

Abstract
Introduction:
Sickle cell disease (SCD) is a debilitating inherited disorder that affects millions worldwide. Four novel SCD therapeutics have been approved, including the hemoglobin (Hb) modulator voxelotor.
Areas Covered:
This review provides an overview of discovery efforts towards modulating Hb allosteric behavior as a treatment for SCD, with a focus on aromatic aldehydes that increase Hb oxygen affinity to prevent the primary pathophysiology of hypoxia-induce erythrocyte sickling.
Expert Opinion:
The quest to develop small molecules, especially aromatic aldehydes, to modulate Hb allosteric properties for SCD began in the 1970s; however, early promise was dogged by concerns that stalled support for research efforts. Persistent efforts eventually culminated in the discovery of the anti-sickling agent 5-HMF in the 2000s, and reinvigorated interest that led to the discovery of vanillin analogs, including voxelotor, the first FDA approved Hb modulator for the treatment of SCD. With burgeoning interest in the field of Hb modulation, there is a growing landscape of intellectual property, including drug candidates at various stages of preclinical and clinical investigations. Hb modulators could provide not only the best chance for a highly effective oral therapy for SCD, especially in the under-developed world, but also a way to treat a variety of other human conditions.
Keywords: sickle cell disease, hemoglobin, oxygen affinity, aromatic aldehydes, Hb modulators, allosteric effectors, anti-sickling agents, red blood cells, polymerization
1. Introduction
1.1. The pathophysiology of sickle cell disease
Sickle Cell Disease (SCD) is the most common inherited hematologic disorder affecting between 80,000 to 100,000 people (mostly of African American origin) in the U.S., and over 15 million individuals worldwide.[1–4] The World Health Organization (WHO) and the United Nations (UN) have declared SCD a public health burden with over 300,000 affected live births every year, with the number of affected individuals projected to increase 30% by 2050.[1–4] The abnormal gene that causes sickle cell disease is most prevalent in sub-Saharan Africa, eastern Saudi Arabia, central India, as well as Caribbean, Central and South American, and Mediterranean nations.[6,7]
SCD results from a single point mutation in the oxygen carrying protein hemoglobin (Hb) that is caused by a substitution of polar glutamic acid (βGlu6) in normal Hb to the hydrophobic valine (βVal6) in sickle Hb (HbS).[8,9] Deoxygenated HbS (deoxyHbS) polymerizes into long, rigid and insoluble 14-stranded fibers, which distorts red blood cells (RBCs) into sickled shape (Figure 1).[10] Polymer formation is initiated by a hydrophobic interaction between the βVal6 residue of one deoxyHbS molecule and a hydrophobic pocket formed by β2Ala70, β2Phe85, and β2Leu88 of a proximate deoxyHbS molecule.[11,12] DeoxyHbS polymerization is exacerbated by the inherent high concentrations of 2,3-diphosphoglycerate (2,3-DPG) and/or sphingosine 1-phosphate (S1P) in sickled red blood cells (RBCs).[13–15] In addition to the primary binding pocket, key secondary interactions between adjacent deoxyHbS molecules in the fiber, such as those mediated by αAsn78 or βAsp73 on a surface-located αF-helix of Hb, are also required to maintain the integrity of the polymer.[10,16–18] There are individuals in Sudan and Congo with rare co-inherited mutations on these secondary contacts that, consequently, demonstrate a reduced tendency for polymerization and RBC sickling. Individuals with these rare variants, such as Hb Stanleyville II or Hb Mobile, have acquired not only the classic (βVal6) SCD mutation, but also a second mutation, αAsn78→Lys (Hb Stanleyville) or βAsp73→Val (Hb Mobile), on the αF-helix of Hb.[16,17,19] As a result of the reduced tendency for polymerization with disruption of secondary contacts, these individuals are known to have mild or even no disease sequelae.[16] In contrast to deoxyHbS, oxygenated HbS (oxyHbS) neither polymerizes nor incorporates into the polymer fibers since the quaternary conformation of oxyHbS molecule does not allow for the pathologic interaction between βVal6 and the hydrophobic acceptor pocket.[11]
Figure 1.
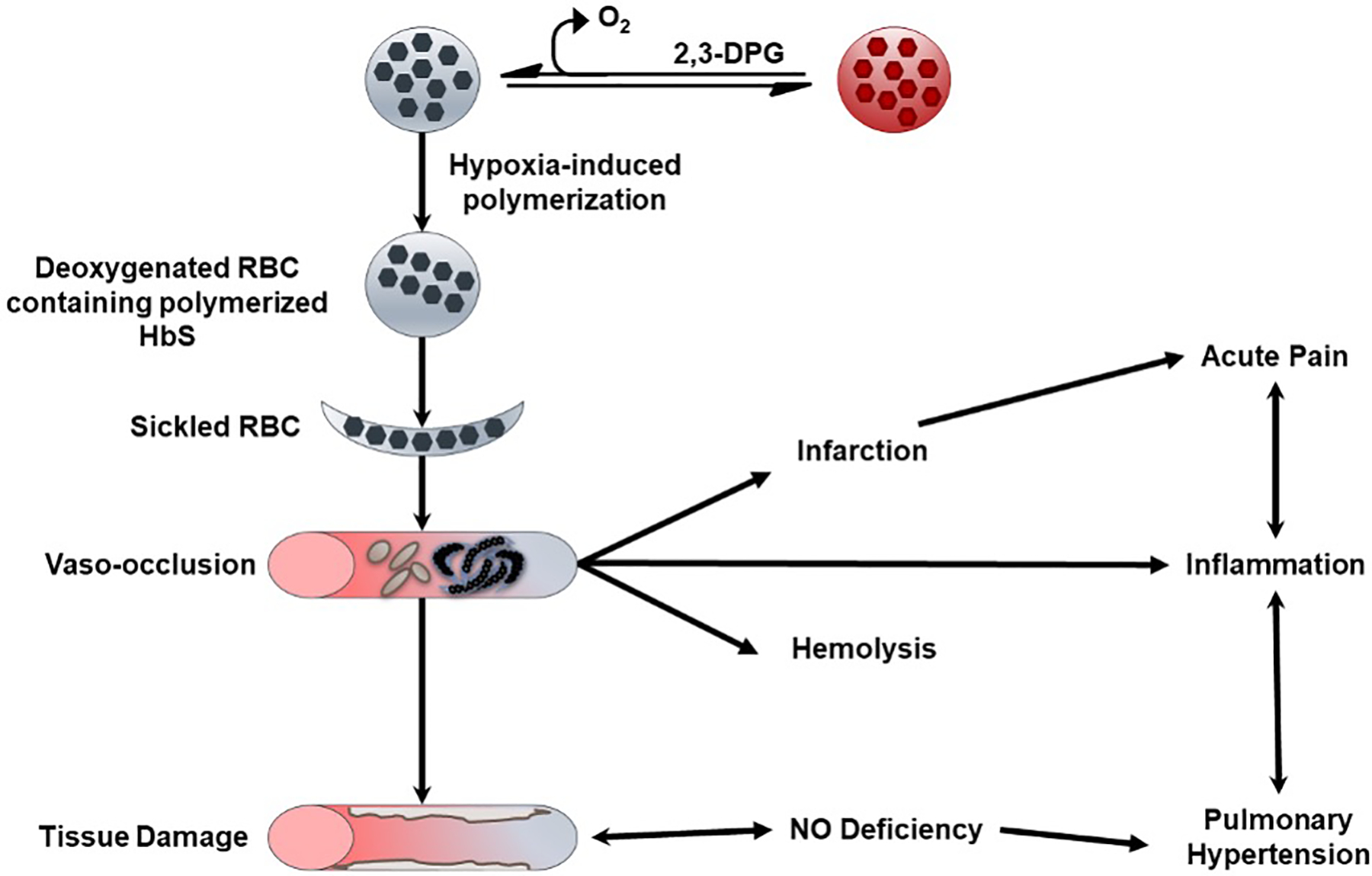
Pathophysiology of sickle cell disease.
Formation of insoluble and rigid sickle RBCs in the micro-vasculature leads to a downstream cascade of RBC adhesion to tissue endothelium, vaso-occlusion, hypoxia-reperfusion injury, and the release of cytokines and micro-infarctions (Figure 1). [1,10] Reperfusion of ischemic tissues generates free radicals and reactive oxygen species (ROS), which cause direct damage and also scavenge free nitric oxide (NO). The rigid and brittle nature of the sickle RBCs results in a chronic hemolytic state with persistent release of free Hb into plasma, which also contributes to NO scavenging and, consequently, increased platelet activation and vascular resistance – all part of a chronic vasculopathy (Figure 1).[2,3,20] Additional sequelae of SCD include pulmonary hypertension, cholelithiasis, leg ulcers, osteonecrosis, acute chest syndrome, priapism and cerebrovascular disease.[21] Ongoing vasculopathy and inflammation eventually results in progressive damage to vital organs leading to a multisystem disease, and a reduced life expectancy. By the fifth decade of life, one half of surviving patients develop some form of irreversible damage to the lungs, kidneys, brain, retina or bones, as well as other co-morbidities like diabetes and systemic hypertension, which further affects quality of life.[20,22,23]
1.2. Hemoglobin - A target for drug discovery
Hemoglobin is a tetrameric allosteric protein (Figure 2), which functions in an equilibrium between its unliganded (deoxyHb), Tense state (T-state) possessing low oxygen affinity and liganded (oxyHb), Relaxed state (R-state) which has a high oxygen affinity (Figure 3A).[24,25] The tetramer consists of two α-subunits (α1 and α2) and two β-subunits (β1 and β2) that interact together, resulting in a large central water cavity, with the α- and β-clefts defining the two entry points into the water cavity (Figure 2). For Hb to function effectively in oxygen transport and delivery, it requires binding of endogenous heterotropic effectors, such as carbon dioxide (CO2), hydrogen ions (H+), chloride ions (Cl−), and 2,3-bisphosphoglycerate (2,3-BPG), to modulate its oxygen affinity and cooperative behavior.[25] These effectors bind to Hb at the surface or central water cavity and preferentially stabilize either the T-state to release oxygen or the R-state to increase oxygen binding affinity (Figure 3A).[24–32] Stabilization of one state over the other can be measured by the oxygen equilibrium curve (OEC); agents stabilizing the R-state shift the curve to the left, whereas those stabilizing the T-state shift the curve to the right (Figure 3B). Notably, the R-state of Hb consists of an ensemble of relaxed quaternary forms that include the classical R, R2, R3, RR2, RR3.[24,25,33–39] The classical R-state was the first to be reported by Max Perutz, followed by the R2-state in 1992 by the Arnone group.[40] The discovery of these two relaxed states initiated a decades-long controversy as to the most physically relevant form of R-state Hb. The Safo group discovered the existence of the relaxed states R3, RR2 and RR3, which provided further structural evidence that Hb function involves an ensemble of tense and relaxed Hb states, and not only the classical T and R states. [24,25,41]
Figure 2.
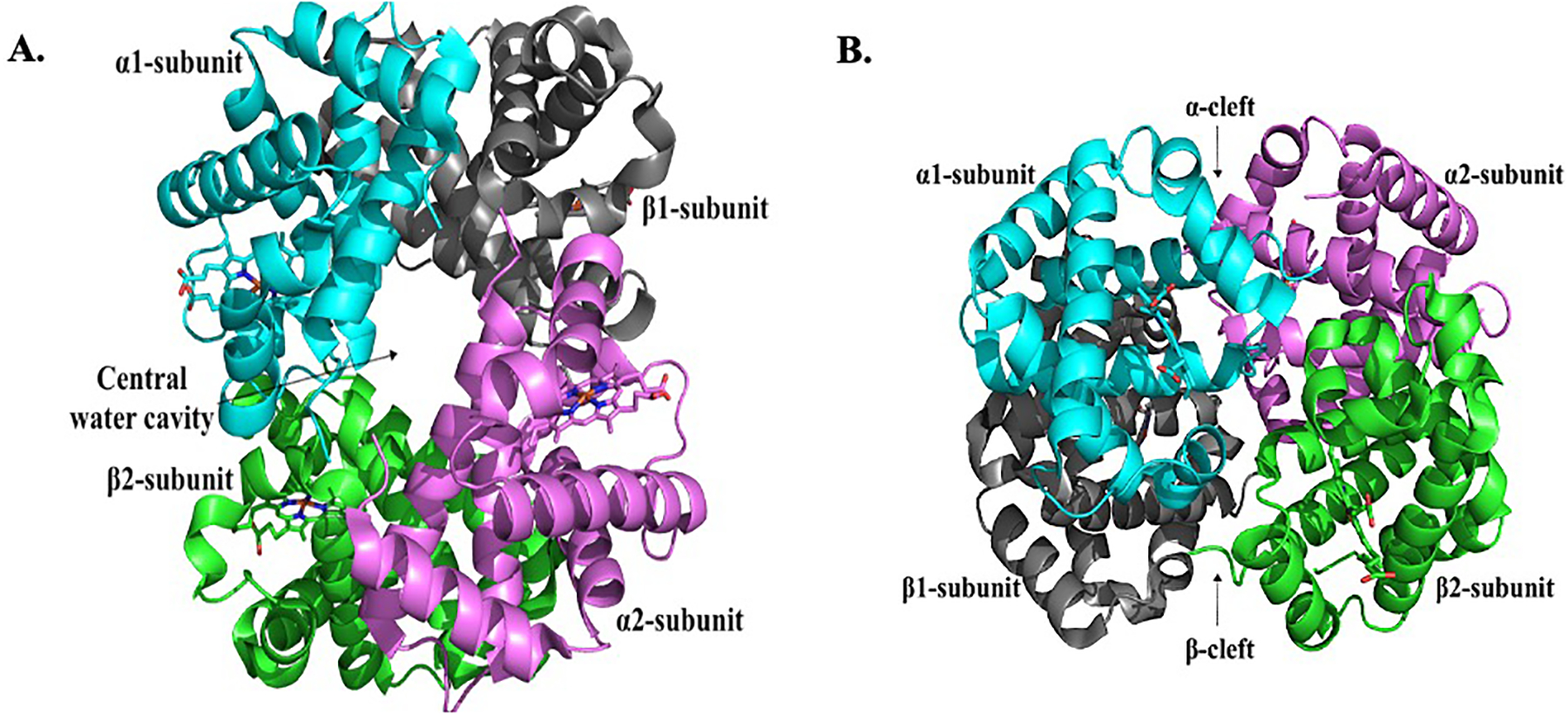
Crystal structure of normal deoxygenated (T-state) Hb (PDB code: 2DN2). (A) Structure showing the characteristic large central water cavity formed by the heterotetramer. (B) A 90° orientation of the structure in (A) showing access into the central water cavity through the α-cleft and β-cleft. The α1-subunit (cyan), α2-subunit (magenta), β1-subunit (grey) and β2-subunit (green) are shown in ribbons, and the hemes in sticks.
Figure 3.
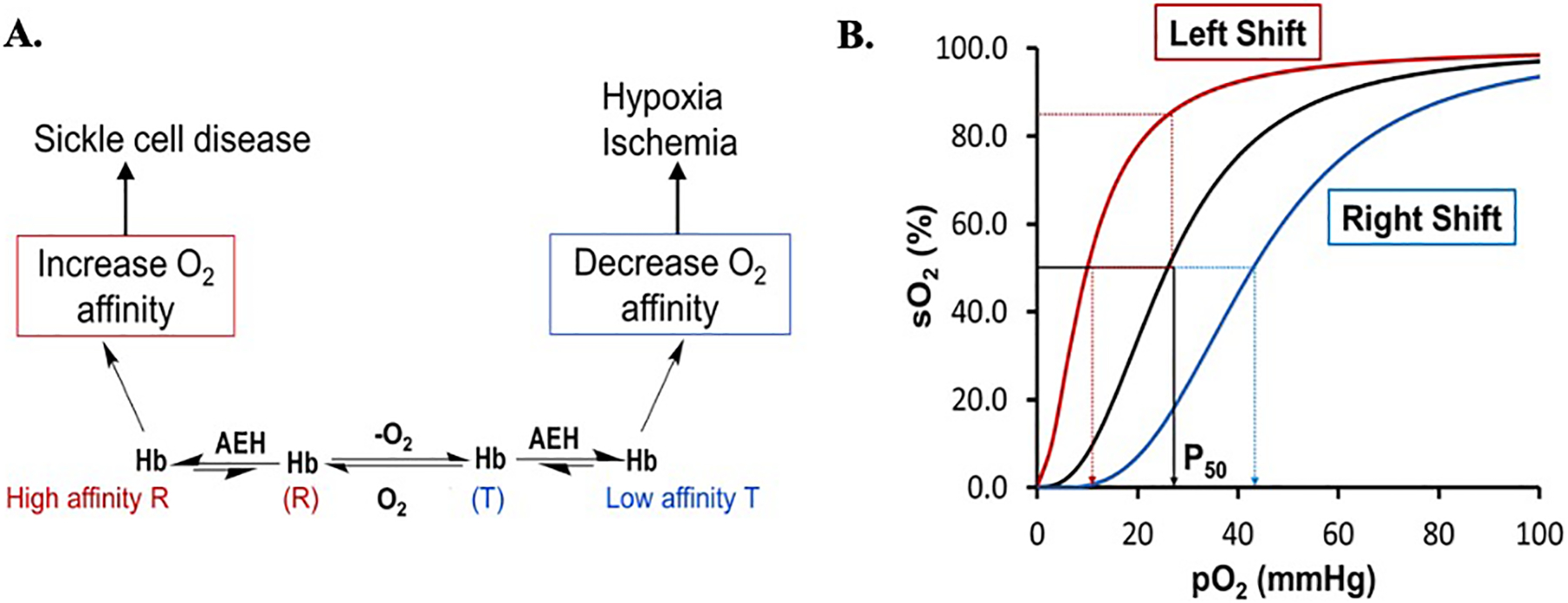
A) Modulation of Hb allosteric activity with synthetic allosteric effectors of Hb (AEHs) and, B) Oxygen equilibrium curve (OEC) of Hb showing P50 (the partial pressure at which 50% of Hb is saturated with oxygen) in black curve. Increasing Hb oxygen affinity with AEH shifts the OEC to the left (red curve) while decreasing it shifts the curve to the right (cyan curve).
Like endogenous effectors, several synthetic allosteric effectors of Hb (AEHs) are also known to bind to the surface, α-cleft or β-cleft, or the middle of the central water cavity of liganded Hb structure (in the R, R2, or R3 state) and/or unliganded Hb structure (in the T state) to allosterically modulate protein activity (Figure 4).[24,25] Scientists have explored the potential of preferentially stabilizing one state over another in the development of therapeutics. For instance, agents stabilizing the T-state have potential therapeutic applications in treating ischemia-related cardiovascular diseases, such as angina, myocardial ischemia, stroke, and trauma, in which more O2 is needed to heal tissues and organs. [42–47] In contrast, effectors that stabilize the R-state increase Hb O2 affinity and are potentially useful for the treatment of sickle cell disease since oxyHbS does not polymerize. [48–62]
Figure 4.
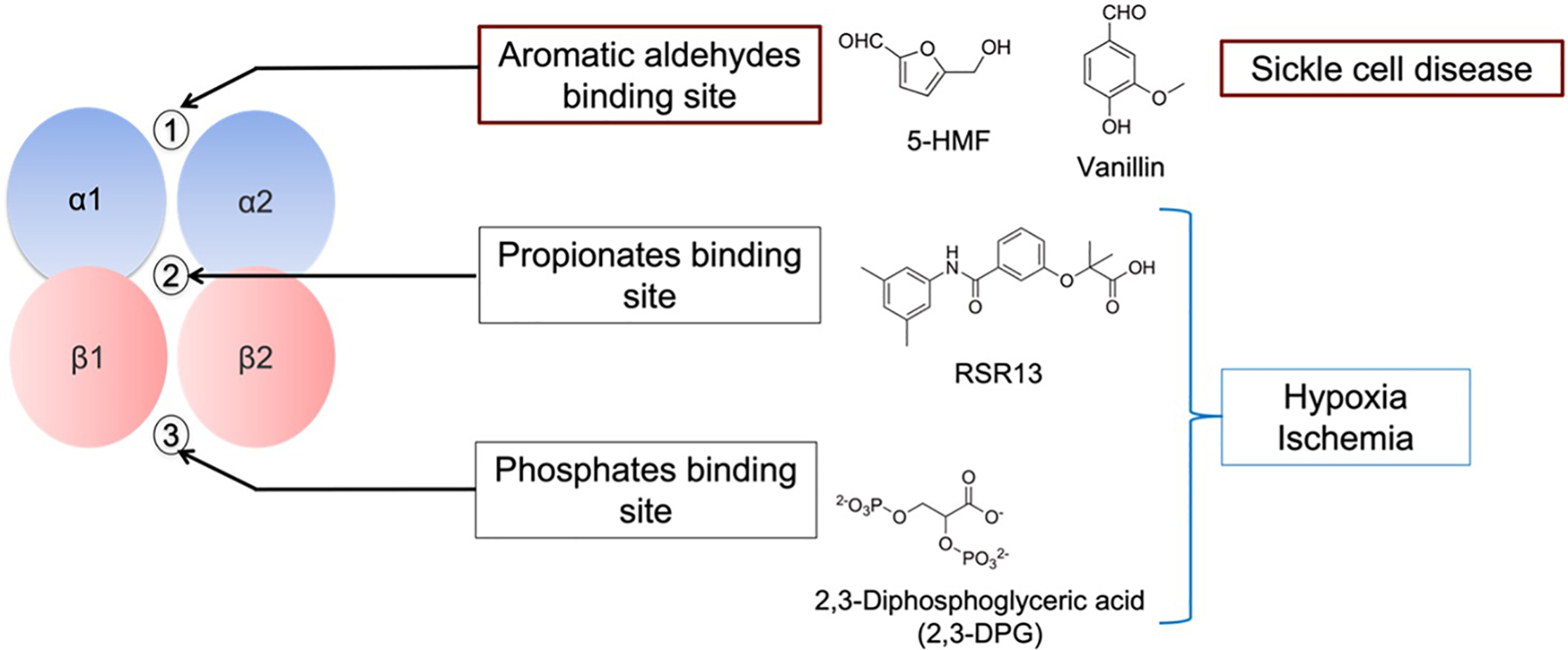
Endogenous and synthetic effectors targeting various allosteric sites of Hb for different therapeutic potential
1.3. Approved drugs for sickle cell disease
Currently, there are four drugs approved for the treatment of SCD, including Hydroxyurea (Droxia and Siklos, sold by Bristol-Myers Squibb Co and Addmedica Laboratories, respectively)[63], L-glutamine (Endari, sold by Emmaus Life Sciences)[64–66], crizanlizumab (Adakveo, sold by Novartis)[67] and, Voxelotor® (Oxbryta, sold by Global Blood Therapeutics)[60–62]. Hydroxyurea, has been used for over 2 decades ago and for a long time was the only approved drug for SCD.[68–70] Hydroxyurea increases the production of fetal hemoglobin (HbF) to prevent polymerization of HbS and may also increase free NO.[71,72] L-glutamine, as the name suggests, is an amino acid that was approved for use in SCD in the US only in 2017.[64,65] L-glutamine increases the amount of reduced form nicotinamide adenine dinucleotide (NADH) in erythrocytes to reduce oxidative stress and potentially result in fewer painful crises.[66] Notably, the regulatory authority in the European Union did not approve L-glutamine for SCD due to lack of substantial efficacy. Crizanlizumab, which was approved in 2019, is a monoclonal antibody targeting P-selectin to reduce erythrocyte adhesion and, consequently, reduce the frequency of VOC crises and complications of SCD.[67] Finally, Voxelotor, an aromatic aldehyde developed to prevent the primary pathophysiology of hypoxia-induced HbS polymerization and erythrocyte sickling by increasing Hb O2-affinity, was approved in 2019.[60,61,73] Voxelotor, a synthetic analogue of vanillin, was developed based on decades of progress in the design and development of aromatic aldehydes since the 1970s [57], including early discovery science performed by our group to identify the natural and non-toxic scaffolds vanillin and 5-hydroxymethyl-2-furfural (5-HMF).[48–51,56,58,59,74,75] The clinical efficacy of Voxelotor for the treatment of SCD has been assessed based on increased Hb levels and reduced hemolysis in patients.[60] The most recent data also suggests that the use of Voxelotor for an extended period of time may reduce the number of VOC events, with the greatest improvement in VOC rates observed in patients with the largest increase in Hb.[76] In a 72-week analysis of the results of the Phase 3 HOPE trial, there was a very modest numerical reduction in the annualized VOC event rate in patients on Voxelotor from 2.8 events per person-year to 2.4 events per person-year. Unexpectedly, the frequency of VOC events was lowest for individuals with a post-treatment Hb level of 12 to 13.3 g/dL (drug responders) followed by those with a post-treatment Hb level of 10 to 12 g/dL. The frequency of VOC events was significantly higher in individuals with a post-treatment Hb levels of < 10 g/dL. In fact, the frequency of VOC appeared to be indirectly correlated with achieved Hb levels. In an analysis of 24-week data from the HOPE trial, the greatest reduction in VOC events also occurred for individuals who had Hb occupancy levels of at least 20% or higher compared to those with occupancy less than 20% or in the placebo cohort. While not achieving statistical significance on the clinical endpoint in the trial, the data is consistent mechanistically with treatment induced inhibition of HbS polymerization leading to a reduction in propensity for VOC events. This is in direct contrast to previous SCD drug failures (e.g. Senicapoc) that demonstrated the unmistakable risk for worsening of VOCs due to a large correction of anemia that increased blood viscosity.[77] In the landmark trial by Ataga et al, SCD patients receiving the Senicapoc, a drug that inhibited potassium efflux through the Gardos channel, demonstrated statistically significant increases in hemoglobin and hematocrit, as well as hemolysis markers, such as LDH and indirect bilirubin, suggesting a meaningful reduction in hemolysis. Unlike patients in the Voxelotor cohort that also experienced reduction in hemolysis, SCD patients in the Senicapoc cohort demonstrated a statistically significant increase in VOC events (0.41 vs. 0.30 incidence per year). A paradoxical reduction in VOCs despite higher HbS levels with Voxelotor suggests that the underlying pathophysiology and secondary downstream effects are directly mitigated by inhibiting HbS polymerization. The clinical results with Voxelotor to-date suggest that aromatic aldehydes may have disease-modifying potential that can mitigate adverse disease effects of RBC sickling in SCD.
2. Body of review
2.1. Historical perspective
Sickle cell disease is one of the few genetic diseases that has been extensively studied at the genetic, cellular, molecular, and atomic levels. It all begun in 1910 when James Herrick discovered sickle cell disease after observing the sickled shape of RBCs from a West Indies student who complained of pain episodes and symptoms of anemia.[78] In 1949, Linus Pauling characterized the disease on a molecular level.[79,80] Another landmark discovery that led or contributed to our understanding of the disease on an atomic level, as well as Hb allostery occurred in 1960s when the Nobel laureate Max Perutz solved the crystal structure of Hb. [29,81–83] This incredible feat made possible a new era of Hb targeted drugs that could be rationally designed to target the protein’s allosteric properties and/or the secondary contacts of polymerization. The first allosteric Hb effectors were developed in the 1970s, e.g. aromatic aldehydes, aspirin derivatives, and thiols that form covalent adducts with Hb, stabilizing the high oxygen affinity R-state Hb.[56,57,84–93]. These compounds directly address the primary cause of the disease by stabilizing the high-O2-affinity R-state that cannot polymerize. Sunshine et. al.[94] were among the first to suggest that increasing the O2 affinity of Hb by 4 mmHg could lead to therapeutically significant inhibition of the intracellular polymerization. Non-covalent Hb binding compounds, e.g. amino acids[95–97], phenoxyacetic/benzyloxyacetic acids, halogenated/methylated aromatic carboxylic acid derivatives[98], proline-salicylates,[99] urea- and amino acid-based derivatives[100–102], oligo-peptides[96], natural products and analogs thereof[103,104], carbohydrate derivatives[105], and aromatic alcohols[96,106,107] were also studied as anti-sickling agents. However, these molecules, unlike the covalent binders, showed weak Hb affinity due to the shallowness of the surface binding cavities.[108] Much of the early discovery research for SCD was performed by Max Perutz, Don Abraham, and Irvine Klotz, and this work laid an important foundation for subsequent drug discovery and development. Don Abraham was a pioneer in the use of molecular modeling and/or X-ray Crystallography to design compounds that bind covalently to Hb.[55,84,109–111] Abraham was also the first to use compounds with non-covalent interactions to directly inhibit the binding of Hb tetramers by designing agents that fit hydrophobic surface cavities on the surface of the protein.[55,84,93,109,111,112]
Although Klotz was the first to draw attention to aromatic aldehydes, including the non-toxic vanillin as potential antisickling agents in 1977,[57] it was not until the 1990s when Abraham’s landmark study with vanillin actually highlighted the potential of aromatic aldehyde as SCD therapeutics,[56] as well as established vanillin as a lead for the discovery of several derivatives with improved pharmacologic effects. Synthetic analogues of vanillin, including Voxelotor, are currently the most promising candidates for the treatment of SCD[49,50,52,53,59,61]. This review article is dedicated to Don Abraham, an incredible scientist who requires credit for being a pioneer in targeting Hb for drug discovery.
2.2. Early development of aromatic aldehydes as anti-sickling agents
The primary pathophysiology of sickle cell disease involves polymerization of deoxyHbS within RBCs, leading RBC sickling. Certain classes of aromatic aldehydes can bind to Hb and stabilize the high oxygen affinity state of the protein to prevent RBC sickling. This allosteric mechanism centers around the aldehyde moiety that forms a reversible covalent Schiff-base interaction with the N-termini αVal1 amines of the two Hb α-chains (Figure 5A). Klotz and coworkers in the late 1970s were the first to report anti-sickling aromatic aldehydes, e.g. vanillin, (Figure 5B) [57], and although some of these compounds show promising anti-sickling activity in vitro, none were advanced beyond simple academic studies to determine their potential as drug therapeutics. Abraham revisited the anti-sickling compound vanillin (Figure 5B), and found that in addition to its primary mechanism of increasing Hb affinity for oxygen, vanillin also exhibited direct inhibition of HbS polymerization by binding to the surface of Hb, close to one of the several secondary contact sites in the polymer.[56] However, Abraham uncovered a key obstacle in the preclinical studies; the aldehyde moiety which is key for bioactivity was rapidly metabolized in vivo into the inactive acid analog, vanillic acid.[75] Consequently, many promising natural and early synthetic vanillin analogue candidates were plagued by a short half-life and limited bioavailability.
Figure 5.
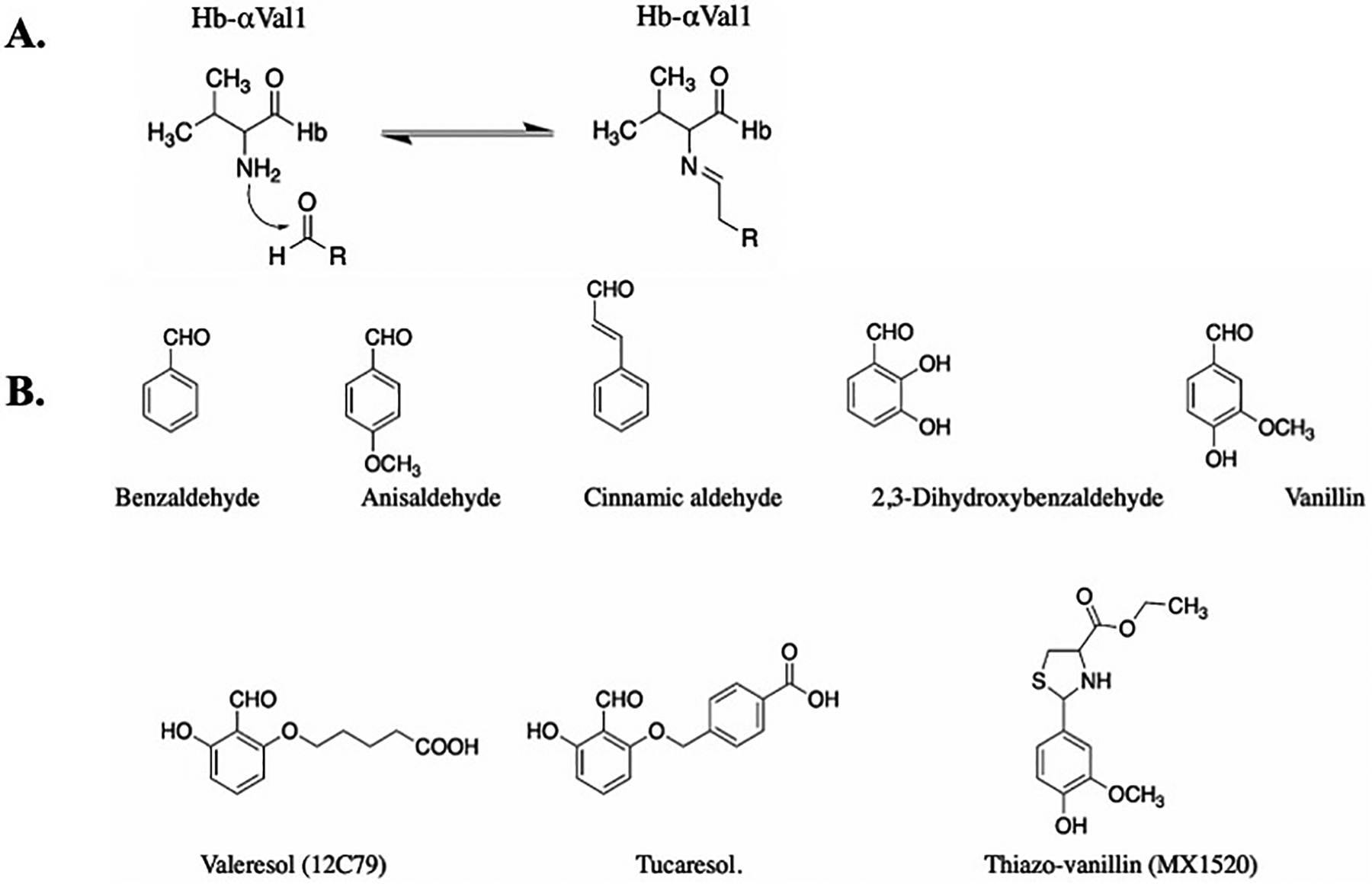
A) Schiff-base adduct formation of aromatic aldehydes with Hb; B) Examples of early aromatic aldehydes as AEHs.
Based on the anticipated Schiff-base interaction of aromatic aldehydes at the α-cleft of Hb, Geoffery Keen and Burroughs Wellcome designed several synthetic aromatic aldehyde-acid effectors of Hb.[110,113] The group postulated that one molecule would bind to classical R-state Hb to crosslink the two α-subunits. Specifically, the aldehyde would form a Schiff-base interaction with the αVal1 amine of one α-subunit and the acid moiety would form a hydrogen-bond interaction with the opposite αVal1 amine of the second α-subunit to stabilize the R-state relative to the T-state, which was expected to increase Hb affinity for oxygen.[110,113] Unexpectedly, a subsequent crystallographic study by Abraham with Valeresol showed that these compounds bind to Hb in 2:1 ratio, with each molecule forming a Schiff-base interaction with each of the two α-subunit αVal1 amine.[55] Subsequent study by the Safo group validated Abraham’s original finding that Hb binds two molecules of aromatic aldehyde.[48] The compounds developed by Keen and Burroughs Wellcome were protected by a composition of matter patent filed in 1985 (US 4,535,185).[114] This patent was likely the first intended to disclose drug candidates targeting Hb for sickle cell disease drug discovery. Two of the most notable compounds were Valeresol and Tucaresol (Figure 5B), which showed significant potency in increasing Hb affinity for oxygen and preventing RBC sickling. [113,115] They were subsequently studied in various clinical trials for the treatment of SCD. Valeresol underwent phase I clinical study in 1985 but was terminated due to poor bioavailability.[116,117] Tucaresol showed even more potent activity, and more importantly was resistant to oxidative metabolism, resulting in improved oral bioavailability and pharmacokinetics. Unfortunately, Tucaresol caused immune-mediated toxicity in phase-II clinical studies, and was therefore terminated.[75,115,118]
With an understanding of the metabolic instability of vanillin, a prodrug of vanillin, thiazovanillin (MX1520) (Figure 5B), in which L-cysteine protected the aldehyde group by forming a thiazolidine complex, was synthesized and studied.[119] Although MX1520 showed improved oral pharmacokinetic properties compared to vanillin, it was not studied in the clinic presumably due to its weak potency. Nonetheless, the discovery shed light on a viable strategy to improve pharmacokinetics and oral bioavailability of aromatic aldehydes as potential anti-sickling drugs. There were no US or international patent filings for MX1520.
The failure to advance early candidate aromatic aldehyde compounds for the treatment of SCD had a chilling effect on enthusiasm for this class of compounds. There were justifiable concerns that the large amount of drug necessary to therapeutically modify one of the most abundant human proteins, Hb (5 mmol) would likely exceed toxicity thresholds, and also the use of R-state Hb stabilizers that inhibit O2 release could cause tissue hypoxia. While we know now that these obstacles can be overcome with clinical evidence with the most recent candidates, skepticism was pervasive after early failures. Nonetheless, these early failures paved the way for innovations that eventually led to the first successful clinical candidates and now more next generation candidates to follow.
2.3. 5-hydroxymethyl-2-furfural (5HMF) – a bridge between early aromatic aldehydes and modern aromatic aldehyde anti-sickling agents
Although research into Hb modifiers for treating SCD slowed down in the mid 1990s to mid 2000s as a result of high-profile failures and other concerns about the viability of this approach, Safo and Abraham saw the potential utility of this therapeutic option and persisted with a focus in this area of research with a focus on natural aromatic aldehydes in the diet with metabolic stability and good toxicity profile. One of the compound studied is 5-HMF, a natural decomposition/dehydration product of sugar, which turned out to be a highly promising candidate (Figure 6).[48,58] 5-HMF is found naturally in all sugar containing foods, including fruits, honey, and caramel.[120,121] 5-HMF showed no in vitro cytotoxic effects on RBCs and exhibited several fold more potent anti-sickling activity compared to vanillin.[48,58,74,75,122,123] A single oral dose of 100 mg/kg of 5-HMF protected transgenic sickle mice from death from acute pulmonary sequestration of sickled cells after a hypoxic challenge[58], and chronic administration was well tolerated for 2 years in rodents at doses of up to 375 mg/kg/day.[124] Unlike vanillin, 5-HMF demonstrated oral bioavailability, although oxidative metabolism still rapidly transformed the aldehyde into its inactivated acid counterpart, 5-hydroxymethyl-2-furoic acid (HMFA). 5-HMF and several of its furfural analogs, as well as their thiazolidine aldehyde protected derivatives, were patented in 2006 (US 7,119,208 B2), 2007 (US 7,160,910 B2), and 2020 (US 10,875,834 B2).
Figure 6.
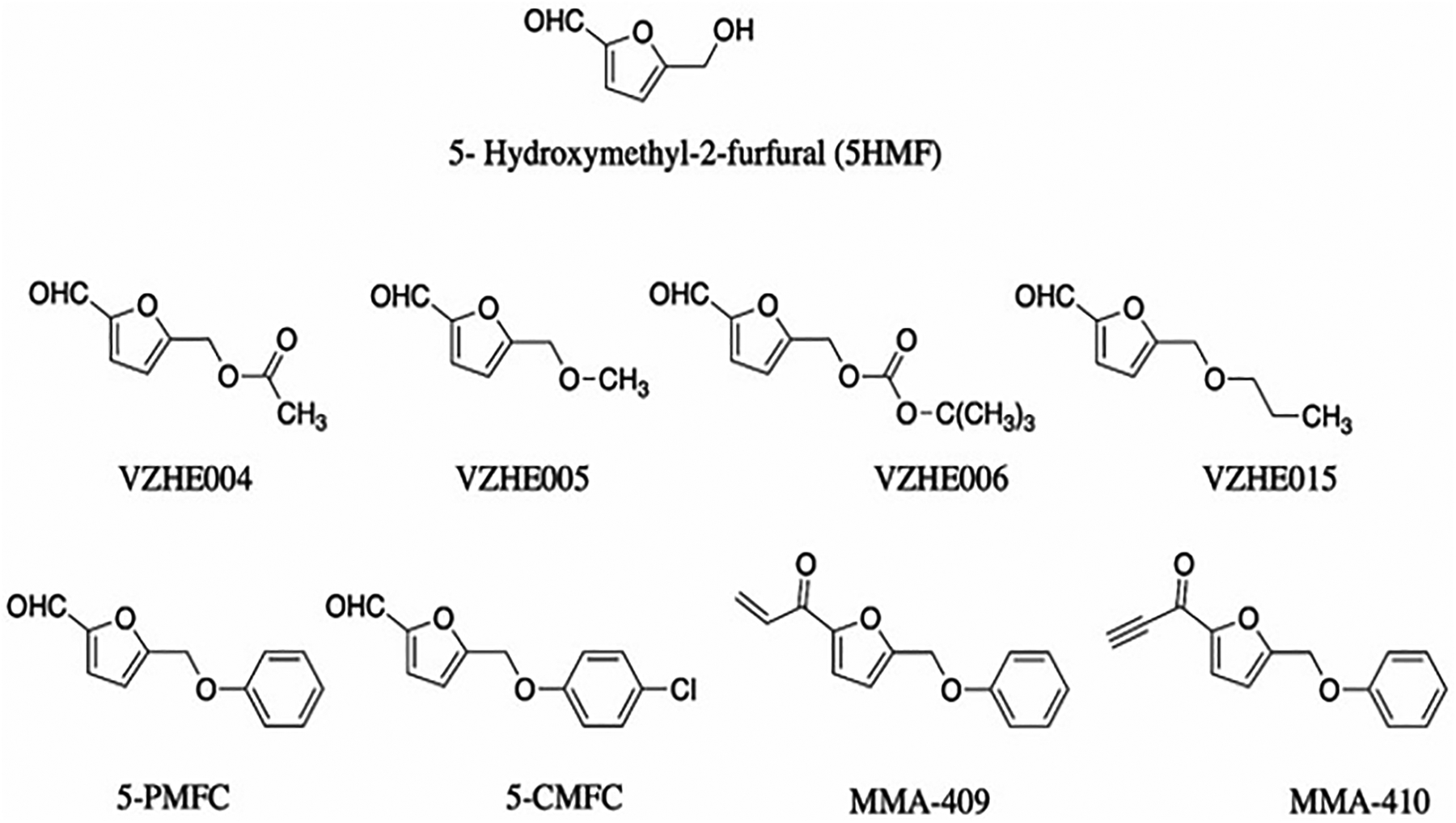
5-HMF and its derivatives
5-HMF was licensed to AesRx LLC, a small company located in Massachusetts, which advanced the drug candidate through pre-clinical development and initial first-in-human clinical trials in close collaboration with scientists from academia and NIH, a prime example of a translational collaboration between academic and start-up company in the research and development of a potentially lifesaving SCD drug therapy. 5-HMF, which was renamed Aes-103, was one of the first molecules to enter the NIH Therapeutics for Rare and Neglected Diseases program. The NIH committed greater than $5 million to support the pre-clinical studies, clinical manufacture, and the initiation of Phase I/II human trials of Aes-103. In Phase I clinical studies, 5-HMF was well tolerated with no safety signals in both normal volunteers and SCD patients (ClinicalTrials.gov identifier NCT01597401),[125], and patients had significant improvements in pain, decreased RBC hemolysis, improved diastolic blood pressures, and increased blood oxygen saturation.[75] Despite the promising phase I results, a phase II study conducted by Baxalta, a company that had acquired the rights to AesRx, was terminated due, in part, to unblinding between the study drug and placebo groups.[126] Moreover, the lead investigators questioned the viability of the drug due to the rapid oxidative metabolism of the key aldehyde in light of newer compounds with potential for prolonged pharmacologic effects.
Although it was known that the allosteric effects of aromatic aldehydes relied on Schiff-base interactions with αVal1 amines of Hb, a more complete understanding of the allosteric effects that lead to increased Hb oxygen affinity became only clear following seminal work on 5-HMF by Safo and Abraham.[48] Peter Goodford had previously proposed that the allosteric activity of aromatic aldehydes occurs due to binding of the compounds to the classical liganded R-state Hb based on in silico model[110]; however, this viewpoint was later countered by x-ray crystallography performed by Abraham who showed that aromatic aldehydes rather bind to deoxygenated Hb to destabilize the T-state and shift the allosteric equilibrium to the high-O2-affinity R-state. The true nature of the interaction was later uncovered by Safo and Abraham in the study of 5-HMF. The scientists showed that the primary target was R2-state Hb (and not the classical R-state Hb as proposed by Peter Goodford), and to a certain extent T-state Hb. As first shown with 5-HMF and subsequently several other aromatic aldehydes, two of these compounds bind in a symmetrical fashion at the α-cleft of R2-state Hb, forming Schiff base adducts with αVal1 nitrogen atoms of the two α-subunits, as well as hydrogen-bond and/or hydrophobic interactions with the protein.[48,53,58,59,127] These interactions lock the two α-subunits together in the R2-state conformation, restricting the transition from the R-state to the T-state. It was also shown that aromatic aldehydes bind to the α-cleft of deoxygenated T-state Hb, although this interaction is weaker than that with R2-state Hb.[48] Binding to the deoxygenated T-state disrupted a water-mediated bridge between αVal1 and the opposite subunit residue αArg141, thus shifting the allosteric equilibrium to the R-state. These findings were a major discovery given that only the classical R-state and T-state structures were known when Peter Goodford and Don Abraham proposed their original binding models.
Although 5-HMF did not advance to licensure, the promising effects of the compound in clinical studies jumpstarted innovation around aromatic aldehydes, more specifically the structure-based approach to improving potency and pharmacokinetics to discover next generation synthetic drug candidates. This renewed interest also brought in scientists from academia and even big pharmaceutical companies looking to identify improved drug candidates. The growth in scientific interest is demonstrated by the large increase in publications on aromatic aldehydes both in medical chemistry and clinical journals.[50–53,59,61,62,127]
2.4. Derivatizing 5-HMF to improve its pharmacologic properties
With 5-HMF established as a lead, Safo and collaborators using structure-based drug discovery developed several 5-HMF derivatives where the alcohol moiety was derivatized as ester or ether (Figure 6). [59,127,128] These derivatives were designed to make increased interactions with the protein that was expected to increase their potency and potentially improve their PK properties. Several of these derivatives, e.g. VZHE004, VZHE006, VZHE005, VZHE015, and 5-PMFC exhibited increased potency, nonetheless none showed any significant improvement in the PK properties.[59,127,128] A patent has been filed for these compounds in 2018 (WO 2018/018035 A1).
Recently, Safo and group also derivatized 5-HMF by replacing the aldehyde moiety with Michael Addition reactive center that is expected to improve the compounds’ metabolic profile. These new derivatives (MMA-compounds, Figure 6), unlike 5HMF bind covalently to Hb through a Michael addition reaction with βCys93 and stabilize the R-state of Hb.[129] It is also expected that these compounds by binding to the βCys93, which is located on the surface of Hb, would directly destabilize polymer formation. Some of the compounds, e.g., MMA-409 showed comparable anti-sickling potency as 5-HMF. Notably, and as expected the in vitro time-dependent OEC with MMA-409 using whole blood showed the biological activity of MMA-409 to persist longer than 5-HMF.[129] A patent titled ‘Metabolically stable 5-HMF derivatives for the treatment of hypoxia’ protecting these compounds was issued in 2020 (US 10,836,729 B1).
2.5. Modern aromatic aldehyde drug discovery - pyridyl-derivatives of vanillin
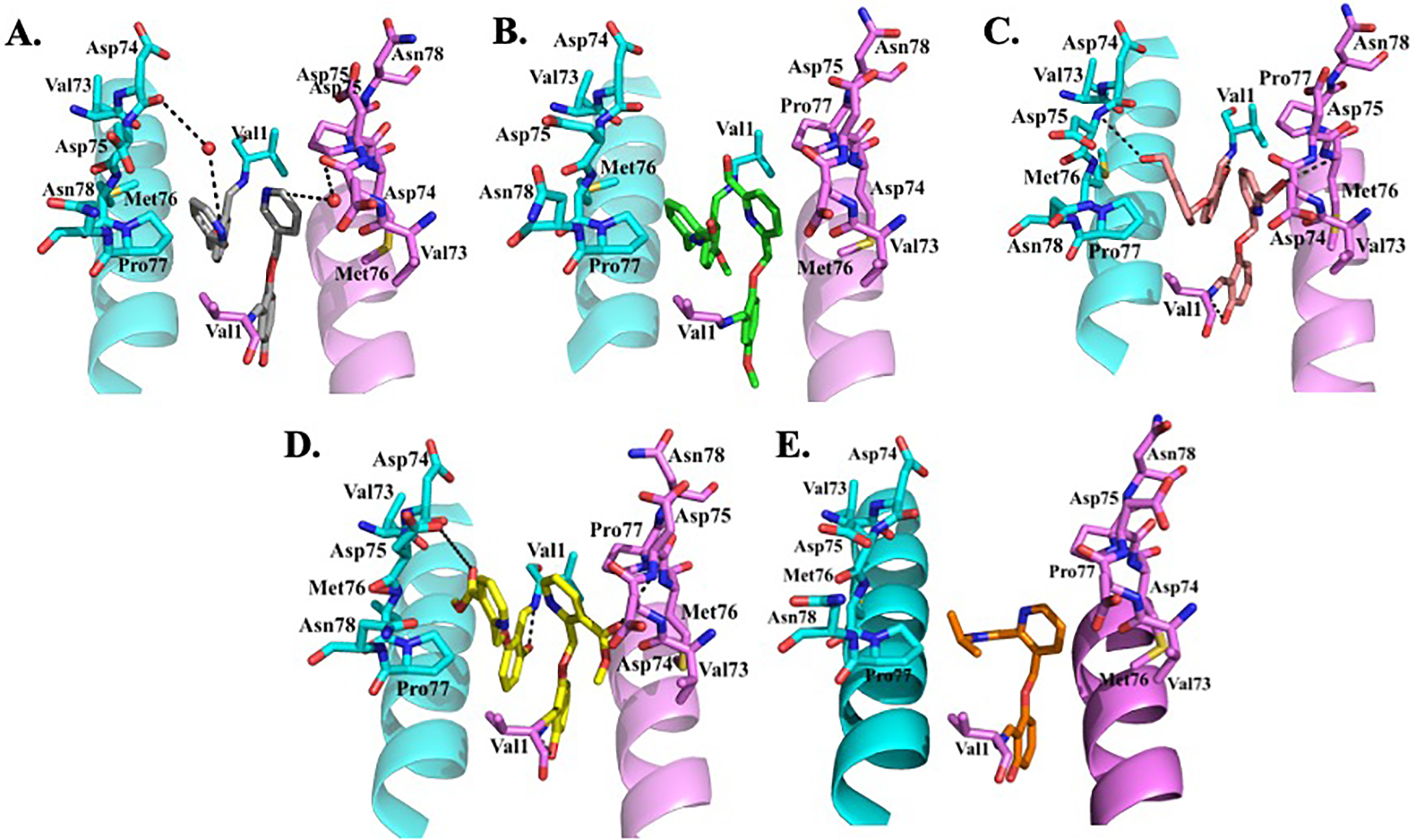
Based on the atomic interactions between Hb and vanillin or 5-HMF, several pyridyl derivatives of vanillin and its analogs were designed by Safo and Abraham.[50,51,54] These compounds (designated as INN and SAJ, Figure 7) like vanillin bind to the αVal1 of Hb to form Schiff-base adducts, which in addition to other protein interactions tie the two α- subunits together to stabilize the R-state and increase Hb oxygen affinity to prevent RBC sickling (Figure 8). The compounds exhibit as much as 2.5–90-fold more potency when compared to vanillin or 5HMF.[50,51,54] As also noted before, these compounds bind to deoxygenated Hb but unlike the R2-state Hb binding, manifest as diffused density.[50,51,54] Interestingly, some of the compounds, INN-312 and SAJ-310 showed inhibition of polymer formation in anoxia (absence of oxygen), an anti-sickling mechanism that is independent of the primary O2-dependent anti-sickling mechanism of aromatic aldehydes.[51] This innovative concept for direct polymer-destabilizing aromatic aldehydes was conceived by crystallographic observation of vanillin binding in close-proximity to the polymer stabilizing αF-helix (~5Å). The investigators surmised that this observation offered a direct polymer-inhibitory potential of perturbing key secondary contacts on the αF-helix of HbS that is well-known based on sentinel work performed by Rhoda et al,[16] as well as other biochemical studies demonstrating markedly reduced gelation and propensity to polymerize.[130–132] By rationally substituting a methoxypyridine ortho to the aldehyde moiety of vanillin and several of its analogs, several compounds were created that were expected to make interaction between the methoxypyridine and the αF-helix.[50,51,54] Indeed structural studies showed that these compounds, e.g. INN-312 and SAJ-310 when bound at the α-cleft dispose their methoxypyridine toward the surface of the Hb tetramer to make hydrophobic contacts (>3.3 Å) with residues of the αF-helix (Figure 8).[50,51] Structure activity studies demonstrated that these first generation pyridyl derivatives of vanillin, unlike earlier aromatic aldehydes could inhibit polymerization and sickling in complete anoxia (oxygen-independent anti-sickling effect), albeit weak. Nonetheless, the finding suggested a potential novel dual therapeutic approach involving the design of compounds that stereospecifically inhibit deoxy-HbS polymer formation while increasing the oxygen affinity of Hb. Based on this novel concept and adopting the structure-based design approach, a second generation of pyridyl derivatives of vanillin with either methylhydroxyl on the pyridine ring (e.g. TD-7[49,133,134] and VZHE-039[52], or ester on the pyridine ring (e.g. PP-6, PP-10 or PP-14) [53,135,136] (Figure 7) were developed to introduce potentially stronger contacts with the protein and αF-helix. All compounds as expected showed significant improvement of biological activity (increase in Hb oxygen affinity, anti-sickling effect under hypoxia) over vanillin, but comparable or slightly better than SAJ-301 or INN-312. Most importantly, while VZHE-039, and the PP compounds demonstrated highly potent anti-sickling effect in anoxia, TD-7 showed only marginal effect, while Voxelotor, which was studied as a comparator appears to completely lose activity under anoxia.[52,53] Structural studies confirmed close interactions between the methylhydroxyl of VZHE-039 or the ester of PP-6 or PP-10 and PP-14 with the αF-helix (Figure 8), while, interestingly, in TD-7 the methylhydroxyl has rotated away from making any interaction with the αF-helix although the pyridine ring still makes weak hydrophobic interaction with the αF-helix (Figure 8).[49,52,53] Voxelotor, with only one molecule bound per tetramer (see below) makes no significant interaction with the αF-helix (Figure 8).[61]
Figure 7.
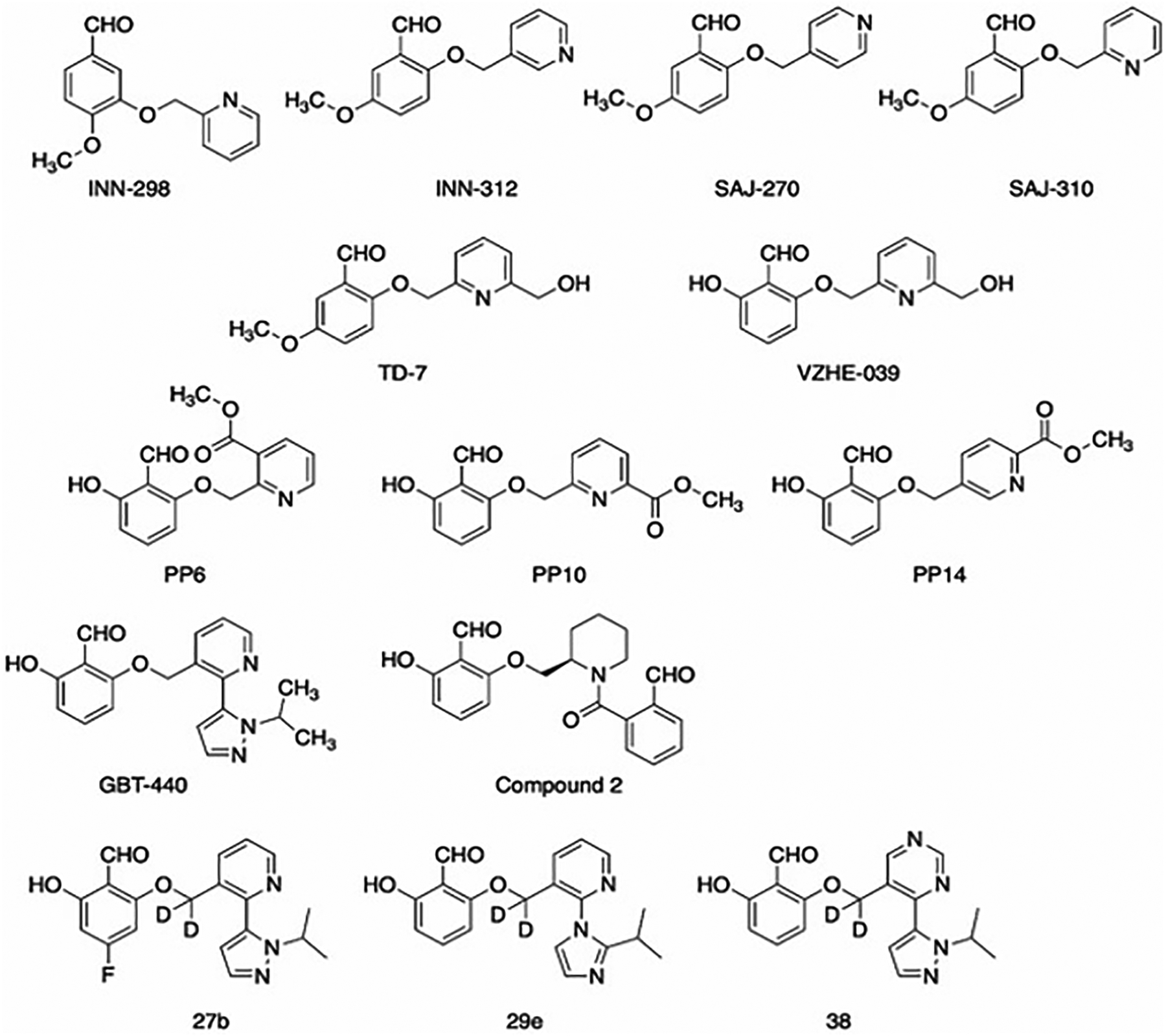
Pyridyl derivatives of vanillin
Figure 8.
A) Mechanism of binding of ECA to Hb; B) ECA and its derivatives as AEHs; C) Representative examples of alkyl and aryl isothiocyanates as AEHs; D) Heteroaryl disulfide compounds as AEHs.
It is notable to point out that TD-7 underwent significant metabolism leading to short duration of biological activity.[49] On the other hand, VZHE-039 or the PP compounds, with the ortho-hydroxy group (relative to the aldehyde moiety) protecting the aldehyde pharmacophore, demonstrated resistance to oxidative metabolism inside the RBC, leading to sustained and improved pharmacologic activities both in vitro and in vivo.[52,53,133,134] Similar metabolic protective behavior by a hydroxy moiety has been previously reported with Tucaresol,[115–118,137] which was also incorporated into Voxelotor to improve these compounds PK properties.[138] The compounds also showed no significant inhibition of several CYPs, as well as exhibiting high GI permeability indicating acceptable oral bioavailability, absent limitations from GI solubility and/or first-pass metabolism.[52,53] VZHE-039 also demonstrated high partitioning (85%) from whole blood into RBCs, which was made possible by the substituted pyridine.[52] Superior partitioning of candidate molecules into the RBC compartment is critical to minimize possible off-target effects, which may likely have derailed clinical development of Tucaresol.[139]
2.6. Development of aromatic aldehydes by Global Blood Therapeutics – First in kind as a SCD drug
Based on the groundwork laid down by the development of vanillin and the INN compounds, a series of bicyclic aryl derivatives of vanillin have been developed by Global Blood Therapeutics, Inc and protected with several patents, including 2017 (US 9,802,900 B2), 2018 (US 10,017,491 B2), 2018 (US 9,957,250 B2), 2019 (US 10,450,269 B1), 2019 (US 10,266,551 B2), 2020 (US 10,806,733 B2), 2020 (US 10,858,317 B2), 2020 (US 10,822,326 B2).[140–147] These compounds like previous aromatic aldehydes form Schiff-base interaction with the αVal1 amines of Hb and prevents hypoxia-induced HbS polymerization and RBC sickling.[61] Voxelotor (Figure 7), which eventually became approved for the treatment of SCD in 2019, showed significant biological potency even at low drug concentrations.[148] In vivo preclinical studies showed Voxelotor to be safe and effective in preventing RBC sickling. It also showed high oral bioavailability, sustained exposure and dramatic RBC partitioning when administered orally.[148–152] As noted above, Voxelotor anti-sickling effect, unlike VZHE-039 was due solely to increasing Hb oxygen affinity. Under anoxic condition, Voxelotor does not show any sickling inhibition, consistent of the structural studies that show the compound to not make significant interaction with the αF-helix.[62] Phase I/2 randomized, placebo-controlled, double-blind, single and multiple ascending dose study of the tolerability and pharmacokinetics of Voxelotor in healthy subjects and patients with SCD resulted in a favorable benefit-risk profile of Voxelotor for the treatment of SCD.[118,138,153,154] It was well tolerated at doses up to 1000 mg for 28 days and 900 mg up to 6 months. The linear, dose-proportional PK and long half-life of supported once-daily dosing. It demonstrated rapid, sustained, and clinically meaningful improvement in anemia, sickling, and clinical laboratory markers of hemolysis. Most adverse events were mild and there were no deaths or adverse events related to tissue hypoxia. In phase 3 randomized, placebo-controlled trial involving participants with SCD, significantly high percentage showed a Hb-response treatment with 1500mg Voxelotor as compared to placebo. At week 24, the 1500mg Voxelotor group showed significant reductions in the indirect bilirubin level and percentage of reticulocytes than the placebo group. Although adverse events of at least grade 3 occurred in ~25% of the participants they were not related to the trial drug or placebo, as determined by the investigators.[60] The FDA granted fast track (2015), orphan drug (2015), rare pediatric disease (2017), and breakthrough therapy designations (2018) to Voxelotor as a result of preliminary clinical evidence demonstrating its potential for substantial improvement over available therapies. The priority review designation was granted on August 22, 2019, and the drug received accelerated approval for the treatment of SCD on November 25, 2019, based on data from the phase 3 Hemoglobin Oxygen Affinity Modulation to Inhibit HbS Polymerization (HOPE) trial that demonstrated efficacy and safety of Voxelotor, compared with placebo, in patients aged ≥12 years with SCD.[5,155] It is also the first in the aromatic aldehydes class of drugs to receive FDA approval.
Unlike previous aromatic aldehydes, e.g., VZHE-039 that bind covalently to Hb in a 2:1 stoichiometry, Voxelotor binds Hb with a 1:1 stoichiometry (Figure 8).[61] The increased steric bulk due to the isopropyl-pyrazole substitution on the pyridine ring precluded two molecules from occupying the α-cleft at the same time. It has been suggested that 1:1 binding as observed with Voxelotor with Hb would be more suitable than 2:1 adducts as observed in VZHE-039 because the former compounds can be administered at effective doses that are lower and potentially safer compared to their counterparts.[61,156] Nonetheless, in vivo it is likely that compounds like VZHE-039 that exhibit dual anti-sickling activity may perform better. The key to understanding the potential of dual anti-sickling drugs in vivo is to recognize the limitations of allosteric hemoglobin modification. There is a limit to increasing the oxygen affinity of Hb, where excessive increase could prevent O2 release, and consequently lead to tissue hypoxia and anaerobic metabolism. Moreover, since the efficacy of anti-sickling drugs depends on the capacity to delay polymerization kinetics just long enough for the RBCs to pass through the circulation back to the lung, polymer-destabilization is potentially a more efficient anti-sickling mechanism because incorporation of a drug-bound tetramer can actually destabilize a growing chain instead of just reducing the number of deoxyHbS tetramers that are available to enter a fiber. Finally, allosteric modification depends on oxygen, and therefore compounds that solely rely on this mechanism to prevent RBC sickling unlike direct polymer destabilizers would not work in areas of tissue hypoxia.
Since the discovery of Voxelotor, several other classes of aromatic aldehydes have been discovered by Global Therapeutics, which have been protected in various patents. These include 2020 (US 10,695,330 B2) and 2020 (WO 2020/072377 A1). The second patent filed in 2020 claimed a series of di-aldehydes derivatives.[157] One notable compound claimed in this patent (compound 2, Figure 7), when administered orally (10 mg/kg) in rats showed a half-life of 187 h and a very high blood/plasma ratio. Crystallographic structure of liganded Hb in complex compound 2 revealed a single compound crosslinking the two α-subunits with each aldehyde group forming Schiff-base interaction with one of the αVal1 amines.[157]
Based on the aromatic aldehyde pharmacophore and adopting a similar approach as Global Blood Therapeutics; FronThera U.S. Pharmaceuticals, LLC (San Diego) recently reported several compounds as potential therapeutic agents to treat SCD.[158] Examples of these compounds are 27b, 29e and 38 (Figure 7). Dose- and time-dependent OEC studies with whole blood at 0.2 mM and 0.5 mM concentrations revealed that most of the compounds showed similar increase in the Hb-O2 affinity as Voxelotor. The compounds also showed comparable anti-sickling effect as, low plasma-protein binding, partitioned efficiently in the RBCs, showed low CYP inhibition and comparable liver microsomal stability in rats and humans as Voxelotor.[158] This invention has been reported in a patent titled ‘Hemoglobin modifier compounds and uses thereof’ in 2020 (US 10,787,430 B2).
2.7. Non-aromatic aldehyde Hb covalent modifiers as potential therapeutic agents
Aromatic aldehydes were one of the first class of compounds to be studied for treating SCD, and for over 2 decades have been the primary focus for SCD drug discovery as it appears to offer the best option in terms of toxicity and efficacy. Nonetheless, other covalent Hb binding compounds have also been studied over the years for the treatment of SCD; some dating as far back as four decades,[84–91,93,112] while others quite recently in response to aromatic aldehyde drawbacks, most notably their metabolic instability and/or relatively weak binding affinity due to reversibility of the Schiff-base covalent interaction.[129,159–163] One of the earliest covalent Hb modifiers was ethacrynic acid (ECA) (Figure 9A), a commercially marketed diuretic compound developed by Merck, for its potential as an anti-sickling agent.[93,164] Abraham and Max Perutz demonstrated that ECA increases the Hb affinity for oxygen and also causes direct polymer inhibition via interaction with the surface-located βCys93 residue of Hb (Figure 9A).[111] However, ECA was too toxic in concentration needed to treat the large amount of HbS. Structural studies of ECA and Hb suggest that small molecules that could form a covalent bond with the βCys93 side chain thiol eliminate a T-state stabilizing salt bridge between βAsp94 and βHis146,[111] resulting in allosteric equilibrium shift to the high O2-affinity Hb that does not polymerize.
Figure 9.
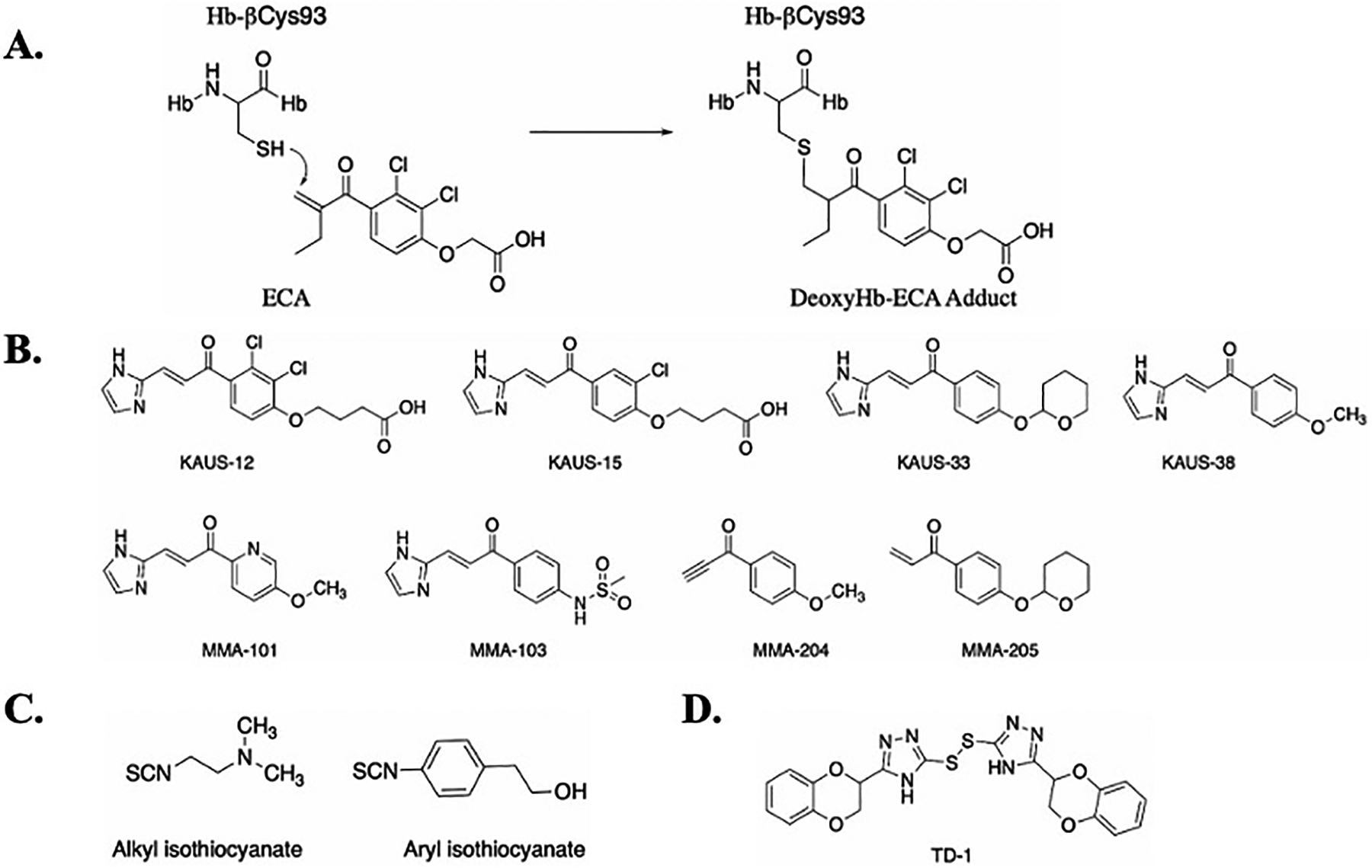
A) Mechanism of binding of ECA to Hb; B) ECA and its derivatives as AEHs; C) Representative examples of alkyl and aryl isothiocyanates as AEHs; D) Heteroaryl disulfide compounds as AEHs.
Based on the initial studies with ECA, Safo, Abdelsattar Omar and Moustafa El-Araby designed a new series of Michael Addition molecules (KAUS compounds, Figure 9B) that were hypothesized to bind covalently to βCys93 of Hb and inhibit RBC sickling, and importantly be more metabolically stable than aromatic aldehydes [163]. These compounds surprisingly showed even weaker allosteric and anti-sickling properties compared to ECA.[163] X-ray studies of Hb in complex with these compounds revealed an unanticipated mode of Michael addition between the β-unsaturated carbon and the N-terminal αVal1 nitrogen at the α-cleft of hemoglobin, with no observable interaction with βCys93.[163] However, a disulfide exchange reaction suggested low level interaction between the KAUS compounds and βCys93,[163] likely explaining why the KAUS compounds were not observed crystallographically at the βCys93 binding site.
Following, a second generation of KAUS-compounds were designed by the group by removing the carboxylate group from the compounds to allow the compounds to bind exclusively or preferentially at βCys93 while preventing binding in the central water cavity.[162,165] The new derivatives (e.g. KAUS-33 and KAUS-38 Figure 9B) with the expected binding to βCys93, exhibited improvement in vitro Hb-O2 affinity and RBC sickling inhibition over the first generation KAUS compounds.[162] However, the compounds showed limited solubility, and their potency was still significantly less than aromatic aldehydes.[162] To improve upon the pharmacologic profiles, the group modified the non-carboxylate azolylacryloyl derivatives to a third generation of compounds (MMA-compounds, Figure 9B), which as expected bind to βCys93.[161] Although these compounds showed significant anti-sickling potency, none showed significant sustained activity when compared to aromatic aldehydes.[161] The compounds have been patented in 2020 (US 10,836,729 B1).
Other class of covalent Hb modifiers are the cyanates and isothiocyanates[88]. Aliphatic isothiocyanates (Figure 9C) form adducts with Hb by binding covalently to βCys93 to disrupt the native T-state salt-bridge interaction between βAsp94 and βHis146 similar to ECA. Aromatic isothiocyanates (Figure 9C) react at the amino terminal amine on the α-chain of Hb to increase the oxygen affinity of Hb similar to aromatic aldehydes.[88] Although isothiocyanates can be administered at low doses and less frequently, like other permanent covalent binders, their lack of specificity could lead to toxicity. Due to these reasons, studies on this class of compounds are limited. Some of the early studies on these compounds have been claimed in 1974 (US 3,833,724).
Another series of compounds that engage in a covalent bond with the surface-located residue βCys93 of Hb to increase Hb affinity for oxygen are thiols; some of the recent compounds developed by the Zapol group at the Massachusetts General Hospital and Harvard Medical School.[159] One of the compounds di(5-(2,3-dihydro-1,4-benzodioxin-2-yl)-4H-1,2,4-triazol-3-yl)disulfide (TD-1, Figure 9D) was identified via a throughput screening of 38,700 compounds for their ability to modulate the Hb-O2 affinity. It was observed that TD-1 induced a greater increase in Hb-O2 affinity compared to 5-HMF and inhibited in vitro hypoxia-induced sickling of RBC without causing hemolysis.[160] TD-1 was claimed in the patent titled ‘heteroaryl disulfide compounds as allosteric effectors for increasing the oxygen binding affinity of hemoglobin’ in 2020 (US 10,758,569 B2).[166] Using X-ray crystallography, the group showed the compounds are capable of forming covalent interactions with βCys93 of the T-state, R-state and/or R3-state Hb to shift the allosteric equilibrium to the high O2-affinity R-state.[159]
2.8. A rare non-covalent Hb allosteric effector
As noted in the introduction, small molecule non-covalent effectors of Hb were studied during the early years of SCD drug discovery. These compounds were mostly designed to target surface cavities of HbS and directly destabilize polymer formation. Examples of these molecules include amino acids and their derivatives [95–97, 100–102], alcohols [96, 106, 107], halogenated/methylated aromatic carboxylic acid derivatives [98], proline salicylates [99], natural products and analogs thereof [103, 104], and carbohydrate derivatives [105]. Unlike covalent binders, e.g., aromatic aldehydes, thiols or isothiocyanates, that bind to well defined binding pockets via covalent interaction, the non-covalent compounds on the other hand bind reversibly to shallow cavities on the surface of the protein with low affinity, explaining their ineffectiveness in blocking polymer formation.[108] Although not universally true, covalent binders, because of their transient or permanent covalent interaction with the protein, provide pharmacological advantages over the non-covalent binders including enhanced potency, selectivity, and prolonged duration of action.
Interestingly, scientists at Pfizer recently published a series of pyrazole containing non-covalent compounds that bind to the α-cleft of Hb to increase its affinity for oxygen with potential for anti-sickling effects. [167,168] This is the first report of compounds with a non-covalent binding mode that attempts to recapitulate the biological activity seen from the covalent interactions of aromatic aldehyde with the α-cleft αVal1 amine. These pyrazole containing compounds have been reported in a patent titled ‘Chemical Compounds’ in 2021(US 11,014,908 B2).[167] A lack of in vitro and ex vivo data using standardized assays makes it quiet challenging to comparatively assess the compounds. But our interpretation of the publicly available data suggests that these compounds, although significantly improved over the previously studied non-covalent Hb binders, may not be as potent as the latest generation of aromatic aldehydes. One of these compounds, PF-07059013, was evaluated in Townes mice at steady state blood concentrations that should have been sufficient to completely occupy the Hb tetramers in RBCs. Nevertheless, the level of anti-sickling activity was similar to that achieved with aromatic aldehydes at lower occupancy levels.
3. Expert opinion
Sickle cell disease is one of the earliest characterized genetic disorders and is still broadly categorized as a ‘rare disease’ by multiple health agencies and organizations. Since its discovery in 1910 by James Herrick, only four drugs, including Hydroxyurea,[68] L-glutamine,[65] Crizanlizumab[67], and the aromatic aldehyde, Voxelotor[5] have been approved to treat SCD. Although the molecular basis of SCD has been very well established, the development of Hb target therapies to treat SCD has been a challenge over the years, underlying the complex nature of the disease, coupled with the fact that historically, large, well-funded pharmaceutical companies have not focused on drug development in SCD. Nonetheless, recent years have seen a dramatic increase in commercial interest in developing drugs for SCD both academia and pharmaceutical companies, although new composition of matter patents on SCD therapeutics still appear to be mainstays of academia and small pharmaceutical businesses. These drug-discovery efforts have contributed to the plethora of information in treating SCD and associated symptoms. A wide range of new therapeutic options to treat SCD is under development; one of these is focused on targeting the primary pathophysiology using aromatic aldehydes that bind to HbS and allosterically shift the oxygen equilibrium to the R-state and consequently increase HbS O2 affinity to inhibit HbS polymerization, and/or directly inhibit interactions between deoxyHbS tetramers. With the FDA approval of Voxelotor,[5] the first small molecule directly targeting Hb, the prospects for new drugs for patients with SCD seems to be growing significantly stronger each year. While non-specific binding and off-target effects are potential liabilities associated with covalent Hb binders, aromatic aldehydes are generally well tolerated in vivo when administered at therapeutically efficacious doses,[169] due in part to their reversible binding nature under physiological conditions, and when unbound easily metabolized to acids for excretion. In the opinion of these authors, aromatic aldehydes thus provide the highest chance of clinical success of any Hb binding agent. These molecules hold great promise that will provide realistic therapeutic options for individuals with SCD. Even though aromatic aldehydes target the primary pathophysiology of the disease and provide realistic therapeutic options for individuals with SCD, ultimately, clinicians may find that combination therapies that involve aromatic aldehydes and other compounds, e.g., HU or Crizanlizumab may provide the best outcomes for addressing the many pathophysiological effects associated with the disease. This will depend on a variety of criteria such as age, frequency of crisis, medication tolerability, and degree of SCD-mediated physical deterioration.
Article Highlights.
Sickle cell disease (SCD), the most common inherited hematologic disorder, occurs because of a single point mutation of βGlu6 in normal hemoglobin (HbA) to βVal6 in sickle hemoglobin (HbS).
SCD is characterized by polymerization of HbS and sickling of red blood cells (RBCs), leading to several other pathophysiologies, including vaso-occlusion, hemolytic anemia, stroke, pain crisis, and organ damage.
Much of the early drug discovery research for SCD was performed by Max Perutz, Don Abraham, and Irvine Klotz, which laid important foundation for subsequent drug discovery and development.
Aromatic aldehydes are promising new clinical candidates that directly prevent the primary pathophysiology of hypoxia-induced HbS polymerization and erythrocyte sickling by increasing Hb oxygen affinity. One such compound, Voxelotor was recently approved for SCD treatment.
In the opinion of these authors, Hb modulators, such as aromatic aldehydes could provide not only the best chance for a highly effective oral therapy for SCD, especially in the under-developed world, but also a way to treat a variety of other human conditions.
Acknowledgements
This article is dedicated to the memory of Don Abraham, a pioneer in sickle cell drug discovery.
Funding
This paper was funded by NIH grants: R61HL156158, R01MD009124, S10OD021756.
Footnotes
Declaration of interests
Virginia Commonwealth University and some of the authors have patents related to several of the molecules mentioned in the review. Some of these compounds have been licensed over the years to several companies, including AesRx, Baxalta, Shire, Takeda, and Illexcor Therapeutics for the development to treat SCD. MK Safo and A Fleischman are co-owners of Illexcor Therapeutics which has licensed and developed some of the compounds. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- [1].Thein MS, Igbineweka NE, Thein SL. Sickle cell disease in the older adult. Pathology 2017;49(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Alrayyes S, Baghdan D, Haddad RY, Compton A-A, Mohama S, Goreishi R, et al. Sickle cell disease; An overview of the disease and its systemic effects. Dis Month. 2018;64(6):283–289. [DOI] [PubMed] [Google Scholar]
- [3].Chaturvedi S, DeBaun MR. Evolution of sickle cell disease from a life-threatening disease of children to a chronic disease of adults: The last 40 years. Am J Hematol. 2016;91(1):5–14. [DOI] [PubMed] [Google Scholar]
- [4].Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376:1561–73. [DOI] [PubMed] [Google Scholar]
- [5].FDA approves voxelotor for sickle cell disease. (accessed April 12, 2021). Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-voxelotor-sickle-cell-disease
- [6].Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 2013;381(9861):142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Data & Statistics on Sickle Cell Disease. (accessed April 29, 2021). Available from: https://www.cdc.gov/ncbddd/sicklecell/data.html
- [8].Ingram VM. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature 1957;180(4581):326–8. [DOI] [PubMed] [Google Scholar]
- [9].Ingram VM. Abnormal human haemoglobins. I. The comparison of normal human and sickle-cell haemoglobins by fingerprinting. Biochim Biophys Acta. 1958;28(3):539–45. [DOI] [PubMed] [Google Scholar]
- [10].Bunn HF (Howard F, 1935-, Forget BG, 1939-. Hemoglobin--molecular, genetic, and clinical aspects W.B. Saunders Co.; 1986. [Google Scholar]
- [11].Ghatge MS, Ahmed MH, Omar ASM, Pagare PP, Rosef S, Kellogg GE, et al. Crystal structure of carbonmonoxy sickle hemoglobin in R-state conformation. J Struct Biol. 2016;194(3):446–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ferrone FA. Polymerization and sickle cell disease: A molecular view. Microcirculation 2004;11(2):115–28. [DOI] [PubMed] [Google Scholar]
- [13].Charache S, Grisolia S, Fiedler AJ, Hellegers AE. Effect of 2,3-diphosphoglycerate on oxygen affinity of blood in sickle cell anemia. J Clin Invest. 1970;49(4):806–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang Y, Berka V, Song A, Sun K, Wang W, Zhang W, et al. Elevated sphingosine-1-phosphate promotes sickling and sickle cell disease progression. J Clin Invest. 2014;124(6):2750–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sun K, D’Alessandro A, Ahmed MH, Zhang Y, Song A, Ko T-P, et al. Structural and functional insight of sphingosine 1-phosphate-mediated pathogenic metabolic reprogramming in sickle cell disease. Sci Rep. 2017;7(1):15281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rhoda M-D, Martin J, Blouquit Y, Garel M-C, Edelstein SJ, Rosa J. Sickle cell hemoglobin fiber formation strongly inhibited by the stanleyville II mutation (α78 Asn → Lys). Biochem Biophy Res Co. 1983;111(1):8–13. [DOI] [PubMed] [Google Scholar]
- [17].Benesch RE, Kwong S, Edalji R, Benesch R. alpha Chain mutations with opposite effects on the gelation of hemoglobin S. J Biol Chem. 1979;254(17):8169–72. [PubMed] [Google Scholar]
- [18].Nagel RL, Johnson J, Bookchin RM, Garel MC, Rosa J, Schiliro G, et al. Beta-chain contact sites in the haemoglobin S polymer. Nature 1980;283:832–4. [DOI] [PubMed] [Google Scholar]
- [19].Burchall G, Maxwell E. Haemoglobin Stanleyville II modifies sickle disease phenotype. Pathology 2010;42:310–2. [DOI] [PubMed] [Google Scholar]
- [20].Sedrak A, Kondamudi NP. Sickle Cell Disease. StatPearls, Treasure Island (FL): StatPearls Publishing; 2018. [PubMed] [Google Scholar]
- [21].Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: Reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21(1):37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010;376(9757):2018–31. [DOI] [PubMed] [Google Scholar]
- [23].Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet 2017;390(10091):311–23. [DOI] [PubMed] [Google Scholar]
- [24].Safo MK, Ahmed MH, Ghatge MS, Boyiri T. Hemoglobin-ligand binding: understanding Hb function and allostery on atomic level. Biochim Biophys Acta. 2011;1814(6):797–809. [DOI] [PubMed] [Google Scholar]; (**)
- [25].Ahmed MH, Ghatge MS, Safo MK. Hemoglobin: Structure, Function and Allostery. In: Hoeger U, Harris JR, editors. Vertebrate and invertebrate respiratory proteins, lipoproteins and other body fluid proteins, Cham: Springer International Publishing; 2020, p. 345–82. [Google Scholar]
- [26].Safo MK, Abraham DJ. The X-ray structure determination of bovine carbonmonoxy hemoglobin at 2.1 A resoultion and its relationship to the quaternary structures of other hemoglobin crystal froms. Protein Sci. 2001;10(6):1091–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Perutz MF. Nature of haem-haem interaction. Nature 1972;237:495–9. [DOI] [PubMed] [Google Scholar]
- [28].Perutz MF, Wilkinson AJ, Paoli M, Dodson GG. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu Rev Biophys Biomol Struct. 1998;27:1–34. [DOI] [PubMed] [Google Scholar]
- [29].Fermi G, Perutz MF, Shaanan B, Fourme R. The crystal structure of human deoxyhaemoglobin at 1.74 A resolution. J Mol Biol. 1984;175(2):159–74. [DOI] [PubMed] [Google Scholar]
- [30].Baldwin J, Chothia C. Haemoglobin: the structural changes related to ligand binding and its allosteric mechanism. J Mol Biol. 1979;129(2):175–220. [DOI] [PubMed] [Google Scholar]
- [31].Minton AP, Imai K. The three-state model: A minimal allosteric description of homotropic and heterotropic effects in the binding of ligands to hemoglobin. Proc Natl Acad Sci U S A 1974;71(4):1418–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Abraham DJ, Safo MK, Boyiri T, Danso-Danquah RE, Kister J, Poyart C. How allosteric effectors can bind to the same protein residue and produce opposite shifts in the allosteric equilibrium. Biochem. 1995;34(46):15006–20. [DOI] [PubMed] [Google Scholar]
- [33].Sawicki CA, Gibson QH. Quaternary conformational changes in human hemoglobin studied by laser photolysis of carboxyhemoglobin. J Biol Chem. 1976;251(6):1533–42. [PubMed] [Google Scholar]
- [34].Samuni U, Dantsker D, Juszczak LJ, Bettati S, Ronda L, Mozzarelli A, et al. Spectroscopic and functional characterization of T state hemoglobin conformations encapsulated in silica gels. Biochem. 2004;43(43):13674–82. [DOI] [PubMed] [Google Scholar]
- [35].Song X, Simplaceanu V, Ho NT, Ho C. Effector-induced structural fluctuation regulates the ligand affinity of an allosteric protein: binding of inositol hexaphosphate has distinct dynamic consequences for the T and R states of hemoglobin. Biochem. 2008;47(17):4907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wilson J, Phillips K, Luisi B. The crystal structure of horse deoxyhaemoglobin trapped in the high-affinity (R) state. J Mol Biol. 1996;264(4):743–56. [DOI] [PubMed] [Google Scholar]
- [37].Schumacher MA, Dixon MM, Kluger R, Jones RT, Brennan RG. Allosteric transition intermediates modelled by crosslinked haemoglobins. Nature 1995;375(6526):84–7. [DOI] [PubMed] [Google Scholar]
- [38].Abraham DJ, Peascoe RA, Randad RS, Panikker J. X-ray diffraction study of di and tetra-ligated T-state hemoglobin from high salt crystals. J Mol Biol. 1992;227(2):480–92. [DOI] [PubMed] [Google Scholar]
- [39].Kavanaugh JS, Rogers PH, Arnone A. Crystallographic evidence for a new ensemble of ligand-induced allosteric transitions in hemoglobin: the T-to-T(high) quaternary transitions. Biochem. 2005;44(16):6101–21. [DOI] [PubMed] [Google Scholar]
- [40].Silva MM, Rogers PH, Arnone A. A third quaternary structure of human hemoglobin A at 1.7-A resolution. J Biol Chem. 1992;267(24):17248–56. [PubMed] [Google Scholar]
- [41].Jenkins JD, Musayev FN, Danso-Danquah R, Abraham DJ, Safo MK. Structure of relaxed-state human hemoglobin: insight into ligand uptake, transport and release. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 1):41–8. [DOI] [PubMed] [Google Scholar]
- [42].Scott C, Suh J, Stea B, Nabid A, Hackman J. Improved survival, quality of life, and quality-adjusted survival in breast cancer patients treated with efaproxiral (Efaproxyn) plus whole-brain radiation therapy for brain metastases. Am J Clin Oncol. 2007;30(6):580–7. [DOI] [PubMed] [Google Scholar]
- [43].Suh JH, Stea B, Nabid A, Kresl JJ, Fortin A, Mercier J-P, et al. Phase III study of efaproxiral as an adjunct to whole-brain radiation therapy for brain metastases. J Clin Oncol. 2006;24(1):106–14. [DOI] [PubMed] [Google Scholar]
- [44].Kunert MP, Liard JF, Abraham DJ. RSR-13, an allosteric effector of hemoglobin, increases systemic and iliac vascular resistance in rats. Am J Physiol. 1996;271(2 Pt 2):H602–613. [DOI] [PubMed] [Google Scholar]
- [45].Khandelwal SR, Randad RS, Lin PS, Meng H, Pittman RN, Kontos HA, et al. Enhanced oxygenation in vivo by allosteric inhibitors of hemoglobin saturation. Am J Physiol. 1993;265(4 Pt 2):H1450–1453. [DOI] [PubMed] [Google Scholar]
- [46].Grinberg OY, Miyake M, Hou H, Steffen RP, Swartz HM. The Dose-Dependent Effect of RSR13, a Synthetic Allosteric Modifier of Hemoglobin, on Physiological Parameters and Brain Tissue Oxygenation in Rats. Adv Exp Med Biol. 2003;530:287–96. [DOI] [PubMed] [Google Scholar]
- [47].Hou H, Khan N, Grinberg OY, Yu H, Grinberg SA, Lu S, et al. The effects of Efaproxyn (efaproxiral) on subcutaneous RIF-1 tumor oxygenation and enhancement of radiotherapy-mediated inhibition of tumor growth in mice. Radiat Res. 2007;168(2):218–25. [DOI] [PubMed] [Google Scholar]
- [48].Safo MK, Abdulmalik O, Danso-Danquah R, Burnett JC, Nokuri S, Joshi GS, et al. Structural basis for the potent antisickling effect of a novel class of five-membered heterocyclic aldehydic compounds. J Med Chem. 2004;47(19):4665–76. [DOI] [PubMed] [Google Scholar]; (**)
- [49].Deshpande TM, Pagare PP, Ghatge MS, Chen Q, Musayev FN, Venitz J, et al. Rational modification of vanillin derivatives to stereospecifically destabilize sickle hemoglobin polymer formation. Acta Crystallogr D Struct Biol. 2018;74(Pt 10):956–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pagare PP, Ghatge MS, Musayev FN, Deshpande TM, Chen Q, Braxton C, et al. Rational design of pyridyl derivatives of vanillin for the treatment of sickle cell disease. Bioorg Med Chem. 2018;26(9):2530–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Abdulmalik O, Ghatge MS, Musayev FN, Parikh A, Chen Q, Yang J, et al. Crystallographic analysis of human hemoglobin elucidates the structural basis of the potent and dual antisickling activity of pyridyl derivatives of vanillin. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 11):920–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Abdulmalik O, Pagare PP, Huang B, Xu GG, Ghatge MS, Xu X, et al. VZHE-039, a novel antisickling agent that prevents erythrocyte sickling under both hypoxic and anoxic conditions. Sci Rep. 2020;10:20277. [DOI] [PMC free article] [PubMed] [Google Scholar]; (*)
- [53].Pagare PP, Ghatge MS, Chen Q, Musayev FN, Venitz J, Abdulmalik O, et al. Exploration of Structure–Activity Relationship of Aromatic Aldehydes Bearing Pyridinylmethoxy-Methyl Esters as Novel Antisickling Agents. J Med Chem. 2020;63:14724–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Nnamani IN, Joshi GS, Danso-Danquah R, Abdulmalik O, Asakura T, Abraham DJ, et al. Pyridyl derivatives of benzaldehyde as potential antisickling agents. Chem Biodivers. 2008;5(9):1762–9. [DOI] [PubMed] [Google Scholar]
- [55].Wireko FC, Abraham DJ. X-ray diffraction study of the binding of the antisickling agent 12C79 to human hemoglobin. Proc Natl Acad Sci U S A 1991;88(6):2209–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Abraham DJ, Mehanna AS, Wireko FC, Whitney J, Thomas RP, Orringer EP. Vanillin, a potential agent for the treatment of sickle cell anemia. Blood 1991;77(6):1334–41. [PubMed] [Google Scholar]; (**)
- [57].Zaugg RH, Walder JA, Klotz IM. Schiff base adducts of hemoglobin. Modifications that inhibit erythrocyte sickling. J Biol Chem. 1977;252(23):8542–8. [PubMed] [Google Scholar]; (*)
- [58].Abdulmalik O, Safo MK, Chen Q, Yang J, Brugnara C, Ohene-Frempong K, et al. 5-hydroxymethyl-2-furfural modifies intracellular sickle haemoglobin and inhibits sickling of red blood cells. Br J Haematol. 2005;128(4):552–61. [DOI] [PubMed] [Google Scholar]
- [59].Xu GG, Pagare PP, Ghatge MS, Safo RP, Gazi A, Chen Q, et al. Design, synthesis, and biological evaluation of ester and ether derivatives of antisickling agent 5-HMF for the treatment of sickle cell disease. Mol Pharma. 2017;14(10):3499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Vichinsky E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, et al. A phase 3 randomized trial of Voxelotor in sickle cell disease. N Engl J Med. 2019;381(6):509–19. [DOI] [PubMed] [Google Scholar]
- [61].Metcalf B, Chuang C, Dufu K, Patel MP, Silva-Garcia A, Johnson C, et al. Discovery of GBT440, an orally bioavailable R-state stabilizer of sickle cell hemoglobin. ACS Med Chem Lett. 2017;8(3):321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (**)
- [62].Oksenberg D, Dufu K, Patel MP, Chuang C, Li Z, Xu Q, et al. GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br J Haematol. 2016;175(1):141–53. [DOI] [PubMed] [Google Scholar]
- [63].Mvalo T, Topazian H, Kamthunzi P, Chen J, Kambalame I, Mafunga P, et al. Increasing hydroxyurea use in children with sickle cell disease at Kamuzu Central Hospital, Malawi. Blood Adv. 2018;2(Suppl 1):30–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].L-glutamine (Endari) for sickle cell disease. Med Lett Drugs Ther. 2018;60(1539):21–2. [PubMed] [Google Scholar]
- [65].Kaufman MB. Pharmaceutical approval update. P T 2017;42(8):620–1. [PMC free article] [PubMed] [Google Scholar]
- [66].Cieri-Hutcherson NE, Hutcherson TC, Conway-Habes EE, Burns BN, White NA. Systematic review of l-glutamine for prevention of vaso-occlusive pain crisis in patients with sickle cell disease. Pharmacotherapy 2019;39(11):1095–104. [DOI] [PubMed] [Google Scholar]
- [67].Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376:429–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].FDA approves hydroxyurea for treatment of pediatric patients with sickle cell anemia. (accessed March 10, 2018). Available from: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm590096.htm
- [69].Goldberg MA, Brugnara C, Dover GJ, Schapira L, Lacroix L, Bunn HF. Hydroxyurea and erythropoietin therapy in sickle cell anemia. Semin Oncol. 1992;19(3 Suppl 9):74–81. [PubMed] [Google Scholar]
- [70].Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med. 2008;358(13):1362–9. [DOI] [PubMed] [Google Scholar]
- [71].Chou Y-C, Chen R-L, Lai Z-S, Song J-S, Chao Y-S, Shen C-KJ. Pharmacological induction of human fetal globin gene in hydroxyurea-resistant primary adult erythroid cells. Mol Cell Biol. 2015;35(14):2541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Charache S, Dover GJ, Moore RD, Eckert S, Ballas SK, Koshy M, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood 1992;79(10):2555–65. [PubMed] [Google Scholar]
- [73].Dufu K, Patel M, Oksenberg D, Cabrales P. GBT440 improves red blood cell deformability and reduces viscosity of sickle cell blood under deoxygenated conditions. Clin Hemorheol Microcirc. 2018;70(1):95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Oder E, Safo MK, Abdulmalik O, Kato GJ. New developments in anti-sickling agents: Can drugs directly prevent the polymerization of sickle haemoglobin in vivo? Br J Haematol. 2016;175(1):24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Safo MK, Kato GJ. Therapeutic strategies to alter the oxygen affinity of sickle hemoglobin. Hematol Oncol Clin North Am. 2014;28(2):217–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Smith W. Improvement in the Clinical Global Impression of Change with Voxelotor in Patients with Sickle Cell Disease in the Phase 3 HOPE Trial. Hemoglobinopathies, Excluding Thalassemia, Virtual: 2020. [Google Scholar]
- [77].Ataga KI, Reid M, Ballas SK, Yasin Z, Bigelow C, James LS, et al. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: a phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapoc (ICA-17043). Br J Haematol. 2011;153(1):92–104. [DOI] [PubMed] [Google Scholar]
- [78].Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. 1910. Yale J Biol Med. 2001;74(3):179–84. [PMC free article] [PubMed] [Google Scholar]
- [79].Pauling L, Itano HA. Sickle cell anemia a molecular disease. Science 1949;110(2865):543–8. [DOI] [PubMed] [Google Scholar]
- [80].Pauling L. Molecular disease and evolution. Bull N Y Acad Med. 1964;40(5):334–42. [PMC free article] [PubMed] [Google Scholar]
- [81].Perutz MF, Muirhead H, Cox JM, Goaman LCG. Three-dimensional fourier synthesis of horse oxyhaemoglobin at 2.8 Å resolution: The atomic model. Nature 1968;219:131–9. [DOI] [PubMed] [Google Scholar]
- [82].Ladner RC, Heidner EJ, Perutz MF. The structure of horse methaemoglobin at 2–0 A resolution. J Mol Biol. 1977;114(3):385–414. [DOI] [PubMed] [Google Scholar]
- [83].Muirhead H, Perutz MF. Structure of hæemoglobin: A three-dimensional fourier synthesis of reduced human haemoglobin at 5.5 Å resolution. Nature 1963;199:633–8. [DOI] [PubMed] [Google Scholar]
- [84].Abraham DJ, Perutz MF, Phillips SE. Physiological and x-ray studies of potential antisickling agents. Proc Natl Acad Sci U S A 1983;80(2):324–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Walder JA, Zaugg RH, Walder RY, Steele JM, Klotz IM. Diaspirins that crosslink beta chains of hemoglobin: bis(3,5-dibromosalicyl) succinate and bis(3,5-dibromosalicyl) fumarate. Biochem. 1979;18(20):4265–70. [DOI] [PubMed] [Google Scholar]
- [86].Walder JA, Zaugg RH, Iwaoka RS, Watkin WG, Klotz IM. Alternative aspirins as antisickling agents: acetyl-3,5-dibromosalicylic acid. Proc Natl Acad Sci U S A 1977;74(12):5499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Zaugg RH, Walder JA, Walder RY, Steele JM, Klotz IM. Modification of hemoglobin with analogs of aspirin. J Biol Chem. 1980;255(7):2816–21. [PubMed] [Google Scholar]
- [88].Park S, Hayes BL, Marankan F, Mulhearn DC, Wanna L, Mesecar AD, et al. Regioselective covalent modification of hemoglobin in search of antisickling agents. J Med Chem. 2003;46:936–53. [DOI] [PubMed] [Google Scholar]
- [89].Garel MC, Domenget C, Galacteros F, Martin-Caburi J, Beuzard Y. Inhibition of erythrocyte sickling by thiol reagents. Mol Pharmacol. 1984;26(3):559–65. [PubMed] [Google Scholar]
- [90].Caburi-Martin J, Garel MC, Domenget C, Prehu C, Beuzard Y. Contact inhibition within hemoglobin S polymer by thiol reagents. Biochim Biophys Acta. 1986;874(1):82–9. [DOI] [PubMed] [Google Scholar]
- [91].Garel MC, Domenget C, Caburi-Martin J, Prehu C, Galacteros F, Beuzard Y. Covalent binding of glutathione to hemoglobin. I. Inhibition of hemoglobin S polymerization. J Biol Chem. 1986;261(31):14704–9. [PubMed] [Google Scholar]
- [92].Abraham DJ. Sickle cell anemia treatment and compound. US4482571A. 1984. [Google Scholar]
- [93].Kennedy PE, Williams FL, Abraham DJ. Design, synthesis, and testing of potential antisickling agents. 3. Ethacrynic acid. J Med Chem. 1984;27(2):103–5. [DOI] [PubMed] [Google Scholar]
- [94].Sunshine HR, Hofrichter J, Eaton WA. Requirements for therapeutic inhibition of sickle haemoglobin gelation. Nature 1978;275(5677):238–40. [DOI] [PubMed] [Google Scholar]
- [95].Poillon WN. Noncovalent inhibitors of sickle hemoglobin gelation: effects of aryl-substituted alanines. Biochem. 1982;21(6):1400–6. [DOI] [PubMed] [Google Scholar]
- [96].Noguchi CT, Schechter AN. Inhibition of sickle hemoglobin gelation by amino acids and related compounds. Biochem. 1978;17(25):5455–9. [DOI] [PubMed] [Google Scholar]
- [97].Kumpati J. Liposome-loaded phenylalanine or tryptophan as sickling inhibitor: A possible therapy for sickle cell disease. Biochem Med Metab Biol. 1987;38(2):170–81. [DOI] [PubMed] [Google Scholar]
- [98].Patwa DC, Abraham DJ, Hung TC. Design, synthesis, and testing of potential antisickling agents. 6. Rheologic studies with active phenoxy and benzyloxy acids. Blood Cells. 1987;12(3):589–601. [PubMed] [Google Scholar]
- [99].Abraham DJ, Gazze DM, Kennedy PE, Mokotoff M. Design, synthesis, and testing of potential antisickling agents. 5. Disubstituted benzoic acids designed for the donor site and proline salicylates designed for the acceptor site. J Med Chem. 1984;27(12):1549–59. [DOI] [PubMed] [Google Scholar]
- [100].McCurdy PR, Mahmood L. Intravenous urea treatment of the painful crisis of sickle-cell disease. N Eng J Med. 1971;285(18):992–4. [DOI] [PubMed] [Google Scholar]
- [101].Pariser S, Katz A. Treatment of sickle cell trait hematuria with oral urea. J Urology 1994;151(2):401–3. [DOI] [PubMed] [Google Scholar]
- [102].Elbaum D, Roth EJ, Neumann G, Jaffe ER, Bookchin RM, Nagel RL. Molecular and cellular effects of antisickling concentrations of alkylureas. Blood 1976;48(2):273–82. [PubMed] [Google Scholar]
- [103].Fall ABK, Toppet M, Ferster A, Fondu P, Vanhaelen-Fastré R, Vanhaelen M. In vitro antisickling activity of cromolyn sodium. Br J Haematol. 1998;103(4):957–9. [DOI] [PubMed] [Google Scholar]
- [104].Toppet M, Fall AB, Ferster A, Fondu P, Mélot C, Vanhaelen-Fastré R, et al. Antisickling activity of sodium cromoglicate in sickle-cell disease. Lancet 2000;356(9226):309. [DOI] [PubMed] [Google Scholar]
- [105].Mehanna AS. Sickle cell anemia and antisickling agents then and now. Curr Med Chem. 2001;8(2):79–88. [DOI] [PubMed] [Google Scholar]
- [106].Klotz IM, Haney DN, King LC. Rational approaches to chemotherapy: antisickling agents. Science 1981;213(4509):724–31. [DOI] [PubMed] [Google Scholar]
- [107].Poillon WN. Noncovalent inhibitors of sickle hemoglobin gelation: effects of aliphatic alcohols, amides, and ureas. Biochem. 1980;19(14):3194–9. [DOI] [PubMed] [Google Scholar]
- [108].Safo MK, Aljahdali A, Burnett J, Abraham DJ, Abdulmalik O. Therapeutic Strategies for the Treatment of Sickle Cell Disease. Burger’s Medicinal Chemistry and Drug Discovery, American Cancer Society; 2021, p. 1–31. [Google Scholar]; (*)
- [109].Abraham DJ, Mokotoff M, Sheh L, Simmons JE. Design, synthesis, and testing of antisickling agents. 2. Proline derivatives designed for the donor site. J Med Chem. 1983;26(4):549–54. [DOI] [PubMed] [Google Scholar]
- [110].Beddell CR, Goodford PJ, Kneen G, White RD, Wilkinson S, Wootton R. Substituted benzaldehydes designed to increase the oxygen affinity of human haemoglobin and inhibit the sickling of sickle erythrocytes. Br J Pharmacol. 1984;82(2):397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Perutz MF, Fermi Giulio, Abraham DJ, Poyart Claude, Bursaux E. Hemoglobin as a receptor of drugs and peptides: x-ray studies of the stereochemistry of binding. J Am Chem Soc. 1986;108:1064–78. [Google Scholar]; (**)
- [112].Sheh L, Mokotoff M, Abraham DJ. Design, synthesis, and testing of potential antisickling agents. 9. Cyclic tetrapeptide homologs as mimics of the mutation site of hemoglobin S. Int J Pept Protein Res. 1987;29(4):509–20. [DOI] [PubMed] [Google Scholar]
- [113].Merrett M, Stammers DK, White RD, Wootton R, Kneen G. Characterization of the binding of the anti-sickling compound, BW12C, to haemoglobin. Biochem J. 1986;239(2):387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Kneen G. Pharmaceutical compounds, preparation, use and intermediates therefor and their preparation. US4535183A. 1985. [Google Scholar]
- [115].Arya R, Rolan PE, Wootton R, Posner J, Bellingham AJ. Tucaresol increases oxygen affinity and reduces haemolysis in subjects with sickle cell anaemia. Br J Haematol. 1996;93(4):817–21. [DOI] [PubMed] [Google Scholar]
- [116].Keidan AJ, Franklin IM, White RD, Joy M, Huehns ER, Stuart J. Effect of BW12C on oxygen affinity of haemoglobin in sickle-cell disease. Lancet 1986;1(8485):831–4. [DOI] [PubMed] [Google Scholar]
- [117].Fitzharris P, McLean AE, Sparks RG, Weatherley BC, White RD, Wootton R. The effects in volunteers of BW12C, a compound designed to left-shift the blood-oxygen saturation curve. Br J Clin Pharmacol. 1985;19(4):471–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Rolan PE, Mercer AJ, Wootton R, Posner J. Pharmacokinetics and pharmacodynamics of tucaresol, an antisickling agent, in healthy volunteers. Br J Clin Pharmacol. 1995;39(4):375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Zhang C, Li X, Lian L, Chen Q, Abdulmalik O, Vassilev V, Lai C-S, Asakura T. Anti-sickling effect of MX-1520, a prodrug of vanillin: an in vivo study using rodents. Br J Hematol. 2004;125(6):788–95. [DOI] [PubMed] [Google Scholar]
- [120].Rosatella AA, Simoenov SP, Frade RFM, Afonso CAM. 5-Hydroxymethylfurfural (HMF) as a building block platform: Biological properties, synthesis and synthetic applications. Green Chem. 2011;13:754–793. [Google Scholar]
- [121].van Putten R-J, van der Waal JC, de Jong E, Rasrendra CB, Heeres HJ, de Vries JG. Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem Rev. 2013;113(3):1499–597. [DOI] [PubMed] [Google Scholar]
- [122].Safo MK. Anti-sickling agents. US7119208B2. 2006. [Google Scholar]
- [123].Safo M. Anti-sickling agents. US20040157801A1. 2004. [Google Scholar]
- [124].Testing status of 5-(Hydroxymethyl)-2-furfural M950006. (accessed June 9, 2021) Available from: https://ntp.niehs.nih.gov/whatwestudy/testpgm/status/ts-m950006.html?utm_source=direct&utm_medium=prod&utm_campaign=ntpgolinks&utm_term=ts-m950006.
- [125].Stern W, Mathews D, McKew J, Shen X, Kato GJ. A phase 1, first-in-man, dose-response study of Aes-103 (5-HMF), an anti-sickling, allosteric modifier of hemoglobin oxygen affinity in healthy norman volunteers. Blood 2012;120(21):3210–3210. [Google Scholar]
- [126].Ataga KI, Desai PC. Advances in new drug therapies for the management of sickle cell disease. Expert Opin Orphan Drugs 2018;6(5):329–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Safo MK. Prodrug and protected forms of 5-hydroxymethylfurfuranal (5-HMF) and its derivatives. US10875834B2. 2020. [Google Scholar]
- [128].Safo MK. Prodrug and protected forms of 5-hydroxymethylfurfuranal (5-hmf) and its derivatives. WO2018018035A1. 2018. [Google Scholar]
- [129].Omar AME. Metabolically stable 5-HMF derivatives for the treatment of hypoxia. US10836729B1. 2020. [Google Scholar]
- [130].Hassan W, Beuzard Y, North ML, Rosa J. Functional studies of the double mutant hemoglobin Stanleyville II/S alpha2 78 Lys beta2 6 Val : identification of a site of intermolecular contact on the alpha chain. Biochem Biophys Res Commun. 1977;77(3):1108–17. [DOI] [PubMed] [Google Scholar]
- [131].Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 1985;65(1):183–9. [PubMed] [Google Scholar]
- [132].Sudha R, Anantharaman L, Sivaram MVS, Mirsamadi N, Choudhury D, Lohiya NK, et al. Linkage of interactions in sickle hemoglobin fiber assembly: Inhibitory effect emanating from mutations in the AB region of the α-chain is annulled by a mutation at its EF corner. J Biol Chem. 2004;279(19):20018–27. [DOI] [PubMed] [Google Scholar]
- [133].Safo M Ester nitrates derivatives of aromatic aldehydes with multiple pharmalogic properties to treat sickle cell disease. US10858331B1. 2020. [Google Scholar]
- [134].Safo M. Ester nitrates derivatives of aromatic aldehydes with multiple pharmalogic properties to treat sickle cell disease. US20210053931A1. 2021. [Google Scholar]
- [135].Pagare P. Rational design of allosteric modulators of hemoglobin as dual acting antisickling agents. Theses and Dissertations 2018. [Google Scholar]
- [136].Safo MK. Aromatic aldehydes with sustained and enhanced in vitro and in vivo pharmacologic activity to treat sickle cell disease. US20210002225A1. 2021. [Google Scholar]
- [137].Rolan PE, Parker JE, Gray SJ, Weatherley BC, Ingram J, Leavens W, et al. The pharmacokinetics, tolerability and pharmacodynamics of tucaresol (589C80; 4[2-formyl-3-hydroxyphenoxymethyl] benzoic acid), a potential anti-sickling agent, following oral administration to healthy subjects. Br J Clin Pharmacol. 1993;35(4):419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Hutchaleelaha A, Patel M, Washington C, Siu V, Allen E, Oksenberg D, et al. Pharmacokinetics and pharmacodynamics of voxelotor (GBT440) in healthy adults and patients with sickle cell disease. Br J Clin Pharmacol. 2019;85(6):1290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Rhodes J. Discovery of immunopotentiatory drugs: current and future strategies. Clin Exp Immunol. 2002;130(3):363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Li Z. Bicyclic heteroaryl compounds and uses thereof for the modulation of hemoglobin. US9802900B2. 2017. [Google Scholar]
- [141].Metcalf BW. Compounds and uses thereof for the modulation of hemoglobin. US10017491B2. 2018. [Google Scholar]
- [142].Metcalf BW. Compounds and uses thereof for the modulation of hemoglobin. US9957250B2. 2018. [Google Scholar]
- [143].Xu Q. Compounds and uses thereof for the modulation of hemoglobin. US10450269B1. 2019. [Google Scholar]
- [144].Li Z. Compounds and uses thereof for the modulation of hemoglobin. US10266551B2. 2019. [Google Scholar]
- [145].Metcalf B. Substituted benzaldehyde compounds and methods for their use in increasing tissue oxygenation. US10806733B2. 2020. [Google Scholar]
- [146].Xu Q. Compounds and uses thereof for the modulation of hemoglobin. US10858317B2. 2020. [Google Scholar]
- [147].Metcalf B. Substituted heteroaryl aldehyde compounds and methods for their use in increasing tissue oxygenation. US10822326B2. 2020. [Google Scholar]
- [148].Patel M, Cabrales P, Dufu K, Metcalf B, Sinha U. GTx011, an anti-sickling compound, improves SS blood rheology by reduction of HbS polymerization via allosteric modulation of O2 affinity. Blood 2014;124(21):1370–1370. [Google Scholar]
- [149].Sinha U. Compositions and methods for the modulation of hemoglobin (s). WO2014150256A1. 2014. [Google Scholar]
- [150].Metcalf B. Substituted benzaldehyde compounds and methods for their use in increasing tissue oxygenation. US9018210B2. 2015. [Google Scholar]
- [151].Dufu K, Oksenberg D, Zhou C, Hutchaleelaha A, Archer DR. GTx011, a potent allosteric modifier of hemoglobin oxygen affinity, prevents RBC sickling in whole blood and prolongs RBC half-life in vivo in a murine model of sickle cell disease. Blood 2014;124(21):217–217. [Google Scholar]
- [152].Dufu K, Oksenberg D, Metcalf B, Sinha U. GTx011, a potent allosteric modifier of hemoglobin oxygen affinity, delays polymerization and prevents sickling. Blood 2013;122(21):316–316. [Google Scholar]
- [153].Ramos EL. Dosing regimens for 2-hydroxy-6-((2-(1-isopropyl-1h-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde. WO2017096230A1. 2017. [Google Scholar]
- [154].Howard J, Hemmaway CJ, Telfer P, Layton DM, Porter J, Awogbade M, et al. A phase 1/2 ascending dose study and open-label extension study of voxelotor in patients with sickle cell disease. Blood 2019;133(17):1865–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Lehrer-Graiwer J, Yokoshima L, Tong B, Love TW. Accelerated approval of Oxbryta® (voxelotor): A case study on novel endpoint selection in sickle cell disease. Contemp Clin Trials. 2020;98:106161. [DOI] [PubMed] [Google Scholar]
- [156].Metcalf BW. 1:1 adducts of sickle hemoglobin. US9248199B2. 2016. [Google Scholar]
- [157].Metcalf BW. Modulators of hemoglobin. US11014884B2. 2021. [Google Scholar]
- [158].Jin B. Hemoglobin modifier compounds and uses thereof. US10787430B2. 2020. [Google Scholar]
- [159].Nakagawa A, Ferrari M, Schleifer G, Cooper MK, Liu C, Yu B, et al. A triazole disulfide compound increases the affinity of hemoglobin for oxygen and reduces the sickling of human sickle cells. Mol Pharm. 2018;15(5):1954–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Nakagawa A, Lui FE, Wassaf D, Yefidoff-Freedman R, Casalena D, Palmer MA, et al. Identification of a small molecule that increases hemoglobin oxygen affinity and reduces SS erythrocyte sickling. ACS Chem Biol. 2014;9(10):2318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Omar AM, Abdulmalik O, Ghatge MS, Muhammad YA, Paredes SD, El-Araby ME, et al. An investigation of structure-activity relationships of azolylacryloyl derivatives yielded potent and long-acting hemoglobin modulators for reversing erythrocyte sickling. Biomolecules 2020;10(11):1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Omar AM, David T, Pagare PP, Ghatge MS, Chen Q, Mehta A, et al. Structural modification of azolylacryloyl derivatives yields a novel class of covalent modifiers of hemoglobin as potential antisickling agents. Med Chem Comm. 2019;10:1900–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].M. Omar A, A. Mahran M, S. Ghatge M, Chowdhury N, A. Bamane FH, E. El-Araby M, et al. Identification of a novel class of covalent modifiers of hemoglobin as potential antisickling agents. Org Biomol Chem. 2015;13:6353–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Abraham DJ. Method of treating sickle cell anemia. US4887995A. 1989. [Google Scholar]
- [165].Safo MK. Azolylacryloyl derivatives as therapeutic agents for sickle cell disease. US10344001B2. 2019. [Google Scholar]
- [166].Zapol WM. Heteroaryl disulfide compounds as allosteric effectors for increasing the oxygen-binding affinity of hemoglobin. US10758569B2. 2020. [Google Scholar]
- [167].Gopalsamy A. Chemical compounds. US11014908B2. 2021. [Google Scholar]
- [168].Gopalsamy A, Aulabaugh AE, Barakat A, Beaumont KC, Cabral S, Canterbury DP, Casimiro-Garcia A, Chang JS, Chen MZ, Dow RL, et al. PF-07059013: A noncovalent modulator of hemoglobin for treatment of sickle cell disease. J Med Chem. 2021;64:326–42. [DOI] [PubMed] [Google Scholar]
- [169].Hutchaleelaha A, Patel M, Silva A, Oksenberg D, Metcalf B. GBT440 demonstrates high specificity for red blood cells in nonclinical species. Blood 2015;126(23):2172. [Google Scholar]