matter | 43.6% |
| 10d | 3 |
|
Light yellow oily matter | 50.8% |
| 10e | 3 |
|
Light yellow oily matter | 45.7% |
| 10f | 3 |
|
Light yellow oily matter | 43.4% |
| 10g | 4 |
|
Light yellow oily matter | 78.2% |
| 10h | 4 |
|
Light yellow oily matter | 83.7% |
| 10i | 4 |
|
Light yellow oily matter | 65.2% |
| 10j | 4 |
|
Light yellow oily matter | 71.8% |
| 10k | 5 |
|
Light yellow oily matter | 49.3% |
| 10l | 5 |
|
Light yellow oily matter | 38.1% |
| 10m | 5 |
|
Light yellow oily matter | 45.2% |
| 10n | 5 |
|
Light yellow oily matter | 80.2% |
| 10o | 5 |
|
Light yellow oily matter | 73.8% |
| 10p | 5 |
|
Light yellow oily matter | 63.7% |
| 10q | 6 |
|
Light yellow oily matter | 78.9% |
| 10r | 6 |
|
Light yellow oily matter | 76.2% |
| 10s | 6 |
|
Light yellow oily matter | 83.4% |
| 10t | 6 |
|
Light yellow oily matter | 80.7% |
| 10u | 6 |
|
Light yellow oily matter | 60.6% |
| 10v | 6 |
|
Light yellow oily matter | 67.9% |
2.2. Biological activity
2.2.1. Antioxidant potency
The Oxygen Radicals Absorbance Capacity by Fluorescence (ORAC-FL) assay was employed to test the antioxidant activity of target compounds22. The skeleton compounds naringenin and apigenin were also evaluated for comparison purposes, and vitamin E analogue Trolox was used as a standard. As shown in Table 4, the skeleton compounds naringenin and apigenin demonstrated excellent antioxidant activity with ORAC values of 5.2 eq and 5.5 eq, while introducing alkylamine into the naringenin and apigenin skeleton, respectively, the antioxidant activity significantly decreased. In short, the number of hydroxy groups remarkably influenced the antioxidant potency, the derivatives (5a–5j) with two hydroxy groups showed better antioxidant activity than the derivatives with one hydroxy group (compounds 7a–7l and 10a–10v). While, both the length of methylene and the N terminal secondary amines did not produce an obvious influence on antioxidant activity.
Table 4.
AChE/BuChE inhibitory activities and oxygen radical absorbance capacity (ORAC, Trolox equivalents) by target compounds 5a–5j, 7a–7l, 10a–10v and the positive compounds naringenin, apigenin, genistein and rivastigmine.
| Compound | ORACa | IC50 ± SDb (μM) |
SIe | IC50 ± SDb (μM) |
SIe | ||
|---|---|---|---|---|---|---|---|
| ratAChEc | ratBChEd | huAChEf | huBuChEg | ||||
| 5a | 2.2 ± 0.03 | 6.7 ± 0.31 | 20.9 ± 0.68 | 3.1 | NTh | NTh | – |
| 5b | 2.1 ± 0.04 | 5.8 ± 0.19 | 17.4 ± 0.52 | 3.0 | NTh | NTh | – |
| 5c | 2.3 ± 0.02 | 3.9 ± 0.44 | 22.6 ± 0.23 | 5.8 | NTh | NTh | – |
| 5d | 2.2 ± 0.02 | 2.0 ± 0.46 | 18.1 ± 0.56 | 9.1 | NTh | NTh | – |
| 5e | 2.5 ± 0.01 | 9.2 ± 0.72 | 13.2 ± 0.43 | 1.4 | NTh | NTh | – |
| 5f | 2.3 ± 0.03 | 1.7 ± 0.08 | 16.9 ± 0.27 | 9.9 | 0.91 ± 0.02 | 6.3 ± 0.74 | 6.9 |
| 5g | 2.1 ± 0.02 | 2.5 ± 0.33 | 18.7 ± 0.33 | 7.5 | NTh | NTh | – |
| 5h | 2.3 ± 0.03 | 3.3 ± 0.09 | 15.5 ± 0.52 | 4.7 | NTh | NTh | – |
| 5i | 2.1 ± 0.04 | 4.9 ± 0.16 | 14.7 ± 0.62 | 3.0 | NTh | NTh | – |
| 5j | 2.2 ± 0.05 | 6.5 ± 0.09 | 15.9 ± 0.37 | 2.9 | NTh | NTh | – |
| 7a | 1.1 ± 0.02 | 10.1 ± 0.41 | 20.2 ± 0.23 | 2.0 | NTh | NTh | – |
| 7b | 1.2 ± 0.04 | 7.4 ± 0.29 | 18.7 ± 0.37 | 2.5 | NTh | NTh | – |
| 7c | 1.2 ± 0.02 | 6.7 ± 0.16 | 19.4 ± 0.28 | 2.9 | NTh | NTh | – |
| 7d | 1.0 ± 0.03 | 9.3 ± 0.37 | 20.3 ± 0.26 | 2.2 | NTh | NTh | – |
| 7e | 1.5 ± 0.02 | 8.2 ± 0.47 | 15.8 ± 0.62 | 1.9 | NTh | NTh | – |
| 7f | 1.1 ± 0.03 | 5.4 ± 0.16 | 20.7 ± 0.55 | 3.8 | NTh | NTh | – |
| 7g | 1.2 ± 0.04 | 1.4 ± 0.08 | 15.7 ± 0.29 | 11.2 | NTh | NTh | – |
| 7h | 1.0 ± 0.04 | 0.86 ± 0.05 | 16.9 ± 0.34 | 19.7 | NTh | NTh | – |
| 7i | 1.4 ± 0.03 | 6.1 ± 0.21 | 12.3 ± 0.16 | 2.0 | NTh | NTh | – |
| 7j | 1.1 ± 0.03 | 1.4 ± 0.08 | 15.7 ± 0.29 | 11.2 | NTh | NTh | – |
| 7k | 1.2 ± 0.02 | 0.79 ± 0.06 | 14.8 ± 0.36 | 18.7 | 0.57 ± 0.02 | 8.2 ± 0.37 | 14.4 |
| 7l | 1.2 ± 0.01 | 2.2 ± 0.13 | 15.1 ± 0.28 | 6.9 | NTh | NTh | – |
| 10a | 1.1 ± 0.01 | 8.1 ± 0.27 | 16.8 ± 0.36 | 2.1 | NTh | NTh | – |
| 10b | 1.0 ± 0.04 | 4.3 ± 0.38 | 18.6 ± 0.24 | 4.3 | NTh | NTh | – |
| 10c | 1.2 ± 0.03 | 5.5 ± 0.17 | 13.6 ± 0.67 | 2.5 | NTh | NTh | – |
| 10d | 1.1 ± 0.02 | 6.4 ± 0.22 | 15.1 ± 0.49 | 2.4 | NTh | NTh | – |
| 10e | 1.1 ± 0.02 | 7.9 ± 0.26 | 13.8 ± 0.43 | 2.3 | NTh | NTh | – |
| 10f | 1.0 ± 0.04 | 8.6 ± 0.33 | 16.7 ± 0.52 | 1.9 | NTh | NTh | – |
| 10g | 1.2 ± 0.03 | 4.6 ± 0.21 | 17.4 ± 0.35 | 3.8 | NTh | NTh | – |
| 10h | 1.1 ± 0.03 | 0.92 ± 0.06 | 12.9 ± 0.51 | 14.0 | 0.16 ± 0.01 | 3.7 ± 0.06 | 23.1 |
| 10i | 1.5 ± 0.02 | 6.9 ± 0.14 | 10.3 ± 0.19 | 1.7 | NTh | NTh | – |
| 10j | 1.2 ± 0.03 | 1.8 ± 0.04 | 15.7 ± 0.24 | 8.7 | NTh | NTh | – |
| 10k | 1.1 ± 0.02 | 0.81 ± 0.05 | 13.3 ± 0.27 | 16.4 | 0.13 ± 0.01 | 4.5 ± 0.12 | 34.6 |
| 10l | 1.3 ± 0.03 | 0.62 ± 0.05 | 14.4 ± 0.32 | 23.2 | 0.09 ± 0.002 | 2.2 ± 0.18 | 24.4 |
| 10m | 1.2 ± 0.02 | 1.3 ± 0.08 | 16.6 ± 0.27 | 12.8 | NTh | NTh | – |
| 10n | 1.1 ± 0.04 | 3.6 ± 0.11 | 17.9 ± 0.46 | 4.9 | NTh | NTh | – |
| 10o | 1.2 ± 0.03 | 4.1 ± 0.63 | 16.1 ± 0.25 | 3.9 | NTh | NTh | – |
| 10p | 1.1 ± 0.02 | 6.2 ± 0.54 | 13.8 ± 0.37 | 2.2 | NTh | NTh | – |
| 10q | 1.0 ± 0.02 | 4.2 ± 0.15 | 12.2 ± 0.28 | 2.9 | NTh | NTh | – |
| 10r | 1.2 ± 0.04 | 1.7 ± 0.21 | 14.1 ± 0.22 | 8.3 | NTh | NTh | – |
| 10s | 1.1 ± 0.03 | 2.6 ± 0.33 | 16.9 ± 0.54 | 6.5 | NTh | NTh | – |
| 10t | 1.0 ± 0.02 | 5.9 ± 0.37 | 19.3 ± 0.67 | 3.3 | NTh | NTh | – |
| 10u | 1.2 ± 0.03 | 6.3 ± 0.21 | 17.2 ± 0.45 | 2.7 | NTh | NTh | – |
| 10v | 1.3 ± 0.03 | 8.1 ± 0.49 | 14.8 ± 0.29 | 1.8 | NTh | NTh | – |
| naringenin | 5.2 ± 0.29 | >50 | >50 | – | NTh | NTh | – |
| apigenin | 5.6 ± 0.52 | >50 | >50 | – | NTh | NTh | – |
| donepezil | NTh | 0.017 ± 0.003 | 16.2 ± 0.31 | 953 | 0.013 ± 0.0004 | 2.7 ± 0.06 | 208 |
aResults are expressed as μM of Trolox equivalent/μM of tested compound. bValues are expressed as the mean ± standard deviation of the mean by 3 independent experiments in triplicate. cFrom 5% rat cortex homogenate. dBuChE from rat serum. eSelectivity Index = IC50 (BuChE)/IC50 (AChE). fFrom human erythrocytes. gFrom human serum. hNT = not tested.
2.2.2. AChE and BChE inhibition of target compounds
The inhibitory potency of the synthesised compounds 5a–5j, 7a–7l and 10a–10v against ratAChE (from rat cortex homogenate) and ratBuChE (from rat serum) were evaluated through the slightly modified Ellman’s method17. Donepezil served as a positive compound, and naringenin and apigenin were also tested for comparative purposes. The results were listed in Table 4. Firstly, the 7-O-modified naringenin derivatives were synthesised and evaluated, the screening data in Table 4 showed that compounds 5a–5j displayed good ratAChE inhibitory activity and weak ratBuChE inhibitory potency, indicating that compound 5a–5j were selective ratAChE inhibitors. In addition, both the length of methylene and the N terminal secondary amines produced significantly influence on the ratAChE inhibitory activity. When the length of methylene was 4, compound 5a with piperidine fragment showed good ratAChE inhibitory activity with IC50 value of 6.7 μM. Replacing piperidine fragment of 5a with 1,2,3,4-tetrahydroisoquinoline to get compound 5b, the ratAChE inhibitory activity slightly increased to 5.8 μM. Then opening the ring of 1,2,3,4-tetrahydroisoquinoline of compound 5b to obtain compound 5c with N-ethylbenzylamine fragment, the ratAChE inhibitory activity significantly increased to 3.9 μM. And then adding methoxy group at 2-position of N-ethylbenzylamine to get compound 5d with N-(2-methoxybenzyl)ethanamine fragment, the ratAChE inhibitory activity remarkably increased to 2.0 μM. While, replacing N-(2-methoxybenzyl)ethanamine of 5d with phenylpiperazine fragment to afford compound 5e, the ratAChE inhibitory activity sharply decreased to 9.2 μM. When the length of methylene was 5, compound 5f with N-ethylbenzylamine fragment presented the best ratAChE inhibitory activity with IC50 value of 1.7 μM, and replacing N-ethylbenzylamine of 5f with benzylpiperidine fragment to get compound 5g, the ratAChE inhibitory activity slightly decreased to 2.5 μM. Correspondingly, when the N terminal secondary amines were fixed and were N-ethylbenzylamine fragments, the potencies to inhibit AChE were in the order n = 4 (5c) < n = 5 (5f) > n = 6 (5h) > n = 10 (5i) > n = 12 (5j), and the optimal length of methylene was 5.
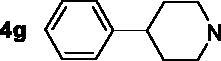
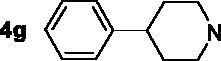
7,4′-O-modified naringenin derivatives 7a–7l were also synthesised and evaluated to explore the structure-activity-relationship (SAR). The data were listed in Table 4, compounds 7a–7l showed good ratAChE inhibitory potency and weak ratBuChE inhibitory potency, displaying that all the compounds 7a–7l were selective ratAChE inhibitors. Similarly, both the length of methylene and the N terminal secondary amines created significantly influence on the ratAChE inhibitory activity. When the length of methylene was 3, compound 7a with 1,2,3,4-tetrahydroisoquinoline fragment showed moderate ratAChE inhibitory activity with an IC50 value of 10.1 μM. Opening the ring of 1,2,3,4-tetrahydroisoquinoline of compound 7a to obtain compound 7b with N-ethylbenzylamine fragment, the ratAChE inhibitory activity significantly increased to 7.4 μM. Replacing N-ethylbenzylamine fragment of 7b with benzylpiperidine to get compound 7c, the ratAChE inhibitory activity slightly increased to 6.7 μM. When replacing benzylpiperidine of compound 7c with 4-phenylpiperidine to obtain compound 7d, the ratAChE inhibitory activity decreased to 9.3 μM. And replacing benzylpiperidine of compound 7c with ethylpiperazine to get compound 7e, the ratAChE inhibitory activity decreased to 8.2 μM. When the length of methylene was 4, a similar phenomenon was also observed, such as compound 7f with 1,2,3,4-tetrahydroisoquinoline (IC50 = 5.4 μM) < compound 7g with N-ethylbenzylamine (IC50 = 1.4 μM) < compound 7j with benzylpiperidine (IC50 = 1.3 μM). In addition, when replacing N-ethylbenzylamine fragment of 7g with N-(2-methoxybenzyl)ethanamine to obtain compound 7h, the ratAChE inhibitory activity increased to 0.86 μM. And replacing benzylpiperidine fragment of 7j with benzylpiperazine to get compound 7i, the ratAChE inhibitory activity sharply decreased to 6.1 μM. Furthermore, when the N terminal secondary amines were fixed and were N-ethylbenzylamine fragment, the potency to inhibit ratAChE was in order n = 3 (7b, 7.4 μM) < n = 4 (7g, 1.4 μM) < n = 5 (7k, 0.79 μM) > n = 6 (7l, 2.2 μM). Thus, the optimal length of methylene was 5. Overall speaking, 7,4′-O-modified naringenin derivatives showed better ratAChE inhibitory potency than 7-O-modified naringenin derivatives, such as, 5b < 7f; 5c < 7g; 5d < 7f; 5e < 7i; 5f < 7k; 5h < 7f.
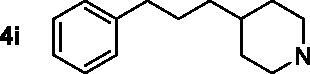
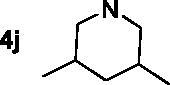
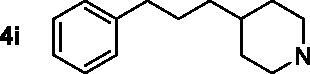
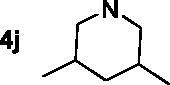
In order to further explore the SAR, a series of 7,4′-O-modified apigenin derivatives 10a–10v were synthesised by replacing the naringenin skeleton of compounds 7a–7l with apigenin skeleton. The data were shown in Table 4, all the 7,4′-O-modified apigenin derivatives 10a–10v showed good selective ratAChE inhibitory potency, which was consistent with agreed with the 7,4′-O-modified naringenin derivatives 7a–7l. Likewise, both the length of methylene and the N terminal secondary amines significantly influenced the ratAChE inhibitory activity. When the length of methylene was 3, the potency of secondary amines to inhibit ratAChE was in order N-ethylbenzylamine (10b, 4.3 μM) > benzylpiperidine (10c, 5.5 μM) > 4-phenylpiperidine (10d, 6.4 μM) > 1,2,3,4-tetrahydroisoquinoline (10a, 7.4 μM) > 4–(3-phenylpropyl)piperidine (10e, 8.6 μM) > 3,5-dimethylpiperidine (10f, 8.6 μM). A similar phenomenon was also observed when the length of methylene was 4, such as N-ethylbenzylamine (10h, 0.92 μM) > benzylpiperidine (10j, 1.8 μM) > 1,2,3,4-tetrahydroisoquinoline (10g, 4.6 μM). Moreover, replacing benzylpiperidine fragment of 10j with benzylpiperazine to get compound 10i, the ratAChE inhibitory activity sharply decreased to 6.9 μM. When the length of methylene was 5, the similar results were also observed, N-ethylbenzylamine (10k, 0.81 μM) > benzylpiperidine (10m, 1.3 μM) > 4-phenylpiperidine (10n, 3.6 μM) > 4–(3-phenylpropyl)piperidine (10o, 4.1 μM) > 3,5-dimethylpiperidine (10p, 6.2 μM), and when replacing N-ethylbenzylamine of 10k with N-(2-methoxybenzyl)ethanamine to obtain compound 10l, the ratAChE inhibitory activity increased to 0.62 μM. When the length of methylene was 6, the potency of secondary amines to inhibit ratAChE was in order N-ethylbenzylamine (10r, 1.7 μM) > benzylpiperidine (10s, 2.6 μM) > 1,2,3,4-tetrahydroisoquinoline (10q, 4.2 μM) > 4-phenylpiperidine (10t, 5.9 μM) > 4–(3-phenylpropyl)piperidine (10u, 6.3 μM) > 3,5-dimethylpiperidine (10v, 8.1 μM). Overall speaking, the derivatives with N-ethylbenzylamine, N-(2-methoxybenzyl)ethanamine and benzylpiperidine fragment presented good ratAChE inhibitory potency. Furthermore, when the the N terminal secondary amines were fixed, the potency of the length of methylene to inhibit ratAChE was in order n = 5 > n = 4 > n = 6 > n = 3, for example, when the N terminal secondary amine was N-ethylbenzylamine, n = 5 (10k, 0.81 μM) > n = 4 (10h, 0.92 μM) > n = 6 (10r, 1.7 μM) > n = 3 (10b, 4.3 μM); when the N terminal secondary amine was benzylpiperidine, n = 5 (10m, 1.3 μM) > n = 4 (10j, 1.8 μM) > n = 6 (10s, 2.6 μM) > n = 3 (10c, 5.5 μM).
Based on the above results, compounds 5f, 7k, 10h, 10k and 10l presented good ratAChE inhibitory potency from the 7-O-modified naringenin derivatives 5a–5j, 7,4′-O-modified naringenin derivatives 7a–7l and 7,4′-O-modified apigenin derivatives 10a–10v, respectively, which were selected to re-evaluate using human erythrocytes AChE (huAChE) and human serum BuChE (huBChE). The results were listed in Table 4, all the representative compounds presented excellent huAChE inhibitory potency and good huBuChE inhibitory potency. As a whole, 7,4′-O-modified apigenin derivatives displayed the best huAChE inhibitory activity, such as 10h (IC50 = 0.16 μM), 10k (IC50 = 0.13 μM) and 10l (IC50 = 0.09 μM). While, in our previous work, we have done much work for the representative 7,4′-O-modified apigenin derivative by the in vitro and in vivo assay. And the 7,4′-O-modified apigenin derivatives were synthesised for comparison purposes to explore the SAR of 7,4′-O-modified naringenin derivatives. So, compounds 5f and 7k were selected to perform further investigation.
2.2.3. HuAChE reversibility of inhibition by 5f and 7k
To determine whether compounds 5f and 7k were reversible huAChE inhibitors, we investigated the recovery of huAChE inhibitors inhibition after dilution (Figure 2A,B)20. In Figure 2(A), when the positive compound donepezil dilute to 0.1 × IC50, the huAChE activity increased to 9.0%, indicating a reversible huAChE inhibitor, which was consistent with our previous work. When the concentration of compounds 5f and 7k was diluted to 0.1 × IC50, the huAChE activity increased to 6.9% and 7.8%, respectively, which was in keeping with donepezil. In order to further explore the reversibility of inhibition, the recovery of huAChE inhibitors inhibition after dilution was carried out with time monitoring. As displayed in Figure 2(B), the huAChE activity was restored to 95.6% with 0.1× IC50 donepezil at 60 min, and the 0.1 × IC50 compounds (5f and 7k) were restored huAChE activity to 89.8% and 96.8%, respectively. The results showed that both 5f and 7k were reversible huAChE inhibitors.
Figure 2.

(A) huAChE recovery after preincubation of compounds 5f and 7k diluted to 1× or 0.1 × IC50, compared to donepezil diluted, and undiluted inhibition. (B) huAChE recovery of donepezil, 5f and 7k diluted to 0.1 × IC50, were monitored with time at room temperature for 120 min. con. = control, done. = donepezil. Data were expressed as the mean ± SEM by three independent experiments.
2.2.4. Enzemye kinetic study on of huAChE
An enzyme kinetic study was applied to determine the mechanism of AChE inhibition for compounds 5f and 7k. The lineweaver-Burk double reciprocal plot was used to define the inhibition by plotting the initial velocities of the substrate at the y-axis and increasing concentrations of the substrate at the x-axis17,19. As shown in Figure 3(A), decreased Vmax and increased Ki values corresponding to increasing concentration of tested compound 5f exhibited a mixed-type of AChE inhibition. As shown in Figure 3(B), replots of the slope versus concentration of compound 5f gave an estimate of the inhibition constant and the Ki value was 0.74 μM. Similarly, Figure 3(C) showed that compound 7k also demonstrated a mixed-type of AChE inhibition, and the Ki value was 0.39 μM (Figure 3(D)). The results exhibited that compounds 5f and 7k were mixed-type of AChE inhibitors and were able to simultaneously bind both catalytic active site (CAS) and peripheral anionic site (PAS) of AChE.
Figure 3.

Enzyme kinetic study on the mechanism of huAChE inhibition by compounds 5f and 7k. (A) Overlaid Lineweaver-Burk reciprocal plots of AChE initial velocity at increasing acetylthiocholine concentration in the absence and in the presence of 5f are displayed. (B) Representing the plots of slope versus the concentration of 5f for determining the inhibition constants Ki. (C) Overlaid Lineweaver-Burk reciprocal plots of AChE initial velocity at increasing acetylthiocholine concentration in the absence and in the presence of 7k are displayed. (D) Representing the plots of slope versus the concentration of 7k for determining the inhibition constants Ki.
2.2.5. Molecular docking of compounds 5f and 7k with AChE
The possible interacting mechanism of compounds 5f and 7k with huAChE (PDB code: 4ey4) was carried out using AUTODOCK 4.2 package20. It had been verified that amino acid residues Tyr72, Asp74, Trp86, Tyr124, Trp286, Tyr337, Phe295 and Phe297 were the key active sites residues of huAChE23. As displayed in 5f-huAChE complex (Figure 4), the carbonyl group at 4-position interacted with the hydroxy group at 5-position via one intramolecular hydrogen bonding, and the O atom of the hydroxy group interacted with Phe295 via one intermolecular hydrogen bonding. The benzene ring of naringenin interacted with Trp286 and Tyr341 via one Pi-Pi interaction, respectively. The benzene ring at 2-position interacted with key residue Trp286 via two Pi-Pi interactions. The benzene ring of the alkylamine side chain at 7-position interacted with key residue Trp86 via two Pi-Pi interactions and one Sigma-Pi interaction. Besides, some hydrophobic interactions were presented between compound 5f and residues (such as Trp86, Tyr124, Tyr337, Phe295, Tyr341 and Trp286). The above interactions offered a possible mechanism for its high AChE inhibitory activity.
Figure 4.

(A) Compound 5f (green stick) acted on residues in the binding site of huAChE (PDB code: 4ey4). (B) 2 D docking model of 5f with huAChE. (C) 3 D docking model of 5f with huAChE.
In 7k-huAChE complex (Figure 5), compound 7k simultaneously occupied the entire huAChE enzymatic catalytic site (CAS), the mid-gorge sites and the peripheral anionic site (PAS). compound 7k interacted with huAChE through multi-interactions. The carbonyl group at 4-position interacted with the hydroxy group at 5-position via one intramolecular hydrogen bonding. The H atom of the hydroxy group interacted with Ser293 via one intermolecular hydrogen bonding, and the O atom of the hydroxy group interacted with Ser293 via two intermolecular hydrogen bonding. The benzene ring at 2-position interacted with key residue Phe297 via one Sigma-Pi interaction. The benzene ring of the alkylamine side chain at 4′-position interacted with key residue Trp286 via two Pi-Pi interactions. The benzene ring of the alkylamine side chain at 7-position interacted with key residue Trp286 via two Pi-Pi interactions and simultaneously interacted with key residue Tyr337 via one Pi-Pi interaction. Furthermore, some hydrophobic interactions could also be observed between the ligand 7k and Tyr341, Trp286, Ser293, Phe297, Tyr124 and Tyr337. In general, the interactions between compound 7k and huAChE provided a reasonable explanation for its potent huAChE inhibitory potency.
Figure 5.

(A) Compound 7k (green stick) acted on residues in the binding site of huAChE (PDB code: 4ey4). (B) 2 D docking model of 7k with huAChE. (C) 3 D docking model of 7k with huAChE.
2.2.6. Molecular dynamics (MD) simulations
The stability of docked binding pose of the compound 5f-AChE complex and 7k-AChE were analysed by molecular dynamics simulation analysis using Amber 1624. Figure 6 showed the root means square deviations (RMSD) analysis of compounds 5f and 7k with the amino acid residues of AChE, respectively. The results demonstrated that the RMSDs of all the replicas for the six simulated systems presented relatively stable fluctuations after 50 ns of the MSMD simulations, displaying that the six simulated systems basically reach equilibrium. The binding free energies of compounds 5f and 7k towards huAChE calculated using MM-PBSA were displayed in Table 5, and have values of −51.98 and −64.42 kcal/mol, respectively, which were mainly contributed by Vander Waals forces, electrostatic interactions and non-polar solvation energies24. Furthermore, Figure 7 showed the key residues and interaction modes of compounds 5f and 7k with huAChE. In Figure 7(A), the hydroxy group of naringenin skeleton formed one intermolecular hydrogen bonding with the key amino acid Phe295 (2.2 Å). Figure 7(B) displayed the interactions modes of 7k with AChE, and three intermolecular hydrogen bonding were observed. The hydroxy group of naringenin skeleton interacted with key amino acid residue Ser293 via two intermolecular hydrogen-bonding interactions with the distance of 1.9 Å and 2.5 Å, respectively. Moreover, the carbonyl of the naringenin skeleton formed one intermolecular hydrogen bonding interaction with the key amino acid Arg296 (2.4 Å).
Figure 6.

RMSD analysis of compounds 5f (black stick) and 7k (red stick).
Table 5.
The binding free energy and components of 5f and 7k (kcal/mol)
| Terms | ΔEvdw | ΔEele | ΔEegb | ΔEesurf | ΔGgas | ΔGsolv | ΔGbind |
|---|---|---|---|---|---|---|---|
| 5f | −59.10 (0.30) | −15.10 (0.42) | 27.40 (0.38) | −5.18 (0.02) | −74.20 (0.57) | 22.22 (0.37) | −51.98 (0.45) |
| 7k | −82.24 (0.42) | −16.34 (0.55) | 40.95 (0.45) | −6.80 (0.03) | −98.57 (0.79) | 34.15 (0.45) | −64.42 (0.85) |
Figure 7.

(A) The docking model for 5f into the protein crystal structure of huAChE (PDB code: 4ey4). (B) The docking model for 7k into the protein crystal structure of huAChE (PDB code: 4ey4).
2.2.7. Propidium iodide displacement assay
From the above results, compounds 5f and 7k interacted with PAS residues of huAChE. Thus, the propidium iodide displacement assay was employed to evaluate the affinity of compounds 5f and 7k (10 and 50 μM), and donepezil was also tested as a positive compound25–27. As listed in Table 6, compound 5f showed higher displacement of propidium iodide (10 μM = 25.7%, 50 μM = 36.9%) than donepezil (10 μM = 20.9%, 50 μM = 33.4%). Correspondingly, compound 7k demonstrated significant displacement of propidium iodide (10 μM = 23.1%, 50 μM = 34.3%). The results were in agreement with the computational study.
Table 6.
The data of propidium iodide displacement assay towards compounds 5f, 7k and doneipezil.
| Compound | Propidium iodide displacement from AChE PAS (% inhibition)a |
|
|---|---|---|
| 10 μM | 50 μM | |
| 5f | 25.7 ± 1.9 | 36.9 ± 3.5 |
| 7k | 23.1 ± 1.2 | 34.3 ± 2.1 |
| Donepezil | 20.9 ± 2.7 | 33.4 ± 2.6 |
aPropidium iodide displacement assay was performed on AChE to test the ability of compounds 5f and 7k to displace propidium with reference to the donepezil at 10 and 50 μM. Data are presented as the mean ± SEM of three independent experiments.
2.2.8. Effects on self-induced Aβ1–42 aggregation
In order to evaluate the inhibition and disaggregation effects of compounds 5f and 7k on self-induced Aβ1–42 aggregation, the Thioflavin T (ThT) fluorescence method was applied17,28. Curcumin was also tested as the positive compound. As listed in Table 7, compounds 5f and 7k significantly inhibited self-induced Aβ1–42 aggregation at 25 μM with 62.1% and 43.8% inhibition rates, respectively, compared with curcumin (45.9%).
Table 7.
Effects on self-induced/Cu2+-induced Aβ1–42 aggregation by compounds 5f, 7k and curcumin.
| Compound | Inhibition of Aβ1–42 aggregationa |
Inhibition of huAChE-induced Aβ1–40 aggregation (%)d | |
|---|---|---|---|
| Self-induced (%)b | Cu2+-induced (%)c | ||
| 5f | 62.1 ± 0.38 | 73.5 ± 0.25 | 51.7 ± 0.75 |
| 7k | 43.8 ± 0.51 | 68.7 ± 0.46 | 43.4 ± 0.43 |
| Donepezil | N.T.e | N.T.e | 28.2 ± 0.29 |
| Curcumin | 45.9 ± 0.67 | 50.6 ± 0.33 | N.T.e |
aThe inhibition effects of Aβ1–42 aggregation was tested using thioflavin-T fluorescence assay, data are expressed as the mean ± SEM by three independent experiments. bInhibition of self-induced Aβ1–42 aggregation, both the concentration of tested compounds and Aβ1–42 were 25 μM. cInhibition of Cu2+-induced Aβ1–42 aggregation, both the tested compounds and Aβ1–42 were 25 μM. d The inhibition huAChE-induced Aβ1–40 aggregation was tested using ThT fluorescence assay, the concentration of tested inhibitor and Aβ1–40 was 100 and 230 μM, respectively, whereas the Aβ1–40/AChE ratio was equal to 100/1. Data are expressed as the mean ± SEM through three independent experiments. e N.T. = not tested.
2.2.9. Effects on huAChE-induced Aβ1–40 aggregation by compounds 5f and 7k
Accumulation of evidence displayed that PAS of AChE could bind to the Aβ, and accelerated the formation of amyloid fibrils. So, inhibiting the PAS of AChE could significantly affect Aβ aggregation3. According to the results from the enzyme kinetic study, docking study and propidium iodide displacement assay compounds 5f and 7k could bind the PAS of AChE. Herein, the inhibition potency of compounds 5f and 7k on huAChE-induced Aβ1–40 was evaluated by employing the ThT assay29. As displayed in Table 7, compounds 5f and 7k significantly inhibited huAChE-induced Aβ1–40 aggregation 51.7% and 43.4%, respectively, which was better than donepezil (28.2%).
2.2.10. Metal-chelating property
The chelating property of compounds 5f and 7k were determined by UV-visual spectrometry and biometals such as Cu2+, Zn2+, Al3+ and Fe2+ were applied19,20. As shown in Figure 8(A), adding CuCl2 and AlCl3 to a solution of 5f, the maximum absorption wavelength shifted from 331 nm to 386 nm and 388 nm, respectively, while, when adding FeSO4 and ZnCl2 to the solution of 5f, respectively, the maximum absorption at 331 nm did not present obvious shift, exhibiting the formation of 5f-Cu2+ and 5f-Al3+ complex. The stoichiometry of the 5f-Cu2+ complex was determined using the molar ratio method by preparing solutions of compound 5f with increasing amounts of CuCl2 at 386 nm. According to the screening data in Figure 8(B), the absorbance linearly increased at first and then tended to be stable. The two straight lines intersected at a mole fraction of 1.1, revealing a 1:1 stoichiometry for complex 5f-Cu2+.
Figure 8.

(A) The UV spectrum of compound 5f (37.5 μM) alone or in the presence of CuCl2, FeSO4, ZnCl2 and AlCl3 (37.5 μM) in methanol; (B) Determination of the stoichiometry of complex-Cu2+ by using molar ratio method at 386 nM.
Furthermore, when adding CuCl2, AlCl3, FeSO4 and ZnCl2 to the solution of 7k, respectively, the results were shown in Figure 9(A). The maximum absorption wavelength shifted from 330 nm to 385 nm after adding Cu2+, while, the maximum absorption wavelength did not show an obvious change after FeSO4, AlCl3 and ZnCl2 to the solution of 7k, respectively, exhibiting the formation of the 7k-Cu2+ complex. Further, the stoichiometry of the 7k-Cu2+ complex was determined using the molar ratio method at 385 nm. As displayed in Figure 9(B), the absorbance linearly increased at first and then tended to be stable. The two straight lines intersected at a mole fraction of 1.1, revealing a 1:1 stoichiometry for complex 7k-Cu2+. Therefore, the results showed that compounds 5f and 7k were selective metal chelation agents.
Figure 9.

(A) The UV spectrum of compound 7k (37.5 μM) alone or in the presence of CuCl2, FeSO4, ZnCl2 and AlCl3 (37.5 μM) in methanol; (B) Determination of the stoichiometry of complex-Cu2+ by using molar ratio method at 385 nM. The final concentration of 7k was 37.5 μM, and the final concentration of Cu2+ ranged from 3.75 to 93.75 μM.
2.2.11. Effects on Cu2+-induced Aβ1–42 aggregation by compounds 5f and 7k
Studies indicated that Cu2+ could cause Aβ aggregation in solution and accelerated Aβ aggregation. Compounds 5f and 7k were selective metal chelators based on the above results, in order to evaluate the effect of compounds 5f and 7k on Cu2+-induced Aβ1–42 aggregation, the ThT fluorescence assay and transmission electron microscopy (TEM) method were employed19,20. Curcumin was also tested as a referenced compound. As listed in Table 7, compounds 5f and 7k significantly inhibited Cu2+-induced Aβ1–42 aggregation with 73.5% and 68.7% inhibition rates, respectively, which were better than that with curcumin (50.6% inhibition rate). Furthermore, the TEM images further supplemented the data of ThT assay. As shown in Figure 10, the fresh Aβ1–42 had aggregated into fibrils after adding Cu2+, while only small fibre aggregates were observed after treating with curcumin. A similar phenomenon was also observed after adding compounds 5f and 7k. Therefore, both the ThT assay and TEM images displayed that the metal chelators 5f and 7k could inhibit Cu2+-induced Aβ1–42 aggregation.
Figure 10.

TEM images of Aβ species from inhibition experiments.
2.2.12. Blood − brain barrier permeation assay in vitro
Blood brain barrier (BBB) permeability played an important role in the development of central nervous system disease drugs, especially AD. In order to investigate the BBB permeability of compounds 5f and 7k, the parallel artificial membrane permeation assay of the blood − brain barrier (PAMPA-BBB) was employed30,31. First of all, 11 reported drugs were collected to verify this assay, and the following ranges of permeability Pe (×10−6 cm/s) were established as our previous work reported: Pe < 1.61 demonstrated weak BBB permeation; 1.61 < Pe < 3.44 exhibited uncertain BBB permeation; Pe > 3.44 displayed high BBB permeation. As listed in Table 8, compounds 5f and 7k showed good BBB permeation with 6.9 × 10−6 cm/s and 11.7 × 10−6 cm/s permeability, respectively, which were similar with testosterone and diazepam, exhibiting that compounds 5f and 7k could cross BBB through passive diffusion.
Table 8.
The predictive permeation of compounds 5f and 7k by PAMPA-BBB assay.
| Compounda | Pe (×10−6 cm/s)b | Prediction |
|---|---|---|
| Testosterone | 17.8 ± 0.93 | CNS+ |
| Verapamil | 16.3 ± 0.82 | CNS+ |
| Clonidine | 5.8 ± 0.21 | CNS+ |
| Norfloxacin | 0.13 ± 0.01 | CNS- |
| 5f | 6.9 ± 0.36 | CNS+ |
| 7k | 11.7 ± 0.54 | CNS+ |
aCompounds 5f and 7k were dissolved in DMSO at 5 mg/mL and diluted with PBS/EtOH (70:30). The final concentration of compounds was 100 μg/mL. bValues are expressed as the mean ± SD of three independent experiments.
2.2.13. Neuroprotective effect
The neuroprotective effect of compounds 5f and 7k on H2O2-induced PC12 cells injury was evaluated using MTT assay20. As presented in Figure 11(A), compounds 5f and 7k did not show any cytotoxicity at 40 μM, respectively, indicating a widely safe range. Further, as shown in Figure 11(B), when PC12 cells were exposed to 100 μM H2O2, the cell viability sharply declined to 54.6% (p < 0.01) compared with the untreated group. When adding 100 μM Vitamine E (VE) into the PC12 cells, the cell viability increased to 67.6% (p < 0.01). Under the same conditions, when adding compound 5f (2.5, 5.0 and 10.0 μM) into PC12 cells, the cell viability significantly increased to 62.9% (p < 0.05), 68.2% (p < 0.01) and 73.4% (p < 0.01), respectively, in a dose-dependant manner. Similarly, when treating with compound 7k (2.5, 5.0 and 10.0 μM), the cell viability increased to 58.3% (p < 0.05), 63.7% (p < 0.05) and 69.8% (p < 0.01), respectively, in a dose-dependant manner. The results showed that compounds 5f and 7k showed significant neuroprotective effects on H2O2-induced PC12 cells injury, and compound 5f displayed a better neuroprotective effect than 7k.
Figure 11.

Cell viability was tested by MTT assay. (A) Cytotoxicity of compounds 5f and 7k on PC12 cells. (B) Attenuation of H2O2-induced PC12 cell injury by compounds 5f and 7k. values were expressed as mean ± SD by three independent experiments. ##p < 0.01 vs control; **p < 0.01, *p < 0.05 vs H2O2 group.
2.2.14. Anti-inflammatory property
Compounds 5f and 7k were selected to test the anti-inflammatory potency by measuring the production of inflammatory mediators TNF-α and NO in LPS-induced BV-2 cells32,33.
Cytotoxicity of compounds 5f and 7k on BV-2 cells. Firstly, the cytotoxicity of compounds 5f and 7k were tested using an MTT assay. As displayed in Figure 12, the cell viability did not show obvious change after adding compounds 5f or 7k (1 μM, 3 μM and 9 μM), showing that compounds 5f and 7k did not any toxic on BV-2 cells.
Evaluation of NO and TNF-α in LPS-stimulated BV-2 cells. In inhibition of LPS-induced, NO production was tested through the Griess reaction method. As shown in Figure 13, the release volume of NO did not produce significant change after adding compounds 5f or 7k (1 μM, 3 μM and 9 μM), showing that compounds 5f and 7k did not produce an effect on the release of NO in BV-2 cells. When BV-2 cells were exposed to 1 μg/mL LPS, the release volume of NO sharply increased. In addition, when pre-treatment with compound 5f (1 μM, 3 μM and 9 μM), leading to a remarkable reduction of LPS-induced NO production with 37.6%, 52.1% and 63.8% inhibition rate, respectively, in a dose-dependent manner. Similarly, when pre-treatment with 7k (1 μM, 3 μM and 9 μM), the percent inhibition rate was 26.7%, 36.2% and 48.5%, respectively, in a dose-dependent manner. In addition, to further investigate the effects of compounds 5f and 7k on LPS-induced TNF-α production in BV-2 cells, the enzyme-linked immunosorbent assay (ELISA) was used. As shown in Figure 14, when BV-2 cells were exposed to 1.0 μg/mL LPS, the levels of TNF-α significantly increased to 1615 pg/mL (p < 0.01) compared with the untreated group (130 pg/mL). When treatment with compound 5f (1 μM, 3 μM and 9 μM), the TNF-α production decreased to 1130 pg/mL (p < 0.05), 728 pg/mL (p < 0.01), 394 pg/mL (p < 0.001), respectively, and the inhibitory rate were 33.5%, 57.2% and 76.8%, respectively, in a dose-dependent manner. When treatment with compound 7k (1 μM, 3 μM and 9 μM), the TNF-α production decreased to 1295 pg/mL (p < 0.05), 915 pg/mL (p < 0.001), 553 pg/mL (p < 0.01), respectively, and the inhibition rates of 7k were 23.8%, 46.2% and 67.5%, respectively, in a dose-dependent manner. Therefore, compounds 5f and 7k reduced the release of NO and suppressed TNF-α production in LPS-induced BV-2 cells, exhibiting that compounds 5f and 7k displayed good anti-neuroinflammatory potency in vitro.
Figure 12.

The cell viability of compounds 5f and 7k on the BV-2 cells was determined using MTT assay. The data are expressed as the mean ± SD by three independent experiments.
Figure 13.

Effects of compounds 5f and 7k on NO release in BV-2 cells and LPS-stimulated BV-2 cells. Data were expressed as mean ± SD through three independent experiments. con. = control; mod. = model. ##p < 0.01 vs control; ***p < 0.01, **p < 0.01, *p < 0.05 vs LPS-induced group.
Figure 14.

Effects of compounds 5f and 7k on TNF-α release in LPS-stimulated BV-2 cells. Data were expressed as mean ± SD through three independent experiments. ##p < 0.01 vs control; ***p < 0.01, **p < 0.01, *p < 0.05 vs LPS-induced group.
2.2.15. Theoretical prediction of the ADME properties
The Molinspiration property program was applied to predict the druglike properties of compounds 5f and 7k34. The items included Log P, MW, TPSA, n-ON, n-OHNH, n-violations n-rotb and volume. The data was listed in Table 9, compound 5f did not break Lipinski’s rule of five, while compound 7k broke Lipinski’s rule of five. Therefore, considering the biological activity in vitro and prediction of druglike property, compound 5f was a promising candidate and deserving further investigation.
Table 9.
Theoretical prediction of the ADME properties of compounds 5f and 7k.
| Compounda | Log P | MW | TPSA (Å2) | n-ON | n-OHNH | n-violations | n-rotb | Volume (Å3) |
|---|---|---|---|---|---|---|---|---|
| 5f | 5.50 | 475.58 | 79.23 | 6 | 2 | 1 | 11 | 449.59 |
| 7k | 8.64 | 678.91 | 71.48 | 7 | 1 | 2 | 21 | 668.93 |
aMW, Molecular weight; TPSA, topological polar surface area; n-ON, number of hydrogen acceptors; n-OHNH, number of hydrogen bond donors.
2.2.16. Hepatotoxicity and hepatoprotective activity by compound 5f
The anti-AD drug tacrine has been forced to withdraw due to severe hepatotoxicity. Thus, the hepatotoxicity of compounds 5f on normal human hepatocytes cell line (LO2) was evaluated using MTT assay22. As displayed in Figure 15(A), LO2 cells were exposed to compound 5f (2.5 μM, 5.0 μM, 10.0 μM, 20.0 μM, 40.0 μM and 80.0 μM), the cell viability did not show an obvious change until the concentration increased to 40.0 μM. Furthermore, the hepatoprotective activity of compound 5f on H2O2-induced live injury was tested using an MTT assay. As presented in Figure 15(B), LO2 cells were exposed to 1000 μM H2O2 for 48 h, cell viability sharply decreased to 55.9% (p < 0.01) compared with the untreated group, when treating with compound 5f (5.0 μM, 10.0 μM and 20.0 μM), the cell viability significantly increased to 65.2% (p < 0.05), 69.7% (p < 0.01) and 79.4% (p < 0.05), respectively, in a dose-dependent manner, exhibiting that compound 5f displayed potent hepatoprotective activity through its antioxidant potency.
Figure 15.

(A) The cell viability of LO2 cell by compound 5f. (B) The protective effect of compound 5f on H2O2-induced hepatic injury, ##p < 0.01 vs control; **p < 0.01, *p < 0.05 vs model group.
2.2.17. In vivo assay
Scopolamine is a muscarinic receptor antagonist that inhibits central cholinergic neuronal activity. Scopolamine-induced mice memory impairment has been widely used to evaluate potential therapeutic agents for the treatment of AD. Herein, compound 5f was selected to perform the experiments in vivo20,22.
Acute Toxicity. The safety profile of compound 5f was evaluated in SPF Kunming mice (half male and half female) at a body weight of 18–20g at doses of 1000, 500, 250 and 100 mg/kg (n = 6 per group) by intragastric administration of compound 5f. After administration, mice were closely observed within 30 min, observation was performed every 15 min from 30 min to 2h, observation was performed every 30 min from 2h to 4h, observation was performed every 1h from 4h to 8h, after that, observation was performed every 1 day, continuous observation for 7 days. The mice were killed after 7 days, and the heart, liver, spleen, lung and kidney were observed. The results showed that the mice at the dose of 1000g/kg and 500 mg/kg presented decreased spontaneous activity and movement, drowsiness, dyspnoea and reduced breathing rate. After 24h, the mice returned to normal. After 7 days, the mice were killed and no abnormalities were observed on the heart, liver, spleen, lung and kidney.
Effect of 5f on Scopolamine-induced mice memory impairment. The step-down passive avoidance task was employed to investigate the effects of 5f on scopolamine-induced memory impairment. As shown in Figure 16, when mice were treated with 3 mg/kg scopolamine, the step-down latency sharply declined to133.7 sec (model group, p < 0.01) compared with saline solution-treated mice (228.3 sec, untreated group). When treating with 5 mg/kg donepezil, the step-down latency significantly improved to (219.4 sec, p < 0.01) compared with the model group, indicating that donepezil clearly reversed scopolamine-induced mice cognitive deficit. Moreover, when treating with compound 5f at concentration of 1.9 mg/kg, 5.7 mg/kg and 17.1 mg/kg, the step-down latency gradually increased to 153.7 sec, 183.4 sec (p < 0.01) and 207.8 sec (p < 0.05), respectively, in a dose-dependent manner. The results showed that compound 5f significantly improved scopolamine-induced memory impairment, and the high dose (17.1 mg/kg) presented a similar effect compared with donepezil.
Figure 16.

Effect of compound 5f (1.9, 5.7 and 17.1 mg/kg) or donepezil (5.0 mg/kg) on scopolamine-induced memory impairment through the step-down passive avoidance assay. Values are expressed as the mean ± SEM (n = 6). ##p < 0.01 vs untreated group. *p < 0.05 and **p < 0.01 vs scopolamine-treated model group.
3. Conclusion
In summary, a series of naringenin-O-alkylamine derivatives were rationally designed as multifunctional agents for treating AD by MTDLs strategy. The target compounds were synthesised and evaluated by antioxidant activity, AChE/BuChE inhibition, inhibition of Aβ aggregation, metal chelation, neuroprotective effect and anti-inflammatory property. The in vitro biological activity results revealed that compounds 5f and 7k showed good antioxidant activity with ORAC values of 2.3eq and 0.57eq, respectively. Compounds 5f and 7k displayed good huAChE inhibitory potency with IC50 values of 0.91 μM and 0.57 μM, respectively, which were supported by molecular docking. Both the kinetic study and propidium iodide displacement assay showed that compounds 5f and 7k could simultaneously bind CAS and PAS of huAChE. Moreover, compounds 5f and 7k could inhibit self-induced Aβ1–42 aggregation with 62.1% and 43.8% inhibition rate, respectively, and significantly inhibited huAChE-Aβ1–40 aggregation with 51.7% and 43.4% inhibition rate, respectively. In addition, compounds 5f and 7k were selective metal chelation agents and remarkably inhibited Cu2+-induced Aβ1–42 aggregation with 73.5% and 68.7% inhibition rates, respectively. Furthermore, compounds 5f and 7k presented good BBB permeability in vitro, good neuroprotective effects and anti-inflammatory properties. Further investigation showed that compound 5f did not show obvious hepatotoxicity and displayed a good hepatoprotective effect by its antioxidant activity. The in vivo study displayed that compound 5f could improve the dyskinesia recovery rate and response efficiency of the AlCl3-induced zebrafish AD model, and further significantly improved scopolamine-induced mice memory impairment. Therefore, compound 5f was a potential multifunctional candidate for the treatment of AD, deserving further investigation.
4. Experimental section
4.1. Chemistry
Unless otherwise noted, all the chemicals and solvents were bought from Sigma-Aldrich and Shanghai Titan Scientific Co., Ltd., and were used without purification. The 1H NMR (400 MHz) and 13 C NMR (100 MHz) spectra were recorded on a Bruker Varian INOVA spectrometer in deuterated solvents (CDCl3) with tetramethylsilane (TMS) as an internal reference. The spectra were measured in chemical shift (δ, ppm) and coupling constant (J, Hz). The high-resolution mass spectra were obtained by Waters Xevo G2-XS-Qtof mass spectrometer. The purity of the final synthesised products was evaluated by HPLC analyses which were conducted with a Waters X-Bridge C18 column (4.6 mm × 150 mm, 5 μm) at a flow ratio of 0.8 ml/min. Mobile phase: A: 0.12%TFA in H2O, B: 0.1% TFA in CH3CN.
4.1.1. Synthesis of intermediates 3a–3e
To a solution of naringenin (10 mmol) in CH3CN (30 ml), powdered K2CO3 (13.0 mmol) was added. After the mixture was stirred at room temperature for 30 min, 12.0 mmol dibromides (1,4-dibromobutane 2b or 1,5-dibromopentane 2c, 1,6-dibromohexane 2d, 1,10-dibromodecane 2e, and 1,12-dibromododecane 2f, respectively) were added into the mixture, and then the reaction mixture was stirred at 65 °C for 8–12 h. The reaction was monitored through TLC, the solvent was evaporated under reduced pressure after reaction completion. The crude residue was dissolved in 50 ml ethyl acetate, washed with water (2 × 50 ml), saturated NaCl (80 ml) and passed through the anhydrous Na2SO4 to remove the residual water. The solvent was evaporated to obtain a crude mixture, which was purified by silica gel chromatography and petroleum ether/acetone = 50:1 as mobile phase to afford intermediates 3a–3e.
7–(4-bromobutoxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (3a). The starting material 1 was treated with 1,4-dibromobutane 2b based on the above procedure to get compound 3a, colourless oily matter, 53.2% yield. 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H, OH), 7.34 (d, J = 6.0 Hz, 2H, 2 × Ar-H), 6.89 (d, J = 8.0 Hz, 2H, 2 × Ar-H), 6.05 (d, J = 2.0 Hz, 1H, Ar-H), 6.02 (d, J = 2.4 Hz, 1H, Ar-H), 5.35 (dd, J1 = 10.4 Hz, J2 = 2.8 Hz, 1H, CH), 4.00 (t, J = 6.4 Hz, 2H, OCH2), 3.47 (t, J = 6.4 Hz, 2H, BrCH2), 3.09 (dd, J1 = 12.8 Hz, J2 = 12.8 Hz, 1H, 1/2 CH2), 2.79 (dd, J1 = 10.4 Hz, J2 = 2.8 Hz, 1H, 1/2 CH2), 2.07–2.00 (m, 2H, CH2), 1.97–1.91 (m, 2H, CH2).
7-((5-bromopentyl)oxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (3b). The starting material 1 was treated with 1,5-dibromopentane 2c based on the above procedure to get compound 3b, colourless oily matter, 46.2% yield. 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H, OH), 7.33 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.89 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 6.04 (dd, J1 = 8.4 Hz, J2 = 2.4 Hz, 2H, 2 × Ar-H), 5.35 (dd, J1 = 10.0 Hz, J2 = 2.8 Hz, 1H, CH), 3.98 (t, J = 6.4 Hz, 2H, OCH2), 3.43 (t, J = 6.8 Hz, 2H, BrCH2), 3.09 (dd, J1 = 12.8 Hz, J2 = 4.8 Hz, 1H, 1/2 CH2), 2.78 (dd, J1 = 14.0 Hz, J2 = 3.2 Hz, 1H, 1/2 CH2), 1.96–1.88 (m, 2H, CH2), 1.84–1.76 (m, 2H, CH2), 1.65–1.55 (m, 2H, CH2).
7-((6-bromohexyl)oxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (3c). The starting material 1 was treated with 1,6-dibromohexane 2d based on the above procedure to get compound 3c, colourless oily matter, 43.5% yield. 1H NMR (400 MHz, CDCl3) δ 12.01 (s, 1H, OH), 7.32 (d, J = 8.0 Hz, 2H, 2 × Ar-H), 6.88 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.04 (d, J = 10.0 Hz, 2H, 2 × Ar-H), 5.35 (dd, J1 = 10.8 Hz, J2 = 2.0 Hz, 1H, CH), 3.97 (t, J = 6.4 Hz, 2H, OCH2), 3.42 (t, J = 6.8 Hz, 2H, OCH2), 3.08 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.78 (dd, J1 = 14.8 Hz, J2 = 2.4 Hz, 1H, 1/2 COCH2), 1.92–1.85 (m, 2H, CH2), 1.82–1.75 (m, 2H, CH2), 1.49–1.47 (m, 4H, 2 × CH2).
7-((10-bromodecyl)oxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (3d). The starting material 1 was treated with 1,10-dibromodecane 2e based on the above procedure to get compound 3d, colourless oily matter, 41.1% yield. 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H, OH), 7.34 (d, J = 8.0 Hz, 2H, 2 × Ar-H), 6.89 (d, J = 8.0 Hz, 2H, 2 × Ar-H), 6.04 (d, J = 9.2 Hz, 2H, 2 × Ar-H), 5.35 (dd, J1 = 10.4 Hz, J2 = 2.4 Hz, 1H, CH), 3.96 (t, J = 6.0 Hz, 2H, OCH2), 3.41 (t, J = 6.4 Hz, 2H, BrCH2), 3.09 (d, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1H, 1/2 CH2), 2.78 (dd, J1 = 14.4 Hz, J2 = 2.4 Hz, 1H, 1/2 CH2), 1.89–1.81 (m, 2H, CH2), 1.79–1.72 (m, 2H, CH2), 1.43–1.37 (m, 4H, 2 × CH2), 1.31–1.27 (m, 8H, 4 × CH2).
7-((12-bromododecyl)oxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (3e). The starting material 1 was treated with 1,12-dibromododecane 2f based on the above procedure to get compound 3e, colourless oily matter, 33.7% yield. 1H NMR (400 MHz, CDCl3) δ 12.01 (s, 1H, OH), 7.32 (d, J = 8.0 Hz, 2H, 2 × Ar-H), 6.88 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.04 (d, J = 9.6 Hz, 2H, 2 × Ar-H), 5.34 (dd, J1 = 10.8 Hz, J2 = 2.0 Hz, 1H, CH), 3.95 (t, J = 6.4 Hz, 2H, OCH2), 3.40 (t, J = 6.8 Hz, 2H, BrCH2), 3.08 (d, J1 = 5.2 Hz, J2 = 4.0 Hz, 1H, 1H, 1/2 CH2), 2.78 (dd, J1 = 14.8 Hz, J2 = 2.4 Hz, 1H, 1/2 CH2), 1.88–1.80 (m, 2H, CH2), 1.77–1.72 (m, 2H, CH2), 1.43–1.38 (m, 4H, 2 × CH2), 1.29–1.27 (m, 12H, 6 × CH2).
4.1.2. Synthesis of target 7-O-modified naringenin derivatives 5a–5j
To a mixture of the corresponding secondary amines 4a–4f (1.3 mmol), anhydrous K2CO3 (1.5 mmol) in CH3CN (6 ml) were added the appropriate intermediates 3a–3e (1.0 mmol). The reaction mixture was heated to 65 °C and stirred for 6–10 h under an argon atmosphere. After complete reaction, the solvent was evaporated under reduced pressure to afford the crude product, which was dissolved in ethyl acetate, washed with water (2 × 30 ml). The combined organic phases were washed with saturated aqueous NaCl (50 ml) and dried over sodium sulphate to remove the residual water. The solvent was evaporated to obtain a crude mixture under reduced pressure, which was purified on a silica gel chromatography using mixtures of CH2Cl2/acetone = 50:1 as mobile phase to afford the target 7-O-modified naringenin derivatives 5a–5j.
5-hydroxy-2–(4-hydroxyphenyl)-7–(4-(piperidin-1-yl)butoxy)chroman-4-one (5a). Compound 3a was treated with piperidine 4a based on the above procedure to obtain target compound 5a, light yellow oily matter, 76.2% yield. 1H NMR (400 MHz, CDCl3) δ 12.03 (s, 1H, OH), 7.34–7.24 (m, 2H, 2 × Ar-H), 6.80 (d, J = 8.0 Hz, 2H, 2 × Ar-H), 6.00 (d, J = 2.0 Hz, 1H, Ar-H), 5.94 (d, J = 2.0 Hz, 1H, Ar-H), 5.27 (dd, J1 = 10.4 Hz, J2 = 2.8 Hz, 1H, OCH), 3.91 (t, J = 6.0 Hz, 2H, OCH2), 3.06 (dd, J1 = 10.8 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.72 (dd, J1 = 14.4 Hz, J2 = 2.8 Hz, 1H, 1/2 COCH2), 2.54–2.47 (m, 4H, 2 × NCH2), 2.44 (t, J = 7.2 Hz, 2H, NCH2), 1.74–1.62 (m, 8H, 4 × CH2), 1.47–1.44 (m, 2H, CH2). HR-ESI-MS: Calcd. for C24H29NO5 [M + H]+: 412.2079, found: 412.2095.
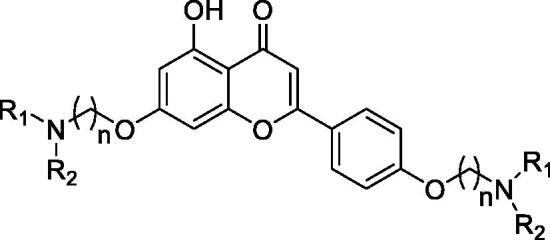
7–(4-(3,4-dihydroisoquinolin-2(1H)-yl)butoxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (5b). Compound 3a was treated with 1,2,3,4-tetrahydroisoquinoline 4b based on the above procedure to obtain target compound 5b, light yellow oily matter, 70.1% yield. 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H, OH), 7.18 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 7.14–7.11 (m, 2H, 2 × Ar-H), 7.08 (t, J = 4.0 Hz, Ar-H), 7.03 (t, J = 3.6 Hz, 1H, Ar-H), 6.70 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.01 (d, J = 2.0 Hz, 1H, Ar-H), 5.95 (d, J = 2.0 Hz, 1H, Ar-H), 5.23 (dd, J1 = 10.4 Hz, J2 = 2.8 Hz, 1H, CH), 3.94 (t, J = 5.2 Hz, 2H, OCH2), 3.73 (s, 2H, phCH2), 3.02 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.94 (t, J = 5.6 Hz, 2H, NCH2), 2.84 (t, J = 5.6 Hz, 2H, NCH2), 2.69 (dd, J1 = 14.4 Hz, J2 = 2.8 Hz, 1H, 1/2 COCH2), 2.62 (t, J = 6.8 Hz, 2H, NCH2), 1.81–1.76 (m, 4H, 2 × CH2). HR-ESI-MS: Calcd. for C28H29NO5 [M + H]+: 460.2079, found: 460.2095.
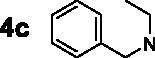
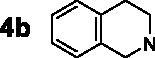
7–(4-(benzyl(ethyl)amino)butoxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (5c). Compound 3a was treated with N-ethylbenzylamine 4c based on the above procedure to obtain target compound 5c, light yellow oily matter, 73.9% yield. 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H, OH), 7.34–7.25 (m, 7H, 7 × Ar-H), 6.90 (t, J = 8.0 Hz, 2H, 2 × Ar-H), 5.99 (d, J = 2.0 Hz, 1H, Ar-H), 5.95 (d, J = 2.0 Hz, 1H, Ar-H), 5.27–5.25 (m, 1H, OCH), 3.90–3.87 (m, 2H, OCH2), 3.59 (s, 2H, phCH2), 3.06 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.72 (d, J = 2.8 Hz, 1H, 1/2 COCH2), 2.61–2.54 (m, 4H, 2 × NCH2), 1.79–1.66 (m, 4H, 2 × CH2), 1.09–1.05 (m, 3H, CH3). HR-ESI-MS: Calcd. for C28H31NO5 [M + H]+: 462.2236, found: 462.2251.
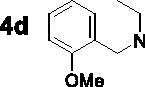
7–(4-(ethyl(2-methoxybenzyl)amino)butoxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (5d). Compound 3a was treated with N-(2-methoxybenzyl)ethanamine 4d based on the above procedure to obtain target compound 5d, light yellow oily matter, 74.6% yield. 1H NMR (400 MHz, CDCl3) δ 12.03 (s, 1H, OH), 7.38 (d, J1 = 6.4 Hz, J2 = 1.2 Hz, 1H, Ar-H), 7.28–7.23 (m, 1H, Ar-H), 7.22 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.92 (t, J = 7.6 Hz, 1H, Ar-H), 6.85 (d, J = 8.4 Hz, 1H, Ar-H), 6.82 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 5.97 (dd, J1 = 18.4 Hz, J2 = 2.0 Hz, 2H, 2 × Ar-H), 5.26 (dd, J1 = 10.4 Hz, J2 = 2.8 Hz, 1H, CH), 3.88 (t, J = 6.4 Hz, 2H, OCH2), 3.78 (s, 3H, OCH3), 3.05 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.74 (d, J = 2.8 Hz, 1H, 1/2 COCH2), 2.71–2.67 (m, 2H, NCH2), 2.66–2.63 (m, 2H, NCH2), 1.75–1.71 (m, 4H, 2 × CH2), 1.16 (t, J = 7.2 Hz, 3H, CH3). HR-ESI-MS: Calcd. for C29H33NO6 [M + H]+: 492.2341, found: 492.2373.
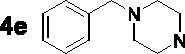
7–(4-(4-benzylpiperazin-1-yl)butoxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (5e). Compound 3a was treated with benzylpiperazine 4e based on the above procedure to obtain target compound 5e, light yellow oily matter, 70.3% yield. 1H NMR (400 MHz, CDCl3) δ 12.03 (s, 1H, OH), 7.30–7.24 (m, 7H, 7 × Ar-H), 6.79 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.00 (d, J = 2.0 Hz, 1H, Ar-H), 5.94 (d, J = 2.0 Hz, 1H), 5.28 (dd, J1 = 10.8 Hz, J2 = 2.4 Hz, 1H, CH), 3.91 (t, J = 6.0 Hz, 2H, OCH2), 3.53 (s, 2H, phCH2), 3.06 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.73 (dd, J1 = 14.4 Hz, 1H, 1/2 COCH2), 2.68–2.50 (m, 8H, 4 × NCH2), 2.46 (t, J = 7.2 Hz, 2H, NCH2), 1.76–1.71 (m, 2H, CH2), 1.70–1.65 (m, 2H, CH2). HR-ESI-MS: Calcd. for C30H34N2O5 [M + H]+: 503.2501, found: 503.2534.
7-((5-(benzyl(ethyl)amino)pentyl)oxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (5f). Compound 3b was treated with N-ethylbenzylamine 4c based on the above procedure to afford target compound 5f, light yellow oily matter, 68.7% yield. 1H NMR 12.01 (s, 1H, OH), 7.36–7.27 (m, 7H, 7 × Ar-H), 6.90–6.87 (m, 2H, 2 × Ar-H), 5.99 (d, J = 2.0 Hz, 1H, Ar-H), 5.94 (d, J = 2.0 Hz, 1H, Ar-H), 5.27 (dd, J1 = 10.8 Hz, J2 = 2.4 Hz, 1H, CH), 3.92–3.88 (m, 2H, OCH2), 3.55 (s, 2H, phCH2), 3.06 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.71 (d, J = 2.8 Hz, 1H, 1/2 COCH2), 2.59–2.53 (m, 4H, 2 × NCH2), 1.77–1.65 (m, 4H, 2 × CH2), 1.51–1.46 (m, 2H, CH2), 1.14–1.09 (m, 3H, CH3). HR-ESI-MS: Calcd. for C29H33NO5 [M + H]+: 476.2393, found: 476.2417.
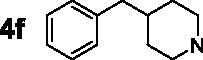
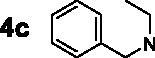
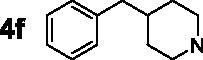
7-((5–(4-benzylpiperidin-1-yl)pentyl)oxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (5g). Compound 3b was treated with benzylpiperidine 4f based on the above procedure to afford target compound 5g, light yellow oily matter, 60.7% yield. 1H NMR 12.00 (s, 1H, OH), 7.26–7.16 (m, 4H, 4 × Ar-H), 7.20–7.16 (m, 1H, Ar-H), 7.11 (d, J = 7.2 Hz, 2H, 2 × Ar-H), 6.87 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 5.98 (d, J = 2.0 Hz, 1H, Ar-H), 5.93 (d, J = 2.0 Hz, 1H, Ar-H), 5.25 (dd, J1 = 10.4 Hz, J2 = 2.8 Hz, 1H, OCH), 3.85 (t, J = 6.4 Hz, 2H, OCH2), 3.19 (d, J = 11.6 Hz, 2H, phCH2), 3.05 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.71 (dd, J1 = 14.0 Hz, J2 = 2.8 Hz, 1H, 1/2 COCH2), 2.58–2.52 (m, 4H, 2 × NCH2), 2.18 (t, J = 7.2 Hz, 2H, NCH2), 1.73–1.64 (m, 6H, 3 × CH2), 1.58–1.42 (m, 2H, CH2), 1.43–1.35 (m, 2H, CH2). HR-ESI-MS: Calcd. for C32H37NO5 [M + H]+: 516.2705, found: 516.2741.
7-((6-(benzyl(ethyl)amino)hexyl)oxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (5h). Compound 3c was treated with N-ethylbenzylamine 4c based on the above procedure to afford target compound 5h, light yellow oily matter, 62.4% yield. 1H NMR (400 MHz, CDCl3) δ 12.01 (s, 1H, OH), 7.35–7.23 (m, 7H, 7 × Ar-H), 6.88 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.00 (d, J = 2.0 Hz, 1H, Ar-H), 5.94 (d, J = 2.0 Hz, 1H, Ar-H), 5.27 (dd, J1 = 10.4 Hz, J2 = 2.8 Hz, 1H, CH), 3.91 (t, J = 6.4 Hz, 2H, OCH2), 3.78–3.73 (m, 2H, phCH2), 3.05 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.72 (d, J = 2.8 Hz, 1H, 1/2 COCH2), 2.60–2.54 (m, 4H, 2 × NCH2), 1.87–1.73 (m, 4H, 2 × CH2), 1.50–1.43 (m, 4H, 2 × CH2), 1.13–1.07 (m, 3H, CH3). HR-ESI-MS: Calcd. for C30H35NO5 [M + H]+: 490.2549, found: 490.2573.
7-((10-(benzyl(ethyl)amino)decyl)oxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (5i). Compound 3d was treated with N-ethylbenzylamine 4c based on the above procedure to afford target compound 5i, light yellow oily matter, 52.6% yield. 1H NMR (400 MHz, CDCl3) δ 12.01 (s, 1H, OH), 7.37–7.25 (m, 7H, 7 × Ar-H), 6.88 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 5.99 (d, J = 2.0 Hz, 1H, Ar-H), 5.94 (d, J = 2.0 Hz, 1H, Ar-H), 5.28 (dd, J1 = 10.4 Hz, J2 = 2.8 Hz, 1H, CH), 3.91 (t, J = 6.4 Hz, 2H, OCH2), 3.80–3.75 (m, 2H, phCH2), 3.06 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.70 (d, J = 2.8 Hz, 1H, 1/2 COCH2), 2.61–2.52 (m, 4H, 2 × NCH2), 1.76–1.70 (m, 4H, 2 × CH2), 1.45–1.39 (m, 4H, 2 × CH2), 1.33–1.25 (m, 8H, 4 × CH2), 1.14–1.06 (m, 3H, CH3). HR-ESI-MS: Calcd. for C34H43NO5 [M + H]+: 546.3175, found: 546.3198.
7-((12-(benzyl(ethyl)amino)dodecyl)oxy)-5-hydroxy-2–(4-hydroxyphenyl)chroman-4-one (5j). Compound 3e was treated with N-ethylbenzylamine 4c based on the above procedure to afford target compound 5j, light yellow oily matter, 51.7% yield. 1H NMR (400 MHz, CDCl3) δ 12.03 (s, 1H, OH), 7.38–7.24 (m, 7H, 7 × Ar-H), 6.90–6.88 (m, 2H, 2 × Ar-H), 6.00 (d, J = 2.0 Hz, 1H, Ar-H), 5.94 (d, J = 2.0 Hz, 1H, Ar-H), 5.29 (dd, J1 = 10.4 Hz, J2 = 2.8 Hz, 1H, CH), 3.93 (t, J = 6.4 Hz, 2H, OCH2), 3.81–3.78 (brs, 2H, phCH2), 3.06 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.71 (d, J = 2.8 Hz, 1H, 1/2 COCH2), 2.60–2.49 (m, 4H, 2 × NCH2), 1.75–1.67 (m, 4H, 2 × CH2), 1.46–1.36 (m, 4H, 2 × CH2), 1.34–1.20 (m, 12H, 6 × CH2), 1.11–1.07 (m, 3H, CH3). HR-ESI-MS: Calcd. for C36H47NO5 [M + H]+: 574.3488, found: 574.3521.
4.1.3. Synthesis of intermediates 6a–6c
The synthesis of intermediates 6a–6c could reference our previous work. Briefly, the starting material 1 (10.0 mmol) was reacted with 55.0 mmol dibromides (2a, 2b and 2d, respectively) in the presence of 30.0 mmol K2CO3 in CH3CN at 65 °C for 10–15 h under an argon atmosphere. After complete reaction, the solvent was treated under universal method to afford the crude product, which was dissolved in CH2Cl2, washed with water (2 × 30 ml). The combined organic phases were washed with saturated aqueous NaCl (50 ml) and dried over Na2SO4. The solvent was evaporated to obtain a crude mixture, which was further purified on a silica gel chromatography using mixtures of petroleum/acetone = 50:1 as mobile phase to get the key intermediates 6a–6c.
7–(3-bromopropoxy)-2–(4-(3-bromopropoxy)phenyl)-5-hydroxychroman-4-one (6a). The starting material 1 was treated with 1,3-dibromopropane 2a based on the above procedure to obtain compound 6a, white solid, 32.8% yield. 1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, CDCl3) δ 12.01 (s, 1H, OH), 7.38 (d, J = 8.6 Hz, 2H, 2 × Ar-H), 6.96 (d, J = 8.7 Hz, 2H, 2 × Ar-H), 6.05 (dd, J = 11.4, 2.2 Hz, 2H, 2 × Ar-H), 5.37 (dd, J = 12.9, 2.8 Hz, 1H, OCH), 4.12 (dd, J = 12.6, 5.9 Hz, 4H, 2 ×OCH2), 3.59 (dt, J = 20.1, 6.4 Hz, 4H, 2 ×BrCH2), 3.09 (dd, J = 17.1, 13.0 Hz, 1H, 1/2COCH2), 2.79 (dd, J = 17.1, 3.0 Hz, 1H, 1/2COCH2), 2.38–2.25 (m, 4H, 2 ×CH2). 13 C NMR (100 MHz, CDCl3) δ 196.1, 167.1, 164.2, 163.0, 159.3, 130.8, 127.9 (2 C), 115.0 (2 C), 103.4, 95.7, 94.7, 79.1, 65.9, 65.5, 43.3, 32.4, 32.0, 30.0, 29.6.
7–(4-bromobutoxy)-2–(4-(4-bromobutoxy)phenyl)-5-hydroxychroman-4-one (6b). The starting material 1 was treated with 1,4-dibromobutane 2b based on the above procedure to obtain compound 6b, colourless oily matter, 43.9% yield. 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H, OH), 7.37 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.94 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 6.04 (d, J = 2.4 Hz, 1H, Ar-H), 6.02 (d, J = 2.4 Hz, 1H, Ar-H), 5.36 (dd, J1 = 10.0 Hz, J2 = 2.8 Hz, 1H, CH), 4.02 (t, J = 6.0 Hz, 2H, OCH2), 4.00 (t, J = 6.0 Hz, 2H, OCH2), 3.50 (t, J = 6.4 Hz, 2H, BrCH2), 3.47 (t, J = 6.8 Hz, 2H, BrCH2), 3.10 (dd, J1 = 12.8 Hz, J2 = 8.0 Hz, 1H, 1/2 CH2), 2.79 (dd, J1 = 14.4 Hz, J2 = 2.8 Hz, 1H, 1/2 CH2), 2.10–2.01 (m, 4H, 2 × CH2), 1.99–1.91 (m, 4H, 2 × CH2).
7-((6-bromohexyl)oxy)-2–(4-((6-bromohexyl)oxy)phenyl)-5-hydroxychroman-4-one (6c). The starting material 1 was treated with 1,6-dibromohexane 2d based on the above procedure to obtain compound 6c, colourless oily matter, 30.4% yield. 1H NMR (400 MHz, CDCl3) δ 1H NMR 12.02 (s, 1H, OH), 7.36 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.94 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.03 (d, J = 9.2 Hz, 2H, 2 × Ar-H), 5.36 (dd, J1 = 11.2 Hz, J2 = 1.6 Hz, 1H, OCH), 4.00–3.94 (m, 4H, 2 × OCH2), 3.45–3.40 (m, 4H, 2 × BrCH2), 3.09 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.78 (dd, J1 = 14.8 Hz, J2 = 2.4 Hz, 1H, 1/2 COCH2), 1.92–1.86 (m, 4H, 2 × CH2), 1.83–1.76 (m, 4H, 2 × CH2), 1.52–1.49 (m, 8H, 4 × CH2).
4.1.4. Synthesis of 7,4’-O-modified naringenin derivatives 7a–7k
The synthesis of 7,4′-O-modified naringenin derivatives 7a–7k could reference our previous work. Briefly, the key intermediates 6a–6c (1.0 mmol) were reacted with secondary amines 4a–4h (3.0 mmol), respectively, in the presence of K2CO3 (3.5 mmol) at 65 °C for 8–12 h. After complete reaction, the solvent was treated by universal method to afford the crude product, which was further purified on a silica gel chromatography using mixtures of petroleum/acetone = 30:1 as mobile phase to get the target 7,4′-O-modified naringenin derivatives 7a–7k.
7–(3-(3,4-dihydroisoquinolin-2(1H)-yl)propoxy)-2–(4-(3–(3,4-dihydroisoquinolin-2(1H)-yl)propoxy)phenyl)-5-hydroxychroman-4-one (7a). Compound 6a was treated with 1,2,3,4-tetrahydroisoquinoline 4b based on the above procedure to afford target compound 7a, light yellow oily matter, 40.9% yield. 1H NMR (400 MHz, CDCl3) δ 12.04 (s, 1H, OH), 7.36 (d, J = 8.3 Hz, 2H, 2 × Ar-H), 7.17 − 7.03 (m, 8H, 8 × Ar-H), 6.96 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.07 (d, J = 9.3 Hz, 2H, 2 × Ar-H), 5.43–5.31 (m, 1H, OCH), 4.10–4.07 (m, 4H, 2 × OCH2), 3.09–3.06 (m, 4H, 2 × NCH2), 2.87–2.65 (m, 10H, COCH2 + 2 × CH2 + 2 × NCH2), 2.13–1.95 (m, 4H, 2 × NCH2), 1.34 (d, J = 8.0 Hz, 4H, 2 × CH2). HR-ESI-MS: Calcd. for C39H42N2O5 [M + H]+: 619.3127, found: 619.3159.
7–(3-(benzyl(ethyl)amino)propoxy)-2–(4-(3-(benzyl(ethyl)amino)propoxy)phenyl)-5-hydroxychroman-4-one (7b). Compound 6a was treated with N-ethylbenzylamine 4c based on the above procedure to afford target compound 7b, light yellow oily matter, 57.2% yield. 1H NMR (400 MHz, CDCl3) δ 12.06 (s, 1H, OH), 7.33–7.28 (m, 12H, 12 × Ar-H), 6.90 (dd, J = 8.2, 4.4 Hz, 2H, 2 × Ar-H), 6.04–5.91 (m, 2H, 2 × Ar-H), 5.36 (d, J = 15.3 Hz, 1H, OCH), 4.09–3.91 (m, 4H, 2 × OCH2), 3.60 (s, 2H, phCH2), 3.58 (s, 2H, phCH2), 2.77 (dd, J1 = 14.8 Hz, J2 = 2.4 Hz, 1H, 1/2 COCH2), 2.59–2.56 (m, 8H, 4 × NCH2), 2.00–1.91 (m, 4H, 2 × CH2), 1.07–1.04 (m, 6H, 2 × CH3). HR-ESI-MS: Calcd. for C39H42N2O5 [M + H]+: 623.3440, found: 623.3466.
7–(3-(4-benzylpiperidin-1-yl)propoxy)-2–(4-(3–(4-benzylpiperidin-1-yl)propoxy)phenyl)-5-hydroxychroman-4-one (7c). Compound 6a was treated with benzylpiperidine 4f based on the above procedure to afford target compound 7c, light yellow oily matter, 47.1% yield. 1H NMR (400 MHz, CDCl3) δ 12.02 (s, 1H, OH), 7.35 (d, J = 8.2 Hz, 2H, 2 × Ar-H), 7.26 (s, 4H, 4 × Ar-H), 7.17 (dd, J = 22.2, 5.7 Hz, 6H, 6 × Ar-H), 6.93 (d, J = 8.1 Hz, 2H, 2 × Ar-H), 6.04 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 5.40–5.33 (m, 1H, OCH), 4.03–4.00 (m, 4H, 2 × OCH2), 3.09–3.07 (m, 1H, CH), 3.00–2.73 (m, 6H, 2 × CH2 + NCH2), 2.54 (t, J = 6.8 Hz, 6H, 3 × NCH2), 2.50–2.43 (m, 2H, NCH2), 1.98–1.95 (m, 8H, NCH2 + 3 × CH2), 1.67–1.65 (m, 4H, 2 × CH2), 1.54 (d, J = 7.4 Hz, 2H, 2 × CH), 1.42 − 1.21 (m, 6H, 3 × CH2). 13 C NMR (100 MHz, CDCl3) δ 196.1, 167.5, 164.2, 163.0, 159.5, 140.7, 140.6, 130.5, 129.2 (4 C), 128.3 (2 C), 128.3 (2 C), 127.8 (2 C), 125.9, 125.9, 114.9 (2 C), 103.2, 95.7, 94.6, 67.0, 66.6, 55.6, 55.3, 54.0 (4 C), 43.3, 43.2 (2 C), 43.2 (2 C), 38.0, 37.9, 32.1 (2 C), 32.0 (2 C), 26.8, 26.6. HR-ESI-MS: Calcd. for C45H54N2O5 [M + H]+: 703.4066, found: 703.4092.
5-hydroxy-7–(3-(4-phenylpiperidin-1-yl)propoxy)-2–(4-(3–(4-phenylpiperidin-1-yl)propoxy)phenyl)chroman-4-one (7d). Compound 6a was treated with 4-phenylpiperidine 4g based on the above procedure to afford target compound 7d, light yellow oily matter, 56.8% yield. 1H NMR (400 MHz, CDCl3) δ 12.03 (s, 1H, OH), 7.37 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 7.29 (d, J = 7.0 Hz, 4H, 4 × Ar-H), 7.26 − 7.20 (m, 6H, 6 × Ar-H), 6.95 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.07 (d, J = 7.2 Hz, 2H, 2 × Ar-H), 5.33 (t, J = 16.5 Hz, 1H, CH), 4.11 − 3.99 (m, 4H, 2 × OCH2), 3.10–3.08 (m, 6H, COCH2, 2 × CH + NCH2), 2.80–2.78 (m, 2H, NCH2), 2.63–2.47 (m, 6H, 3 × NCH2), 2.15–2.04 (m, 6H, NCH2 + 2 × CH2), 1.84 (d, J = 8.6 Hz, 8H, 4 × CH2). 13 C NMR (100 MHz, CDCl3) δ 196.1, 167.6, 164.2, 163.0, 159.6, 146.3, 146.3, 130.5, 128.6 (2 C), 128.5 (2 C), 127.8 (2 C), 127.0 (4 C), 126.3, 126.3, 114.9 (2 C), 103.2, 95.7, 94.7, 79.1, 67.0, 66.6, 55.6, 55.3, 54.5 (4 C), 43.3, 42.7, 42.7, 33.5 (2 C), 33.4 (2 C), 26.9, 26.7. HR-ESI-MS: Calcd. for C43H50N2O5 [M + H]+: 675.3753, found: 675.3769.
7–(3-(4-ethylpiperazin-1-yl)propoxy)-2–(4-(3–(4-ethylpiperazin-1-yl)propoxy)phenyl)-5-hydroxychroman-4-one (7e). Compound 6a was treated with 1-ethylpiperazine 4h based on the above procedure to afford target compound 7e, light yellow oily matter, 52.3% yield. 1H NMR (400 MHz, CDCl3) δ 12.04 (s, 1H, OH), 7.31 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.88 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 5.98 (d, J = 6.0 Hz, 2H, 2 × Ar-H), 5.30 (d, J = 10.8 Hz, 1H, OCH), 3.96 (d, J = 4.6 Hz, 4H, 2 × OCH2), 3.54 (s, 1H, 1/2CH2), 3.04 (d, J = 3.9 Hz, 1H, 1/2CH2), 2.80 − 2.22 (m, 24H, 12 × NCH2), 1.93 (dd, J = 13.6, 7.0 Hz, 4H, 2 × CH2), 1.07–1.05 (m, 6H, 2 × CH3). HR-ESI-MS: Calcd. for C33H48N4O5 [M + H]+: 581.3658, found: 581.3679.
7–(4-(3,4-dihydroisoquinolin-2(1H)-yl)butoxy)-2–(4-(4–(3,4-dihydroisoquinolin-2(1H)-yl)butoxy)phenyl)-5-hydroxychroman-4-one (7f). Compound 6b was treated with 1,2,3,4-tetrahydroisoquinoline 4b based on the above procedure to afford target compound 7f, light yellow oily matter, 61.3% yield. 1H NMR (400 MHz, CDCl3) δ 12.04 (s, 1H, OH), 7.36 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 7.13–7.10 (m, 6H, 6 × Ar-H), 7.04–7.00 (m, 2H, 2 × Ar-H), 6.95 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.04 (d, J = 10.0 Hz, 2H, 2 × Ar-H), 5.36 (dd, J1 = 10.8 Hz, J2 = 2.0 Hz, 1H, OCH), 4.04–3.99 (m, 4H, 2 × OCH2), 3.67 (s, 2H, phCH2), 3.65 (s, 2H, phCH2), 3.10 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H, 1/2 COCH2), 2.93–2.90 (m, 4H, 2 × NCH2), 2.80–2.74 (m, 5H, 2 × NCH2 + 1/2 COCH2), 2.63–2.56 (m, 4H, 2 × NCH2), 1.93–1.69 (m, 8H, 4 × CH2). HR-ESI-MS: Calcd. for C41H46N2O5 [M + H]+: 647.3440, found: 647.3461.
7–(4-(ethyl(2-methoxybenzyl)amino)butoxy)-2–(4-(4-(ethyl(2-methoxybenzyl)amino)butoxy)phenyl)-5-hydroxychroman-4-one (7h). Compound 6b was treated with N-(2-methoxybenzyl)ethanamine 4d based on the above procedure to afford target compound 7h, light yellow oily matter, 43.9% yield. 1H NMR (400 MHz, CDCl3) δ 1H NMR 12.02 (s, 1H, OH), 7.52 (d, J = 8.0 Hz, 1H, Ar-H), 7.48 (d, J = 7.2 Hz, 1H, Ar-H), 7.36 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 7.31–7.24 (m, 2H, 2 × Ar-H), 6.96 (t, J = 7.6 Hz, 2H, 2 × Ar-H), 6.91 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 6.89 (d, J = 3.2 Hz, 1H, Ar-H), 6.87 (d, J = 3.2 Hz, 1H, Ar-H), 6.01 (dd, J1 = 6.4 Hz, J2 = 2.4 Hz, 2H, 2 × Ar-H), 5.36 (dd, J1 = 10.0 Hz, J2 = 2.8 Hz, 1H, OCH), 3.99–3.93 (m, 4H, 2 × OCH2), 3.87 (s, 2H, phCH2), 3.84 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.81 (s, 2H, phCH2), 3.09 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.79 (dd, J1 = 14.0 Hz, J2 = 3.2 Hz, 1H, 1/2 COCH2), 2.78–2.63 (m, 8H, 4 × NCH2)1.84–1.81 (m, 4H, 2 × CH2), 1.78–1.76 (m, 4H, 2 × CH2), 1.24–1.16 (m, 6H, 2 × CH3). HR-ESI-MS: Calcd. for C43H54N2O7 [M + H]+: 711.3965, found: 711.3987.
7–(4-(4-benzylpiperazin-1-yl)butoxy)-2–(4–(4-(4-benzylpiperazin-1-yl)butoxy)phenyl)-5-hydroxychroman-4-one (7i). Compound 6b was treated with benzyl piperazine 4e based on the above procedure to afford target compound 7i, light yellow oily matter, 51.9% yield. 1H NMR (400 MHz, CDCl3) δ 12.03 (s, 1H, OH), 7.35–7.22 (m, 12H, 12 × Ar-H), 6.92 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.02 (dd, J1 = 9.2 Hz, J2 = 2.4 Hz, 2H, 2 × Ar-H), 5.33 (dd, J1 = 10.4 Hz, J2 = 2.8 Hz, 1H, OCH), 3.99–3.94 (m, 4H, 2 × OCH2), 3.51 (s, 2H, phCH2), 3.50 (s, 2H, phCH2), 3.08 (dd, J1 = 12.8 Hz, J2 = 4.4 Hz, 1H, 1/2 COCH2), 2.76 (dd, J1 = 14.4 Hz, J2 = 2.8 Hz, 1H, 1/2 COCH2), 2.49–2.30 (m, 20H, 10 × NCH2), 1.83–1.73 (m, 4H, 2 × CH2), 1.71–1.58 (m, 4H, 2 × CH2). HR-ESI-MS: Calcd. for C45H56N4O5 [M + H]+: 733.4284, found: 733.4297.
7-((6-(benzyl(ethyl)amino)hexyl)oxy)-2–(4-((6-(benzyl(ethyl)amino)hexyl)oxy)phenyl)-5-hydroxychroman-4-one (7k). Compound 6c was treated with N-ethylbenzylamine 4c based on the above procedure to afford target compound 7k, light yellow oily matter, 40.1% yield. 1H NMR (400 MHz, CDCl3) δ 1H NMR 12.02 (s, 1H, OH), 7.34–7.25 (m, 12H,12 × Ar-H), 6.92 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.04 (d, J = 10.0 Hz, 2H, 2 × Ar-H), 5.36 (dd, J1 = 11.2 Hz, J2 = 2.0 Hz, 1H, OCH), 3.98–3.93 (m, 4H, 2 × OCH2), 3.62 (s, 2H, phCH2), 3.59 (s, 2H, phCH2), 3.09 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H, 1/2 COCH2), 2.78 (dd, J1 = 14.8 Hz, J2 = 2.4 Hz, 1H, 1/2 COCH2), 2.58–2.54 (m, 8H, 4 × NCH2), 1.95–1.84 (m, 4H, 2 × CH2), 1.81–1.73 (m, 4H, 2 × CH2), 1.50–1.43 (m, 8H, 4 × CH2), 1.09–1.03 (m, 6H, 2 × CH3). HR-ESI-MS: Calcd. for C45H58N2O5 [M + H]+: 707.4379, found: 707.4413.
4.1.5. Synthesis of intermediates 9a–9d
The synthesis of intermediates 9a–9d could reference our previous work. Briefly, the starting material 8 (10.0 mmol) was treated with 55.0 mmol dibromides (2a–2d) in the presence of 30.0 mmol K2CO3 in CH3CN at 65 °C for 10–15 h. The reaction was monitored using TLC, after the reaction completed, the mixture was treated through under universal method to afford the crude product, which was further purified on a silica gel chromatography using mixtures of petroleum/ethyl acetate = 50:1 as mobile phase to get the key intermediates 9a–9d.
7–(3-bromopropoxy)-2–(4-(3-bromopropoxy)phenyl)-5-hydroxy-4H-chromen-4-one (9a). The starting material 8 was treated with 1,3-dibromopropane 2a based on the above procedure to obtain compound 9a, colourless oily matter, 56.9% yield. 1H NMR (400 MHz, CDCl3) δ 12.70 (s, 1H, OH), 7.75 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 6.94 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 6.50 (s, 1H, Ar-H), 6.42 (d, J = 2.4 Hz, 1H, Ar-H), 6.29 (d, J = 2.0 Hz, 1H, Ar-H), 4.13–4.10 (m, 4H, 2 × BrCH2), 3.57–3.52 (m, 4H, 2 × OCH2), 2.31–2.71 (m, 4H, 2 × CH2).
7-((5-bromopentyl)oxy)-2–(4-((5-bromopentyl)oxy)phenyl)-5-hydroxy-4H-chromen-4-one (9c). The starting material 8 was treated with 1,5-dibromopentane 2c based on the above procedure to obtain compound 9c, colourless oily matter, 52.7% yield. 1H NMR (400 MHz, CDCl3) δ 12.81 (s, 1H, OH), 7.82 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 6.99 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 6.57 (s, 1H, Ar-H), 6.47 (d, J = 2.0 Hz, 1H, Ar-H), 6.34 (d, J = 2.0 Hz, 1H), 4.07–4.02 (m, 4H, 2 × OCH2), 3.46 (t, J = 6.8 Hz, 4H, 2 × BrCH2), 1.98–1.93 (m, 4H, 2 × CH2), 1.88–1.83 (m, 4H, 2 × CH2), 1.68–1.64 (m, 4H, 2 × CH2).
4.1.6. Synthesis of 7,4’-O-modified naringenin derivatives 10a–10v
The synthesis of 7,4′-O-modified naringenin derivatives 7a–7k could reference our previous work. Briefly, the key intermediates 9a–9d (1.0 mmol) were reacted with secondary amines 4a–4j (3.0 mmol), respectively, in the presence of K2CO3 (3.5 mmol) at 65 °C for 8–12 h. After complete reaction, the mixture was treated through the universal method to afford the crude product, which was further purified on a silica gel chromatography using mixtures of CH2Cl2/acetone = 50:1 as mobile phase to get the target 7,4′-O-modified naringenin derivatives 10a–10v.
7–(3-(3,4-dihydroisoquinolin-2(1H)-yl)propoxy)-2–(4-(3–(3,4-dihydroisoquinolin-2(1H)-yl)propoxy)phenyl)-5-hydroxy-4H-chromen-4-one (10a). Compound 9a was treated with 1,2,3,4-tetrahydroisoquinoline 4b based on the above procedure to afford target compound 10a, light yellow oily matter, 41.3% yield. 1H NMR (400 MHz, CDCl3) δ 12.72 (s, 1H, OH), 7.74 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 7.21 (t, J = 5.2 Hz, 4H, 4 × Ar-H), 7.13 (d, J = 7.0 Hz, 2H, 2 × Ar-H), 7.06 (d, J = 7.3 Hz, 4H, 4 × Ar-H), 6.91 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 6.49 (s, 1H, Ar-H), 6.40 (d, J = 1.9 Hz, 1H, C = CH), 6.26 (d, J = 1.9 Hz, 1H, Ar-H), 4.02 (t, J = 5.3 Hz, 4H, 2 × OCH2), 3.00 (s, 4H, 2 × NCH2), 2.56 (dd, J = 15.6, 9.2 Hz, 4H, 2 × NCH2), 2.03–2.00 (m, 8H, 2 × CH2, 2 × NCH2), 1.53–1.49 (m, 4H, 2 × CH2). HR-ESI-MS: Calcd. for C39H40N2O5 [M + H]+: 617.2971, found: 617.2986.
7–(3-(benzyl(ethyl)amino)propoxy)-2–(4-(3-(benzyl(ethyl)amino)propoxy)phenyl)-5-hydroxy-4H-chromen-4-one (10b). Compound 9a was treated with N-ethylbenzylamine 4c based on the above procedure to afford target compound 10b, light yellow oily matter, 56.8% yield. 1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, CDCl3) δ 12.80 (d, J = 7.8 Hz, 1H, OH), 7.80 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 7.29 (dt, J = 7.7, 5.8 Hz, 8H, 8 × Ar-H), 7.24–7.18 (m, 2H, 2 × Ar-H), 6.95 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 6.56 (s, 1H, Ar-H), 6.42 (d, J = 2.0 Hz, 1H, C = CH), 6.30 (d, J = 1.9 Hz, 1H, Ar-H), 4.06 (d, J = 3.2 Hz, 4H, 2 × OCH2), 3.61–3.58 (m, 4H, 2 × NCH2), 2.64–2.48 (m, 8H, 4 × NCH2), 1.97–1.94 (m, 4H, 2 × CH2), 1.06 (t, J = 7.0 Hz, 6H, 2 × CH3). 13 C NMR (100 MHz, CDCl3) δ 182.5, 165.0, 164.1, 162.2, 162.1, 157.8, 139.9, 139.9, 128.9 (2 C), 128.9 (2 C), 128.3 (2 C), 128.3 (2 C), 128.0 (2 C), 126.9 (2 C), 123.4, 115.1 (2 C), 105.5, 104.3, 98.6, 93.1, 77.5, 77.2, 76.8, 66.8, 66.5, 58.4, 58.3, 49.5, 49.4, 47.6, 47.6, 27.0, 27.0, 12.0, 12.0. HR-ESI-MS: Calcd. for C39H44N2O5 [M + H]+: 621.3284, found: 621.3297.
7–(3-(4-benzylpiperidin-1-yl)propoxy)-2–(4-(3–(4-benzylpiperidin-1-yl)propoxy)phenyl)-5-hydroxy-4H-chromen-4-one (10c). Compound 9a was treated with benzylpiperidine 4f based on the above procedure to afford target compound 10c, light yellow oily matter, 43.6% yield. 1H NMR (400 MHz, CDCl3) δ 12.71 (s, 1H, OH), 7.73 (d, J = 8.3 Hz, 2H, 2 × Ar-H), 7.21 − 7.17 (m, 4H, 4 × Ar-H), 7.12 (d, J = 7.2 Hz, 2H, 2 × Ar-H), 7.06 (d, J = 7.4 Hz, 4H, 4 × Ar-H), 6.90 (d, J = 8.3 Hz, 2H, 2 × Ar-H), 6.47 (s, 1H, Ar-H), 6.39 (s, 1H, C = CH), 6.25 (s, 1H, Ar-H), 4.01 (s, 4H, 2 × OCH2), 3.01 (s, 4H, 2 × NCH2), 2.56 (t, J = 13.8 Hz, 4H, 2 × NCH2), 2.11–2.08 (m, 10H, 2 × NCH2 + 2 × CH2 + 2 × CH), 1.51–1.47 (m, 4H, 2 × CH2), 1.22–1.19 (m, 8H, 4 × CH2). HR-ESI-MS: Calcd. for C45H52N2O5 [M + H]+: 701.3910, found: 701.3947.
5-hydroxy-7–(3-(4-phenylpiperidin-1-yl)propoxy)-2–(4-(3–(4-phenylpiperidin-1-yl)propoxy)phenyl)-4H-chromen-4-one (10d). Compound 9a was treated with 4-phenylpiperidine 4g based on the above procedure to afford target compound 10d, light yellow oily matter, 50.8% yield. 1H NMR (400 MHz, CDCl3) δ 12.80 (s, 1H, OH), 7.83 (d, J = 8.7 Hz, 2H, 2 × Ar-H), 7.33 − 7.28 (m, 4H, 2 × Ar-H), 7.22 (dd, J = 17.9, 7.5 Hz, 6H, 2 × Ar-H), 7.00 (d, J = 8.7 Hz, 2H, 2 × Ar-H), 6.57 (s, 1H, Ar-H), 6.51 (d, J = 1.6 Hz, 1H, C = CH), 6.36 (d, J = 1.6 Hz, 1H, Ar-H), 4.13 (d, J = 1.8 Hz, 4H, 2 × OCH2), 3.16 (d, J = 6.1 Hz, 4H, 2 × NCH2), 2.67–2.64 (m, 4H, 2 × NCH2), 2.56 (dd, J = 14.9, 7.0 Hz, 2H, 2 × CH), 2.17–2.12 (m, 8H, 2 × NCH2 + 2 × CH2), 1.91–1.87 (m, 8H, 4 × CH2). HR-ESI-MS: Calcd. for C43H48N2O5 [M + H]+: 673.3597, found: 673.3618.
5-hydroxy-7–(3-(4–(3-phenylpropyl)piperidin-1-yl)propoxy)-2–(4-(3–(4–(3-phenylpropyl)piperidin-1-yl)propoxy)phenyl)-4H-chromen-4-one (10e). Compound 9a was treated with 4-(3-phenylpropyl)piperidine 4i based on the above procedure to afford target compound 10e, light yellow oily matter, 45.7% yield. 1H NMR (400 MHz, CDCl3) δ 12.75 (s, 1H, OH), 7.76 (d, J = 8.3 Hz, 2H, 2 × Ar-H), 7.23 (d, J = 7.0 Hz, 5H, 5 × Ar-H), 7.13 (d, J = 7.6 Hz, 5H, 5 × Ar-H), 6.94 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.51 (s, 1H, Ar-H), 6.42 (s, 1H, C = CH), 6.27 (s, 1H, Ar-H), 4.06 (d, J = 5.2 Hz, 4H, 2 × OCH2), 3.20 (s, 4H, 2 × NCH2), 2.78 (s, 4H, 2 × NCH2), 2.56 (t, J = 7.5 Hz, 4H, 2 × NCH2), 2.32–2.15 (m, 8H, 4 × CH2), 1.76 (d, J = 13.5 Hz, 4H, 2 × CH2), 1.62–1.57 (m, 10H, 2 × CH + 4 × CH2), 1.30 (d, J = 6.0 Hz, 4H, 2 × CH2). 13 C NMR (100 MHz, CDCl3) δ 182.4, 164.5, 164.0, 162.1, 161.7, 157.7, 142.4 (2 C), 128.4 (4 C), 128.4 (4 C), 128.1 (2 C), 125.8 (2 C), 123.6, 115.0 (2 C), 105.7, 104.3, 98.6, 92.9, 66.5, 66.2, 55.1, 55.0, 53.6 (2 C), 53.6 (2 C), 36.1 (2 C), 35.6, 35.6, 30.7, 30.6, 29.8, 29.7, 28.6 (4 C), 25.4, 25.4. HR-ESI-MS: Calcd. for C49H60N2O5 [M + H]+: 757.4536, found: 757.4571.
7–(3-(3,5-dimethylpiperidin-1-yl)propoxy)-2–(4-(3–(3,5-dimethylpiperidin-1-yl)propoxy)phenyl)-5-hydroxy-4H-chromen-4-one (10f). Compound 9a was treated with 3,5-dimethylpiperidine 4j based on the above procedure to afford target compound 10f, light yellow oily matter, 43.4% yield. 1H NMR (400 MHz, CDCl3) δ 12.78 (s, 1H, OH), 7.80 (d, J = 8.6 Hz, 2H, 2 × Ar-H), 6.99 (d, J = 8.7 Hz, 2H, 2 × Ar-H), 6.55 (s, 1H, Ar-H), 6.47 (d, J = 1.7 Hz, 1H, C = CH), 6.33 (s, 1H, Ar-H), 4.08 (t, J = 5.3 Hz, 4H, 2 × OCH2), 3.01 − 2.85 (m, 4H, 2 × NCH2), 2.57–2.54 (m, 4H, 2 × NCH2), 2.07 (dd, J = 11.4, 8.0 Hz, 4H, 2 × NCH2), 1.73 (d, J = 12.3 Hz, 4H, 2 × CH2), 1.60–1.48 (m, 4H, 2 × CH2), 1.30–1.20 (m, 4H, 4 × CH), 0.87 (d, J = 6.2 Hz, 12H, 4 × CH3). 13 C NMR (100 MHz, CDCl3) δ 182.5, 164.9, 164.1, 162.2, 162.1, 157.8, 128.1 (2 C), 123.6, 115.1 (2 C), 105.6, 104.4, 98.7, 93.1, 67.1, 66.7, 61.5 (2 C), 61.4 (2 C), 55.4, 55.2, 42.0, 42.0, 30.9 (2 C), 30.8 (2 C), 26.4, 26.4, 19.7 (4 C). HR-ESI-MS: Calcd. for C35H48N2O5 [M + H]+: 577.3597, found: 577.3622.
7–(4-(3,4-dihydroisoquinolin-2(1H)-yl)butoxy)-2–(4-(4–(3,4-dihydroisoquinolin-2(1H)-yl)butoxy)phenyl)-5-hydroxy-4H-chromen-4-one (10g). Compound 9b was treated with 1,2,3,4-tetrahydroisoquinoline 4b based on the above procedure to afford target compound 10g, light yellow oil, 78.2% yield, 98.0% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 12.86 (s, 1H, OH), 7.79 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 7.17–7.10 (m, 8H, 8 × Ar-H), 6.96 (d, J = 8.0 Hz, 2H, 2 × Ar-H), 6.56 (s, 1H, Ar-H), 6.44 (s, 1H, Ar-H), 6.32 (s, 1H, Ar-H), 4.08–4.02 (m, 4H, 2 × OCH2), 3.81 (s, 2H, phCH2), 3.77 (s, 2H, phCH2), 2.97–2.95 (m, 4H, 2 × phCH2), 2.93–2.86 (m, 4H, 2 × NCH2), 2.72–2.67 (m, 4H, 2 × NCH2), 1.87–1.83 (m, 8H, 4 × CH2). HR-ESI-MS: Calcd. for C41H44N2O5 [M + H]+: 645.3284, found: 645.3306.
7–(4-(benzyl(ethyl)amino)butoxy)-2–(4-(4-(benzyl(ethyl)amino)butoxy)phenyl)-5-hydroxy-4H-chromen-4-one (10h). Compound 9b was treated with N-ethylbenzylamine 4c based on the above procedure to afford target compound 10h, light yellow oil, 83.7% yield, 99.5% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 12.80 (s, 1H, OH), 7.79 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 7.35 (d, J = 7.2 Hz, 4H, 4 × Ar-H), 7.30 (t, J = 7.6 Hz, 4H, 4 × Ar-H), 7.23 (t, J = 7.6 Hz, 2H, 2 × Ar-H), 6.94 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.54 (s, 1H, Ar-H), 6.42 (s, 1H, Ar-H), 6.30 (s, 1H, Ar-H), 3.99–3.95 (m, 4H, 2 × OCH2), 3.61 (s, 4H, 2 × phCH2), 2.59–2.50 (m, 8H, 4 × NCH2), 1.84–1.80 (m, 4H, 2 × CH2), 1.70–1.64 (m, 4H, 2 × CH2), 1.08 (t, J = 7.2 Hz, 6H, 2 × CH3). 13 C NMR (100 MHz, CDCl3) δ 182.4, 164.9, 164.0, 162.1, 157.7, 139.3, 129.0 (5 C), 128.2 (5 C), 128.0 (2 C), 127.0 (2 C), 123.3, 115.0 (2 C), 105.4, 98.5, 93.0 (2 C), 68.4, 68.0, 58.0 (2 C), 52.5, 52.4, 47.3 (2 C), 26.9, 26.7, 23.4, 23.3, 11.6 (2 C). HR-ESI-MS: Calcd. for C41H48N2O5 [M + H]+: 649.3597, found: 649.3612.
7–(4-(4-Benzylpiperazin-1-yl)butoxy)-2–(4–(4-(4-benzylpiperazin-1-yl)butoxy)phenyl)-5-hydroxy-4H-chromen-4-one (10i). Compound 9b was treated with benzylpiperazine 4e based on the above procedure to afford target compound 10i, light yellow oil, 65.2% yield, 98.1% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 12.80 (s, 1H, OH), 7.80 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 7.31–7.24 (m, 10H, 10 × Ar-H), 6.97 (d, J = 8.8 Hz, 2H, 2 × Ar-H), 6.52 (s, 1H, Ar-H), 6.45 (d, J = 2.0 Hz, 1H, Ar-H), 6.33 (d, J = 2.4 Hz, 1H, Ar-H), 4.06–4.01 (m, 4H, 2 × OCH2), 3.51 (s, 4H, 2 × phCH2), 2.53–3.40 (m, 20 H, 10 × NCH2), 1.86–1.80 (m, 4H, 2 × CH2), 1.73–1.64 (m, 4H, 2 × CH2). HR-ESI-MS: Calcd. for C45H54N4O5 [M + H]+: 731.4128, found: 731.4143.
7–(4-(4-Benzylpiperidin-1-yl)butoxy)-2–(4–(4-(4-benzylpiperidin-1-yl)butoxy)phenyl)-5-hydroxy-4H-chromen-4-one (10j). Compound 9b was treated with benzylpiperidine 4f based on the above procedure to afford target compound 10j, light yellow oil, 71.8% yield, 98.2% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 12.79 (s, 1H, OH), 7.80 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 7.29–7.25 (m, 4H, 4 × Ar-H), 7.20–7.12 (m, 6H, 6 × Ar-H), 6.97 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.55 (s, 1H, Ar-H), 6.45 (s, 1H, Ar-H), 6.32 (s, 1H, Ar-H), 4.06–4.02 (m, 4H, 2 × OCH2), 2.98 (d, J = 10.8 Hz, 4H, 2 × phCH2), 2.54 (d, J = 6.8 Hz, 4H, 2 × NCH2), 2.43 (t, J = 7.2 Hz, 4H, 2 × NCH2), 1.95 (t, J = 11.2 Hz, 4H, 2 × NCH2), 1.84–1.80 (m, 4H, 2 × CH2), 1.72–1.64 (m, 8H, 4 × CH2), 1.57–1.53 (m, 2H, 2 × CH), 1.42–1.31 (m, 4H, 2 × CH2). HR-ESI-MS: Calcd. for C47H56N2O5 [M + H]+: 729.4223, found: 729.4267.
7-((5-(benzyl(ethyl)amino)pentyl)oxy)-2–(4-((5-(benzyl(ethyl)amino)pentyl)oxy)phenyl)-5-hydroxy-4H-chromen-4-one (10k). Compound 9c was treated with N-ethylbenzylamine 4c based on the above procedure to afford target compound 10k, light yellow oily matter, 49.3% yield. 1H NMR (400 MHz, CDCl3) δ 12.81 (s, 1H, OH), 7.82 (d, J = 8.6 Hz, 2H, 2 × Ar-H), 7.32 (q, J = 7.9 Hz, 7H, 7 × Ar-H), 7.25 (d, J = 10.6 Hz, 3H, 3 × Ar-H), 6.98 (d, J = 8.6 Hz, 2H, 2 × Ar-H), 6.57 (s, 1H, Ar-H), 6.46 (s, 1H, C = CH), 6.34 (s, 1H, Ar-H), 4.00 (dd, J = 10.1, 6.0 Hz, 4H, 2 × OCH2), 3.57 (s, 4H, 2 × NCH2), 2.56 − 2.42 (m, 8H, 4 × NCH2), 1.79 (s, 4H, 2 × CH2), 1.60–1.51 (m, 4H, 2 × CH2), 1.47 (d, J = 5.9 Hz, 4H, 2 × CH2), 1.05 (t, J = 7.0 Hz, 6H, 2 × CH3). 13 C NMR (100 MHz, CDCl3) δ 182.6, 165.1, 164.2, 162.3, 162.2, 157.8, 129.0 (4 C), 128.3 (6 C), 128.1 (2 C), 126.9 (2 C), 123.5, 115.1 (2 C), 105.6, 104.3, 98.6, 93.1, 68.7, 68.4, 58.2 (2 C), 53.1 (2 C), 47.5 (2 C), 29.1, 29.0, 26.9, 26.9, 24.0, 23.9, 11.8 (2 C). HR-ESI-MS: Calcd. for C43H52N2O5 [M + H]+: 677.3910, found: 677.3943.
7-((5-(ethyl(2-methoxybenzyl)amino)pentyl)oxy)-2–(4-((5-(ethyl(2-methoxybenzyl)amino)pentyl)oxy)phenyl)-5-hydroxy-4H-chromen-4-one (10l). Compound 9c was treated with N-(2-methoxybenzyl)ethanamine 4d based on the above procedure to afford target compound 10l, light yellow oily matter, 38.1% yield. 1H NMR (400 MHz, CDCl3) δ 12.82 (s, 1H, OH), 7.83 (d, J = 8.0 Hz, 2H, 2 × Ar-H), 7.50 (d, J = 7.0 Hz, 2H, 2 × Ar-H), 7.28 (s, 2H, 2 × Ar-H), 6.98 (t, J = 8.8 Hz, 4H, 4 × Ar-H), 6.89 (d, J = 8.0 Hz, 2H, 2 × Ar-H), 6.57 (s, 1H, Ar-H), 6.47 (s, 1H, C = CH,), 6.34 (s, 1H, Ar-H), 4.03 (s, 4H, 2 × OCH2), 3.85 (s, 6H, 2 × OCH3), 3.80 (d, J = 8.8 Hz, 4H, 2 × NCH2), 2.78 − 2.55 (m, 8H, 4 × NCH2), 1.83 (s, 4H, 2 × CH2), 1.71 (s, 4H, 2 × CH2), 1.50 (d, J = 5.8 Hz, 4H, 2 × CH2), 1.18 (t, J = 6.2 Hz, 6H, 2 × CH3). 13 C NMR (100 MHz, CDCl3) δ 182.5, 165.0, 164.1, 162.2, 162.1, 157.9 (2 C), 157.8, 131.0, 131.0 (2 C), 128.9, 128.8, 128.7, 128.1 (2 C), 123.4, 120.6 (2 C), 115.0 (2 C), 110.5 (2 C), 105.5, 104.3, 98.6, 93.0, 68.5, 68.2, 65.6, 55.5 (2 C), 53.0, 51.1 (2 C), 47.6 (2 C), 30.6, 29.0, 28.8, 23.9, 23.9, 19.3, 13.8, 11.2. HR-ESI-MS: Calcd. for C45H56N2O7 [M + H]+: 737.4121, found: 737.4143.
7-((5–(4-benzylpiperidin-1-yl)pentyl)oxy)-2–(4-((5–(4-benzylpiperidin-1-yl)pentyl)oxy)phenyl)-5-hydroxy-4H-chromen-4-one (10m). Compound 9c was treated with benzylpiperidine 4f based on the above procedure to afford target compound 10m, light yellow oily matter, 45.2% yield. 1H NMR (400 MHz, CDCl3) δ 12.76 (s, 1H, OH), 7.77 (d, J = 8.6 Hz, 2H, 2 × Ar-H), 7.23 (d, J = 4.1 Hz, 4H, 4 × Ar-H), 7.17 (d, J = 7.1 Hz, 2H, 2 × Ar-H), 7.10 (d, J = 7.3 Hz, 4H, 4 × Ar-H), 6.94 (d, J = 8.6 Hz, 2H, 2 × Ar-H), 6.52 (s, 1H, Ar-H), 6.42 (s, 1H, C = CH), 6.28 (s, 1H, Ar-H), 3.99 (d, J = 2.3 Hz, 4H, 2 × OCH2), 3.12 (d, J = 10.8 Hz, 4H, 2 × NCH2), 2.53 (s, 8H, 4 × NCH2), 2.11 (s, 4H, 2 × CH2), 1.81 (s, 4H, 2 × CH2), 1.68 (s, 8H, 4 × CH2), 1.56 (s, 6H, 2 × CH, 2 × CH2), 1.47 (d, J = 5.8 Hz, 4H, 2 × CH2). 13 C NMR (100 MHz, CDCl3) δ 182.5, 164.9, 164.1, 162.1 (2 C), 157.8, 140.2 (2 C), 129.2 (4 C), 128.4 (4 C), 128.1 (2 C), 126.1 (2 C), 123.4, 115.0 (2 C), 105.6, 104.3, 98.6, 93.0, 68.3, 68.0, 58.3, 58.3, 53.6 (4 C), 42.7 (2 C), 37.4 (2 C), 30.8 (4 C), 28.9, 28.8, 25.6, 25.6, 24.0, 23.9. HR-ESI-MS: Calcd. for C49H60N2O5 [M + H]+: 757.4536, found: 757.4559.
5-hydroxy-7-((5–(4-phenylpiperidin-1-yl)pentyl)oxy)-2–(4-((5–(4-phenylpiperidin-1-yl)pentyl)oxy)phenyl)-4H-chromen-4-one (10n). Compound 9c was treated with 4-phenylpiperidine 4g based on the above procedure to afford target compound 10n, light yellow oily matter, 80.2% yield. 1H NMR (400 MHz, CDCl3) δ 12.83 (s, 1H, OH), 7.83 (d, J = 8.6 Hz, 2H, 2 × Ar-H), 7.31 (dd, J = 14.1, 6.8 Hz, 5H, 5 × Ar-H), 7.25 − 7.14 (m, 5H, 5 × Ar-H), 7.00 (d, J = 8.6 Hz, 2H, 2 × Ar-H), 6.57 (s, 1H, Ar-H), 6.48 (s, 1H, C = CH), 6.35 (s, 1H, Ar-H), 4.05 (s, 4H, 2 × OCH2), 3.14 (d, J = 11.1 Hz, 4H, 2 × NCH2), 2.55 (dd, J = 15.0, 6.9 Hz, 2H, 2 × CH), 2.51 − 2.41 (m, 4H, 2 × NCH2), 2.13 (t, J = 12.7 Hz, 4H, 2 × NCH2), 1.88 (s, 12H, 6 × CH2), 1.67 (d, J = 6.1 Hz, 4H, 2 × CH2), 1.54 (d, J = 6.2 Hz, 4H, 2 × CH2). 13 C NMR (100 MHz, CDCl3) δ 182.4, 165.0, 164.1, 162.2, 162.1, 157.7, 146.1, 146.1, 128.5 (4 C), 128.1 (2 C), 126.9 (4 C), 126.3 (2 C), 123.4, 115.0 (2 C), 105.5, 104.2, 98.5, 93.0, 68.5, 68.1, 58.9, 58.9, 54.4 (4 C), 42.6 (2 C), 33.2 (4 C), 29.1, 28.9, 26.6, 26.6, 24.2, 24.1. HR-ESI-MS: Calcd. for C47H56N2O5 [M + H]+: 729.4223, found: 729.4249.
5-hydroxy-7-((5–(4-(3-phenylpropyl)piperidin-1-yl)pentyl)oxy)-2–(4-((5–(4–(3-phenylpropyl)piperidin-1-yl)pentyl)oxy)phenyl)-4H-chromen-4-one (10o). Compound 9c was treated with 4-(3-phenylpropyl)piperidine 4i based on the above procedure to afford target compound 10o, light yellow oily matter, 73.8% yield. 1H NMR (400 MHz, CDCl3) δ 12.80 (s, 1H, OH), 7.82 (d, J = 8.5 Hz, 2H, 2 × Ar-H), 7.26 (s, 5H, 5 × Ar-H), 7.17 (t, J = 7.2 Hz, 5H, 5 × Ar-H), 6.98 (d, J = 8.6 Hz, 2H, 2 × Ar-H), 6.56 (s, 1H, Ar-H), 6.46 (s, 1H, C = CH), 6.33 (s, 1H, Ar-H), 4.02 (s, 4H, 2 × OCH2), 3.05 (d, J = 10.8 Hz, 4H, 2 × NCH2), 2.58 (t, J = 7.7 Hz, 4H, 2 × NCH2), 2.50–2.39 (m, 4H, 2 × NCH2), 2.04 (t, J = 10.3 Hz, 4H, 2 × CH2), 1.84 (s, 4H, 2 × CH2), 1.76–1.55 (m, 12H, 6 × CH2), 1.49 (d, J = 5.3 Hz, 4H, 2 × CH2), 1.32–1.25 (m, 10H, 2 × CH + 4 × CH2). 13 C NMR (100 MHz, CDCl3) δ 182.6 165.0, 164.2, 162.2 (2 C), 157.8, 142.7 (2 C), 128.5 (4 C), 128.4 (4 C), 128.1 (2 C), 125.8 (2 C), 123.5, 115.1 (2 C), 105.6, 104.4, 98.6, 93.1, 68.4, 68.1, 58.7, 58.7, 53.9 (4 C), 36.2 (4 C), 36.0, 35.5, 31.6, 31.6, 29.1, 28.9, 28.8 (4 C), 26.2, 26.1, 24.1, 24.1. HR-ESI-MS: Calcd. for C53H68N2O5 [M + H]+: 813.5162, found: 813.5187.
7-((5–(3,5-dimethylpiperidin-1-yl)pentyl)oxy)-2–(4-((5–(3,5-dimethylpiperidin-1-yl)pentyl)oxy)phenyl)-5-hydroxy-4H-chromen-4-one (10p). Compound 9c was treated with 3,5-dimethylpiperidine 4j based on the above procedure to afford target compound 10p, light yellow oily matter, 80.2% yield. 1H NMR (400 MHz, CDCl3) δ 12.80 (s, 1H, OH), 7.82 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.99 (d, J = 8.5 Hz, 2H, 2 × Ar-H), 6.57 (s, 1H, Ar-H), 6.47 (s, 1H, C = CH), 6.34 (s, 1H, Ar-H), 4.03 (s, 4H, 2 × OCH2), 2.95 (s, 4H, 2 × NCH2), 2.46 (s, 4H, 2 × NCH2), 1.85 (s, 8H, 2 × NCH2, 2 × CH2), 1.68 (s, 12H, 4 × CH, 4 × CH2), 1.50 (s, 4H, 2 × CH2), 0.87 (d, J = 6.5 Hz, 12H, 4 × CH3). HR-ESI-MS: Calcd. for C39H56N2O5 [M + H]+: 633.4223, found: 633.4251.
7-((6–(3,4-dihydroisoquinolin-2(1H)-yl)hexyl)oxy)-2–(4-((6–(3,4-dihydroisoquinolin-2(1H)-yl)hexyl)oxy)phenyl)-5-hydroxy-4H-chromen-4-one (10q). Compound 9d was treated with 1,2,3,4-tetrahydroisoquinoline 4b based on the above procedure to afford target compound 10q, light yellow oily matter, 78.9% yield. 1H NMR (400 MHz, CDCl3) δ 12.81 (s, 1H, OH), 7.82 (d, J = 8.7 Hz, 2H, 2 × Ar-H), 7.11 (d, J = 4.1 Hz, 6H, 6 × Ar-H), 7.01 (dd, J = 12.1, 7.2 Hz, 4H, 4 × Ar-H), 6.57 (s, 1H, Ar-H), 6.47 (d, J = 1.8 Hz, 1H, C = CH), 6.35 (d, J = 1.8 Hz, 1H, Ar-H), 4.08–4.01 (m, 4H, 2 × OCH2), 3.66–3.63 (m, 4H, 2 × NCH2), 2.92 (t, J = 5.6 Hz, 4H, 2 × NCH2), 2.75 (t, J = 5.8 Hz, 4H, 2 × CH2), 2.60–2.51 (m, 4H, 2 × NCH2), 1.87 (d, J = 5.7 Hz, 8H, 4 × CH2), 1.73–1.69 (m, 4H, 2 × CH2), 1.58–1.55 (m, 4H, 2 × CH2). 13 C NMR (100 MHz, CDCl3) δ 182.6, 165.1, 164.2, 162.3 (2 C), 157.8, 134.8 (2 C), 134.4 (2 C), 128.8 (2 C), 128.1 (2 C), 126.7 (2 C), 126.2 (2 C), 125.7 (2 C), 123.5, 115.1 (2 C), 105.6, 104.4, 98.6, 93.2, 68.6, 68.3, 58.4 (2 C), 56.3 (2 C), 51.1 (2 C), 29.2 (4 C), 29.0 (2 C), 27.0 (2 C), 24.1 (2 C). HR-ESI-MS: Calcd. for C45H52N2O5 [M + H]+: 701.3910, found: 701.3938.
7-((6-(benzyl(ethyl)amino)hexyl)oxy)-2–(4-((6-(benzyl(ethyl)amino)hexyl)oxy)phenyl)-5-hydroxy-4H-chromen-4-one (10r). Compound 9d was treated with N-ethylbenzylamine 4c based on the above procedure to afford target compound 10r, light yellow oily matter, 76.2% yield. 1H NMR (400 MHz, CDCl3) δ 12.74 (s, 1H, OH), 7.74 (d, J = 8.2 Hz, 2H, 2 × Ar-H), 7.21 − 7.18 (m, 3H, 3 × Ar-H), 7.13 (dd, J = 17.8, 7.1 Hz, 7H, 7 × Ar-H), 6.91 (d, J = 8.2 Hz, 2H, 2 × Ar-H), 6.48 (s, 1H, Ar-H), 6.39 (s, 1H, C = CH), 6.26 (s, 1H, Ar-H), 3.97–3.95 (m, 4H, 2 × OCH2), 3.06 (d, J = 10.6 Hz, 4H, 2 × NCH2), 2.40 − 2.30 (m, 4H, 2 × NCH2), 2.04 (t, J = 9.7 Hz, 4H, 2 × NCH2), 1.81–1.77 (m, 10H, 2 × CH2 + 2 × CH3), 1.58–1.55 (m, 4H, 2 × CH3), 1.46–1.42 (m, 4H, 2 × CH3), 1.34 (d, J = 6.1 Hz, 4H, 2 × CH3). 13 C NMR (100 MHz, CDCl3) δ 182.5, 165.1, 164.1, 162.3, 162.2, 157.8, 129.2 (4 C), 128.3 (6 C), 128.1 (2 C), 127.1 (2 C), 123.4, 115.0 (2 C), 105.5, 104.3, 98.6, 93.1, 68.6, 68.3, 57.9 (2 C), 52.9 (2 C), 47.3 (2 C), 29.2, 29.0, 27.2, 27.2, 26.7, 26.6, 26.0, 25.9, 11.5 (2 C). HR-ESI-MS: Calcd. for C45H56N2O5 [M + H]+: 705.4223, found: 705.4250.
7-((6–(4-benzylpiperidin-1-yl)hexyl)oxy)-2–(4-((6–(4-benzylpiperidin-1-yl)hexyl)oxy)phenyl)-5-hydroxy-4H-chromen-4-one (10s). Compound 9d was treated with benzylpiperidine 4f based on the above procedure to afford target compound 10s, light yellow oily matter, 83.4% yield. 1H NMR (400 MHz, CDCl3) δ 12.74 (s, 1H, OH), 7.74 (d, J = 8.5 Hz, 2H, 2 × Ar-H), 7.20 (s, 4H, 4 × Ar-H), 7.11 (dd, J = 13.5, 6.5 Hz, 2H, 2 × Ar-H), 7.06 (d, J = 7.2 Hz, 4H, 4 × Ar-H), 6.91 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.49 (s, 1H, Ar-H), 6.39 (s, 1H, C = CH), 6.25 (s, 1H, Ar-H), 3.95–3.92 (m, 4H, 2 × OCH2), 3.09 (d, J = 10.5 Hz, 4H, 2 × NCH2), 2.50–2.47 (m, 8H, 4 × NCH2), 2.09–2.06 (m, 4H, 2 × CH2), 1.76–1.73 (m, 4H, 2 × CH2), 1.64–1.62 (m, 8H, 4 × CH2), 1.55–1.51 (m, 6H, 2 × CH2 + 2 × CH), 1.43–1.41 (m, 4H, 2 × CH2), 1.36–1.28 (m, 4H, 2 × CH2). 13 C NMR (100 MHz, CDCl3) δ 182.5, 165.0, 164.1, 162.2, 162.2 157.8, 140.2 (2 C), 129.2 (4 C), 128.4 (4 C), 128.1 (2 C), 126.1 (2 C), 123.4, 115.0 (2 C), 105.5, 104.3, 98.6, 93.1, 68.5, 68.2, 58.3, 58.3, 53.5 (4 C), 42.8 (2 C), 37.5 (2 C), 30.8 (2 C), 29.8, 29.8, 29.0, 28.9, 27.2, 27.2, 25.9, 25.8, 25.7, 25.6. HR-ESI-MS: Calcd. for C51H64N2O5 [M + H]+: 785.4849, found: 785.4871.
5-hydroxy-7-((6–(4-phenylpiperidin-1-yl)hexyl)oxy)-2–(4-((6–(4-phenylpiperidin-1-yl)hexyl)oxy)phenyl)-4H-chromen-4-one (10t). Compound 9d was treated with 4-phenylpiperidine 4g based on the above procedure to afford target compound 10t, light yellow oily matter, 80.7% yield. 1H NMR (400 MHz, CDCl3) δ 12.81 (s, 1H, OH), 7.82 (d, J = 8.3 Hz, 2H, 2H, 2 × Ar-H), 7.31 (dd, J = 20.5, 13.4 Hz, 10H, 10 × Ar-H), 6.98 (d, J = 8.3 Hz, 2H, 2 × Ar-H), 6.56 (s, 1H, Ar-H), 6.46 (s, 1H, C = CH), 6.34 (s, 1H, Ar-H), 4.01–3.99 (m, 4H, 2 × OCH2), 3.64–3.61 (m, 4H, 2 × NCH2), 2.56 (d, J = 6.7 Hz, 4H, 2 × NCH2), 2.52 − 2.45 (m, 4H, 2 × NCH2), 1.82–1.78 (m, 4H, 2 × CH2), 1.56–1.54 (m, 4H, 2 × CH2), 1.46–1.44 (m, 4H, 2 × CH2), 1.37–1.34 (m, 4H, 2 × CH2), 1.26–1.24 (m, 4H, 2 × CH2), 1.08 (t, J = 6.7 Hz, 6H, 2 × CH, 2 × CH2). 13 C NMR (100 MHz, CDCl3) δ 182.5, 165.0, 164.1, 162.2, 162.1, 157.7, 146.0 (2 C), 128.5 (4 C), 128.0 (2 C), 126.9 (4 C), 126.3 (2 C), 123.3, 115.0 (2 C), 105.5, 104.2, 98.5, 93.0, 68.6, 68.2, 58.9 (2 C), 54.3 (4 C), 42.6 (2 C), 33.1 (4 C), 29.1, 28.9, 27.4, 27.4, 26.7, 26.6, 26.0, 25.9. HR-ESI-MS: Calcd. for C49H60N2O5 [M + H]+: 757.4536, found: 757.4555.
5-hydroxy-7-((6–(4-(3-phenylpropyl)piperidin-1-yl)hexyl)oxy)-2–(4-((6–(4–(3-phenylpropyl)piperidin-1-yl)hexyl)oxy)phenyl)-4H-chromen-4-one (10u). Compound 9d was treated with 4-(3-phenylpropyl)piperidine 4i based on the above procedure to afford target compound 10u, light yellow oily matter, 60.6% yield. 1H NMR (400 MHz, CDCl3) δ 12.81 (s, 1H, OH), 7.82 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 7.27 (s, 3H, 3 × Ar-H), 7.17 (d, J = 7.2 Hz, 7H, 7 × Ar-H), 6.99 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.56 (s, 1H, Ar-H), 6.46 (s, 1H, C = CH), 6.33 (s, 1H, Ar-H), 4.03–4.01 (m, 4H, 2 × OCH2), 3.05–3.04 (m, 4H, 2 × NCH2), 2.59 (t, J = 7.4 Hz, 4H, 2 × phCH2), 2.46–2.35 (m, 4H, 2 × NCH2), 2.00 (t, J = 10.4 Hz, 4H, 2 × NCH2), 1.84–1.82 (m, 4H, 2 × CH2), 1.71 (d, J = 11.5 Hz, 4H, 2 × CH2), 1.63–1.60 (m, 8H, 4 × CH2), 1.51–1.47 (m, 4H, 2 × CH2), 1.41–1.36 (m, 6H, 2 × CH2, 2 × CH), 1.332–1.28 (m, 8H, 4 × CH2). 13 C NMR (100 MHz, CDCl3) δ 182.5, 165.0, 164.1, 162.2, 162.1, 157.7, 142.7 (2 C), 128.4 (4 C), 128.3 (4 C), 128.1 (2 C), 125.7 (2 C), 123.4, 115.0 (2 C), 105.5, 104.2, 98.5, 93.1, 68.5, 68.2, 58.8 (2 C), 53.9 (4 C), 36.2 (2 C), 36.1, 35.5, 31.7 (4 C), 29.1, 28.9, 28.8 (4 C), 27.4, 27.3, 26.4, 26.4, 26.0, 25.9. HR-ESI-MS: Calcd. for C55H72N2O5 [M + H]+: 841.5475, found: 841.5498.
7-((6–(3,5-dimethylpiperidin-1-yl)hexyl)oxy)-2–(4-((6–(3,5-dimethylpiperidin-1-yl)hexyl)oxy)phenyl)-5-hydroxy-4H-chromen-4-one (10v). Compound 9d was treated with 3,5-dimethylpiperidine 4j based on the above procedure to afford target compound 10v, light yellow oily matter, 67.9% yield. 1H NMR (400 MHz, CDCl3) δ 12.79 (s, 1H, OH), 7.80 (d, J = 8.2 Hz, 2H, 2 × Ar-H), 6.97 (d, J = 8.2 Hz, 2H, 2 × Ar-H), 6.55 (s, 1H, Ar-H), 6.45 (s, 1H, C = CH), 6.32 (s, 1H, Ar-H), 4.01–3.99 (m, 4H, 2 × OCH2), 2.99 (d, J = 10.3 Hz, 4H, 2 × NCH2), 2.50 − 2.37 (m, 4H, 2 × NCH2), 1.80–1.77 (m, 8H, 2 × NCH2, 2 × CH2), 1.63–1.57 (m, 8H, 4 × CH, 2 × CH2), 1.50–1.46 (m, 4H, 2 × CH2), 1.38–1.34 (m, 4H, 2 × CH2), 1.25–1.21 (m, 4H, 2 × CH2), 0.86 (d, J = 6.2 Hz, 12H, 4 × CH3). 13 C NMR (100 MHz, CDCl3) δ 182.5, 165.0, 164.1, 162.2, 162.1, 157.8, 128.1 (2 C), 123.4, 115.0 (2 C), 105.5, 104.2, 98.6, 93.1, 68.5, 68.2, 60.8 (4 C), 58.6 (2 C), 41.6 (2 C), 30.3 (4 C), 29.8, 29.0, 28.9, 27.3, 27.3, 25.9 (2 C), 25.9, 19.5 (4 C). HR-ESI-MS: Calcd. for C41H60N2O5 [M + H]+: 661.4536, found: 661.4572.
4.2. Biological activity
4.2.1. Antioxidant activity
The antioxidant potency of target compounds was tested using an oxygen radical absorbance capacity fluorescein (ORAC-FL) assay. The detailed procedure was reported in our previous work17,22.
4.2.2. Inhibition experiments of AChE and BuChE
The AChE and BuChE inhibitory activities of the synthesised compounds were evaluated using the Ellman assay with slight modification. AChE from 5% rat cortex homogenate or human erythrocytes (Sigma Co.). BuChE from rat serum or human serum (Sigma Co.). The detailed procedure referenced our previous work17,20.
4.2.3. Inhibitory effect on self-induced/Cu2+-induced Aβ1-42 aggregation
The Thioflavin T-based flurometric assay was employed to test the inhibition potency on self-induced/Cu2+-induced Aβ1-42 aggregation. The detailed procedure referenced our previous work20,22,28.
4.2.4. Metal chelation property
The metal chelation property was investigated by UV absorption in a Shimadzu UV-2450 spectrophotometer. The detailed procedure referenced our previous work20,22.
4.2.5. Transmission electron microscopy (TEM) assay
For the Cu2+-induced experiment, the Aβ stock solution was diluted with HEPES buffer (20 mM, pH 6.6, 150 mM NaCl). The sample preparation was the same as for the ThT assay. Aliquots (10 μL) of the samples were placed on a carbon-coated copper/rhodium grid for 2 min at room temperature. The excess sample was removed using filter paper followed by washing twice with ddH2O. Each grid was incubated with uranyl acetate (1% w/v ddH2O). Upon removal of excess uranyl acetate, the grids were dried for 15 min at room temperature. Images from each sample were taken on a transmission electron microscope20,22.
4.2.6. Neuroprotective effects on H2O2-induced PC12 cell injury
The neuroprotective effects of 5f and 7k were evaluated on H2O2-induced PC12 cell injury. The cell viability was measured with MTT assay and the detailed procedure referenced our previous work20,22.
4.2.7. In vitro anti-neuroinflammatory activity evaluation
The anti-neuroinflammatory property of compounds 5f and 7k was assessed using BV-2 cells by evaluating the level of NO production and TNF-α production32,33. Firstly, the cytotoxicity of compounds 5f and 7k was tested on BV-2 cells using an MTT assay. BV-2 cells were seeded in DMEM (100 μL) and cultured in a 96-well cell culture microplate and incubated in a humidified incubator containing 5% CO2 at 37 °C for 24 h. Then, 10 μL of 5f or 7k were added to cells in triplicate wells, followed by incubation for another 30 min. Subsequently, the treated cells were exposed to LPS (1.0 μg/mL, 10 μ L) and continued the culture for 24 h, and MTT solution was added to each well and incubated for 4 h at 37 °C. Finally, the crystals obtained were dissolved in DMSO (200 μ L) and OD values were measured at 490 nm. The results were recorded and expressed as the percent cell viability compared to the control group.
Griess reagent system was used to evaluate the inhibition of LPS-induced NO production. The methods of culturing BV-2 cells, setting blank control and adding tested compounds 5f or 7k (1 μM, 3 μM and 9 μM) and LPS were the same as those in the cytotoxicity assay. After that, the cell culture supernatants and different concentrations of NaNO2 as a standard were added to a 96-well plate since the NaNO2 concentration in the supernatant could act as an indicator of NO production. Then, to each well was added the same volume of Griess reagent and incubated at room temperature for 10 min. Finally, the absorbance was monitored at 540 nm using an ELISA plate reader to measure the results of NO production.
Inhibition of LPS-induced TNF-α production was evaluated with an enzyme-linked immunosorbent assay (ELISA). The methods of BV-2 cells culture, adding different concentrations of tested compounds 5f or 7k (1 μM, 3 μM and 9 μM) (10 μL) and LPS (1.0 μg/mL, 10 μL) and setting blank control group were the same as above in cytotoxicity assay. Then, the supernatant of the cell culture solution (50 μL) was added to a 96-well plate and the release of cytokine TNF-α was measured by ELISA according to the instructions of the ELISA kit.
4.2.8. Hepatoprotective activity20,22
Materials. LO2 cells purchased from Wuhan Punosi Life Technology Co., Ltd. 30% H2O2, Trichloroacetic acid (TCA), SRB, DMEM medium (HyClone), Foetal bovine serum (LNSER, article number: S711-001S), Penicillin streptomycin mixture (double antibiotic) (Solarbio), 0.25% trypsin-EDTA digestive juice (Solarbio), DMSO (Solarbio), Potassium dihydrogen phosphate (Xiyu Science Co., Ltd., CAS: 7778-77-0); Ultra-clean workbench (Wuxi Easy Purification Equipment Co., Ltd., model: SW-CJ-VS2); Carbon dioxide incubator (Esco, model: 2014-88759); Full-wavelength microplate reader (ThermoFisher, USA, model: 1510-01871);
Experiment method. (a) Cell culture: cultured in DMEM medium containing 10% FBS and 1% double-antibody, placed in 37 °C, 5% CO2 constant temperature and humidity incubator, changed every 2 days, subcultured when cells were 90% confluent. (b) Effect of compounds on LO2 proliferation by SRB assay: Discard the culture medium, fix the cells with 10% (m/v) trichloroacetic acid (TCA) 100 μL, and place in a refrigerator at 4 °C for 1 h. The fixing solution was discarded, washed 5 times with distilled water, and dried in an oven. Adding 100 μL SRB solution to each well after drying, and leave it at room temperature for 10-30 min. Discard the supernatant, wash it 5 times with 1% glacial acetic acid water, air dry. Finally, add 100 μL/well of 10 mmol/L Tris-based buffer solution and oscillate on a plate shaker. The OD value was measured at 515 nm using a microplate reader. Cell viability (%) = OD test well/OD blank control well x 100%. (c) Effect of compounds on H2O2-induced LO2 cell injury by SRB assay. The LO2 cells in the logarithmic growth phase were passaged, and the single-cell suspension with a cell density of 4 × 105/mL was adjusted, and the cells were seeded on a 96-well plate at 100 μL per well, and a blank control group, H2O2 was set. Group, 10 micromolar compounds group, each group of 3 duplicate wells, after 24h of culture, the old culture solution was discarded, the blank well and H2O2 group were replaced with complete medium, and the monomer compound group was replaced with a drug containing 10 micromolar. Complete medium. After 48h of culture, the H2O2 group and the monomer compound group were added with hydrogen peroxide prepared in a complete medium, and the final concentration of hydrogen peroxide was 1 mmol. The blank group and the H2O2 group were added with an equal volume of complete medium, and the culture was continued for 6h. After treatment with H2O2 for 6h, the survival rate was measured by the SRB method.
4.2.9. In vivo assay
4.2.9.1. Acute toxicity
Kunming mice at a body weight of 18–22 g (six weeks old, either gender) were supplied by the Centre of Experimental Animals of Sichuan Academy of Chinese Medicine Sciences (eligibility certification no. SCXK-Chuan2018-19). Mice were maintained under standard conditions with a 12 h:12 h light-dark cycle at 20−22 °C with a relative humidity of 60 − 70%. Sterile food and water were provided according to institutional guidelines. Prior to each experiment, mice were fasted overnight and allowed free access to water. Compound 5f at doses of 1000, 500, 250 and 100 mg/kg (n = 6 per group) by intragastric administration. After the administration of the compound 5f, the mice were observed continuously for the first 4 h for any abnormal behaviour and mortality changes, intermittently for the next 24 h, and occasionally thereafter for 14 days for the onset of any delayed effects. All animals were sacrificed on the 14th day after drug administration and were macroscopically examined for possible damage to the heart, liver, and kidneys.
4.2.10. Assay method
The step-down passive avoidance task was employed to investigate the effects of 5f on scopolamine-induced memory impairment17,20. Sixty mice were randomly divided into six groups. They were compound 5f group (1.9 mg/kg, 5.7 mg/kg and 17.1 mg/kg), same volume of water (untreated group), model group (3 mg/kg scopolamine), 5 mg/kg donepezil. After 30 min, memory impairment was induced by administering scopolamine (3 mg/kg). Then 30 min later, the learning and memory capacity of the mouse were measured by the Y-maze test. The maze was made of black-colored acryl and positioned at equal angles. Rats were placed at the end of the arm and allowed to move freely through the maze during 8 min sessions. Arm entry sessions were recorded when the hind paws of the rat were completely placed in the arm. Consecutive entry into three arms in alternative order was defined as successive entries on overlapping triplet sets and the alternation percentage was calculated as the ratio of actual to possible alternations (defined as the total number of arm entries minus 2), multiplied by 100.
The representative 1H and 13 C NMR spectra for the synthesised compounds are available as Supplementary material.
Supplementary Material
Funding Statement
This work was supported by funding from the State Key Laboratory of Functions and Applications of Medicinal Plants, Guizhou Medical University (Grant number FAMP202107K); Guizhou Science and Technology Platform Talents (QKHRCPT[2019]5627). And the Special Project of Nanyang Normal University (2022QN003 and 2020ZX015).
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Disclosure statement
The authors declare no competing financial interest.
References
- 1.World Alzheimer Report 2019: Attitudes to Dementia, Alzheimer’s Disease International; 2019, pp. 1–13.
- 2.Jagust W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat Rev Neurosci 2018;19:687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anand P, Singh B.. A review on cholinesterase inhibitors for Alzheimer's disease. Arch Pharm Res 2013;36:375–99. [DOI] [PubMed] [Google Scholar]
- 4.Hebscher M, Voss JL.. Testing network properties of episodic memory using non-invasive brain stimulation. Curr Opin Behav Sci 2020;32:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez A, Opazo C, Alarcón R, et al. Acetylcholinesterase promotes the aggregation of amyloid-beta-peptide fragments by forming a complex with the growing fibrils. J Mol Biol 1997;272:348–61. [DOI] [PubMed] [Google Scholar]
- 6.Hardy J, Selkoe DJ.. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297: 353–6. [DOI] [PubMed] [Google Scholar]
- 7.Calsolaro V, Edison P.. Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimers Dement 2016;12:719–32. [DOI] [PubMed] [Google Scholar]
- 8.Cheignon C, Tomas M, Bonnefont-Rousselot D, et al. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol 2018;14:450–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lane DJR, Ayton S, Bush AI.. Iron and Alzheimer's disease: an update on emerging mechanisms. J Alzheimers Dis 2018;64:S379–S395. [DOI] [PubMed] [Google Scholar]
- 10.Cavalli A, Bolognesi ML, Minarini A, et al. Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem 2008;51:347–72. [DOI] [PubMed] [Google Scholar]
- 11.Zhang P, Xu S, Zhu Z, Xu J.. Multi-target design strategies for the improved treatment of Alzheimer's disease. Eur J Med Chem 2019;176:228–47. [DOI] [PubMed] [Google Scholar]
- 12.de Freitas Silva M, Dias KST, Gontijo VS, et al. Multi-target directed drugs as a modern approach for drug design towards Alzheimer's disease: an update. Curr Med Chem 2018;25:3491–525. [DOI] [PubMed] [Google Scholar]
- 13.Huang W, Liang M, Li Q, et al. Development of the "hidden" multifunctional agents for Alzheimer's disease. Eur J Med Chem 2019;177:247–58. [DOI] [PubMed] [Google Scholar]
- 14.Joshi R, Kulkarni YA, Wairkar S.. Pharmacokinetic, pharmacodynamic and formulations aspects of Naringenin: an update. Life Sci 2018;215:43–56. [DOI] [PubMed] [Google Scholar]
- 15.Ghofrani S, Joghataei MT, Mohseni S, et al. Naringenin improves learning and memory in an Alzheimer's disease rat model: insights into the underlying mechanisms. Eur J Pharmacol 2015;764:195–201. [DOI] [PubMed] [Google Scholar]
- 16.Wu J, Kou X, Ju H, et al. Design, synthesis and biological evaluation of naringenin carbamate derivatives as potential multifunctional agents for the treatment of Alzheimer's disease. Bioorg Med Chem Lett 2021;49:128316. [DOI] [PubMed] [Google Scholar]
- 17.Sang Z, Qiang X, Li Y, et al. Design, synthesis and evaluation of scutellarein-O-alkylamines as multifunctional agents for the treatment of Alzheimer's disease. Eur J Med Chem 2015;94:348–66. [DOI] [PubMed] [Google Scholar]
- 18.Bai P, Wang K, Zhang P, et al. Development of chalcone-O-alkylamine derivatives as multifunctional agents against Alzheimer's disease. Eur J Med Chem 2019;183:111737. [DOI] [PubMed] [Google Scholar]
- 19.Qiang X, Sang Z, Yuan W, et al. Design, synthesis and evaluation of genistein-O-alkylbenzylamines as potential multifunctional agents for the treatment of Alzheimer's disease. Eur J Med Chem 2014;76:314–31. [DOI] [PubMed] [Google Scholar]
- 20.Sang Z, Wang K, Shi J, et al. The development of advanced structural framework as multi-target-directed ligands for the treatment of Alzheimer's disease. Eur J Med Chem 2020;192:112180. [DOI] [PubMed] [Google Scholar]
- 21.Li HQ, Shi L, Li QS, Liu PG, et al. Synthesis of C(7) modified chrysin derivatives designing to inhibit beta-ketoacyl-acyl carrier protein synthase III (FabH) as antibiotics. Bioorg Med Chem 2009;17:6264–9. [DOI] [PubMed] [Google Scholar]
- 22.Sang Z, Wang K, Shi J, et al. Apigenin-rivastigmine hybrids as multi-target-directed liagnds for the treatment of Alzheimer's disease. Eur J Med Chem 2020;187:111958. [DOI] [PubMed] [Google Scholar]
- 23.Košak U, Brus B, Knez D, et al. The magic of crystal structure-based inhibitor optimization: development of a butyrylcholinesterase inhibitor with picomolar affinity and in vivo activity. J Med Chem 2018;61:119–39. [DOI] [PubMed] [Google Scholar]
- 24.Wang LL, Du Y, Li SM, et al. Design, synthesis and evaluation of tetrahydrocarbazole derivatives as potential hypoglycemic agents. Bioorg Chem 2021;115:105172. [DOI] [PubMed] [Google Scholar]
- 25.Sharma P, Tripathi A, Tripathi PN, et al. Design and development of multitarget-directed N-Benzylpiperidine analogs as potential candidates for the treatment of Alzheimer's disease. Eur J Med Chem 2019;167:510–24. [DOI] [PubMed] [Google Scholar]
- 26.Tripathi A, Choubey PK, Sharma P, et al. Design and development of molecular hybrids of 2-pyridylpiperazine and 5-phenyl-1,3,4-oxadiazoles as potential multifunctional agents to treat Alzheimer's disease. Eur J Med Chem 2019;183:111707. [DOI] [PubMed] [Google Scholar]
- 27.Tripathi PN, Srivastava P, Sharma P, et al. Biphenyl-3-oxo-1,2,4-triazine linked piperazine derivatives as potential cholinesterase inhibitors with anti-oxidant property to improve the learning and memory. Bioorg Chem 2019;85:82–96. [DOI] [PubMed] [Google Scholar]
- 28.Sang Z, Wang K, Han X, et al. Design, synthesis, and evaluation of novel ferulic acid derivatives as multi-target-directed ligands for the treatment of Alzheimer's disease. ACS Chem Neurosci 2019;10:1008–24. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Qiang X, Li Y, et al. Pterostilbene-O-acetamidoalkylbenzylamines derivatives as novel dual inhibitors of cholinesterase with anti-β-amyloid aggregation and antioxidant properties for the treatment of Alzheimer's disease. Bioorg Med Chem Lett 2016;26:2035–9. [DOI] [PubMed] [Google Scholar]
- 30.Di L, Kerns EH, Fan K, et al. High throughput artificial membrane permeability assay for blood-brain barrier. Eur J Med Chem 2003;38:223–32. [DOI] [PubMed] [Google Scholar]
- 31.Sang Z, Wang K, Bai P, et al. Design, synthesis and biological evaluation of novel O-carbamoyl ferulamide derivatives as multi-target-directed ligands for the treatment of Alzheimer's disease. Eur J Med Chem 2020;194:112265. [DOI] [PubMed] [Google Scholar]
- 32.Tian C, Qiang X, Song Q, et al. Flurbiprofen-chalcone hybrid Mannich base derivatives as balanced multifunctional agents against Alzheimer's disease: design, synthesis and biological evaluation. Bioorg Chem 2020;94:103477. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z, Song Q, Cao Z, et al. Design, synthesis and evaluation of flurbiprofen-clioquinol hybrids as multitarget-directed ligands against Alzheimer's disease. Bioorg Med Chem 2020;28:115374. [DOI] [PubMed] [Google Scholar]
- 34.http://www.molinspiration.com/cgi-bin/properties
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.