

Abstract
Chemically recyclable solid polymeric materials with commercializable properties only using CO2 and inexpensive bulk chemicals as chemical feedstock can open a brand-new avenue to economically viable, large-scale fixation of CO2 over a long period of time. Despite previous great advancements, development of such a kind of CO2-based polymers remains a long-term unsolved research challenge of great significance. Herein, we reported the first methodology to polymerize six-membered lactone with two substituents vicinal to the ester group (HL), a compound previously found to be non-polymerizable. The present methodology enables the first synthesis of chemically recyclable solid polyesters (polyHL) with a high CO2 content (28 wt %) and large molecular weights (Mn up to 613.8 kg mol−1). Transparent membranes with promising pressure-sensitive adhesive (PSA) properties comparable with their commercial counterparts can be conveniently fabricated from the polyesters. Mechanistic studies indicate that rigorous removal of water impurity is the key to the successful polymerization of the relatively inert disubstituted six-membered lactone. A complete monomer recovery from polyHL was also successfully achieved under mild catalytic conditions. The synthesis of polyHL only requires CO2 and two inexpensive bulk chemicals, H2 and 1,3-butadiene, as the starting materials, thus providing a new strategy for potential scalable chemical utilization of CO2 with desirable economic values and concomitant mitigation of CO2 emissions. This work should inspire future research to make useful new solid CO2-based polymers that can meaningfully increase the scale of chemical utilization of CO2 and promote the contribution of chemical utilization of CO2 to global mitigation of CO2 emissions.
Keywords: chemical recyclability, CO2 utilization, CO2-based polyester, ring-opening polymerization, circular economy
Graphical abstract
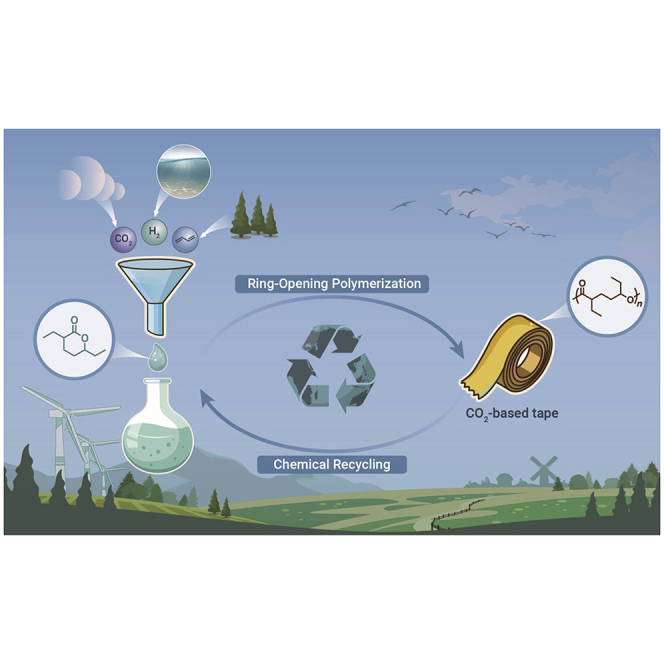
Public summary
-
•
CO2-based recyclable polymers are promising in reducing CO2 emission and pollution
-
•
Disubstituted δ-lactone, a previously non-polymerizable monomer, was polymerized
-
•
Complete monomer recovery was successfully achieved via chemical recycling process
-
•
CO2 constitutes 28% of the weight of newly designed chemically recyclable polymers
-
•
The polymers show pressure-sensitive adhesive property comparable to commercial tapes
Introduction
The scale of annual global production of synthetic polymers, mostly carbonaceous solid materials, is ca. 0.4 gigaton currently and is projected to reach ca. 1.2 gigaton in 2050.1 Utilization of CO2 as a main chemical feedstock to synthesize commercial solid polymers has great potentials in mitigation of CO2 emissions.2 To date, however, only ca. 0.001 gigaton of CO2 has been used as a chemical feedstock in the production of commercial CO2-based polymers, primarily polycarbonates and polyols.3,4 Thus, development of scalable new CO2-based polymers represents a significant challenge in the area of mitigating CO2 emissions.
We reason that three requirements should be met to achieve large-scale production of new CO2-based polymers. Firstly, the co-feedstock to synthesize the polymers should be inexpensive bulk chemicals in order to meet both economic and scalable requirements. Secondly, the polymers can also be conveniently processed into desirable solid shapes with commercializable properties. Finally, to address the aggravating pollution issues from increasing polymer wastes, the polymers should be recyclable. Unfortunately, CO2-based polymers satisfying all the three requirements, which can potentially fix large-scale CO2 in solid polymeric materials for a long period of time, are still unprecedented.
Polymers containing heteroatoms in their backbones,5, 6, 7 especially polyesters,8, 9, 10 featured with readily cleavable carboxylic ester backbone linkages, are excellent candidates for recycling. Therefore, making new polyesters from CO2 and cheap bulk chemicals as co-feedstocks, especially large-volume olefins, such as ethylene and 1,3-butadiene, that can be derived from biomass, has long been pursued over the past several decades.11, 12, 13, 14, 15, 16 Although ester-containing solid polyolefins from CO2 and 1,3-butadiene have been previously achieved in multiple-step fashions, the lack of processability, commercializable properties, and recyclability limits its potential large-scale utilization.14,15,17,18
Herein, we report the first successful synthesis of polyesters using only CO2 and cheap bulk chemicals as the co-feedstock (Figure 1). The synthesis was achieved via ring-opening polymerization (ROP) of an intermediate derived from CO2, H2, and 1,3-butadiene—a diethyl-substituted six-membered lactones (HL) that was previously thought to be non-polymerizable.16 Solid polyesters with high molecular weight (MW) (Mn up to 613.8 kg mol−1) and high CO2 content (28 wt %) were obtained. Remarkably, the polyesters can be conveniently processed into transparent and flexible membranes with pressure-sensitive adhesive (PSA) properties comparable with their commercial counterparts. A series of catalytic methods were also developed for clean chemical recycling of the polyesters back to their starting monomer HL.
Figure 1.

Synthesis of the first chemically recyclable polyester using only CO2 and cheap bulk chemicals as the starting materials
Results
Methodology development for the synthesis of polyesters from CO2, H2, and 1,3-butadiene
As shown in Figure 1, a two-step palladium-catalyzed procedure was used to synthesize HL from CO2, H2, and 1,3-butadiene according to the literature.19,20 The resultant HL is a 63/37 diastereomeric mixture according to various NMR spectroscopies (Figures S1–S4). To generate polyHL through ROP of HL, catalysts including tin(II)2-ethylhexanoate [Sn(Oct)2] and dibutyltin dilaurate (DBTDL) associated with benzyl alcohol (BnOH) were initially attempted at varied conditions ([HL]/[Cat.]/[BnOH] = 40/1/1 or 40/0.5/1 at different temperatures), but no polymer was obtained (Table S1, runs 1–4). Then diphenyl phosphate (DPP) was also employed, but no reaction was detected either (Table S1, runs 5–7, [HL]/[DPP]/[BnOH] = 30/1/1 at −25°C, 25°C, and 80°C in bulk for 24 h, respectively). Subsequently, organic bases, such as 1,8-diazabicyclo[5.4.0]undec-7-ene and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), were tested at a ratio of [HL]/[Cat.]/[BnOH] = 40/1/1 at 30°C for 96 h in tetrahydrofuran (THF) (Table S1, runs 8–13). Upon numerous experimental trials, we found that TBD/BnOH can catalyze the ROP of HL at room temperature in bulk or THF after 96 h, resulting in liquid polyHL with moderate conversion (Table S1, run 9: Conv. = 54%, Mn = 6,010 g mol−1, Ð = 1.14; run 10: Conv. = 37%, Mn = 5,596 g mol−1, Ð = 1.18, respectively).
Subsequently, we conjectured that increasing the basicity of the organocatalyst might promote the polymerization reactivity. Thus, three common phosphazene bases (PBs) in association with BnOH were investigated. For a 50/1 ratio of [HL]/[BnOH] with 1 mol % of tBu-P1 (tert-butyliminotris(dimethylamino)phosphorane) or tBu-P2 (1-tert-butyl-2,2,4,4,4-pentakis-(dimethylamino)-2λ5,4λ5-catenadi(phosphazene)) at −25°C in THF ([HL]0 = 5.3 M), no polymer was obtained for tBu-P1 after 72 h (Table 1, run 1), whereas a 41% conversion was observed for tBu-P2 after 120 h (Table 1, run 2; Mn = 5,304 g mol−1, Ð = 1.07). Encouragingly, the ROP process was dramatically improved when tBu-P4 (1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)phosphoranylide-namino]-2λ5,4λ5-catenadi(phosphazene)) was employed, leading to 87% conversion after 12 h (Table 1, run 3). Corresponding polyHL with Mn = 19,880 g mol−1 and moderate Ð = 1.90 was obtained. The difference in catalytic reactivities might be a consequence of the large basic differences among the three PBs21 (pKa = 26.9, 33.5, and 42.7 in acetonitrile22 for tBu-P1, tBu-P2, and tBu-P4, respectively). Next, altering the tBu-P4 loading from 2 mol % to 0.2 mol % led to a much more controlled polymerization (Table 1, runs 4–7). Particularly, polyHL with Mn = 9,154 g mol−1 and a much narrower Ð = 1.09 was obtained when the tBu-P4 loading was reduced to 0.2 mol % (Table 1, run 7), suggesting a living polymerization behavior. However, as the system was gradually diluted ([HL]0 = 2.0, 1.6 and 1.3 M in THF), the ROP of HL became less controlled: the conversion and Mn were significantly decreased, along with a broader Ð (Table 1, runs 8–10). Besides, elevating the reaction temperature from −25°C to 41°C also led to a less controlled polymerization (Table 1, runs 11–13). It should be emphasized that very careful drying of all reaction agents is a prerequisite for the success of the ROP methodology in our hands. In the absence of drying, no polymerization was detected even using highly active catalysts, including PBs (Table S11, run 1, condition: [HL]/[tBu-P4]/[BnOH] = 40/1/0, [HL]0 = 5.3 M in THF at −25°C for 12 h).
Table 1.
Results of ROP of HL by phosphazene bases/BnOH systems
| Run | HL/Catalyst/BnOH | Catalyst | Temp. (°C) | Time (h) | [HL]0 (mol L−1) | Conva. (%) | Mn, calb (g mol−1) | Mnc (g mol−1) | Đc |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 50/0.5/1 | tBu-P1 | −25 | 72 | 5.3 | ND | – | – | – |
| 2 | 50/0.5/1 | tBu-P2 | −25 | 120 | 5.3 | 41 | 3,308 | 5,304 | 1.07 |
| 3 | 50/0.5/1 | tBu-P4 | −25 | 12 | 5.3 | 87 | 6,899 | 19,880 | 1.90 |
| 4 | 50/1/1 | tBu-P4 | −25 | 12 | 5.3 | 88 | 6,977 | 18,921 | 1.69 |
| 5 | 50/0.25/1 | tBu-P4 | −25 | 12 | 5.3 | 88 | 6,977 | 13,116 | 1.35 |
| 6 | 50/0.2/1 | tBu-P4 | −25 | 12 | 5.3 | 88 | 6,977 | 12,885 | 1.23 |
| 7 | 50/0.1/1 | tBu-P4 | −25 | 8 | 5.3 | 88 | 6,977 | 9,154 | 1.09 |
| 8 | 50/0.1/1 | tBu-P4 | −25 | 12 | 2.0 | 67 | 5,338 | 6,888 | 1.74 |
| 9 | 50/0.1/1 | tBu-P4 | −25 | 12 | 1.6 | 58 | 4,635 | 6,834 | 1.39 |
| 10 | 50/0.1/1 | tBu-P4 | −25 | 12 | 1.3 | 37 | 2,996 | 4,678 | 1.60 |
| 11 | 50/0.1/1 | tBu-P4 | −9 | 12 | 5.3 | 82 | 6,509 | 10,290 | 1.64 |
| 12 | 50/0.1/1 | tBu-P4 | 28 | 12 | 5.3 | 65 | 5,182 | 5,726 | 2.29 |
| 13 | 50/0.1/1 | tBu-P4 | 41 | 12 | 5.3 | 52 | 4,167 | 5,479 | 2.18 |
| 14 | 25/0.1/1 | tBu-P4 | −25 | 12 | 5.3 | 87 | 3,504 | 4,127 | 1.11 |
| 15 | 100/0.2/1 | tBu-P4 | −25 | 12 | 5.3 | 88 | 13,846 | 19,601 | 1.08 |

Conditions: HL = 0.104 g, (0.67 mmol) in THF; HL were added to a tBu-P4/BnOH mixture.
Monomer conversion were measured by 1H NMR.
Mn,cal = ([HL]0/[BnOH]0) × Conv.% × MHL + MBnOH.
Mn and Đ were determined by GPC at 40°C in THF relative to PMMA standards.
To verify the living polymerization behavior, the ROP of HL at a feed ratio of [HL]/[tBu-P4]/[BnOH] = 50/0.1/1 at −25°C in THF was further investigated (Table S2). The polymerization kinetics data strongly support that the polymerization is living: the monomer conversion grows linearly with the reaction time (Figure 2A); the ln[M]0/[M] versus time plot showed a clear first-order kinetic character relative to the monomer concentration [M] (Figure 2B); a linear correlation of the Mn of polyHL with the monomer conversion was observed, and the dispersity Ð of the obtained polyHL remained low (Ð ∼ 1.1) during the chain propagation stage (Figure 2C). In addition, The GPC curves for polyHL obtained at varied times were all confirmed to be unimodal (Figure 2D). In addition, The Mn of the resultant polymers increased linearly with increased [HL]/[BnOH] ratio, and the Ð of the polymers remained low, which further confirmed the living polymerization behavior (Table 1, runs 14 and 15).
Figure 2.

ROP of HL catalyzed by tBu-P4/BnOH
(A) Plot of HL conversion versus time.
(B) First-order kinetic plots of ln([M]/[M]0) versus time.
(C) Plot of molecular weight (Mn) and Ð versus HL conversion.
(D) GPC traces of polyHL obtained at different polymerization time. [HL]/[tBu-P4]/[BnOH] = 50/0.1/1, [HL]0 = 5.3 M in THF, T = −25°C.
Matrix-assisted laser desorption ionization time of flight mass spectrometer (MALDI-TOF MS) was carried out to interrogate the chain-end fidelity of the resultant polyHL produced at a feed ratio of [HL]/[tBu-P4]/[BnOH] = 25/0.1/1 at −25°C in THF. The MS signals of polyHL (Table 1, run 14) showed only one group of molecular ion peaks with the same spacing of 156.1 g mol−1 (Mn = n∙156.1 + 131.5), which could be assigned to the linear-polyHL initiated by BnOH (Figure 3A). The NMR spectroscopies showed that both the methylene signal of BnO− (δ 5.12 ppm) and methine signal (δ 3.49 ppm) next to the hydroxyl end group were observed in the 1H NMR spectrum, and corresponding 13C NMR signals were also detected (Figures 3B and S5).
Figure 3.

Structural analyses of polyHL obtained by tBu-P4 and BnOH at a ratio of 0.1/1
(A) MALDI-TOF mass spectrum of the resulting polyHL shows only one group of molecular ion peaks, and the enlarged MALDI-TOF mass spectrum confirms that the molecular ion peaks correspond to the BnOH end-capped product.
(B) 1H NMR spectrum of linear-polyHL end-capped with BnOH.
Inspired by the excellent chain-end fidelity of polyHL sample, telechelic polyHL catalyzed by tBu-P4 and 1,4-benzenedimethanol (1,4-BDM) was obtained at a feed ratio of [HL]/[tBu-P4]/[1,4-BDM] = 15/0.15/1. MALDI-TOF MS signals showed only one group of molecular ion peaks (Mn = n∙156.1 + 161.1), which perfectly matched the expected telechelic polymer structure (Figure S6).
Mechanistic studies for the ROP of HL to polyHL
To investigate the possible mechanism in our system, ROPs at a feed ratio of [HL]/[BnOH] = 50/1 with varied loadings of tBu-P4 (2, 1, 0.5, 0.4, and 0.2 mol %; Table 1, runs 3–7) were performed. Notably, the Mn of the polyHL produced increased significantly as the loading of tBu-P4 increased, and the trends of bimodal distribution of the GPC traces have become more apparent as the tBu-P4 loading increased, which might be due to the existence of other competitive initiation mechanisms (Figure S7). MALDI-TOF spectrum of low MW analogs catalyzed by 1 mol % of tBu-P4 disclosed three sets of molecular ion peaks with the same MW spacing of 156 g mol−1. The signals were assignable to BnOH end-capped, with no chain end, and water end-capped polyHL (Figure S8), respectively. As the tBu-P4 loading increased to 2 mol % relative to HL, the major signal set changed from BnOH chain end to no chain end, and the observed MW increased significantly, and the signals of the oligomers initiated by residual water became negligible (Figure S9).
We next investigated the feasibility for PBs to directly catalyze the ROP of HL without the addition of any alcohol initiator. The ROP reactions were carried out at −25°C in THF with 2 mol % of tBu-P1, tBu-P2, and tBu-P4 (Table S3, run 1–3). After 12 h of reaction, no polymer was generated for tBu-P1 and tBu-P2. To our surprise, maximum conversion of 88% was achieved for tBu-P4 after 12 h and a solid polyHL sample with unexpected ultrahigh MW of Mn = 613.8 kg mol−1 and moderate Ð = 1.45 was obtained (Table S3, run 3). Adjusting the concentrations of phosphazene base only had small effects on Mn and Ð of the polyHL samples (Table S3, runs 4–6).
To test the controllability of the ROP of HL catalyzed by tBu-P4 alone, kinetic experiments were performed at −25°C in THF ([HL]/[tBu-P4] = 50/1, [M]0 = 4.0 M) (Figure S10; Table S4). The ln[M]0/[M] versus time plot revealed that the monomer conversion achieved up to 63% in 4 h, then the polymerization rate tended to slow down and the conversion reached 84% in the subsequent 6–8 h (Figures S10A and S10B). Notably, the Mn of polyHL has a clear linear correlation with the monomer conversion throughout the polymerization process, but the dispersity Ð significantly broadened after half monomer conversion (Figure S10C). The GPC curves also exhibited a gradually emerging bimodal distribution, presumably due to inevitable transesterification reactions at higher monomer conversion (Figure S10D). These data indicated that relatively controlled ROP manners could be realized at which the reactions were quenched within the first 4 h.
MALDI-TOF spectrum for polyHL generated by tBu-P4 alone only showed one set of mass peaks assignable to cyclic-polyHL mass peak (Figure S11). No characteristic signals of chain end could be seen in the 1H and 13C NMR spectra either, further supporting the idea that the polymer obtained might have cyclic components (Figures S12 and S13). Notably, high abundance of water-initiated signals could only be detected by MALDI-TOF when the residual water was deliberately reserved in the ROP system (Figure S14).
The topologies of putative cyclic- and linear-polyHLs were further studied by intrinsic viscosity measurements. According to the Mark-Houwink plot (Figure S15), the ratio of [η]cyclic/[η]linear was estimated to be 0.79, which is slightly higher than the theoretically predicted value.10,23 This phenomenon might be attributed to the presence of minor linear-polyHL in the putative cyclic products. Besides, the Mark-Houwink exponent α values were 0.70 for cyclic-polyHL and 0.72 for linear-polyHL, suggesting that both polymers were random coils in THF solution.
Plausible chain initiation mechanisms for ROP of HL catalyzed by tBu-P4/BnOH and tBu-P4 alone were studied through NMR spectroscopy. Monitoring the stoichiometric reaction between tBu-P4 and BnOH at RT in 1H NMR exhibited the formation of complex [tBu-P4H+···OBn]. The disappearance of the hydroxyl proton signal at δ 0.92 ppm and chemical shift changes of tBu-P4 and BnOH were also observed (Figure S16). In addition, in situ NMR tests were performed to gain further mechanistic evidence. By lowering the ratio of HL/BnOH to 1/1, the dominant product observed is consistent with the ring-opened intermediate generated in the initiation step (Figure 5, path 2, and Figure S17). In contrast, only a trace amount of BnO− was observed (Figure S17). These results suggest that chain propagation is slower than the initiation step, thus leading to the accumulation of the initial intermediate with only one repeat unit. The disfavored ring-opening steps might be attributable to the increased steric hindrance surrounding the proposed propagating secondary alkoxides relative to the initiating primary BnO−.
Figure 5.

A proposed multiple competitive initiating mechanism for the ROP of HL to polyHL
Subsequently, the ability of tBu-P4 to extract an acidic H from HL was also verified. HL and tBu-P4 were mixed at ratios of 1/1, 2/1, 4/1, and 8/1, respectively, in a J.-Young tube with added toluene-d8 at RT (no polymerization occurred under these conditions), and corresponding 1H and 31P NMR spectra were taken after sufficient oscillations (Figures S18–S20). Characteristic signals of [tBu-P4H]+ (δ 7.92–8.07 ppm 1H NMR; δ 12.48 ppm and −23.64 ppm in 31P NMR) were clearly observed. However, as the HL/tBu-P4 feed ratio increased, the intensity of the [tBu-P4H]+ signals increased slightly. Meanwhile, the characteristic signals of tBu-P4 (δ 1.72, 2.70, and 2.72 ppm in 1H NMR; δ 4.96 and −25.31 ppm in 31P NMR) remained unchanged; that is, tBu-P4 could not be fully consumed even in the presence of a great excess of HL. These data clearly indicated that tBu-P4 was only able to deprotonate a tiny portion of HL, which might account for the lack of direct correlation between tBu-P4 concentration and the Mn of cyclic-polyHL produced in this system (vide supra).
Density functional theory (DFT) calculations indicate that deprotonation of the acidic HL monomer at the α position to generate the [>C−-C(O)-O-R] carbanion species is preferred over δ-H abstracted alkyl anion [>C−-O-C(O)-R] (Table S13; Figure S21). The free energy for the formation of α-H abstracted acylated anion is 26.5 kcal mol−1 less than that of the formation of δ-H abstracted alkyl anion in THF. We also performed quantum-mechanical calculations to evaluate the Gibbs free energy for proton abstraction from BnOH, HL, and H2O by tBu-P4 (see supplemental information and Figure S22). The Gibbs free energy for the deprotonations follows the order of BnOH (3.9 kcal mol−1) < HL (5.7 kcal mol−1) < H2O (9.3 kcal mol−1). Such order suggests that BnO− is the most readily generated active initiating species, while deprotonation of HL is slightly disfavored.
The feasibility of BnO− being the active initiating species was further supported by the observation that alkali metal alkoxides, such as KOMe, NaOMe, KOEt, NaOEt, KOtBu, and NaOtBu, could also initiate the ROP of HL at −25°C, giving solid polyHL with a MW of up to 475.7 kg mol−1 and narrow dispersity Ð (see in Table S5). It is also worth mentioning that the ROPs initiated by alkali metal alkoxides were less rapid compared with tBu-P4 catalyst, which might be attributed to the observed poor solubility of the alkali metal alkoxides in the reactant mixture.
The presence of water impurity can presumably interfere with the above-mentioned process. HL with approximately 100 ppm water was prepared for the ROP with tBu-P4/BnOH at a ratio of [HL]/[tBu-P4]/[BnOH] = 100/1/1 at −25°C for 12 h, [HL]0 = 5.3 M in THF (Table S12, run 2). Intriguingly, the HL conversion dramatically dropped to 21%, and the polyHL produced had a lower MW of Mn = 17,938 g mol−1 with a greatly broadening dispersity of Ð = 2.258.
Physical properties of polyHL
We next investigated the physical properties of polyHL. The thermostability of polyHL produced by tBu-P4 and tBu-P4/BnOH systems were also analyzed through thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC). Both the linear- and cyclic-polyHL exhibited high thermal stability (Td,5% > 325°C). The TGA and derivative thermogravimetry (DTG) curves of cyclic-product showed Td,5% = 332.3°C and Tmax = 367.3°C (Figure S23), which were 6°C and 12°C higher than those of the linear polymer with a similar MW (Figure S24). These results are consistent with conclusions from another study that the thermal stability of cyclic polymers are generally higher than that of their linear analogs.9,10,24,25 The DSC curves for the second heating scan curves (5°C min−1) of two polyHL specimens displayed two similar glass-transition temperatures (Tg) of −29.7°C and −30.6°C for cyclic- and linear-polyHL, respectively, and no crystalline peaks were observed (Figures S25 and S26). All the data confirmed that both the cyclic- and linear-polyHL are amorphous polymeric materials with excellent thermal stability.
Broadly speaking, polyHL is a new type of polyhydroxyalkanoate. Previous polyhydroxyalkanoates, such as poly(γ-butyrolactone), poly(δ-valerolactone), poly(ε-valerolactone), and poly(lactic acid), have attracted widespread attention due to their degradability. However, their drawbacks, such as relatively high brittleness and low impact strength9,26,27 limited their potential applications. In contrast, polyHL has relatively high flexibility, which provides new opportunities in applications, such as PSAs and polyester polyols used to produce polyurethanes.
The ability to obtain high MW solid polymers via the ROP of HL by tBu-P4 alone provides a promising avenue to obtain PSAs with potential utilities. A simple 180° peel test was carried out to measure the peel strength of polyHL. Cyclic samples were employed here because our current synthetic methodology can give much higher MW for cyclic-polyHL than a linear one. Glass slides were utilized as the rigid substrate, and a sheet of A4 paper (15 × 2.6 cm) was used as the face substrate (Figure 4A). The polyHL samples were evenly coated on the glass slides by using a coating blade. Cross-sectional scanning electron microscope (SEM) showed that the thin film had a fair uniform thickness of 36.9 ± 1.2 μm (Figure 4B). The test was performed at 25°C on an Instron 5966 universal testing instrument at 180° peel angle at a rate of 10 mm min−1. To our delight, polyHL_319, polyHL_562, and polyHL_160 (the numbers refer to the sample with an Mn of 319, 562, and 160 kg mol−1, respectively) exhibited adhesion with peel strength of 3.8 ± 0.12, 3.5 ± 0.20, and 1.5 ± 0.65 N cm−1, respectively (Figure 4C; Table S6). The peel strength of polyHL_319 and polyHL_562 were relatively higher than those of commercialized 3M Scotch tapes tested under the same test conditions (3M 665, 2.4 ± 0.40 N cm−1; 3M 810, 1.9 ± 0.31 N cm−1) and vinyl electrical tape (3M 1,600, 0.8 ± 0.17 N cm−1). PolyHL_160 exhibited a peel strength of 1.5 ± 0.65 N cm−1, comparable with that of the 3M 810 Scotch tape. In addition, a high MW polymer sample (Mn = 613.8 kg mol−1, Ð = 1.45; Table S3, run 3) was solvent-cast into a PTFE mold to form a transparent and colorless polymer film with good flexibility and viscoelasticity (Figure 4D).
Figure 4.

The pressure-sensitive adhesive properties of high MW cyclic-polyHL and images of a polyHL thin film
(A) Diagram of the 180° peel test.
(B) Cross-sectional SEM image of the polyHL thin film to determine the average thickness.
(C) Results of the 180° peel test: the peel strength of polyHL samples with varied MW (red) and three commercialized tapes (blue) are shown.
(D) Images of the polyHL thin film to show that the film is colorless with excellent light permeability.
Development of methodology for chemical recycling of polyHL to HL
Van ’t Hoff analysis was performed to calculate the thermodynamic parameters of the polymerization (Figures S27 and S28; Tables S7–S9). According to Dainton’s equation,28 the change in enthalpy and entropy were calculated to be −13.12 kJ mol−1 and −49.09 J mol−1 K−1, respectively, which further gave a Tc of −6°C in THF at [HL]0 = 1.0 mol L−1.
To test the chemical recyclability, cyclic-polyHL with Mn in the range of 300–400 kg mol−1 were employed. Initially, several trifluoromesylate metal salts, including AgCF3SO3, Cu(CF3SO3)2, Fe(CF3SO3)3, Sc(CF3SO3)3, and Y(CF3SO3)3 were employed to catalyze the depolymerization of polyHL in a sealed tube in toluene ([HL]0 = 0.5 M) at 120°C for 24 h (Table S10, runs 1–5). However, only Fe(CF3SO3)3 and Sc(CF3SO3)3 gave 53% and 27% recovery of HL monomer, respectively (Table S10, runs 3 and 4). FeCl2, Fe(acac)2, Sn(Oct)2, DBTDL, and tBu-P4 exhibited no obvious reactivity even at a higher temperature of 150°C in mesitylene for 12 h (Table S10, runs 6–10). We next examined ZnCl2 in toluene at 130°C, 140°C, and 150°C for 12 h, respectively, and found that the monomer conversion increased with an elevated temperature (Table S10, runs 11–13, 31%, 39%, and 54%, respectively). Particularly, when employing more polar 1,2-dichlorobenzene (o-DCB) at 150°C and 160°C, the monomer recovery significantly increased to 91% and 100%, respectively (Table S10, runs 14 and 15), thus achieving a complete chemical recycling procedure by zinc chloride catalyst.
To further cut down the energy input during the chemical recycling process, La (La[N(SiMe3)2]3) were also tested. At 50°C with [HL]0 = 0.5 M in toluene, excellent HL recovery was detected: 47% in 3 h, 81% in 12 h, and 88% in 24 h (Table S11, runs 1–3), and no significant improvement of recovery rate was observed even in a system diluted to 0.1 M (Table S11, runs 4 and 5). To achieve a complete recovery of HL monomer, the temperature was elevated to 80°C for the reaction with [HL]0 = 0.5 M in toluene, the recovery of HL monomer reached 85% within 3 h and remained constant over 12 h (Table S11, runs 6 and 7), indicating a much more rapid depolymerization process. When the system was diluted to 0.1 M at 80°C, 93% recovery of HL monomer was achieved in 3 h (Table S11, run 8), and 100% recovery was achieved in 12 h (Figure S29; Table S11, run 9). Finally, the validity of our catalytic method to chemically recycle linear-polyHL was confirmed with ZnCl2, giving 100% recovery of HL in o-DCB at 160°C in 12 h (Figure S30).
Discussion
Our mechanistic studies may rationalize why HL was observed to be non-polymerizable previously.16 Firstly, for the initiation step, our data support a multiple competitive initiating mechanism (Figure 5), including (path 1) direct abstraction of the proton from the C-H bond vicinal to the carbonyl group of HL (the most acidic proton in the lactone, Figure S21) to generate highly reactive species, (path 2) hydroxyl deprotonation of alcohols to form alkoxides, and (path 3) deprotonation of residual water to form hydroxide. Compared with lactones without a substituent at the carbon vicinal to the carbonyl group, the ethyl substituent at the same carbon lowers the acidity of the C-H bond and also sterically hinders the approach of bulky bases, especially tBu-P4, thus disfavoring the polymerization process through path 1. Secondly, for the subsequent nucleophilic ring-opening step, both ethyl substituents are vicinal to the ester group, which should sterically hinder the approach to the ester by any nucleophiles, thus increasing corresponding activation barriers no matter which initiating mechanism leads to the ring-opening nucleophile. The experimentally measured thermodynamic data for ROP of HL also clearly indicated that the two ethyl substituents of HL greatly increase the entropic penalty compared with non-/mono-substituted δ-lactones,28,29 thus resulting in significantly less thermodynamically favorable polymerization than the less-substituted counterparts (Table S9). Therefore, the ROPs of HL are both thermodynamically and kinetically challenging, requiring judiciously selected reaction conditions to allow for polymerization, including sub-zero temperatures, prolonged reaction times, and highly concentrated conditions (vide supra).
Our work subsequently showed that the polymerization of HL is highly sensitive to water impurity in the reaction system. In essence, the H2O-initiated oligomers contain a carboxylic acid end group, which should have a strong tendency to quench the active alkoxide species for the chain propagation process and form dormant species featured by a terminal carboxylate anion (Figure 5, path 3). Potassium benzoate and cesium formate were tested as potential ROP initiators under our optimized conditions (Table S12, run 3 and 4, T = −25°C, t = 24 h, [HL]0 = 5.0 M in THF). At elevated temperature T = 30°C, potassium acetate (KOAc) was also employed at a ratio of [HL]/[KOAc]/[BnOH] = 50/1/1 and 50/1/0 (Table S12, runs 5 and 6). However, no HL conversion was detected for all three species. These observations confirm that carboxylate is a dormant species. As such, rigorous drying is the key to the present success of ROP of HL, or the formation of carboxylate would inhibit the reaction.
Conclusion
In conclusion, we have developed the first methodology to polymerize six-membered lactone with two substituents vicinal to the ester group, which enables the first synthesis of chemically recyclable solid polyesters with a high CO2 content, high MW, and promising adhesive properties comparable with commercial counterparts by only using CO2 and bulk chemicals as the starting materials. This work provides a brand-new avenue to potential large-scale utilization of CO2 as a main chemical feedstock, while the chemical recyclability may allow for fixation of CO2 in solid polymeric materials over a long period of time.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (no. U2032132). We thank the Analytical Instrumentation Center (AIC) of the School of Physical Science and Technology, ShanghaiTech University, for their technical support. We thank Conger Li of ShanghaiTech University for the SEM test of polyHL samples. We thank Dr. Hua Liu of AIC, ShanghaiTech University, for technical support in in situ NMR tests, and Dr. Wenbin Yao of Dow Chemical Company for helpful discussions about polyols.
Author contributions
B.L. conceived the project and analyzed the data. Y.L. performed experiments, analyzed the data, and drafted the manuscript. L.X. conducted the DFT calculations and wrote sections of the manuscript. N.G. and Y.-S. performed part of the syntheses of the monomer and samples. All authors discussed the experimental and theoretical results and commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Published Online: February 5, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xinn.2022.100216.
Lead contact website
https://spst.shanghaitech.edu.cn/spst_en/2018/0301/c2939a51331/page.htm.
Supplemental information
References
- 1.Geyer R., Jambeck J.R., Law K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017;3:e1700782. doi: 10.1126/sciadv.1700782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meys R., Kätelhön A., Bachmann M., et al. Achieving net-zero greenhouse gas emission plastics by a circular carbon economy. Science. 2021;374:71–76. doi: 10.1126/science.abg9853. [DOI] [PubMed] [Google Scholar]
- 3.Muthuraj R., Mekonnen T. Recent progress in carbon dioxide (CO2) as feedstock for sustainable materials development: co-polymers and polymer blends. Polymer. 2018;145:348–373. [Google Scholar]
- 4.Michael C. Carbon dioxide (CO2) as chemical feedstock for polymers – already nearly 1 million tonnes production capacity installed! Nova Institute. 2021. http://nova-institute.eu/press/?id=236
- 5.Brooks A.A., Snyder R.L., Coates G.W. Chemically recyclable thermoplastics from reversible-deactivation polymerization of cyclic acetals. Science. 2021;373:783–789. doi: 10.1126/science.abh0626. [DOI] [PubMed] [Google Scholar]
- 6.Shi C., McGraw M.L., Li Z.-C., et al. High-performance pan-tactic polythioesters with intrinsic crystallinity and chemical recyclability. Sci. Adv. 2020;6:eabc0495. doi: 10.1126/sciadv.abc0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan J., Xiong W., Zhou X., et al. 4-Hydroxyproline-derived sustainable polythioesters: controlled ring-opening polymerization, complete recyclability, and facile functionalization. J. Am. Chem. Soc. 2019;141:4928–4935. doi: 10.1021/jacs.9b00031. [DOI] [PubMed] [Google Scholar]
- 8.Shi C., Li Z.-C., Caporaso L., et al. Hybrid monomer design for unifying conflicting polymerizability, recyclability, and performance properties. Chem. 2021;7:670–685. [Google Scholar]
- 9.Zhu J.B., Watson E.M., Tang J., et al. A synthetic polymer system with repeatable chemical recyclability. Science. 2018;360:398–403. doi: 10.1126/science.aar5498. [DOI] [PubMed] [Google Scholar]
- 10.Hong M., Chen E.Y.X. Completely recyclable biopolymers with linear and cyclic topologies via ring-opening polymerization of gamma-butyrolactone. Nat. Chem. 2016;8:42–49. doi: 10.1038/nchem.2391. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki Y., Inoue Y., Hashimoto H. Reaction of carbon dioxide with butadiene catalysed by palladium complexes. Synthesis of 2-ethylidenehept-5-en-4-olide. J. Chem. Soc. Chem. Commun. 1976;15:605–606. [Google Scholar]
- 12.Haack V., Dinjus E., Pitter S. Synthesis of polymers with an intact lactone ring structure in the main chain. Die Angew. Makromol. Chem. 1998;257:19–22. [Google Scholar]
- 13.Price C.J., Reich B.J.E., Miller S.A. Thermodynamic and kinetic considerations in the copolymerization of ethylene and carbon dioxide. Macromolecules. 2006;39:2751–2756. [Google Scholar]
- 14.Nakano R., Ito S., Nozaki K. Copolymerization of carbon dioxide and butadiene via a lactone intermediate. Nat. Chem. 2014;6:325–331. doi: 10.1038/nchem.1882. [DOI] [PubMed] [Google Scholar]
- 15.Liu M., Sun Y., Liang Y., et al. Highly efficient synthesis of functionalizable polymers from a CO2/1,3-butadiene-derived lactone. ACS Macro Lett. 2017;6:1373–1378. doi: 10.1021/acsmacrolett.7b00774. [DOI] [PubMed] [Google Scholar]
- 16.Duparc V.H., Shakaroun R.M., Slawinsky M., et al. Ring-opening (co)polymerization of six-membered substituted δ-valerolactones with alkali metal alkoxides. Eur. Polym. J. 2020;134:109858. [Google Scholar]
- 17.Tang S., Zhao Y., Nozaki K. Accessing divergent main-chain-functionalized polyethylenes via copolymerization of ethylene with a CO2/butadiene-derived lactone. J. Am. Chem. Soc. 2021;143:17953–17957. doi: 10.1021/jacs.1c08578. [DOI] [PubMed] [Google Scholar]
- 18.Chen L., Li Y., Yue S., et al. Chemoselective RAFT polymerization of a trivinyl monomer derived from carbon dioxide and 1,3-butadiene: from linear to hyperbranched. Macromolecules. 2017;50:9598–9606. [Google Scholar]
- 19.Sharif M., Jackstell R., Dastgir S., et al. Efficient and selective palladium-catalyzed telomerization of 1,3-butadiene with carbon dioxide. ChemCatChem. 2017;9:542–546. [Google Scholar]
- 20.Behr A., Brehme V.A. Homogeneous and heterogeneous catalyzed three-step synthesis of 2-ethylheptanoic acid from carbon dioxide, butadiene and hydrogen. J. Mol. Cat. A Chem. 2002;187:69–80. [Google Scholar]
- 21.Liu S., Ren C., Zhao N., et al. Phosphazene bases as organocatalysts for ring-opening polymerization of cyclic esters. Macromol. Rapid Comm. 2018;39:1800485. doi: 10.1002/marc.201800485. [DOI] [PubMed] [Google Scholar]
- 22.Zhao N., Ren C., Li H., et al. Selective ring-opening polymerization of non-strained γ-butyrolactone catalyzed by a cyclic trimeric phosphazene base. Angew. Chem. Int. Ed. Engl. 2017;56:12987–12990. doi: 10.1002/anie.201707122. [DOI] [PubMed] [Google Scholar]
- 23.Roovers J. In: Cyclic Polymers. Semlyen J.A., editor. Springer; 2002. Organic cyclic polymers; pp. 347–384. [Google Scholar]
- 24.Kaitz J.A., Diesendruck C.E., Moore J.S. End group characterization of poly(phthalaldehyde): surprising discovery of a reversible, cationic macrocyclization mechanism. J. Am. Chem. Soc. 2013;135:12755–12761. doi: 10.1021/ja405628g. [DOI] [PubMed] [Google Scholar]
- 25.Hong M., Chen E.Y.X. Towards truly sustainable polymers: a metal-free recyclable polyester from biorenewable non-strained -butyrolactone. Angew. Chem. Int. Ed. Engl. 2016;55:4188–4193. doi: 10.1002/anie.201601092. [DOI] [PubMed] [Google Scholar]
- 26.Aubin M., Prud'homme R.E. Preparation and properties of poly(valerolactone) Polymer. 1981;22:1223–1226. [Google Scholar]
- 27.Domenek S., Fernandes-Nassar S., Ducruet V. In: Synthesis, Structure and Properties of Poly(lactic Acid) Di Lorenzo M.L., Androsch R., editors. Springer International Publishing; 2018. Rheology, mechanical properties, and barrier properties of poly(lactic acid) pp. 303–341. [Google Scholar]
- 28.Schneiderman D.K., Hillmyer M.A. Aliphatic polyester block polymer design. Macromolecules. 2016;49:2419–2428. [Google Scholar]
- 29.Olsen P., Odelius K., Albertsson A.C. Thermodynamic presynthetic considerations for ring-opening polymerization. Biomacromolecules. 2016;17:699–709. doi: 10.1021/acs.biomac.5b01698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.