

Abstract
Stem and stem-cell-derived cells have immense potential as a regenerative therapy for various degenerative diseases. DNA is the storehouse of genetic data in all cells, including stem cells, and its integrity is fundamental to its regenerative ability. Stem cells undergo rapid propagation in labs to achieve the necessary numbers for transplantation. Accelerated cell growth leads to the loss of DNA integrity by accumulated metabolites, such as reactive oxygen, carbonyl, and alkylating agents. Transplanting these cells would result in poor engraftment and regeneration of the deteriorating organ. Moreover, transplanting DNA-damaged cells leads to mutations, DNA instability, cellular senescence, and possibly, life-threatening diseases such as cancer. Therefore, there is an immediate need for a quality control method to evaluate the cell’s suitability for transplantation. Here, we provide step-by-step protocols for the assessment of the DNA integrity of stem cells prior to cell transplantation.
Keywords: DNA integrity, DNA damage, iPS cells, comet assay, γH2A.X, cardiomyocytes, cell transplantation
Introduction
Experimental and clinical evidence demonstrates that cell transplantation can moderately improve the left ventricle contractile performance of failing hearts1,2,3,4,5,6,7,8,9. Recent advances have opened further appealing opportunities for cardiovascular regeneration; these include the forced expression of reprogramming factors in somatic cells to induce pluripotency and differentiate these induced pluripotent stem cells (iPSC) into different cardiac lineages, importantly cardiomyocyte (CM)10,11,12,13. The hereditary material in every cell, including the artificially generated iPSCs and iPSC-derived CM (iPS-CM), is DNA. The genetic instructions stored in DNA dictates the growth, development, and function of cells, tissues, organs, and organisms. DNA is not inert; cell metabolites, such as reactive oxygen, carbonyl, and nitrogen species, and alkylating agents can cause DNA damage in vitro and in vivo14,15,16,17. Importantly, DNA damage occurs intuitively in every cell, with a significant frequency. If these damages are not corrected, it will lead to DNA mutation, cellular senescence, the loss of DNA and cell integrity, and possibly, diseases, including life-threatening cancers. Therefore, retaining DNA integrity is essential to any cell, especially iPSCs, that has enormous potential in the clinic.
For assessing the quantity and integrity of isolated genomic DNA, expensive equipment is available on the market. However, there are no simple and cost-effective methods to assess the DNA integrity in cells without isolating the cells. Moreover, user-induced DNA degradation during DNA isolation is one of the major drawbacks in using these methods. The single-cell gel electrophoresis (known as comet assay)18,19 and γH2A.X immunolabeling8 techniques are fundamental approaches in research labs for assessing DNA damage. These two methods do not require expensive equipment or isolated genomic DNA to analyze DNA integrity8,20,21. Since, these techniques have been performed with whole cells; user-induced DNA/RNA/protein degradation during the sample preparation will not affect these protocols. Here, to assess the DNA damage and DNA damage response in stem and stem-cell-derived cells, we provide step-by-step protocols to perform both the comet assay and γH2A.X immunolabeling. Moreover, combining these two approaches, we propose a naive assessment that can be used to evaluate the cell’s suitability for transplantation.
The comet assay, or single-cell gel electrophoresis, measures the DNA breaks in cells. Cells embedded in low-melting agarose are lysed to form nucleoids containing supercoiled DNA. Upon electrophoresis, small pieces of fragmented DNA and broken DNA strands migrate through the agarose pores, whereas the intact DNA, due to their enormous size and their conjugation with the matrix protein, will have a restricted migration. The pattern of stained DNA under a fluorescence microscope mimics a comet. The comet head contains intact DNA and the tail is composed of fragments and broken DNA strands. The fraction of DNA damage can be measured by the fluorescence intensity of the damaged DNA (comet tail) relative to the intact DNA (comet head) intensity. The parameter tail moment can be calculated as shown in Figure 1.
Figure 1: Schematic of the comet assay.
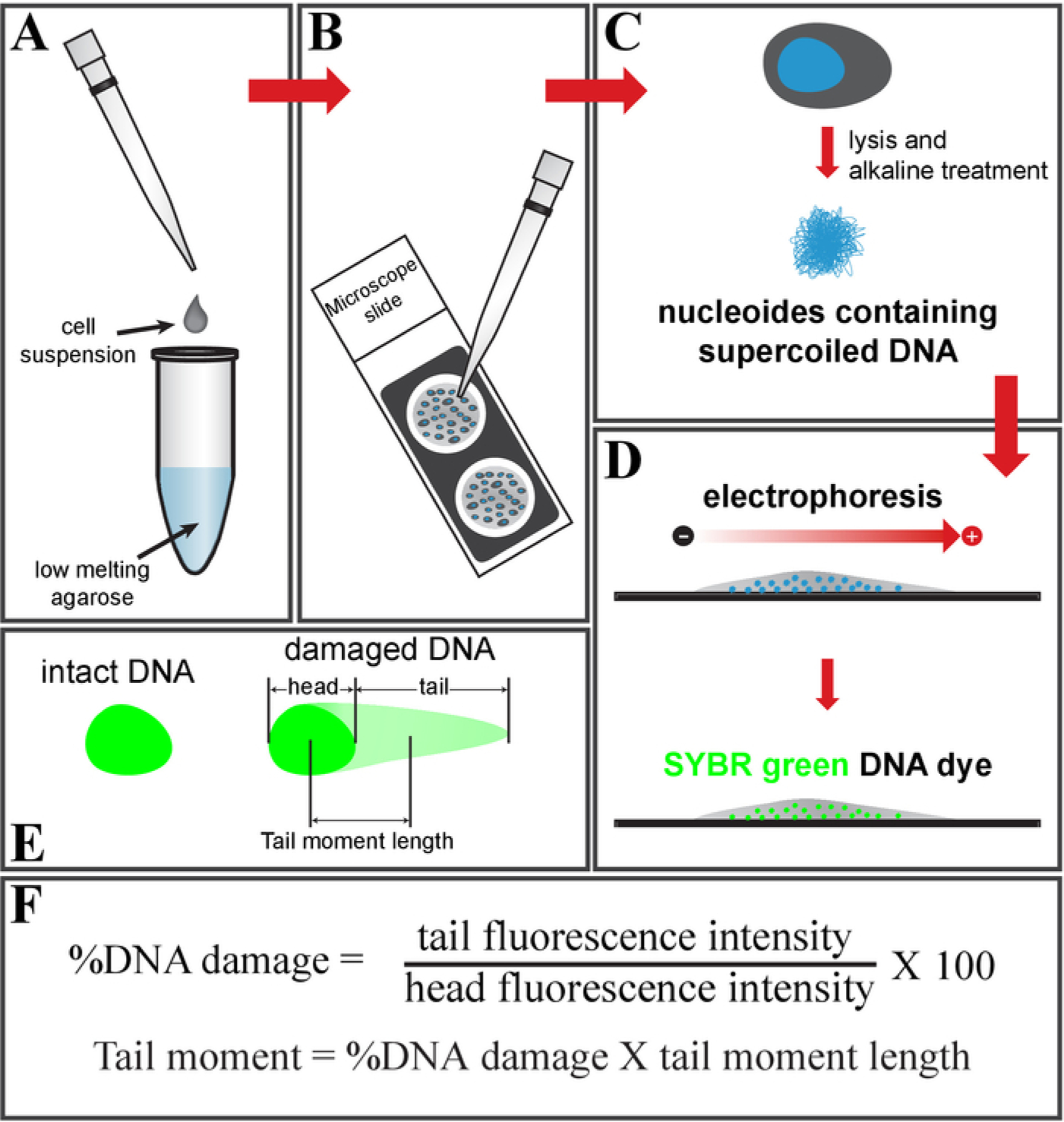
(A) Mix the cell suspension with low-melting-point agarose and (B) place it on a glass slide. (C) Treat it with cell lysis buffer, followed by an alkaline solution, to get nucleoids containing supercoiled DNA. (D) Electrophorese and stain the DNA using SYBR green DNA dye. (E) Schematic of intact (left) and damaged DNA (right, comet shape). (F) Formula to calculate a fraction of the DNA damage and tail moment.
DNA damage induces the phosphorylation of histone H2A.X (γH2A.X) at Ser139 by ATM, ATR, and DNA-PK kinases. The phosphorylation and recruitment of H2A.X at DNA strand breaks is called DNA damage response (DDR) and happens rapidly after DNA is damaged. Following this process, checkpoint-mediated cell cycle arrest and DNA repair processes are initiated. After the successful completion of DNA repair, γH2A.X is dephosphorylated and inactivated by phosphatases. Prolonged and multiple DNA strand breaks lead to the accumulation of γH2A.X foci in DNA. This indicates the cell’s inability to repair the DNA damage and the loss of DNA integrity. These γH2A.X foci in DNA can be identified by and the number of DDR foci can be counted using the protocol in section 2.
Protocol
1. Comet Assay
1. Reagent preparation
-
For low-melting-point agarose, place 500 mg of low-melting agarose in 100 mL of DNA-/RNA-free water. Heat the bottle in a microwave oven until the agarose dissolves. Place the bottle in a 37 °C water bath until needed.
NOTE: For further reading, see Azqueta et al., who studied the effects of agarose concentration on the DNA-unwinding time and several electrophoresis factors23.
Dilute SYBR green DNA dye in Tris-EDTA (TE) Buffer at a 1:10,000 ratio to obtain a 1x working stock dye solution. This dye is light-sensitive, so store it in a dark place.
For the cell lysis buffer, dissolve 100 mL of lysis buffer (2.5 M NaCl, 0.1 M EDTA, 10 mM Tris, 1% Triton X-100) in deionized water (DI H2O). Then, add 10 M NaOH to adjust the pH to 10.0. Cool the buffer to 4 °C.
For the alkaline DNA-unwinding/electrophoresis running buffer, dissolve 1 L of alkaline solution (0.3 M NaOH, 1 mM EDTA) in DI H2O. Cool the buffer to 4 °C. Make sure the pH >13.
-
For precoated comet slides, place a few drops of 0.5% agarose on a glass slide. Immediately, using the flat surface of another slide, spread the agarose to form a thin layer of agarose coating.
NOTE: Dry the slides before using.
-
For iPS cell culture, briefly culture iPS cells in basement-membrane-matrix-coated (see Table of Materials) culture plates in mTESR1 culture medium until confluency is reached.
NOTE: Human-cardiac-fibroblast-derived iPS cells were used in this protocol. The reprogramming of iPS cell derivation and culture conditions is described in recent articles from our research group12,13,21.
Table Of Materials
| Nmae of Material/Equipment | Company | Catalog Number |
|---|---|---|
| UltraPure™ Low Melting Point Agarose | Invitrogen | 16520050 |
| SYBR® Green I nucleic acid gel stain | Sigma-Aldrich | S9430 |
| mTeSR™1 | Stemcell Technologies | 85850 |
| Accutase | Stemcell Technologies | 7920 |
| RPMI | ||
| Gibco™ B-27™ Supplement, Serum Free, | ||
| 8-well chamber slide | ||
| Matrigel | ||
| 0.25% Trypsin | ||
| Triton X-100 | ||
| BSA | ||
| Phospho-Histone H2A.X (Ser139) Antibody | Cell Signaling Technology | #2577 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Rabbit IgG | Jackson | 711-545-152 |
| DAPI | ||
| phalloidinA488 | ||
| Rabbit-Cy3 | ||
| Vectashield |
2. Sample preparation
Discard the culture medium and wash the cells. Dissociate the cells using detachment solution (see Table of Materials). Wash the cell pellet with phosphate-buffered saline (PBS) and resuspend the cells in PBS at 1 x 105 cells/mL.
3. Sample slide preparation
NOTE: Perform the following steps under low-light conditions to avoid ultraviolet-light-induced cell damage.
Mix 10 μL of cell suspension and 90 μL of agarose solution (1:10 ratio) in a tube. Place the tube in a 37 °C water bath to prevent the agarose from solidifying.
Mix the solutions without introducing air bubbles and pipette 70 μL/spot onto the precoated comet slide. Using a pipette tip, spread the cell agarose mixture to form a thin layer. Place the slide in 4 °C for 15 min.
In a container, completely submerge the slide in cold lysis buffer. Place the container in the dark at 4 °C for 1 h.
Replace the lysis buffer with cold alkaline solution. Make sure the slides are completely submerged. Place the container in the dark at 4 °C for 30 min.
4. Electrophoresis
-
Place the slide in a horizontal electrophoresis tank. Fill the tank with cold alkaline electrophoresis buffer until the slides are completely submerged. Apply voltage at 1 V/cm for 15 to 30 min.
NOTE: Total voltage = distance between the electrodes x 1 V
In a container, completely submerge the slide in cold DI H2O. After 2 min, remove the DI H2O and add fresh DI H2O. Repeat this step.
Replace the DI H2O with cold 70% ethanol. Wait 5 min. Gently remove the slide, do not tilt it, and let it air-dry.
Once dried, add 100 μL of diluted DNA dye to each spot. Wait 15 min at room temperature. Using a FITC filter in an epifluorescence microscope, take images of 50 to 100 comets in total per sample.
5. Image analysis and calculation of the results
-
For scoring the comets, use the National Institutes of Health’s (NIH) ImageJ with comet assay plugin (download ImageJ here: https://imagej.nih.gov/ij/download.html; download the plugin here: https://www.med.unc.edu/microscopy/resources/imagej-plugins-and-macros/comet-assay/).
NOTE: The plugin comes with a PDF instruction which contains all details to perform the analysis. Additionally, screenshots of the comet analysis are shown in Figure 2A–C. Representative comet images of control and doxorubicin (Doxo)-treated iPS cells and the quantification of the comets are shown in Figure 3A–C.
Figure 2: DNA damage and tail moment quantification by ImageJ.
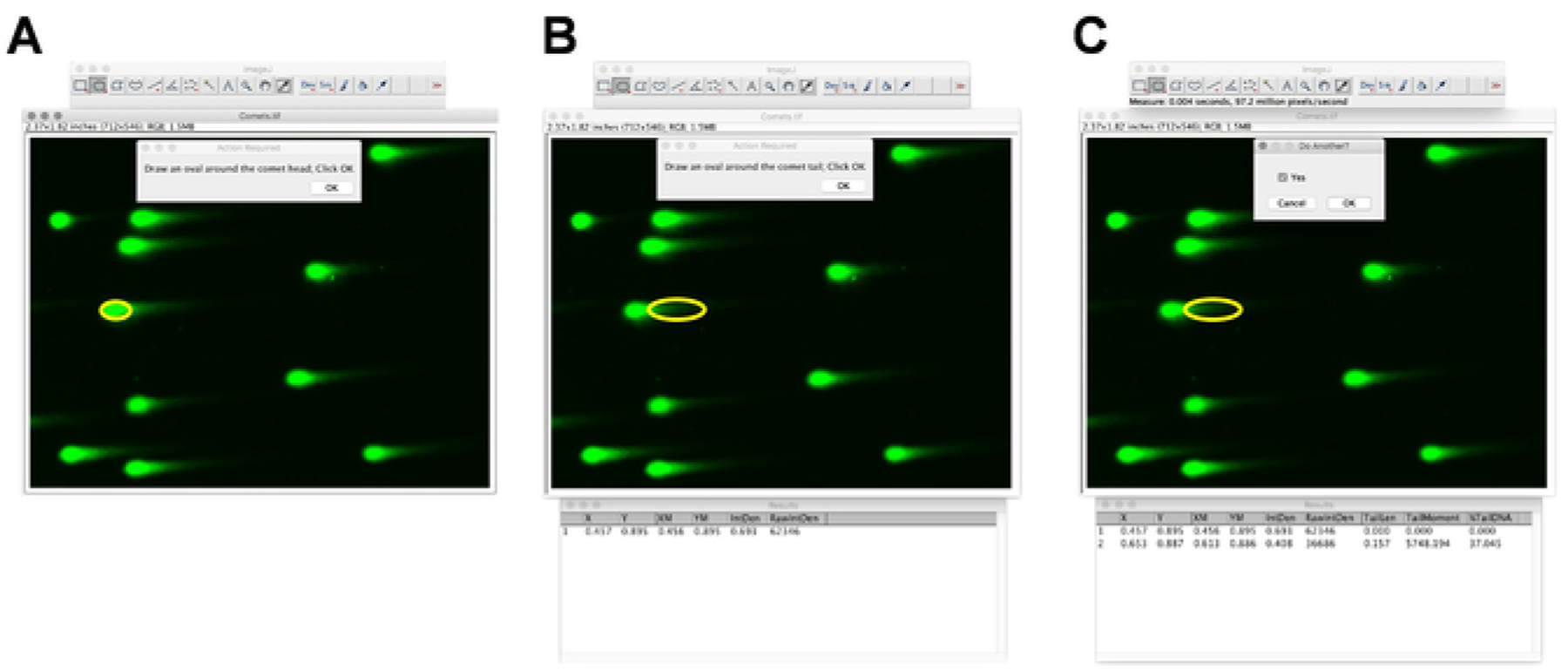
(A-C) Screenshots of ImageJ, with the comet analysis plugin showing a selection of the comet head and tail.
Figure 3: Doxorubicin induces DNA damage in human iPS cells.
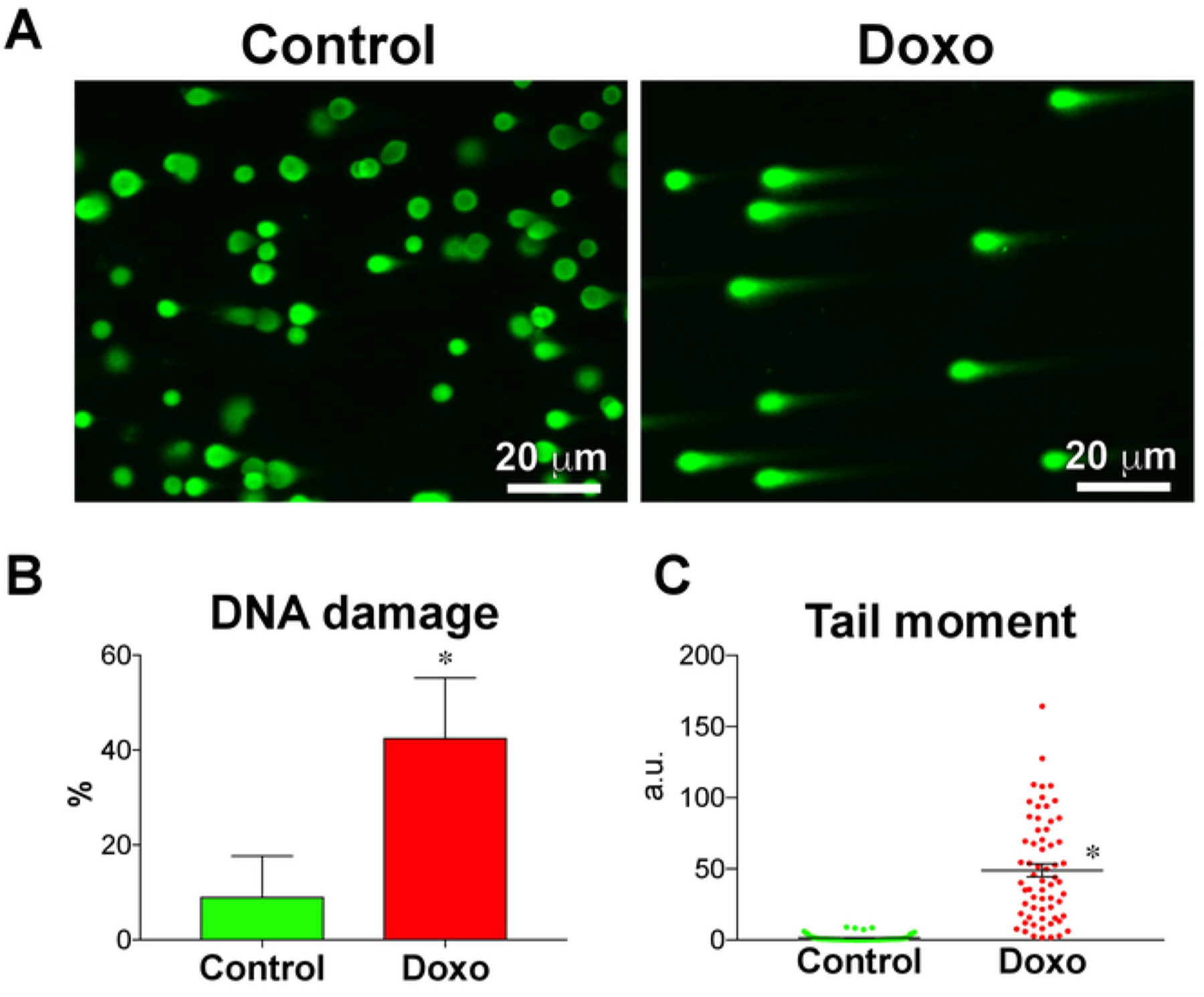
(A) DNA damage in doxorubicin-treated (Doxo) and nontreated (control) iPS cells, analyzed using the comet assay. (B) Fraction of DNA damage (n = 3) and (C) tail moment (n = 63 comets) quantified using the comet assay. Treatment with doxorubicin, a DNA-intercalating agent, significantly increased the fraction of DNA damage and tail moment in iPS cells. Data are means ± SEM. *P < 0.05, Student’s unpaired t-test.
2. DNA Damage Response
1. Sample and reagent preparation
-
For an iPS-CM cell culture, culture iPS-CM cells in basement-membrane-matrix-coated (see Table of Materials) culture plates in RPMI medium supplemented with B-27 (CMM) until confluency is reached.
NOTE: Human-iPS-cell-derived cardiomyocytes were used in this protocol. The protocol for iPS cell differentiation into cardiomyocytes can be found in recent articles from our research group12,13,21.
- Seed iPS-CMs in chamber slides.
- Discard the culture medium from the iPS-CM wells. Wash the cells three times with PBS.
- Add 1 mL of 0.25% trypsin per well and incubate at 37 °C for 5 min. Check the plate under a microscope for cell detachment. Add 1 mL of CMM to stop the trypsin.
- Place cell suspension in a 15 mL tube and centrifuge for 5 min at 4 °C and 200 x g. Discard the medium, add 1 mL of CMM, and resuspend the cells. Count the cells and adjust the cell count to obtain 20 x 105 cells in 1.6 mL of CMM.
- Seed 200 µL of cell suspension per well of the 8-well chamber slide (see Table of Materials). Tap the slide gently to spread the cells around the well and incubate at 37 °C.
For the iPS-CM doxorubicin treatment, discard the culture medium from the iPS-CM wells. Wash the cells with PBS. Treat the cells with 1 μM doxorubicin and CMM for 4 h. Aspirate the medium and wash the cells with PBS.
For the blocking buffer, prepare 10 mL of bovine serum albumin (BSA) solution containing 3% BSA and 0.05% Triton X-100. To prepare 10 mL of blocking buffer, add 1 mL of normal donkey serum to 9 mL of prepared BSA solution.
For the primary antibody solution, add 1 μL of anti-rabbit phospho-histone H2A.X (Ser139) antibody to 200 μL of blocking buffer.
For the secondary antibody mix, add 1 μL each of fluorochrome-conjugated anti-rabbit secondary antibody, DAPI, and fluorochrome-conjugated phalloidin to 200 μL of blocking buffer.
2. Immunolabeling
Wash the cells three times with PBS and add 200 μL of freshly prepared 4% paraformaldehyde (PFA) to each well. Fix the cells for 20 min in the dark at room temperature. Wash the cells with PBS.
Remove the PBS and add 200 μL of blocking buffer per well and block for 30 min in a humidified chamber at room temperature. Remove the blocking buffer and add 200 μL of primary antibody solution. Incubate for 45 min in a dark, humidified chamber at room temperature. Then, wash the wells three times with PBS.
Remove the PBS and add 200 μL of secondary antibody mix. Incubate for 45 min in a dark, humidified chamber at room temperature. Then, wash the wells three times with PBS. Wash with DI H2O before mounting them with antifade. Place a coverglass and store the slides in the dark at 4 °C.
Using an epifluorescence microscope, take three to five images of random fields per sample, as shown in Figure 4A, and proceed to image analysis.
Figure 4: DNA damage response in iPS-derived cardiomyocytes.
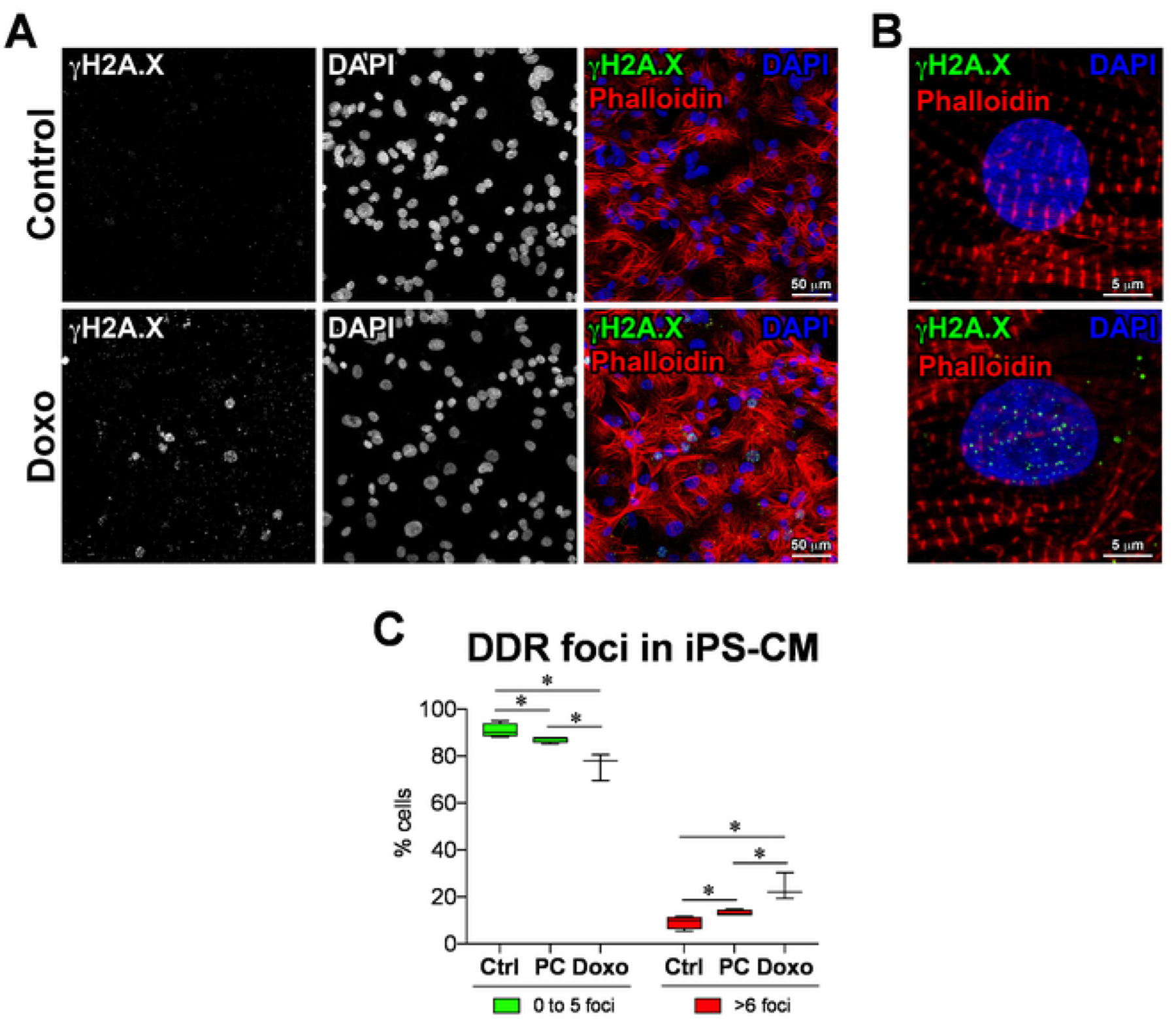
(A) DNA damage response marker γH2A.X identified by immunofluorescence in doxorubicin-treated (Doxo) and nontreated (control) iPS-cardiomyocytes as well as prolonged culture iPS-cardiomyocytes (PC). (B) Higher magnification of γH2A.X (green punctae) labeling at the sites of DNA damage in iPS-CMs. (C) Quantification of DDR foci, analyzed using CellProfiler with custom pipeline modules. Data are means ± SD; n = 3 to 4. *P < 0.05, Student’s unpaired t-test.
3. Counting of the DDR foci
Download and install CellProfiler24 (http://cellprofiler.org).
NOTE: Image processing in this study was performed using CellProfiler version 2.1.1. Tutorials for CellProfiler that were used can be found elsewhere (https://www.youtube.com/watch?v=PvhRL9xpduk&list=PL7CC87670239B4D10). If a newer version of CellProfiler is being used, the pipeline might require minor adaptations.
Download the pipeline punctae_gH2AX.cpipe. Select File > Import > Pipeline from the file and choose punctae_gH2AX.cpipe.
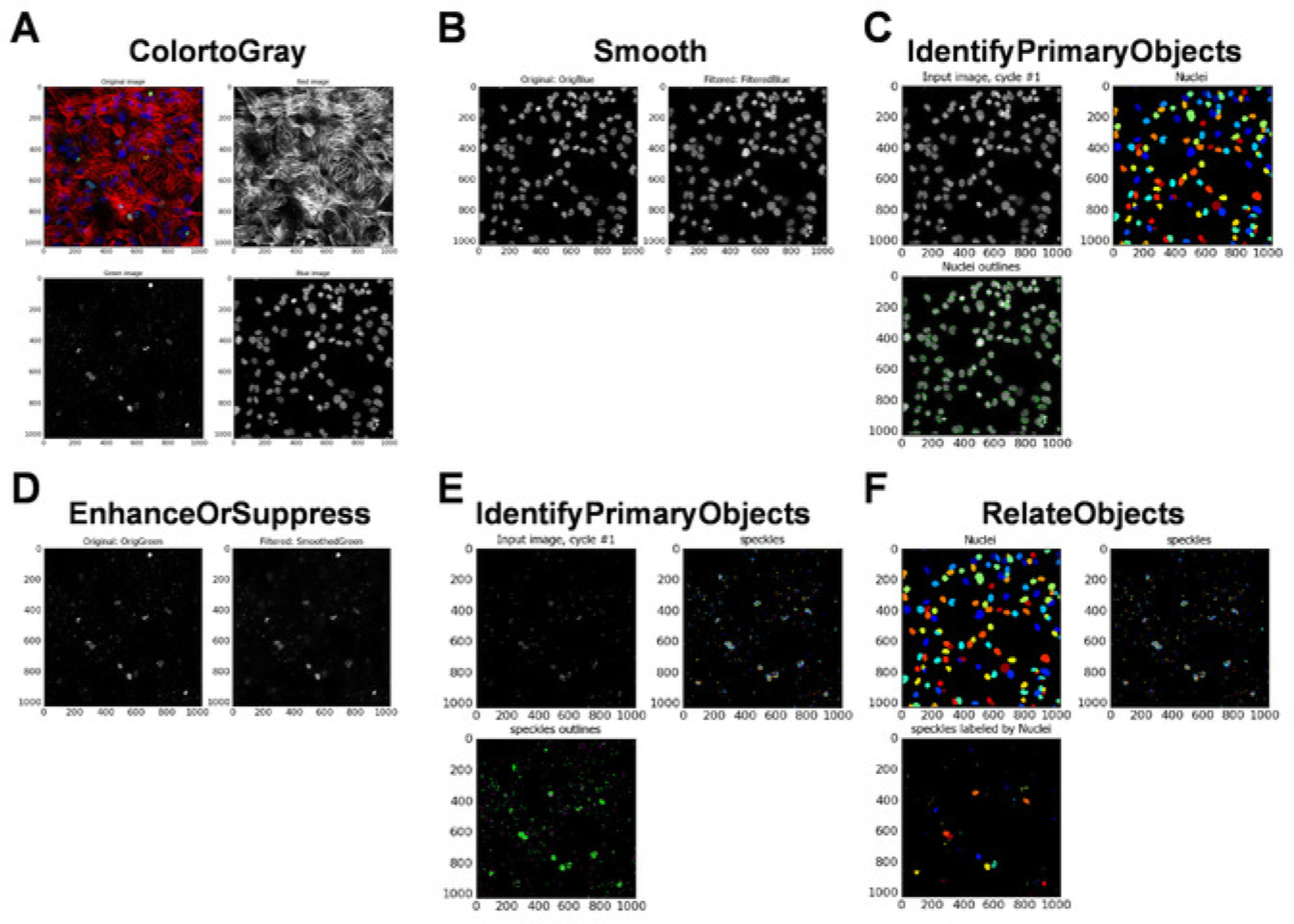
Drag-and-drop the folder containing the acquired images of γH2A.X foci to the File list window in the Input modules/Images section for analysis (Figure 5A). Then, follow the instructions as shown in Figure 5A–L.
- Set the following parameters for the images:
- For Groups, select no. In Analysis modules, select ColorToGray (see Figure 5D for module settings).
-
In Analysis modules, select Smooth (see Figure 5E for module settings).
NOTE: This value may need to be optimized, depending on the image resolution.
-
In Analysis modules, select IdentifyPrimaryObjects (see Figure 5F for module settings).
NOTE: Optimize these values to make sure the whole nucleus is selected. After this step, the computer may lag.
-
In Analysis modules, select Smooth (see Figure 5G for module settings).
NOTE: This value may need to be optimized, depending on the image resolution.
In Analysis modules, select EnhanceOrSuppressFeatures (see Figure 5H for module settings).
-
In Analysis modules, select IdentifyPrimaryObjects (see Figure 5I for module settings).
NOTE: Optimize these values to make sure the punctae are selected. After this step, the computer may lag again.-
Click Analyze Images at the bottom left panel to perform image analysis. At the successful completion of the analysis, several images are generated (Figure 6A–F).NOTE: Users should save these images for future reference.
- Note the .csv file containing the quantitative data for further analysis that is generated and saved in the appropriate location on the computer. In the spreadsheet called MyExpt_Image, columns B and D will have the number of nuclei that have up to five and more punctae, respectively. Add columns B and D to get the total number of nuclei.
Repeat step 2.3.3 – 2.3.8 for all images (change the MyExpt_filename before every analysis). Plots can be made for assessing DNA integrity, as shown in Figure 4C.
Figure 5: Automated DNA damage response analysis by CellProfiler.

(A-L) Screenshots of CellProfiler with specified settings for importing the images, and the automated identification and quantification of γH2A.X punctae in iPS-CMs.
Figure 6: Automated DNA damage response analysis by CellProfiler.
(A-F) Data images generated by CellProfiler at the completion of the image analysis.
Representative Results
Human induced pluripotent stem cells were cultured, and the DNA damage and the tail moment, which were used as a measure of DNA integrity, were analyzed by comet assay. iPS cells were embedded in low-melting-point agarose and placed on a glass slide. The cells were, then, treated with lysis buffer, followed by an alkaline solution, to obtain supercoiled DNA. Nucleoids were electrophoresed and comets were visualized by DNA dye (Figure 1A–D). The comets were, then, analyzed with ImageJ, using the comet assay plugin (Figure 2A–C).
Human iPS cells were treated with Doxo to induce DNA damage and to be used as a positive control. Representative micrographs of comets from Doxo-treated and nontreated iPS cells are shown in Figure 3A. A basal amount of DNA damage was found in iPS cells, expressed as a fraction of DNA damage and tail moment. However, the Doxo treatment increased the DNA damage in iPS cells as expected (Figure 3B,C). This shows that the comet assay can be used to assess DNA integrity not only in somatic cells18,19 but also in pluripotent stem cells21.
Freshly differentiated iPS cardiomyocytes (iPS-CMs), iPS-CMs cultured for 6 months (prolonged culture [PC]), and iPS-CMs treated with Doxo were subjected to γH2A.X immunolabeling. Representative micrographs of γH2A.X immunolabelling are shown in Figure 4A (lower-magnification) and Figure 4B (higher-magnification). The number of γH2A.X foci (DDR foci) (punctae) in each nucleus are quantified, using Cell Profiler with custom pipeline modules (Figure 5A–L). The percentage of cells that are positive for γH2A.X are classified into nuclei with zero to five punctae and nuclei with more than 10 punctae. In the control iPS-CM, more than 90% of the cells had less than five DDR foci per nuclei, and a total of less than 10% of the cells had more than six DDR foci per nuclei (Figure 4C, control [Ctrl]). iPS-CMs cultured for 6 months had less than 90% cells with less than five DDR foci per nuclei, and a total of more than 13% of the cells had more than six DDR foci per nuclei (Figure 4C, PC), whereas the Doxo-treated iPS-CM showed less than 80% of the cells with less than five DDR foci per nuclei, and a total of about 24% of the cells had more than six DDR foci per nuclei (Figure 4C, Doxo). This data clearly shows that prolonged cell culture and Doxo treatment induce significant DNA damage in iPS-CMs and are not suitable for cell transplantation.
Discussion
DNA integrity portrays cell integrity. Cells with damaged DNA are frequently in stress and eventually lose their integrity. The integrity of stem and stem-cell-derived cells that are being propagated for the purpose of transplantation is principal for the cells to perform their desired function. Transplanting cells with damaged DNA would result in a poor engraftment rate and performance of the cell8,20. Therefore, examining the DNA integrity prior to cell transplantation is a necessary quality control protocol. Here we describe two cost-effective approaches, namely the comet assay and γH2A.X immunofluorescence labeling, to assess the DNA damage and quality of stem and stem-cell-derived cells.
A major cause of cellular aging is thought to be the accumulation of DNA damage in cells. Using the comet assay, Al-Baker et al. analyzed the DNA damage in young and senescent human dermal fibroblasts22. Previously, we have shown that transplanting DNA damage-free cardiac progenitor cells isolated from a p53 transgenic mouse increased the rate of engraftment in the host organ8. Recently, we have shown the role of the p53 transactivation domain in DNA damage repair mechanisms in human iPS cells21. In both studies, we have employed both the comet assay and γH2A.X immunofluorescence labeling methodologies to assess the DNA damage in stem cells. Both these methods are cost-effective and can be performed with basic lab equipment. An annoying critical problem that we often experienced is agarose solidifying in the pipette and tubes. We overcome this issue by prewarming all the tubes and tips prior to pipetting agarose. While the comet assay takes up to 2 days to complete, the optimized γH2A.X immunofluorescence labeling procedure can be completed in under 4 h.
Human iPS cell cultures with more than 10% DNA damage, measured by the comet assay, did not efficiently differentiate into cardiomyocytes (data not shown) in vitro. The comet assay is sensitive and dynamic in assessing DNA damage in stem cells. However, DNA integrity assessments in certain cells, such as iPS-derived cardiomyocytes, are cumbersome by comet assay due to the nature of the cells. The cardiomyocytes are enriched with troponin, sarcomeric proteins, and the secreted extracellular matrix proteins. Denaturing these complex structures to make supercoiled DNA and its reproducibility are questionable. Most importantly, 25% to 40% of human cardiomyocytes are binucleated25, and these percentages vary in iPS-derived cardiomyocytes. Since the cell wall is disrupted in the comet assay, the precise assessment of the percentage of DNA-damaged cells is impossible. Therefore, in the case of iPS-CMs, the γH2A.X immunofluorescence labeling procedure to assess DNA damage is a simplistic alternative for the comet assay.
Based on these results and those from previous publications by our group8,20,21, we suggest that, if there is more than 10% DNA damage, assessed by the comet assay, or less than 90% of the cells have zero to five DDR foci, then the culture should be disqualified for cell transplantation.
Acknowledgements
This work was supported in part by National Heart, Lung, and Blood Institute Grants RO1-HL-99507, HL-114120, HL-131017, HL138023, and UO1-HL-134764 (to J. Zhang) and by American Heart Association Scientific Development Grant 17SDG33670677 (to R. Kannappan).
Footnotes
Video Link
The video component of this article can be found at https://www.jove.com/video/58971/
Disclosures
The authors have nothing to disclose.
References
- 1.Bolli R et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. The Lancet 378 (9806), 1847–1857 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 2.Makkar RR et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. The Lancet 379 (9819), 895–904 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomita Y et al. Cardiac neural crest cells contribute to the dormant multipotent stem cell in the mammalian heart. Journal of Cell Biology 170 (7), 1135–1146 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oyama T et al. Cardiac side population cells have a potential to migrate and differentiate into cardiomyocytes in vitro and in vivo. Journal of Cell Biology 176 (3), 329–341 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fischer KM et al. Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing Pim-1 kinase. Circulation 120 (21), 2077–2087 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cottage CT et al. Cardiac progenitor cell cycling stimulated by pim-1 kinase. Circulation Research 106 (5), 891–901 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Angert D et al. Repair of the injured adult heart involves new myocytes potentially derived from resident cardiac stem cells. Circulation Research 108 (10), 1226–1237 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kannappan R et al. p53 Modulates the Fate of Cardiac Progenitor Cells Ex vivo and in the Diabetic Heart In vivo. EBioMedicine 16, 224–237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatzistergos KE et al. Bone marrow mesenchymal stem cells stimulate cardiac stem cell proliferation and differentiation. Circulation Research 107 (7), 913–922 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okita K, Yamanaka S Induced pluripotent stem cells: opportunities and challenges. Philosophical Transactions of the Royal Society B Biological Sciences 366 (1575), 2198–2207 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circulation Research 104 (4), e30–41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye L et al. Cardiac repair in a porcine model of acute myocardial infarction with human induced pluripotent stem cell-derived cardiovascular cells. Cell Stem Cell 15 (6), 750–761 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L et al. Derivation and high engraftment of patient-specific cardiomyocyte sheet using induced pluripotent stem cells generated from adult cardiac fibroblast. Circulation: Heart Failure 8 (1), 156–166 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirakawa K, Midorikawa K, Oikawa S, Kawanishi S Carcinogenic semicarbazide induces sequence-specific DNA damage through the generation of reactive oxygen species and the derived organic radicals. Mutation Research 536 (1–2), 91–101 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Martinez GR et al. Oxidative and alkylating damage in DNA. Mutation Research 544 (2–3), 115–127 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biological Interactions 160 (1), 1–40 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Wiseman H, Halliwell B Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochemical Journal 313 (Pt 1), 17–29 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ostling O, Johanson KJ Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochemical and Biophysical Research Communications 123 (1), 291–298 (1984). [DOI] [PubMed] [Google Scholar]
- 19.Singh NP, McCoy MT, Tice RR, Schneider EL A simple technique for quantitation of low levels of DNA damage in individual cells. Experimental Cell Research 175 (1), 184–191 (1988). [DOI] [PubMed] [Google Scholar]
- 20.Goichberg P et al. Age-associated defects in EphA2 signaling impair the migration of human cardiac progenitor cells. Circulation 128 (20), 2211–2223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Kannappan R, Mattapally S, Wagle PA, Zhang J Transactivation domain of p53 regulates DNA repair and integrity in human iPS cells. American Journal of Physiology-Heart and Circulatory Physiology (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Al-Baker EA, Oshin M, Hutchison CJ, Kill IR Analysis of UV-induced damage and repair in young and senescent human dermal fibroblasts using the comet assay. Mechanisms of Ageing and Development 126 (6–7), 664–672 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Azqueta A, Gutzkow KB, Brunborg G, Collins AR Towards a more reliable comet assay: optimising agarose concentration, unwinding time and electrophoresis conditions. Mutation Research 724 (1–2), 41–45 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Carpenter AE et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biology 7 (10), R100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paradis AN, Gay MS, Zhang L Binucleation of cardiomyocytes: the transition from a proliferative to a terminally differentiated state. Drug Discovery Today 19 (5), 602–609 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]