

Abstract
Pueraria lobata var. thomsonii (hereinafter abbreviated as Podalirius thomsonii), a member of the legume family, is one of the important traditional Chinese herbal medicines, and its puerarin extract is widely used in the health and pharmaceutical industry. Here, we assembled a high-quality genome of P. thomsonii using long-read single-molecule sequencing and Hi-C technologies. The genome assembly is ~1.37 Gb in size and consists of 5145 contigs with a contig N50 of 593.70 kb, further clustered into 11 pseudochromosomes. Genome structural annotation resulted in ~869.33 Mb (~62.70% of the genome) repeat regions and 45 270 protein-coding genes. Genome evolution analysis revealed that P. thomsonii is most closely related to soybean and underwent two ancient whole-genome duplication events; one was in the common ancestor shared by legume species and the other occurred independently at around 7.2 million years ago, after its speciation. A total of 2373 gene families were found to be unique in P. thomsonii compared with five other legume species. Genes and metabolites related to puerarin content in tuberous tissues were characterized. A total of 572 genes that were upregulated in the puerarin biosynthesis pathway were identified, and 235 candidate genes were further enriched by omics data. Furthermore, we identified six 8-C-glucosyltransferase (8-C-GT) candidate genes significantly involved in puerarin metabolism. Our study filled a key genomic gap in the legume family, and provided valuable multi-omic resources for the genetic improvement of P. thomsonii.
Introduction
Pueraria lobata (2n = 2x = 22) is a semiwoody, perennial liana that belongs to the Leguminosae family and is widely distributed throughout Asia, including China, Japan, Korea, and other regions in Southeast Asia, as well as in North and South America. As an economic crop, it contains puerarin and other functional components and is used in the production of both pharmaceuticals and health foods. The roots of both P. lobata (hereinafter abbreviated as P. lobata) and P. lobata var. thomsonii (Benth.) (hereinafter abbreviated as Podalirius thomsonii) have long been used for treating fever, toxicosis, indigestion, and liver damage from alcohol abuse in traditional Chinese medicine [1], which was recorded in The Divine Husbandman’s Classic of Materia Medica (Shen Nong Ben Cao Jing) compiled in the Eastern Han Dynasty (25–250 AD) [2]. Since P. thomsonii has much higher starch content and milder therapeutic effects, its root extract or powder is commonly consumed as a dietary supplement or an ingredient in sweet or savory dishes in Asian countries [3].
The classification of genus Pueraria is still largely in dispute. There are currently 20 accepted species, with synonyms crossing over in other genera, such as Dolichos and Glycine [4]. P. lobata var. thomsonii, sometimes called Thomson’s kudzu, or fenge, was recognized as one species in the Pueraria genus, along with P. lobata var. montana (hereinafter abbreviated as P. montana) and P. lobata. Synonyms such as P. thomsonii Benth., P. lobata var. thomsonii (Benth.) Maesen and P. lobata var. chinensis Ohwi have been used in various mentions [5]. P. montana is morphologically and genetically distinct from lobata and thomsonii [3]. Genetic markers, such as RAPD (random amplified polymorphic DNA), SSR (simple sequence repeat), and SNP (single nucleus polymorphism) markers, and sRNA probes have been used to characterize genetic variations in the species P. thomsonii and its close relatives [6–9]. However, most of the basic molecular and genetic analyses remain inconclusive in Pueraria due to the lack of genomic information. High-quality genome assemblies and their annotation in P. thomsoniiwould greatly improve the value of the resources available to distinguish and understand the genomic compositions.
The pharmaceutical values of P. lobata and P. thomsonii are mostly attributed to their isoflavones, specifically daidzein and its glycosylated derivatives, daidzin and puerarin [1, 3, 10]. Puerarin is the chemotaxonomic marker and the major bioactive compound isolated from the roots of Podalirius thomsonii. The isoflavones and other metabolic compounds differ among Pueraria species, resulting in the distinct therapeutic characters of Pueraria species [11, 12]. Previous studies identified possible enzymes involved in the isoflavone biosynthesis pathway by comparing gene expressions or metabolite contents in different tissues of P. lobata [13–16]. Due to the complications attributable to the historical genome duplication as well as gene family expansions and contractions [8], little is known about the genes involved in isoflavone biosynthesis. A chromosome-level genome assembly with detailed function annotations could pave the way to elucidating the key players in the isoflavone biosynthesis pathway.
In this study, we generated a high-quality chromosome-level genome assembly of P. thomsonii, the first genome assembly in the Pueraria genus, by combining short and long-read DNA sequencing and Hi-C scaffolding technologies. We structurally and functionally annotated the genome with clustered gene families related to diverse biological processes. Comparative genomics analysis supported the idea that P. thomsonii is most closely related to soybean and experienced two historical whole-genome duplication (WGD) events. The key gene families and functional gene members in the isoflavonone and puerarin biosynthesis pathways, especially the UDP-glycosyltransferase gene family, were characterized based on a combination of genomic, transcriptomic, and metabolic data. The chromosome-level assembly contributes a new genome to the legume family, and lays a solid foundation for the genomic utilization of P. thomsonii both for legume science and genetic improvement of medicinal germplasm resources.
Results
Genome assembly and annotation
We firstly generated 78.95 Gb of 250 bp paired-end Illumina sequences. Genome complexity estimation based on K-mer analysis revealed that the diploid genome of P. thomsonii is ~1.35 Gb in size and has around 0.8% heterozygosity (Supplementary Fig. 1). We then de novo produced 91.6 Gb PacBio single-molecule long polymerase reads and assembled the genome using Falcon followed by assembly polishing using both the long and short reads. The total length of the PacBio assembly is 1.38 Gb with contig N50 of 598 kb. The assembly was further scaffolded into 281 scaffolds using Hi-C technology (Supplementary Fig. 2), 11 of which were at the chromosome level, accounting for 99.3% of the total assembly size (Fig. 1a, Supplementary Table 1). BUSCO analysis found that 92.9% of the core eukaryotic gene set were complete in our assembly, the majority of them being single-copy genes (Supplementary Table 3). These results suggest that the quality of the genome assembly is relatively high in contiguity and completeness.
Figure 1.

Genome landscape of assembly, annotation, and gene family clusters of P. thomsonii. a Overview of the P. thomsonii genomic features. Track I is the circular representation of the pseudomolecule. Tracks II–V are repetitive sequence density (bin = 1 Mb), gene density (bin = 1 Mb), GC content (bin = 1 Mb), and genome collinearity, respectively. b Distribution of single-copy, multiple-copy, unique, other orthologous and unclustered genes in the 17 plant species used in this study: Oryza sativa, Vitis vinifera, Pisum sativum, Phaseolus vulgaris, Vigna unguiculata, Vigna angularis, Glycine max, Pueraria lobata, Malus domestica, Cucumis melo, Momordica charantia, Populus alba, Salix brachista, Arabidopsis thaliana, Corchorus capsularis, Theobroma cacao, and Solanum lycopersicum. c Number of overlapped gene families among the six legume species visualized in the UpSet plot. PSAT, P. sativum; VANG, V. angularis, PVUL, P. vulgaris, GMAX, G. max; PTHO, P. thomsonii; VUNG, V. unguiculata.
The predicted repeat regions account for 62.70% of the genome assembly. Long terminal repeats (LTRs) were the main type of repetitive element, accounting for ~14.22% of the repeat regions. DNA transposons and long interspersed nuclear elements (LINEs) accounted for 4.03% and 2.47% of the assembly, respectively (Supplementary Table 3).
Structural annotation of the genome assembly yielded 45 270 gene models. On average, the gene length is 2446 bp, with the length of coding sequences 933 bp and an average of four exons per gene. Out of all gene models, 42 735 (~94.4% of the predicted protein-coding genes of P. thomsonii) were functionally annotated with known genes, conserved domains, or gene ontology (GO) terms (Supplementary Table 4, Supplementary File).
Identification and evolution of gene families
We identified orthologs using the whole-genome gene sets of P. thomsonii and 16 other representative plant species, including five other species of the legume family (Glycine max, Vigna angularis, Pisum sativum, Phaseolus vulgaris, and Vigna unguiculata), 10 non-leguminous dicots (Salix brachista, Corchorus capsularis, Arabidopsis thaliana, Cucumis melo, Malus domestica, Momordica charantia, Populus alba, Solanum lycopersicum, Vitis vinifera, and Theobroma cacao), and one monocot species (Oryza sativa) as outgroup. In the P. thomsonii genome, 39 270 genes were clustered into 16 546 ortholog groups (Fig. 1b). The six legume species shared a core set of 11 204 gene families (Fig. 1c). Compared with G. max, 4743 gene families were expanded and 3099 gene families were contracted in P. thomsonii (Supplementary Fig. 3). These expanded gene families are functionally diverse and are functionally enriched in pathways such as flavonoids, alkaloids, sterols, and terpenoid biosynthesis (Supplementary Fig. 4). A total of 2373 gene families were unique in P. thomsonii in comparison with other leguminous species. These genes were enriched in functions related to nitrogen metabolism, streptomycin biosynthesis, ubiquinone, and other terpenoid-quinone biosynthesis (Supplementary Fig. 5).
Positive selection scanning of the 201 single-copy orthologous genes found 34 significant genes. Enrichment analysis of the functions of these positively selected genes indicated that they were enriched in functions such as circadian rhythm, homologous recombination, and starch and sucrose metabolism (Supplementary Fig. 6).
Whole-genome duplication
A total of 201 single-copy orthologous genes shared across the 17 species were used to construct the phylogenetic tree. P. thomsonii was most closely related to G. max, giving an estimate of the divergence from soybean at about 20.1 million years ago (MYA), after the separation of Leguminosae and Orchidaceae at ~102.5 MYA (Fig. 2a). Both Ks and 4-fold debatable third-codon transversion (4DTv) values were calculated based on homologous gene pairs in P. vulgaris and three other representative plant species, G. max, A. thaliana, and O. sativa. The distribution of Ks or 4DTv values in P. thomsonii showed two peaks at Ks values of around 0.05 (4DTv ~0.04) and 0.46 (4DTv ~0.2; Fig. 2b, Supplementary Fig. 7), which is consistent with published research findings [17, 18] that the Ks range is between 0.4 and 1.2. Calculated from the r value of 5.17 × 10−3 in published studies [19], the first peak occurred after the divergence of P. thomsonii from G. max and was dated at ~4.8 Mya, later than the onset of the most recent WGD in G. max. The second peak, shared between P. thomsonii and G. max, resulted from a much older WGD, which happened in the common ancestor of the legume species, at ~44.5 Mya.
Figure 2.
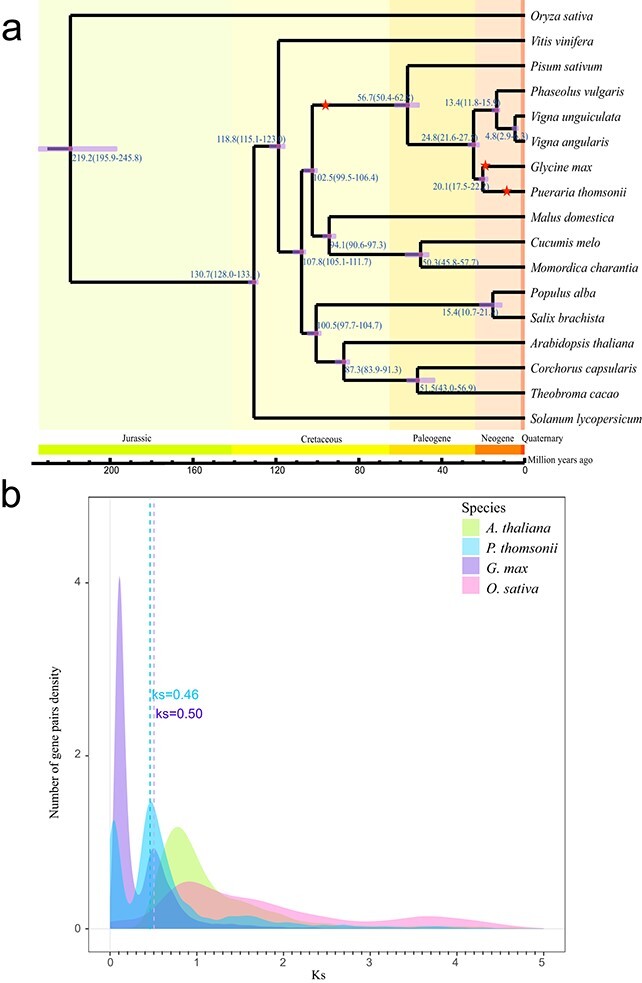
Evolution and WGD in P. thomsonii and 16 other species. a Phylogenetic trees and divergence times of the 17 species. The red stars on the branches represent occurrences of WGD events. b Distribution of Ks values of A. thaliana, P. thomsonii, G. max and O. sativa.
Combined metabolic and gene expression analyses reveal isoflavone biosynthesis pathway
We performed untargeted metabolic profiling using the roots of high- and low-puerarin genotypes, ZG-19 and ZG-39, respectively. A total of 614 metabolites were detected. Both principal component analysis (PCA) and correlation analysis showed that biological repeat samples are much more closely related within genotypes than between genotypes (Fig. 3a, Supplementary Fig. 8). A total of 225 differential metabolites (DMs) were identified between the two genotypes, of which 172 were up-regulated and 53 were down-regulated in ZG19 relative to ZG-39 (Fig. 3b, Supplementary File ). GO enrichment analysis of the DMs showed significant differences in the synthetic pathways of flavonoids, flavonol, and isoflavones between ZG19 and ZG-39 (Fig. 3c).
Figure 3.

General overview of metabolome and transcriptome. a PCA of metabolites. b Volcano plot of differential metabolites between the roots of ZG-19 and ZG-39. c KEGG enrichment analysis of the DMs between the roots of ZG-19 and ZG-39. d Volcano plot of DEGs between the roots of ZG-19 and ZG-39. e KEGG enrichment pathway with both DMs and DEGs. The red bars are the negative log10 and the green color represents the enrichment P-value of DMs, which is expressed as −log(P-value), and higher vertical coordinates represent stronger enrichment. f Canonical correlation analysis between the DMs and DEGs in the isoflavonoid biosynthesis pathway. Gene IDs are in green and metabolite names are in purple; within the same region, the further from the origin and the closer to each other the higher the correlation.
A total of 1814 differentially expressed genes (DEGs) were identified between the two genotypes, of which 464 were upregulated and 1350 were downregulated in ZG19 relative to ZG-39 (Fig. 3d). The enriched function categories of the DMs and DEGs overlap; the results showed that they were all genes or metabolites relevant to flavonoids, isoflavones, and ATP-binding cassette transport (Fig. 3e). We identified a large number of metabolites and gene pairs that were highly correlated across the samples with |r| > 0.8 (Supplementary Figs. 9 and 10). Pairwise correlation coefficients were calculated between the identified metabolites and genes. Sixty percent of all the significant correlations involved upregulated metabolites and downregulated or unchanged genes. In 15% of the significant correlations, metabolites and gene expressions changed in the same direction (Supplementary Fig. 9). Moreover, we identified a large number of DEGs and DMs in the isoflavone biosynthesis pathway (Supplementary Fig. 11). Canonical correlation analysis of the DEGs and DMs in the isoflavone biosynthesis pathway showed that the expression of gene chr11g3_854.1, encoding 2-hydroxyisoflavanone synthase, was highly correlated with the content of daidzein in the tubers, while the expression of chr11g1_1158.17, which encodes UDP-glycosyltransferase, was highly correlated with the pratensein content in the tubers (Fig. 3f).
Puerarin synthesis pathway
We identified 572 genes homologous to nine gene families putatively encoding the enzymes in the puerarin synthesis pathway (Supplementary File 3). All nine gene families in the P. thomsonii genome were expanded compared with the other five legume species (Supplementary Fig. 12). We predicted that out of all the homologous genes, 40 genes differentially expressed between the high- and low-puerarin roots were functionally involved in the puerarin biosynthesis pathway in P. thomsonii (Fig. 4, Supplementary File 4). We further analyzed the genes in the glycosyltransferase family catalyzing the glycosylation modification. A total of 104 glucosyltransferase (GT) genes were identified in P. thomsonii. There were 13 genes homologous to 8-C-glucosyltransferase (8-C-GT), 6 of which were homologous to the previously studied PIUGT43 gene catalyzing the C-glucosylation of daidzein to puerarin [14]. The DEGs in the GT family with the top three largest gene expression changes in the roots of ZG-19 and ZG-39 were chr11g8_848.22, chr11g8_844.18, and chr11g7_666.8. We also identified 179 genes significantly correlated with the puerarin content in the roots (Supplementary File 5).
Figure 4.

Expression profile of genes encoding enzymes of the puerarin biosynthesis pathway in the roots of ZG-19 and ZG-39 genotypes. Gene expression values (FPKM) were scaled by log2(FPKM+1). Red and blue asterisks represent significant up- and downregulation of gene expression in root tissue of ZG-19 relative to ZG-39, respectively. Expression profile of all members of the glucosyltransferase (GT) gene family in P. lobata var. thomsonii are presented; genes in red and orange are homologous to 8-C-GT genes. Moreover, genes in red are the homologous genes of PIUGT43, which was previously found to catalyze the C-glucosylation of daidzein to puerarin [16]. PAL, phenylalanine ammonialyase; C4H, cinnamate-4-hydroxylase; 4CL, 4-coumarate-CoA ligase; CHS, chalcone synthase; CHR, chalcone reductase; CHI, chalcone isomerase; IFS, 2-hydroxyisoflavanonesynthase; 8-C-GT, 8-C-glucosyltransferase; HID, 2-hydroxyisoflavanone dehydratase.
The gene (chr11g3_854.1) encoding isoflavone synthase, which catalyzes the synthesis of daidzein, was identified to be highly associated with the synthesis pathway of puerarin. Moreover, daidzein was detected as an intermediate metabolite in puerarin synthesis, implying that gene chr11g3_854.1 likely promotes daidzein synthesis and accumulates in the roots. The synthesis of puerarin is also likely to be dependent on daidzein via glycosyltransferase chr11g8_848.22 or chr11g8_844.18.
Discussion
Pueraria has great ecological and economical impact globally, yet genomic resources and understanding are still limited at the level of molecular markers and transcriptomes. Our study provided a foundational resource by providing the first high-quality, chromosome-level genome of P. thomsonii by combining long-read sequencing and Hi-C scaffolding technologies. The genome sizes estimated using short reads and from the assembly were very close, at 1.35–1.38 Gb, which is relatively large, with a high percentage of repeat regions compared with other legume species.
We found that P. thomsonii was most closely related to soybean based on genomic phylogeny; both went through a shared ancient WGD as well as a lineage-specific WGD. Large genomic blocks of G. max were found in synteny with much more fragmented chromosome regions in P. thomsonii (Supplementary Fig. 13). This suggested that the P. thomsonii genome had undergone extensive chromosome rearrangement despite its estimated more recent WGD compared with G. max, which coincided with the smaller number of chromosomes in P. thomsonii [15].
Analyzing the annotated gene set revealed that, compared with five other leguminous species, P. thomsonii is unique in that ~14.3% of the gene families were enriched in pathways related to sugar and starch metabolism (Supplementary File 6), which coincided with the characteristically high starch and sugar content in P. thomsonii roots. These unique genes could shed light on the genomics of starch accumulation in root tissues in the legume family.
Puerarin is the most abundant bioactive compound in the Pueraria genus. Although the genes and enzymes involved in the puerarin synthesis pathway have been studied [13, 16, 20], our study for the first time identified and characterized the genes of the puerarin biosynthesis pathway using multi-omic data collected at the genomic, transcriptomic, and metabolic levels. The results showed that all gene families in the puerarin biosynthesis pathway were expanded compared with other legume species and 40 out of all 235 homologous genes were functionally involved in the pathway.
Moreover, we analyzed the GT gene family, encoding a key enzyme that catalyzes glycosylation modifications at different stages downstream of flavone or isoflavone biosynthesis and confers biological and pharmochemical properties in isoflavonoids such as puerarin. We constructed a phylogenetic tree using the 104 glucosyltransferase genes filtered and identified in this study along with previously characterized UGTs. We then classified these genes into seven clusters/subfamilies according to their classification in the previous study [16] (Supplementary Table 5, Supplementary Fig. 14). There were six genes in P. thomsonii that belonged to cluster 6, characterized as a plant isoflavonoid C-glycosyltransferase (CGT) group. However, our phylogeny analysis of the UGT genes placed the six genes as well as their homologous genes from P. lobata within the (iso)flavone 7-OH glycosylation group (cluster 5), which was inconsistent with the previous classification, and suggested further verification was needed.
The reference genome of P. thomsonii not only reveals the genes of the puerarin synthesis pathway more comprehensively, but also provides the possibility of gene localization, screening and evolutionary studies. This is very important for the cultivation of P. thomsonii as a herbal medicine.
Conclusions
We report the first high-quality chromosome-scale genome of P. thomsonii. The genome size is ~1.37 GB, with contig N50 of 593.70 kb. Approximately 99% of the assembled sequences were represented by 11 pseudochromosomes. In total, we identified ~62.70% of the repetitive sequences and 45 270 protein-coding genes. The P. thomsonii genome experienced two WGD events, the first at ~44.5 Mya and the most recent one at ~4.8 Mya after divergence from G. max. Using multi-omics data, genes and metabolites were found associated with puerarin content in root tissues and were enriched in isoflavone biosynthesis and sugar and starch metabolic pathways. Nine gene families involved in the puerarin biosynthesis pathway were characterized, and 235 candidate genes and 13 key glycosyltransferase candidate genes were further selected. Our study developed the important multi-omic resources for exploiting and improving P. thomsonii as an economic crop for edible starch and traditional Chinese medicine.
Materials and methods
Genome sequencing
For genome sequencing, we grew and labeled plants of P. thomsonii from the Lijian Scientific Research Base of Guangxi Academy of Agricultural Sciences (GXAAS). Fresh, young, and healthy leaves were harvested from well-growing individuals, and were immediately frozen in liquid nitrogen and stored at −80°C for DNA extraction. High-quality genomic DNA was extracted from the harvested samples using a modified cetyltrimethyl ammonium bromide (CTAB) method [21]. For third-generation genome sequencing, the SMRT Bell library was prepared using SMRTbell Express Template Prep Kit 2.0, which was loaded into a Pacific Biosciences PacBios loaded into a Prep Kit 2.0 Sequel II instrument on PacBio SMRT cells 8 M (Pacific Biosciences, Menlo Park, CA, USA), acquiring one movie of 30 hours per SMRT cell. For short-read sequencing, the Illumina library with average insert sizes of 397 bp was generated using a Genomic DNA Sample Preparation Kit (Illumina), which was loaded into an Illumina 6000 instrument with PE250.
Hi-C sequencing
To construct chromosome-level superscaffolds, Hi-C sequencing data were produced as previously described [22]. Briefly, fresh samples were cut into small segments and cross-linked by immersion in 3% formaldehyde for 15 min. Then the material was crushed to a fine powder, which was used to isolate the nuclei. The isolated nuclei were processed by purifying, digesting with MboI, blunt-end repairing, and labeling with biotin. After processing, the DNA was re-ligated with T4 DNA ligase. After digestion with proteinase K and cross-linking reduction using formaldehyde, biotin-containing DNA fragments were captured and used to construct Hi-C libraries. We obtained 127.5 Gb of sequencing data by sequencing the final library using the Illumina X Ten sequencing platform.
Genome survey
Short reads produced by the Illumina 6000 platform were quality-filtered by HTQC [23] (version v1.92.310) using the following method. Adaptors were firstly removed from the sequencing reads, and read pairs with any one end having an average quality <20 were discarded. Ends of reads were trimmed if average quality was <20 in the sliding window size of 5 bp, and read pairs with any end shorter than 75 bp were removed. The quality-filtered reads were used for estimation of genome size. The 17-mer occurrence distribution of sequencing reads was generated from short libraries with Jellyfish [24] (v2).
Genome assembly
Subreads generated with the above long-read third-generation sequencing were used for genome assembly of P. lobata. The draft assembly of the genome was made using Falcon (v0.3.0) [25]. To correct errors in the primary assembly, Racon (v1.44) tools were used to polish the genome [26]. Illumina-derived short reads were used to correct any remaining errors by NextPolish (v1.1.0) [27].
To scaffold the contigs, 425 011 698 clean-read pairs were sequenced from the Hi-C library. Fastp (0.19.5) [28] was used to filter Hi-C data, including removing adaptors and low-quality reads. HiC-Pro Proto was used to obtain effective reads [29]. After filtering, 6% of high-quality and effective Hi-C data was retained. The resulting effective reads were mapped to the assembled and polished P. thomsonii genome using BWA (bwa-0.7.17) [30] with default parameters. ALL-HiC software was used to anchor scaffolds to chromosomes to obtain the chromosome-level P. thomsonii assembly.
Genome structural and functional annotation
We used LTR_finder (v1.07) [31], repeatscont (v1.0.5) [32], and trf (v4.09) [33] tools to identify repetitive elements in the P. thomsonii genome assembly, then used these sequences as the library, and finally used RepeatMasker (open-4.0.9) [34] to conduct repeat annotation. Maker (v3.01.02) [35] was used to annotate gene structures, using the protein sets of Medicago sativa, Lotus japonicus, and G. max retrieved from the NCBI database as homology evidence.
Gene functions were inferred according to the best match of alignments to NCBI Swiss-Prot [36] protein databases using BLASTP (NCBI BLAST v2.6.0+) [37, 38] and the KEGG database [39] with an e-value threshold of 1e−5. GO [40] IDs were obtained by using Blast2GO [41].
Gene family identification
To identify ortholog groups of the protein-coding genes, whole-genome protein sequences were pairwise aligned using BLASTP programs (NCBI BLAST+ v2.6.0) with a maximal e-value of 1e−5 from P. thomsonii and other 16 species, including O. sativa, G. max, S. brachista, C. capsularis, A. thaliana, C. melo, M. domestica, M. charantia, P. vulgaris, P. alba, S. lycopersicum, V. angularis, V. vinifera, P. sativum, T. cacao, and V. unguiculata. To exclude putatively fragmented genes, identity <30%, coverage <50% and g protein-encoding sequences shorter than 50 bp amino acids were filtered out. OrthoMCL (v14–137) [42] was used to cluster genes into ortholog groups with the parameter of inflation set as 1.5.
Phylogenetic analysis
Single-copy orthologs were used to construct a phylogenetic tree for P. thomsonii and the other 16 species. Protein sequences were aligned with MUSCLE (v3.8.31) [43], and alignments of the corresponding coding sequences were generated and concatenated with the guidance of protein alignment. The maximum likelihood method was used to construct the phylogenetic tree with RAxML (v8.2.11) [44].
Gene family expansion and contraction analysis
Based on the identified gene families and the phylogenetic tree with predicted divergence times of these species, CAFÉ [45] was used to analyze gene family expansion and contraction, by using a random birth and death model. A conditional P-value was calculated for each gene family, and families with a conditional P-value <.05 were considered to have accelerated rates of gene gain or loss. The significantly expanded and contracted gene families in P. thomsonii were mapped to KEGG pathways for enrichment analysis, using a hypergeometric test with false discovery rate (FDR)-adjusted P-value (Q-value) <0.05 (https://github.com/StoreyLab/qvalue).
Positively selected genes
Based on the phylogenetic tree, the rate ratio (ω) of non-synonymous (Ka) to synonymous (Ks) nucleotide substitutions was estimated using the PAML (v4.9e) package [46] to scan the selective constraints on the candidate genes that were single-copy across all 17 species. After high-quality alignments of related sequences were obtained as described above, we compared a series of evolutionary models in the likelihood framework using the species trees. A branch-site model was used to estimate average ω across the tree (ω0), ω of the appointed branch to test (ω2), and ω of all other branches (ω1). Genes under positive selection were identified when the adjusted P-value was <.05.
Whole-genome duplication analysis
We used the synonymous substitution rate (Ks) to detect WGD events. First, gene pairs used in the Ks and 4DTv calculations were acquired using JCVI [47] between A. thaliana, O. sativa, G. max, and P. thomsonii. Specific methodology was consistent with previous studies [48]. Ks values were calculated using KaKs_Calculator [49] (v2.0). Finally, the Ks distribution was used to evaluate WGD events. WGD events were dated using the local multi-rate clock and t = Ks/2r in which r = 5.17 × 10−3 refers to the nucleotide substitution rate [19].
RNA sequencing and expression analysis
We chose two genotypes for RNA-Seq: ZG-19 with high puerarin content and ZG-39 with low puerarin content. Freeze-dried root samples were collected and immediately frozen in liquid nitrogen. For each genotype, three biological replicates were collected in parallel. RNA was isolated using the Plant RNA Kit (R6827, Omega), and first-strand cDNA was synthesized using random primers with TransScript II All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (One-Step gDNA Removal) (AT341, Transgen Biotech). One microgram of RNA per sample was used as input material for RNA-Seq. RNA-Seq libraries were prepared using the Illumina mRNA-Seq Library Preparation Kit and sequenced using an Illumina HiSeq 2000 with 150 bp paired-end read sequencing.
Low-quality reads, such as adaptors, reads containing >5% unknown nucleotides, and reads with Q20 <20% (percentage of sequences with sequencing error rates <1%), were removed by fastqc. The clean reads were mapped to P. thomsonii reference genome using HISAT2 [50]. We predicted new transcripts from the genome alignment using StringTie [51] and mapped the reads to the merged transcriptome set using Bowtie2 [52] and quantified the normalized expression level [reads per kilobase of transcript per million reads mapped (FPKM)] of each gene and transcript using RSEM [53]. DESeq2 [54] (version 1.22.2) was used to identify differentially expressed genes (DEGs) with |log2(fold change)| ≥1 and the adjusted FDR <0.05. DEGs were functionally annotated using the KEGG database. Enrichment analysis was performed for annotated DEGs.
Untargeted metabolic profiling
In addition to RNA extraction samples, another set of six samples from ZG-39 and ZG-19 were collected for metabolic profiling. They were freeze-dried and crushed using a mixer mill (MM 400, Retsch) with a zirconia bead for 1.5 min at 30 Hz. For each sample, 100 mg powder was weighed and extracted overnight at 4°C with 1.2 ml 70% aqueous methanol. Following centrifugation at 12 000 rpm for 10 min, the extracts were filtered with an SCAA-104 (0.22 μm pore size; Anpel, Shanghai, China, http://www.anpel.com.cn/) before UPLC–MS/MS analysis.
The extracts were analyzed using an UPLC–ESI–MS/MS system (UPLC, Shimadzu Nexera X2, www.shimadzu.com.cn/; MS, Applied Biosystems 4500 Q TRAP, sciex.com). Except for the Analyst v1.6.3 software and ion spray voltage 5500 V (positive ion mode)/−4500 V (negative ion mode), other HPLC conditions, linear ion trap and triple quadrupole (QQQ) scans and experiments were the same as previously described [55].
Differential metabolic analysis
PCA was performed by the function prcomp in R (www.r-project.org) before scaling the data by unit variance [56]. Results for samples and metabolites [hierarchical cluster analysis (HCA)] are presented as heat maps and dendrograms, while Pearson’s correlation coefficient (PCC) between samples was calculated by the cor function in R and presented as heat maps only. Both HCA and PCC were conducted using the R package pheatmap [57]. Metabolites differentially regulated between genotypes were defined by the variable importance in projection (VIP) > = 1 and |log2(fold change)| ≥ 1. VIP values were extracted from the orthogonal partial least squares discrimination analysis (OPLS-DA) results, which also contained score maps and permutation maps, generated using the R package MetaboAnalystR [58]. The data were log-transformed (log2) and mean-centered before OPLS-DA analysis. A permutation test (200 permutations) was carried out to avoid overfitting. Identified metabolites were annotated using the KEGG compound database (http://www.kegg.jp/kegg/compound/), then mapped to the KEGG pathway database (http://www.kegg.jp/kegg/pathway.html). The mapped pathways with significantly regulated metabolites were fed into metabolite sets enrichment analysis (MSEA) and significance was determined by the P-value of the hypergeometric test.
Comparison between differential metabolites and differentially expressed genes
PCCs were calculated between DMs and DEGs across the six samples. The DMs and DEGs from comparison pairs with PCC >0.8 or <−0.8 were retrieved for downstream analysis. Pairwise PCC analysis was performed on the kept metabolites and genes and plotted in a heatmap to visualize their correlation patterns.
Identification of genes in puerarin biosynthesis pathways
Sequences of a list of known gene families encoding enzymes that are involved in puerarin biosynthesis were aligned against the genome-wide gene set of P. thomsonii using BLASTP with e-value ≤1e−5 and identity ≥40%. The key structural domains from pfam [59] and InterPro [60] of the enzymes were queried by HMMER [61] to filter for the bona fide genes in P. thomsonii. Genes possibly involved in puerarin biosynthesis, including the chalcone isomerase (CHI), phenylalanine ammonialyase (PAL), cinnamate-4-hydroxylase (C4H), 4-coumarate-CoA ligase (4CL), chalcone synthase (CHS), chalcone reductase (CHR), 2-hydroxyisoflavanone synthase (IFS/2-HIS), glucosyltransferase (GT), and 2-hydroxyisoflavanone dehydratase (HID), were identified by the same approach in the P. thomsonii genome.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31960420, 31870275), the Guangxi Key R&D Program Project (Guike AB1850028), the Guangxi Natural Science Foundation Project (2018GXNSFBA294001, 2019GXNSFBA245093), and the Special Project for Basic Scientific Research of Guangxi Academy of Agricultural Sciences (Guinongke 2021YT057). We thank Dr Shiyu Chen and Prof. Qiusheng Kong for proofreading the manuscript.
Author contributions
X.H.S., X.X.Y., and H.B.Y. conceived and designed the study. K.P.O, W.D.Z., S.C., and L.Y.L. contributed to sample preparation. L.X., Y.S.Z., D.H., Z.B.X., R.H.M., and D.Q.W. participated in data analysis and substantively revised the manuscript. All authors read and approved the final manuscript.
Data availability
This Whole Genome Shotgun project and Transcription project of P. lobata var. thomsonii has been deposited in DDBJ/ENA/GenBank and SRA under the umbrella of BioProject Accession PRJNA723378. The genome accession described in this paper is JAGTWZ000000000.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary data
Supplementary data is available at Horticulture Research Journal online.
Supplementary Material
References
- 1. Wang S, Zhang S, Wang Set al. A comprehensive review on Pueraria: insights on its chemistry and medicinal value. Biomed Pharmacother2020;131:110734. [DOI] [PubMed] [Google Scholar]
- 2. Zhao Z, Guo P, Brand E. A concise classification of bencao (materia medica). Chin Med. 2018;13:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Egan AN. Economic and ethnobotanical uses of tubers in the genus Pueraria DC. Legume2020;19:24. [Google Scholar]
- 4. WFO . Pueraria montana var. chinensis (Ohwi) Sanjappa & Pradeep. Date accessed 21 May 2021. http://www.worldfloraonline.org/taxon/wfo-0000193855. 2021.
- 5. Li S, Xu L, Chen Det al. Fabaceae (Leguminosae). Flora of China2010;41:226. [Google Scholar]
- 6. Heider B, Fischer E, Berndl Tet al. Analysis of genetic variation among accessions of Pueraria montana (Lour.) Merr. var. lobata and Pueraria phaseoloides (Roxb.) Benth. based on RAPD markers. Genet Resour Crop Evol. 2007;54:529–42. [Google Scholar]
- 7. Jewett D, Jiang C, Britton Ket al. Characterizing specimens of kudzu and related taxa with RAPD's. Castanea. 2003;68:254–60. [Google Scholar]
- 8. Haynsen MS, Vatanparast M, Mahadwar Get al. De novo transcriptome assembly of Pueraria montana var. lobata and Neustanthus phaseoloides for the development of eSSR and SNP markers: narrowing the US origin(s) of the invasive kudzu. BMC Genomics. 2018;19:439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang G, Liu J, Gao Met al. Tracing the edible and medicinal plant Pueraria montana and its products in the marketplace yields subspecies level distinction using DNA barcoding and DNA metabarcoding. Front Pharmacol 2020;11:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou YX, Zhang H, Peng C. Puerarin: a review of pharmacological effects. Phytother Res 2014;28:961–75. [DOI] [PubMed] [Google Scholar]
- 11. Chen YG, Song YL, Wang Yet al. Metabolic differentiations of Pueraria lobata and Pueraria thomsonii using (1)H NMR spectroscopy and multivariate statistical analysis. J Pharm Biomed Anal. 2014;93:51–8. [DOI] [PubMed] [Google Scholar]
- 12. Chen SB, Liu HP, Tian RTet al. High-performance thin-layer chromatographic fingerprints of isoflavonoids for distinguishing between Radix Puerariae lobate and Radix Puerariae Thomsonii. J Chromatogr A. 2006;1121:114–9. [DOI] [PubMed] [Google Scholar]
- 13. Wang X, Li S, Li Jet al. De novo transcriptome sequencing in Pueraria lobata to identify putative genes involved in isoflavones biosynthesis. Plant Cell Rep. 2015;34:733–43. [DOI] [PubMed] [Google Scholar]
- 14. He X, Blount JW, Ge Set al. A genomic approach to isoflavone biosynthesis in kudzu (Pueraria lobata). Planta. 2011;233:843–55. [DOI] [PubMed] [Google Scholar]
- 15. Wang X, Li C, Zhou Zet al. Identification of three (iso)flavonoid glucosyltransferases from Pueraria lobata. Front Plant Sci2019;10:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X, Li C, Zhou Cet al. Molecular characterization of the C-glucosylation for puerarin biosynthesis in Pueraria lobata. Plant J2017;90:535–46. [DOI] [PubMed] [Google Scholar]
- 17. Kreplak J, Madoui MA, Capal Pet al. A reference genome for pea provides insight into legume genome evolution. Nat Genet. 2019;51:1411–22. [DOI] [PubMed] [Google Scholar]
- 18. Vanneste K, Baele G, Maere Set al. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the cretaceous-Paleogene boundary. Genome Res. 2014;24:1334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmutz J, Cannon SB, Schlueter Jet al. Genome sequence of the palaeopolyploid soybean. Nature 2010;463:178–83. [DOI] [PubMed] [Google Scholar]
- 20. Han R, Takahashi H, Nakamura Met al. Transcriptomic landscape of Pueraria lobata demonstrates potential for phytochemical study. Front Plant Sci 2015;6:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–5. [Google Scholar]
- 22. Belton JM, McCord RP, Gibcus Jet al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods2012;58:268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang X, Liu D, Wu Jet al. HTQC: a fast quality control toolkit for Illumina sequencing data. BMC Bioinformatics 2013;14:33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics2011;27:764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chin CS, Peluso P, Sedlazeck FJet al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods. 2016;13:1050–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vaser R, Sovic I, Nagarajan Net al. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27:737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu J, Fan J, Sun Zet al. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 2020;36:2253–5. [DOI] [PubMed] [Google Scholar]
- 28. Chen S, Zhou Y, Chen Yet al. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Servant N, Varoquaux N, Lajoie BRet al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM[J]. 2013; 1–3. arXiv preprint arXiv:1303.3997.
- 31. Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Price AL, Jones NC, De Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:i351–8. [DOI] [PubMed] [Google Scholar]
- 33. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 2009;25:4.10.1–4.10.14. [DOI] [PubMed] [Google Scholar]
- 35. Cantarel BL, Korf I, Robb SMCet al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2007;18:188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boeckmann B, Bairoch A, Apweiler Ret al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Altschul SF, Madden TL, Schäffer AAet al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Camacho C, Coulouris G, Avagyan Vet al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanehisa M, Goto S, Sato Yet al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ashburner M, Ball CA, Blake JAet al. Gene ontology: tool for the unification of biology. Nature Genet. 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Conesa A, Gotz S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;2008:619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Han MV, Thomas GW, Lugo-Martinez Jet al. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol. 2013;30:1987–97. [DOI] [PubMed] [Google Scholar]
- 46. Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. [DOI] [PubMed] [Google Scholar]
- 47. Tang H, Bowers JE, Wang Xet al. Synteny and collinearity in plant genomes. Science. 2008;320:486–8. [DOI] [PubMed] [Google Scholar]
- 48. Qing Z, Liu J, Yi Xet al. The chromosome-level Hemerocallis citrina Borani genome provides new insights into the rutin biosynthesis and the lack of colchicine. Hortic Res. 2021;8:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang D, Zhang Y, Zhang Zet al. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics. 2010;8:77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pertea M, Pertea GM, Antonescu CMet al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen W, Gong L, Guo Zet al. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: application in the study of rice metabolomics. Mol Plant. 2013;6:1769–80. [DOI] [PubMed] [Google Scholar]
- 56. Chen Y, Zhang R, Song Yet al. RRLC-MS/MS-based metabonomics combined with in-depth analysis of metabolic correlation network: finding potential biomarkers for breast cancer. Analyst. 2009;134:2003–11. [DOI] [PubMed] [Google Scholar]
- 57. Kolde R, Kolde MR. Package ‘pheatmap’. R package. 2015;1:790. [Google Scholar]
- 58. Chong J, Xia J. MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data. Bioinformatics. 2018;34:4313–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Finn RD, Bateman A, Clements Jet al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hunter S, Apweiler R, Attwood TKet al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This Whole Genome Shotgun project and Transcription project of P. lobata var. thomsonii has been deposited in DDBJ/ENA/GenBank and SRA under the umbrella of BioProject Accession PRJNA723378. The genome accession described in this paper is JAGTWZ000000000.