

Abstract
Thromboembolic stroke remains a major cause of neurological disability and death. Current stroke treatments (aspirin, tissue plasminogen activator) are significantly limited by timing and risks for hemorrhage which have driven researchers to explore other approaches. Stem cell‐based therapy appears to be an effective option for ischemic stroke. Besides trans‐differentiation into neural cells, stem cells also provide acute protection via paracrine signaling pathways through which releasing neuroprotective factors. We previously reported that intraperitoneal administration of human placenta mesenchymal stem cell (hPMSC) therapy upon reperfusion significantly protected the brain against middle cerebral artery occlusion (MCAO)‐induced injury. In the present study, we specifically investigated the role of hPMSC‐derived angiotensin converting enzyme‐2 (ACE‐2) in protection of MCAO‐induced brain injury by measurement of brain tissue viability, cerebral blood flow, and neurological score. Here, we report for the first time that hPMSC expressing substantial amount of ACE‐2, which mediates hPMSC protection in the MCAO model. Strikingly, we found that the protective effects of hPMSC in MCAO‐induced brain injury could be attenuated by pretreatment of hPMSCs with MLN‐4760, a specific inhibitor of ACE‐2 activity, or by transfection of hPMSCs with ACE‐2‐shRNA‐lentivirus. The hPMSC‐derived ACE‐2 specific protective mechanism was further demonstrated by administration of PD123319, an Angiotensin type‐2 receptor antagonist, or A779, a MasR antagonist. Importantly, our study demonstrated that the protective effects of hPMSC in experimental stroke are ACE‐2/MasR dependent and this signaling pathway represents an innovative and highly promising approach for targeted stroke therapy.
Keywords: angiotensin converting enzyme 2, cerebral blood flow, human placenta mesenchymal stem cells, infarction, ischemic stroke, MasR
Angiotensin converting enzyme‐2 (ACE‐2) contributes to human placenta mesenchymal stem cell (hPMSC)‐induced protection against ischemic injury through MasR pathway.
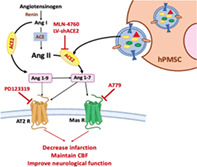
Significance statement.
This study demonstrated that human placenta mesenchymal stem cell (hPMSC)‐derived angiotensin converting enzyme‐2 (ACE‐2) is required to protect against progressive loss of cerebral blood perfusion following ischemic stroke. This effect is also mediated through vascular endothelial Mas receptor but not angiotensin type‐2 receptor. Consequently, the results of the authors' experimental stroke studies with human derived stem cells describe new paracrine function of hPMSC in which ACE2 and Mas receptor are critical, therefore, providing a novel therapeutic strategy for stroke in near future.
1. INTRODUCTION
Ischemic strokes represent 87% of the total incidence of stroke and are the leading cause of neurologically based morbidity in the elderly. 1 The frequency of ischemic strokes among “younger” (<65 years) individuals has also increased reflecting hypertension 2 and pregnancy, as well as oral contraceptive use and behavioral risks (sedentary lifestyle, alcohol, tobacco, and recreational drug abuse) which increase risk. 2 , 3 , 4 Most recently Kolikonda et al have reported that as many as 5% of patients with COVID‐19 infections may go on to develop acute ischemic strokes which may be a common clinical feature of COVID disease. 5
Current therapies for ischemic stroke include tissue plasminogen activator (t‐PA) and antithrombotics (aspirin/heparin) which both have a very narrow time‐window (<4.5 hours) for t‐PA administration after the onset of stroke symptoms. Both carry significant risks for hemorrhage. 6 , 7 The absence of effective and safe therapies for the critical acute phase of stroke has motivated researchers to seek alternative therapeutic approaches to reduce cerebral injury before and beyond this time frame. 8 , 9
Stem cell‐based therapies (SCTs) represent an innovative approach for treating cardiovascular‐related diseases, including and especially ischemic stroke. Although stem cells have long been assumed to provide their therapeutic benefits by engrafting and differentiating within the post‐stroke brain tissue to restitute damaged cells 10 ; we and others have reported that stem cells protect against acute brain injury in stroke through paracrine signaling to limit initial tissue injury. 11 , 12 , 13 , 14 , 15 Stem cells secrete several paracrine neurotrophic factors 1 , 16 that shield the brain against acute inflammatory, immune, and vascular stresses in stroke. 10 , 13 , 17 Such factors are being increasingly recognized for their potential contributions to acute postischemic neuroprotection. 13 , 14 We recently reported that in the setting of stroke, ischemia triggers a progressive and devastating vasoconstriction within the brain which begins after 4 hours to cause infarction at 24 hours. 15 We found that placenta derived stem cells and extracellular vesicles (EVs) derived from these stem cells were able to prevent the development of progressive infarction by maintaining cerebral perfusion, however, in that report we did not establish the mechanistic basis of this phenomenon.
One candidate pathway that might explain the basis of stem cell mediated‐preservation of blood flow in the middle cerebral artery occlusion (MCAO) model of stroke may include “angiotensin converting enzyme‐2 (ACE‐2).” ACE‐2, a recently recognized component of the renin angiotensin system (RAS), may improve outcomes in chronic experimental models of stroke. 18 , 19 ACE‐2 enzymatic activity catabolizes the vasoconstrictor Angiotensin‐II (derived from the vasoconstrictor Angiotensin‐II) to form Angiotensin 1‐7 and Angiotensin 1‐9 (derived from Ang ‐I) which activate the Mas (masR) and Angiotensin type‐2 (AT2) receptors, respectively. By binding to these two receptors, these ACE‐2 pathway products exert vasodilator, anti‐inflammatory, and antiproliferative effects 20 , 21 , 22 which may be beneficial in the setting of stroke. We reported that intraperitoneal administration of human placenta mesenchymal stem cells (hPMSCs) was highly protective against brain injury and neurological disability in the MCAO model of stroke. 15
In our current study, we report for the first time that hPMSCs express substantial amounts of ACE‐2. Using pharmacological and genetic methods, we demonstrate that ACE‐2 expressed by hPMSCs mediates the protective effects of stem cells in ischemic stroke. We further evaluated the vascular receptors (AT2R vs masR) through which hPMSCs‐derived ACE‐2 products protect the brain against acute MCAO injury. We conclude that ACE‐2 expressed by hPMSCs and in particular the ACE‐2 product of Angiotensin‐II, Angiotensin 1‐7 and its cognate receptor (the MasR) play critical roles in maintaining cerebral perfusion after reperfusion insults. By preserving brain perfusion, these mechanisms and mediators prevent development of severe chronic structure and functional brain injury in the setting of experimental stroke. These findings may lead to the introduction of novel and powerful approaches for the treatment of stroke injury.
2. MATERIAL AND METHODS
2.1. Animals
All animal protocols were approved by the LSUHSC‐S Institutional Animal Care and Use Committee (IACUC) according to NIH guidelines. We used male C57BL/6J mice (Jackson Laboratories, Bar Harbor, Maine) in all studies at 9 to 16 weeks of age. Animals were housed in a barrier facility and maintained on a normal diet.
2.2. Surgery for MCAO model
Male mice (25‐30 g) were anesthetized with ketamine (200 mg/kg i.p.)/xylazine (10 mg/kg i.p.). Once under deep anesthesia, MCAO was induced by creating a midline incision at the neck to expose the right carotid bifurcation. The right external carotid artery branch was isolated and ligated, and a microclip placed on the common carotid artery. A silicone‐coated 6‐0‐nylon microfilament was introduced into the common carotid artery and the microclip released to allow advancement of the filament through the artery until the bulb‐tip occluded the origin of the middle cerebral artery. This filament was left in place for 1 hour (ischemia). Reperfusion was initiated by withdrawal of the filament. For sham groups, the right external carotid artery was isolated without further manipulation. The wounds were closed using surgical sutures (6‐0) and mice were allowed to recover from anesthesia. Postoperative monitoring of eating, drinking, and movement was performed at 4 and 24 hours following recovery.
2.3. Neurological testing
Neurological outcomes were evaluated at 24 hours after reperfusion using a 24‐point scale as described previously. 15 Briefly, mice were given positive scores (0‐3) for each of the following parameters: 5 minutes of spontaneous activity, symmetry of movement and forelimbs (outstretching while tail is held), response to vibrissae contact, floor and beam walking, wire cage wall climbing, and reaction to touch on either side of the trunk.
2.4. hPMSCs isolation and culture
hPMSCs cells used in this study were isolated as described previously. 23 Tissue procurement was approved by the Institutional Review Board at Louisiana State University of Health Sciences Center‐Shreveport (LSUHSC‐S). Placental mesenchymal stem cells (PMSCs) were isolated from freshly delivered human placenta. Briefly, placentas delivered by normal pregnant women were collected immediately after delivery. Villous tissue was separated by sterile dissection from different cotyledons, excluding chorionic and basal plates. After extensive washing with ice‐cold phosphate‐buffered saline (PBS), villous tissue was digested with trypsin and DNase I in Dulbecco's Modified Eagle's Medium (DMEM) at 37°C for 90 minutes. Digested microvillus tissue was collected and cultured in DMEM supplemented with 10% fetal bovine serum (FBS). hPMSCs started to grow in 3 to 5 days. At ~80% confluence, the cells were passaged with TrypLE Express (Invitrogen, Carlsbad, California). hPMSCs were characterized with mesenchymal stem cells markers including positive expression of CD44, CD73 and CD90, and negative expression of CD34 and HLA‐DR. Primary isolated hPMSCs also expressed Oct3/4 and were able to differentiate into adipocytes, chondrocytes, and osteocytes. hPMSCs were subcultivated at a 1:3 ratio at confluence and passages 3 to 10 were used in the present study.
2.5. Intraperitoneal injection of hPMSCs
Trypsinized hPMSCs were washed twice with Ca++/Mg++ free Hanks Balanced Salt Solution (HBSS) and centrifuged (1500 rpm, 5 minutes, 25°C). 5 × 105 hPMSCs were suspended in 500 μL HBSS solution without Ca++/Mg++ and injected intraperitoneally (IP) into MCAO‐treated mice at reperfusion.
2.6. ACE‐2 inhibition in hPMSCs
2.6.1. MLN‐4760 treatment of hPMSCs
To investigate contributions of ACE‐2 in hPMSC‐mediated stroke protection, hPMSCs were treated with 10 μM MLN‐4760 (Sigma Aldrich), to selectively inhibit ACE‐2 activity. 24 MLN‐treated hPMSC were PBS washed after 48 hours treatment and injected (5 × 105 cells in 500 μL HBSS) into the MCAO mice.
2.6.2. Lentivirus transduction of hPMSCs
Two hours prior to transfection, the medium of HEK293FT at their 90% confluence, was changed to antibiotic free DMEM supplemented with 10 vol%/vol% FBS. Production of third generation lentivirus was performed combining the transfer vector pLV [shRNA]‐EGFP‐hACE‐2 (purchased from VectorBuilder) with packaging plasmid (pMDEL/pRRE, pRSV/REV, and pMD2G), and Lipofectamine3000 enhancer reagent. The mixture was briefly vortexed and incubated at room temperature for 20 minutes, and then added dropwise to the HEK293FT cells. Flask was agitated gently to distribute the precipitates and then incubated at 37°C, 5% CO2. Four hours late, cell culture media was gently replaced with fresh medium. At 24 hours post‐transfection, the medium was replaced with DMEM supplemented with 10% FBS and antibiotics, then incubated at 37°C, 5% CO2. The collection of viral supernatants was made after 48 hours, centrifuged at 1000 rpm for 5 minutes at 4°C, and passed through a 0.45 mm pore filter to remove cellular debris. Lentivirus was added as droplets to hPMSC (40% confluent) cultured in DMEM supplemented with 10% FBS and antibiotics. After overnight incubation, lentivirus was removed, and fresh media added. Following 48 hours, transduction efficiency was determined either by visualizing for expression of fluorescent marker GFP, or Western blotting for downregulated expression of ACE‐2 in hPMSCs.
2.7. Laser speckle measurement of cerebral blood flow
The Perimed Laser Speckle Imaging system (Pericam PSI HR; Sweden) was used to measure cerebral blood perfusion within the brains of the different experimental groups. Twenty‐four hours after reperfusion, anesthesia was induced and maintained with 3% isoflurane, and mice were placed on a warm pad. The coronal skin was removed, and perfusion recordings accomplished using a high‐resolution laser speckle camera (Perimed Laser Speckle Imager) at a working distance of 10 cm. “Perfusion” reflects total cerebral flow signal measured in selected tissue regions of interest. Measurements are expressed as perfusion units (PU), using a fixed scale (arbitrary units). “Perfusion” was measured in arbitrary units normalized for the selected region of interest, then perfusion of either ipsilateral or contralateral was normalized to baseline levels (values obtained from averaged sham total cerebral blood flow [CBF]).
2.8. 2,3,5‐Triphenyltetrazolium chloride staining of infarcted tissue
Twenty‐four hours after reperfusion, mice were deeply anesthetized with isoflurane and decapitated. The extent and severity of MCAO was evaluated after removal of the brain and staining of brain slices with 2,3,5‐Triphenyltetrazolium chloride (TTC; Sigma) to measure infarct size. After dissection, the brain was immersed in cold PBS for 10 m and sliced into 2.0 mm‐thick sections using an anatomical slicer. Brain slices were incubated in 2% TTC/PBS for 30 m at 37°C. Areas of infarction in each brain slice were recorded (Nikon 990) and measured using Image‐J program (NIH). The infarcted area was adjusted for edema using Reglodi's method: Edema adjusted‐infarct volume: infarct volume × (contralateral hemisphere/ipsilateral hemisphere). 25 Cumulative dead (white‐stained) regions were combined from each brain to generate a total brain tissue infarcted volume score for each mouse.
2.9. Western blot analysis
In addition to preparing hPMSCs for injection, separate samples of these cells were tested for ACE‐2 expression. After the treatments described above, culture media were discarded, and hPMSCs were washed with ice‐cold PBS and cells collected in Laemmli sample buffer (Bio‐Rad) with 10% 2‐mercaptoethanol. The lysates were scraped using cell scraper and collected in microfuge tubes, and sonicated at 50% power for 15 seconds, boiled at 95°C for 15 minutes and stored at −80°C. Twenty microliters of protein was separated via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE), then immunoblotted to polyvinylidene fluoride (PVDF) and incubated at 4°C with rabbit anti‐ACE‐2 (1:1000, Invitrogen; USA). Membranes were incubated with goat anti‐rabbit IgG‐HRP antibodies (1:2500, Sigma) for 2 hours at 25°C. Signal was detected using ChemiDocTM MP imaging system (Bio‐Rad) and results analyzed with NIH Image‐J software.
2.10. Statistical analysis
Statistical analysis was performed using GraphPad Prism software. For all experiments, data are expressed as mean ± SEM. The statistical significance of the differences between groups was calculated using Student's t test, one‐way or two‐way analysis of variance (ANOVA) with Bonferroni post hoc tests where appropriate and indicated in the figure legend. A P value <.05 was considered statistically significant.
3. RESULTS
3.1. ACE‐2 inhibition eliminates hPMSC protection in the MCAO stroke model
By Western blotting, we found that hPMSCs express more than three times more ACE‐2 (3.37 ± 0.54) than human brain endothelial cells (hBMEC‐D3) (1.10 ± 0.59, P = .04; Figure 1A). We previously demonstrated that intraperitoneal (IP) administration of hPMSCs at the time of reperfusion in the MCAO model of ischemic stroke produced highly significant preservation of the ipsilateral hemisphere characterized by almost complete inhibition of cerebral infarction, significant preservation of CBF within the post‐MCAO brain, and improvement of neurological function. 15 To evaluate contributions of hPMSCs‐derived ACE‐2 in brain protection after ischemic stroke, we inhibited ACE‐2 activity in hPMSCs using 10 μM MLN‐4760, a highly potent and selective ACE‐2 inhibitor. 24 While IP injected hPMSC (nontreated 5 × 105 cells in 500 mL HBSS) into the MCAO mice at the time of reperfusion improved the neurological function of MCAO mice (14.5 ± 1.42, P < .0001), MLN‐treated hPMSCs (5 × 105 cells in 500 mL HBSS) (1 hour following ischemia) did not provide significant protection against neurological deficits (4 ± 0.68) (Figure 1B). Infarcted areas in MLN‐hPMSCs treated MCAO mice were more than six times larger (24.19 ± 2.48) than in mice treated with hPMSCs‐treated MCAO group (3.84 ± 0.80, P < .0001) (Figure 1C). The infarct size of the MCAO mice group injected with MLN‐treated hPMSC (ACE‐2 inhibited) were similar to that of MCAO mice not receiving hPMSC therapy (22.62 ± 2.03, P = .89).
FIGURE 1.
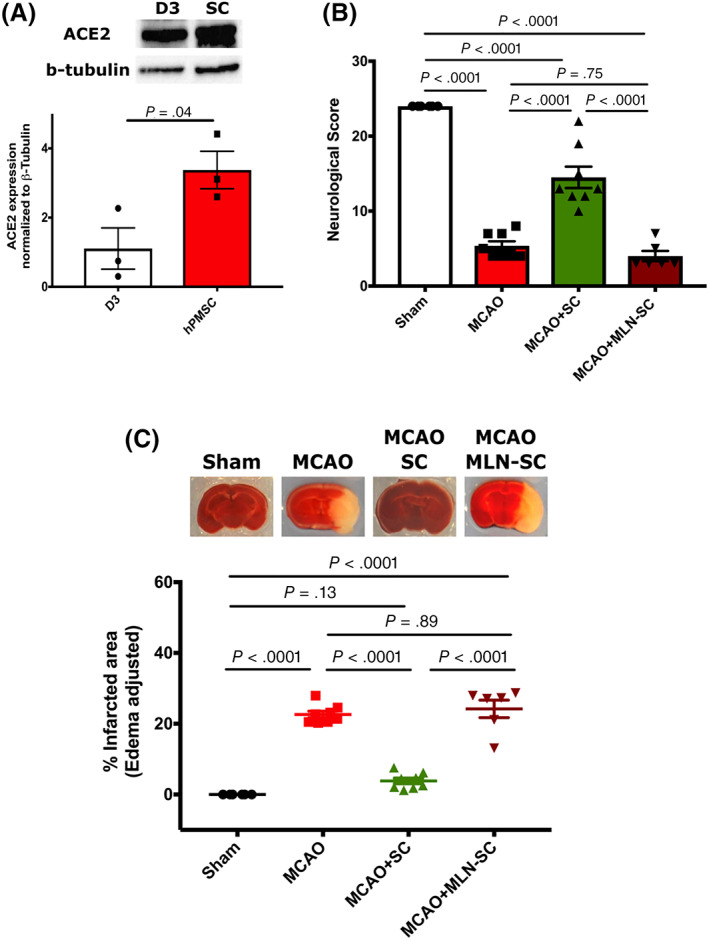
ACE‐2 contributes to the hPMSCs‐based protection in the MCAO model. A, Western blot for expression of ACE‐2 in hPMSCs and hBMEC‐D3 cells. Quantification analysis of ACE‐2 expression normalized to β‐tubulin protein expression showed significantly higher levels of ACE‐2 in the hPMSCs compared to hBMEC‐D3 cells (*P = .04). B, Neurological scores calculated based on 24 scale evaluation tests. 15 Significant differences in neurological scores were seen between MCAO (n = 8) and sham groups (****P < .0001). hPMSC treatment (n = 8) significantly improved neurological scores vs MCAO group (****P < .0001). One‐way analysis of variance showed no significant differences in neurological functions between the MCAO group and MCAO mice treated with MLN‐hPMSC (not significant [NS], P = .74, n = 6). Neurological scores were significantly reduced in MLN‐hPMSC treated MCAO vs hPMSC‐treated MCAO mice (****P < .0001). C, Infarcted areas were assessed by TTC staining. Significant increase in infarcted volume was observed in MCAO+MLN‐treated hPMSC vs MCAO+hPMSC mice (****P < .0001). No significant differences of infarction were observed between MCAO+MLN‐treated hPMSC mice vs MCAO (NS, P = .89). In all graphs, data represent means ± SEM. ACE‐2, angiotensin converting enzyme‐2; hPMSCs, human placenta mesenchymal stem cells; MCAO, middle cerebral artery occlusion; TTC, 2,3,5‐triphenyltetrazolium chloride
Next, to determine whether and to what extent MLN‐treated hPMSC might affect CBF compared to untreated hPMSC following MCAO‐induced strokes, we measured cerebral perfusion using Laser Speckle Contrast Imaging. 26 We found that total CBF was significantly reduced in MLN‐hPMSCs treated MCAO mice (6.25 ± 0.45, P < .0001) compared to hPMSC‐treated MCAO (10.99 ± 0.66) and sham groups (13.42 ± 1.01, P < .0001; Figure 2A) similar to untreated MCAO mice.
FIGURE 2.
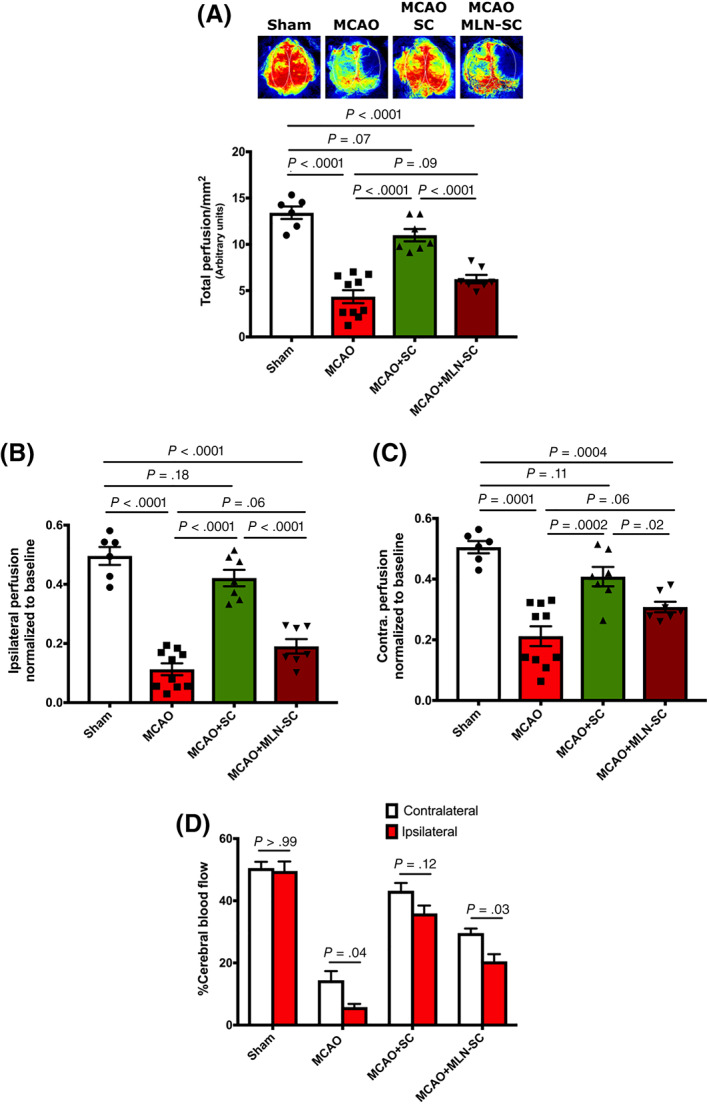
ACE‐2 contributes to the hPMSCs‐based preservation of blood flow in the MCAO model. A, Total cerebral blood flow measured using Laser Speckle imaging. “Perfusion” reflects total cerebral flow signal measured in selected tissue areas. Measurements are expressed as perfusion units (PU), using a fixed scale (arbitrary units). Significant reduction was observed in the perfusion of MCAO+MLN‐treated hPMSCs (n = 7) compared to MCAO+hPMSC (n = 7) (****P < .0001). One‐way ANOVA analysis showed no significant differences in the cerebral blood flow between MCAO+MLN‐treated hPMSC and MCAO (n = 10) groups (not significant [NS], P = .1). B,C, Cerebral perfusion in each pair of ipsilateral (C) and contralateral (D) brain hemispheres were normalized to the average sham total CBF as the reference point. B, A significant decrease in normalized CBF was observed in the ipsilateral hemisphere of both MCAO (78%; ****P < .0001) and MLN‐hPMSC treated MCAO (61%; ****P < .0001) groups was observed compared to the sham group. C, A significant decrease in normalized CBF in the contralateral hemisphere of both MCAO (60%; ****P < .0001) and MLN‐hPMSC treated MCAO (40%; ***P = .0004) groups was observed compared to the sham group. Significant differences were observed in both (B) ipsilateral (****P < .0001) and (C) contralateral (*P = .02) hemispheres of MCAO+MLN‐hPMSCs group compared to MCAO+hPMSCs mice. D, No significant differences were observed in the relative distribution of normalized CBF of contralateral vs ipsilateral hemisphere of both sham (NS; P > .99) and hPMSC‐treated MCAO groups (NS; P = .13). Two‐way analysis of variance (ANOVA) revealed significant differences between contralateral and ipsilateral perfusion of both MCAO (*P = .04) and MLN‐hPMSC treated MCAO (*P = .03) mice. All graph data show the means ± SEM. ACE‐2, angiotensin converting enzyme‐2; CBF, cerebral blood flow; hPMSCs, human placenta mesenchymal stem cells; MCAO, middle cerebral artery occlusion
Normalizing ipsilateral or contralateral blood flow to baseline levels (values obtained from averaged sham total CBF), we found significant decreases in both ipsilateral (0.19 ± 0.30 vs 0.49 ± 0.07 in sham group, P < .0001; Figure 2B) and contralateral (0.31 ± 0.19 vs 0.5 ± 0.09 in sham group, P = .0004; Figure 2C) hemispheres of MLN‐hPMSC treated MCAO mice which was comparable to untreated MCAO group (ipsilateral: 0.11 ± 0.03, P = .06 [Figure 2B]; contralateral: 0.21 ± 0.04, P = .06 [Figure 2C]).
As is shown in Figure 2D, cerebral blood perfusion in both hemispheres of the sham (50% in contralateral vs 49.6% in ipsilateral; P > .99) and hPMSC‐treated MCAO (43% in contralateral vs 36% in ipsilateral; P = .13) groups were comparable (Figure 2D). This normal perfusion significantly shifted towards the contralateral hemispheres in both MCAO (14.5% in contralateral vs 6% in ipsilateral; P = .04) and MLN‐hPMSC treated MCAO (30% in contralateral vs 20% in ipsilateral; P = .03) groups (Figure 2D), suggesting that hPMSC lose their ability to maintain brain blood flow post‐MCAO after MLN treatment to inhibit ACE‐2 activity in hPMSC.
To further validate our findings for a role for hPMSC‐derived ACE‐2 in protection against stroke injury, hPMSCs were transfected with lentivirus‐shACE‐2‐EGFP which reduces expression of ACE‐2 in hPMSCs. Lentiviral transfection efficiency was confirmed by both fluorescent microscopy (Figure 3A) and by Western blot analysis (Figure 3B). Similar to our pharmacological inhibition of ACE‐2 expression by MLN‐4760 (Figures 1 and 2), IP injection of shACE‐2‐hPMSCs (5 × 105 cells in 500 mL HBSS) failed to provide the significant protection against tissue injury (TTC staining; 27.18 ± 2.93) or neurological impairment (5.5 ± 0.96) that was observed in hPMSCs‐treated MCAO mice (infarction: 20.66 ± 3.23, P < .0001 [Figure 3C]; neurological score: 18.33 ± 1.05, P < .0001 [Figure 3D]).
FIGURE 3.
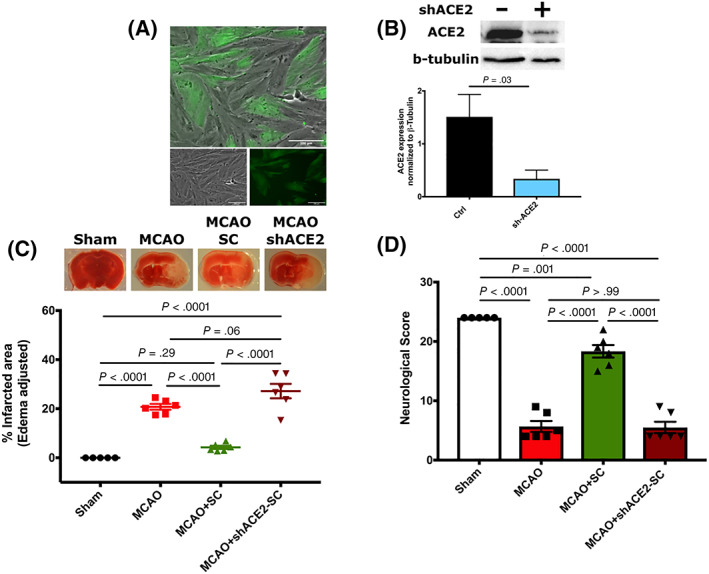
Downregulation of ACE‐2 expression eliminates hPMSCs‐induced protection in the MCAO model. A, Representative image of hPMSCs transfected with lentivirus‐shACE‐2‐GFP. Scale bars = 100 μm. B, Western blot analysis showed significant reduction in protein expression of ACE‐2 in lentiviral‐GFP+‐shACE‐2‐transfected hPMSCs compared to the control hPMSCs (***P = .002). C, TTC staining of brain slices revealed a significant increase in infarcted volume in the ipsilateral hemisphere of MCAO+shACE‐2‐hPMSCs (n = 6) vs MCAO+hPMSC (n = 6) mice (****P < .0001). No significant differences of infarction were observed between MCAO+shACE‐2‐hPMSCs mice vs MCAO (n = 6) (not significant [NS], P = .06). D, Neurological scores were significantly reduced in shACE‐2‐hPMSC treated MCAO (n = 6) vs hPMSC‐treated MCAO (n = 6) mice (****P < .0001). One‐way analysis of variance (ANOVA) showed no significant differences in neurological functions between MCAO group (n = 6) and MCAO mice treated with shACE‐2‐hPMSC (NS, P > .99). In all graphs, data represent means ± SEM. ACE‐2, angiotensin converting enzyme‐2; hPMSCs, human placenta mesenchymal stem cells; MCAO, middle cerebral artery occlusion; TTC, 2,3,5‐triphenyltetrazolium chloride
Furthermore, injection of shACE‐2‐hPMSCs did not correct blood flow disturbances induced by MCAO (Figure 4A‐C). As seen with Laser Speckle imaging, total cerebral perfusion was reduced to 6.94 ± 0.47 in shACE‐2‐hPMSCs treated MCAO group compared to 10.87 ± 0.61 in hPMSCs treated group (P = .0005; Figure 4A). Normalizing this value to baseline level, we found a significant reduction in brain blood flow in both ipsilateral (0.21 ± 0.0 vs 0.46 ± 0.03, P < .0001; Figure 4B) and contralateral (0.44 ± 0.03 vs 0.56 ± 0.03, P = .02; Figure 4C) hemispheres of shACE‐2‐hPMSCs treated MCAO compared to MCAO mice treated with control hPMSC. shACE‐2‐hPMSCs also failed to protect against CBF imbalances observed between ipsilateral (20% perfusion) and contralateral (44%, P < .0001; Figure 4D) hemispheres compared to control hPMSCs given to mice with MCAO (45% in ipsilateral and 54% in contralateral, P = .052).
FIGURE 4.
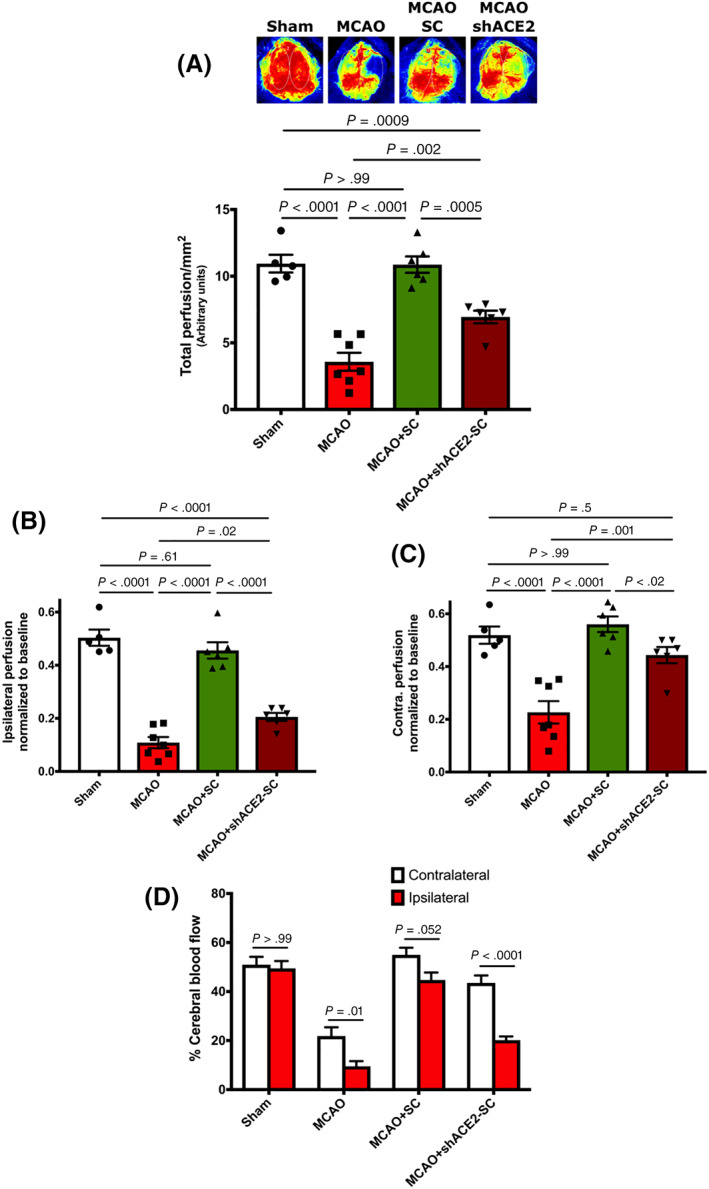
Downregulation of ACE‐2 expression abolishes hPMSCs‐based preservation of blood flow in the MCAO model. A, Laser Speckle imaging revealed a significant reduction in the cerebral blood flow of MCAO+shACE‐2‐hPMSCs compared to MCAO+hPMSC (***P = .0005). B, Significant decrease in blood flow into the ipsilateral hemisphere of MCAO (****P < .0001, n = 7) and MCAO+shACE‐2‐hPMSCs (****P < .0001, n = 6) compared to hPMSC‐treated MCAO mice (n = 6). C, Significant decrease in blood flow into the contralateral hemisphere of MCAO (****P < .0001) and MCAO+shACE‐2‐hPMSCs (*P = .02) brains was detected compared to the MCAO+hPMSC groups. D, Two‐way analysis of variance (ANOVA) revealed significant changes between ipsilateral and contralateral perfusion in MCAO (*P = .01) and MCAO+shACE‐2‐hPMSCs (****P < .0001) groups. No significant differences were detected in ipsilateral and contralateral perfusion of sham (not significant [NS], P > .99) and MCAO+hPMSC (NS, P = .053). All graph data show the means ± SEM. ACE‐2, angiotensin converting enzyme‐2; hPMSCs, human placenta mesenchymal stem cells; MCAO, middle cerebral artery occlusion
In summary, our data are consistent with stem cell derived ACE‐2 playing a critical role in mediating the maintenance of brain perfusion and protection following intraperitoneal hPMSC administration after stroke.
3.2. hPMSC‐derived ACE‐2 products protect the brain against ischemic injury independent of the AT2R receptor
While our data supported a significant role for hPMSC‐derived ACE‐2 activity in the protection afforded by hPMSCs against MCAO (Figures 1, 2, 3, 4), it was still unclear which ACE‐2 derived factors and receptor(s) (AT2R or masR) actually contributed to ACE‐2‐mediated protection offered by hPMSCs.
To investigate the relative contributions of ACE‐2‐Ang 1‐7 axis receptors (AT2R vs MasR) involved in the protection mediated by hPMSC in the MCAO stroke model, first, we blocked AT2R by administering PD123319 (10 mg/kg intravenously) 21 , 27 to mice 1 hour before MCAO surgery.
TTC staining, neurological scores, and laser speckle imaging of these experimental groups showed that AT2R does not appear to contribute to any beneficial effects of hPMSC‐derived ACE‐2. As illustrated in Figure 5A, infarcted areas in PD‐pretreated MCAO mice (25.8%) were comparable to untreated MCAO group (28.5%, P = .36). Furthermore, hPMSCs administered to PD‐pretreated MCAO mice were still able to provide significant protection against ischemic stroke injury (MCAO), as shown by the significant reduction in infarction size (reduced to 3.47 ± 0.98, P < .0001). Neurological function in PD‐pretreated MCAO mice (7.3 ± 1.25) was also significantly improved to 18.17 ± 1.35 in hPMSC treated PD‐MCAO mice (P = .0002; Figure 5B).
FIGURE 5.
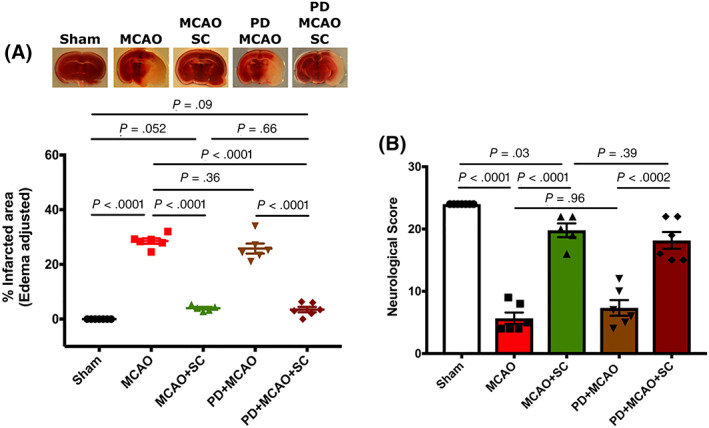
hPMSCs‐derived ACE‐2 products protect against ischemic injury independent of AT2 receptor. The AT2R antagonist, PD 123319 (10 mg/kg i.v.), injected into C57BL/6 mice 1 hour before MCAO surgery to block the AT2R. A, TTC staining of brain slices. No significant differences were found in infarct sizes of PD‐pretreated MCAO and untreated MCAO mice (not significant [NS], P = .36; n = 6). IP injection of hPMSC (5 × 105 cells in 500 mL Hanks Balanced Salts Solution [HBSS]) significantly reduced the infarction in PD‐pretreated MCAO mice (****P < .0001; n = 6). B, Neurological scores were significantly improved in hPMSC‐injected PD‐MCAO group vs untreated PD‐MCAO mice (***P = .0002; n = 6). One‐way analysis of variance (ANOVA) showed no significant differences in neurological functions between MCAO and PD‐pretreated MCAO mice ( NS, P = .39). In all graphs, data represent means ± SEM. ACE‐2, angiotensin converting enzyme‐2; hPMSCs, human placenta mesenchymal stem cells; MCAO, middle cerebral artery occlusion; TTC, 2,3,5‐triphenyltetrazolium chloride
Additionally, hPMSCs produced recovery of CBF, after PD‐pretreatment in MCAO mice (9.95 ± 0.96), to the level observed in hPMSC‐treated MCAO group (12.38 ± 0.70, P = .08). This was significantly higher than brain perfusion observed in the PD‐pretreated MCAO group (5.04 ± 0.69, P = .001; Figure 6A). The decreased levels of blood flow seen in the ipsilateral (0.14 ± 0.02) and contralateral (0.19 ± 0.05) hemispheres of PD‐pretreated MCAO mice were not seen after hPMSC administration (ipsilateral: 0.33 ± 0.03, P = .001; contralateral: 0.41 ± 0.05, P = .01) (Figure 6B,C). Administration of hPMSC further normalized blood distribution between the ipsilateral (30%) and contralateral (36%) hemispheres of PD‐pretreated MCAO group (P = .58) compared to untreated PD‐MCAO mice (ipsilateral: 14%, contralateral: 25%; P = .03) (Figure 6D).
FIGURE 6.

hPMSCs‐derived ACE‐2 products maintain blood perfusion in MCAO mice independent of AT2 receptors. A, Laser speckle measurement of brain perfusion showed no significant differences in total perfusion between PD‐pretreated MCAO (n = 5) and MCAO (n = 6) mice (not significant [NS], P > .99). Significant increases in blood flow into the brain of PD‐pretreated MCAO+hPMSCs (n = 5) mice were observed compared to PD‐pretreated MCAO (**P = .001). B, Significant elevation of blood perfusion observed in the ipsilateral hemisphere of PD‐MCAO+hPMSC mice compared to PD‐MCAO group (**P = .001). C, Significant increase of blood flow into the contralateral hemisphere of hPMSC‐treated PD‐MCAO (*P = .01) observed in comparison with untreated PD‐MCAO mice. There were no significant changes in (B) ipsilateral (NS, P > .99) and (C) contralateral (NS, P = .79) perfusion of MCAO vs PD‐MCAO mice. Two‐tailed Student's t test analysis did not show any significant differences in blood flow between the ipsilateral (NS, P = .55; C) and contralateral (NS, P = .55; D) hemispheres of PD‐pretreated MCAO, regardless of hPMSCs treatment. D, Two‐way analysis of variance (ANOVA) revealed significant disturbances in blood perfusion between ipsilateral and contralateral hemispheres of MCAO (*P = .01) and PD‐MCAO (*P = .03) groups. No significant differences were detected between ipsilateral and contralateral perfusion of sham (NS, P > .99), MCAO+hPMSC (NS, P = .75), and PD‐MCAO+hPMSC (NS, P = .58). All graph data show the means ± SEM. ACE‐2, angiotensin converting enzyme‐2; hPMSCs, human placenta mesenchymal stem cells; MCAO, middle cerebral artery occlusion
3.3. hPMSCs‐ACE‐2 product‐based protection is mediated by masR receptors in the MCAO stroke model
To test how the masR pathway might contribute to the protection provided by ACE‐2, we pretreated mice with masR antagonist A779 (80 mg/kg i.p) 28 1 hour before MCAO surgery. Interestingly, TTC staining revealed no significant protection against infarct development in A779‐pretreated MCAO mice (22.58 ± 2.67) after IP injection of hPMSCs (25.49 ± 1.22, P = .36; Figure 7A). Total CBF (7.05 ± 0.88, P = .78), ipsilateral perfusion (0.16 ± 0.01, P = .57), and contralateral perfusion (0.28 ± 0.02, P = .47) in A779‐pretreated MCAO mice were found to be comparable to their respective hPMSCs treated groups (6.78 ± 0.39, 0.14 ± 0.02, and 0.31 ± 0.04; Figure 7B‐D). In addition, hPMSCs injection failed to protect against the perfusion imbalances that occurred between ipsilateral and contralateral hemispheres of A779‐pretreated MCAO group. As shown in Figure 7E, relative perfusion to the contralateral (32%) remains significantly greater compared to ipsilateral (15%) (P = .0008) hemispheres of A779‐pretreated MCAO mice even after hPMSC administration. Neurological scores were similar in both A779‐pretreated MCAO (5 ± 1) and hPMSC injected A779‐pretreated MCAO groups (6 ± 1.08, P = .52; Figure 7F).
FIGURE 7.
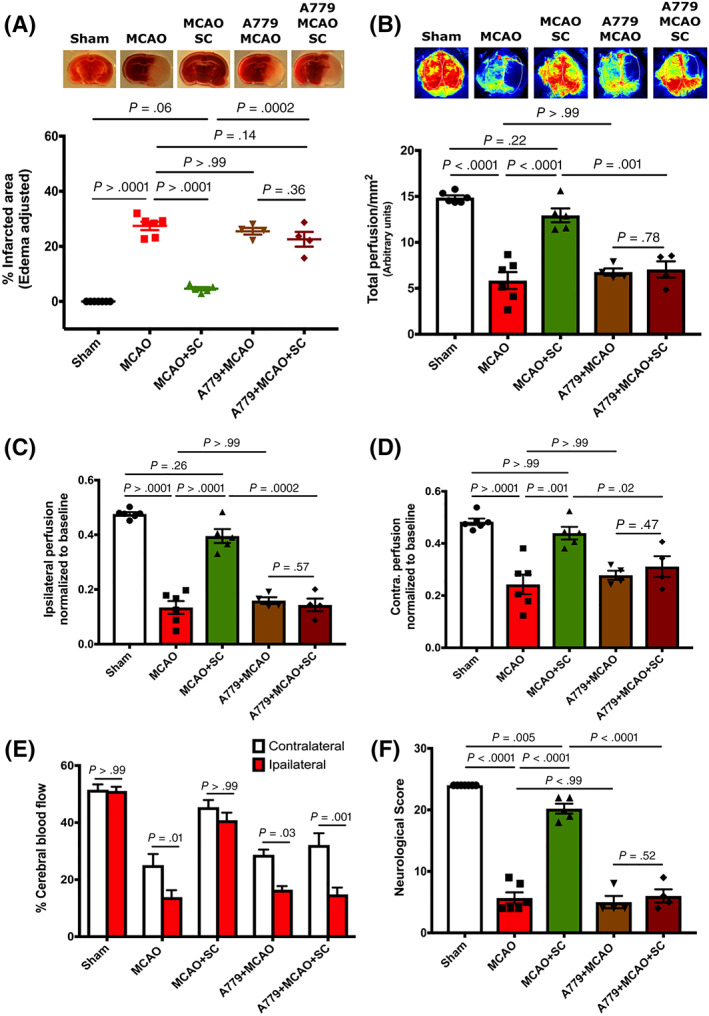
The masR pathway contributes to the ACE‐2‐mediated protection in the MCAO model. Protective potential of hPMSC via masR pathway was tested in the MCAO mice pretreated with masR antagonist (A779; 80 mg/kg i.p). No significant differences were observed in the (A) infarction area (TTC staining; not significant [NS], P = .36) of the MCAO+A779 treated IP with hPMSC (n = 4) compared to untreated MCAO+A779 (n = 4) group. Laser Speckle imaging did not reveal any significant changes in (B) total (NS, P = .79), (C) ipsilateral (NS, P = .57), and (D) contralateral (NS, P = .47) blood perfusion of the MCAO+A779 treated IP with hPMSC (n = 4) compared to untreated MCAO+A779 (n = 4) group. Two‐way analysis of variance (ANOVA) showed significant disturbances in blood perfusion between ipsilateral and contralateral hemispheres of MCAO (*P = .01), A779‐MCAO (*P = .03), and hPMSC‐treated A779‐MCAO (***P = .0008) groups (E). There were no significant differences in the neurological scores (NS, P = .52) of the MCAO+A779 treated IP with hPMSC (n = 4) compared to untreated MCAO+A779 (n = 4) group (F). All graph data show the means ± SEM. ACE‐2, angiotensin converting enzyme‐2; hPMSCs, human placenta mesenchymal stem cells; MCAO, middle cerebral artery occlusion; TTC, 2,3,5‐triphenyltetrazolium chloride
Cumulatively, these data are consistent with hPMSCs‐associated ACE‐2 mediating its protection via the masR pathway, since the masR antagonist (A779), but not AT2R antagonist (PD 123319), eliminated the beneficial effects of hPMSCs in protecting against tissue injury and blood flow dysregulation after stroke.
4. DISCUSSION
Thromboembolic strokes make up 87% of all stroke incidence and remain the leading cause of neurologically based morbidity in adults. 1 Stroke treatment is limited therapeutically to thrombolytics (t‐PA) and anti‐thrombotics. 6 , 7 Mechanical thrombectomy for large vessel occlusion, in susceptible patients, is now also a mainstay of therapy. However, because of the acute, and typically irreversible nature of stroke injury, thrombolytics like t‐PA must be administered within the presently accepted 4.5‐hour therapeutic window to achieve any benefits. However, despite achieving reperfusion, injury after reperfusion includes a devastating progressive vasoconstriction which by 24 hours “strangles” the brain, to produce large infarcts. Stroke therapies that can limit such deleterious effects of I/R injury are desperately needed.
Presently, SCTs are being intensively studied as powerful and promising approaches for stroke therapy. 8 , 29 Historically, stem cells have been presumed to engraft within the poststroke brain and differentiate into cells, which may restitute damaged tissue. 10 More recent studies suggest that the acute benefits of stem cell therapy instead reflect “paracrine signaling actions”. 11 , 12 , 13 , 14 Paracrine mediators released by stem cells, also known as “secretome” include a wide range of neurotrophic factors (eg, cytokines, chemokines, growth factors) and EVs. 11 , 16 , 30 Thus, there are diverse immunomodulatory, angiogenic, antiapoptotic, and antioxidant potential benefits in stroke. 11 In our recently published paper, we demonstrated that intraperitoneal (IP) administration of hPMSC, at the time of reperfusion, produced highly significant protection against injury in our filament induced MCAO model. When we used cell tracer to follow IP injected hPMSC within the peritoneum, blood, and brain, we found that the greatest number of cells appeared in the vascular compartment at 6 hours following administration; however, this corresponded to only <1% of injected hPMSC (~5000 cells in the 2.5 mL of blood when 5 × 105 hPMSCs were administered). As we have not yet successfully observed any hPMSC cells penetrating into the brain, 15 our previous data appear to be most consistent with the paracrine function of hPMSCs related to the release of EVs from the stem cells. We further reported that manipulations which suppress and increase EV formation, also decrease and increase the protection afforded by IP‐injected hPMSCs. In this sense, IP‐injected hPMSCs apparently provides a continuous source of EVs into the circulation which helps to maintain cerebrovascular perfusion and protect the brain tissue against ischemic injury. 15
While we demonstrated that beneficial effects of hPMSCs appear to be mediated by the release of EVs, it has also been difficult to track the distribution of EVs upon their release from hPMSCs. Several studies have introduced methods for labeling and imaging EVs 31 , 32 which might provide a possible tool to study EVs trafficking in vitro and in vivo in future studies to define EVs biodistribution, and the relationship to their therapeutic activity.
Several prior studies supporting ACE2/Ang 1‐7/Mas axis activation of RAS as a therapeutic target in stroke therapy. It has been shown that intracerebroventricular (ICV) injection of ACE2 or the ACE2 product (Ang 1‐7) for 7 days prior to stroke induction (using the endothelin‐1 stroke model) was able to suppress stroke injury. 18 , 19 , 24 , 33 These studies did suggest that ACE‐2 could provide stroke protection, reduce infarct size, and improve neurological function, but they do not demonstrate a rational approach for treating stroke since stroke therapies in human patients are not administered before stroke onset, but later after stroke onset. 19 , 24 , 34 Additionally, because protective effects of ACE‐2/Ang1‐7 in those studies required extended (7d) preadministration of ACE‐2 directly into the brain ventricular space 17 such approaches address ventricular rather than vascular targets and have limited applicability for the treatment of strokes. 30 Other minimally invasive strategies including systemic administration of an ACE2 activator (diminazene aceturate [DIZE]) or oral administration of Ang 1‐7 have been recently developed, but still have several potential limitations. For example, although prestroke systemic injection of DIZE demonstrated protection against stroke‐induced infarction, the efficacy of DIZE might be reduced by binding to albumin in the bloodstream. 30 As a peptide molecule, Ang 1‐7 has been shown to undergo rapid degradation in the stomach when administered orally and as a small molecule it is cleared rapidly from the circulation.
Regarding the multifactorial nature of stroke, in addition to developing promising therapies, novel delivery approaches also need to be identified. In this regard, hPMSCs could be considered as a potential candidate to elevate the levels of neuroprotective molecules such as ACE2 or Ang 1‐7 in the bloodstream or at the site of injury when administered following stroke. Here, we report for the first time that hPMSC abundantly express ACE‐2 protein (Figure 1A). Compared to endothelial cells, a cell type which specifically expresses ACE‐2, we found greater than threefold greater expression in hPMSCs. Furthermore, mice injected IP with hPMSC, which were pretreated with the specific ACE‐2 inhibitor (10 mM) MLN‐476035 or lentivirus‐shRNA‐ACE‐2, no longer showed protection against MCAO, with tissue injury and neurological behavior similar to that seen in untreated MCAO (Figures 1 and 3). We also demonstrated that CBF and infarct size in stroke were significantly improved by IP administration of hPMSC in a vascular MasR dependent fashion (Figure 7). To our knowledge, these in vivo data provide initial evidence, which supports our hypothesis that hPMSC‐based protection in MCAO is mediated through the ACE‐2/Ang 1‐7/MasR axis of RAS, but it is likely that the EVs are providing a multifactorial protection. Based on our evidence, hPMSC and EVs based therapy in stroke appears to actively convert vasoconstrictor mediators, for example, Angiotensin II into vasodilatory and anti‐inflammatory substances. Therefore, hPMSC/EVs actions are unique in not being directly active, but rather enzymatically change the vasoconstrictor/vasodilator ratio to achieve these benefits. Further studies are required to determine what other factors may be involved, and whether it is critical for specific levels of some factors to be released at certain times while others are more important at different times, which would make drug targeting more difficult. Related more specifically to repair mechanisms, we can speculate (based on what is known about stem cells from multiple sources) that hPMSCs release neurotropic factors that promote angiogenesis, neuromodulation, and neurogenesis. Future work will focus on identifying potential repair pathways induced by hPMSCs and EVs.
With respect to COVID‐19 pathophysiology, because the SARS‐CoV‐2 virus infects endothelial cells using ACE2 as a binding receptor and triggers internalization of ACE2, the normal catabolism of Ang II to Ang1‐7/Ang1‐9 in the vascular compartment is inhibited. 35 , 36 Recent studies have suggested that the endothelial ACE2 deficiency activates and intensifies hyper‐coagulation pathways due to the dysregulation between ACE‐Ang II‐AT1R axis and the ACE2‐Ang 1‐7/1‐9‐masR/AT2R axis. 37 , 38 , 39 , 40 Verdecchia et al showed that ACE2 catalytic capacity is lost upon SARS‐CoV‐2 penetration of endothelial cells 14 leading to derangement of several endothelial regulated functions including vasoregulation, inflammation, and thrombosis.
Such loss of ACE2 activity may increase stroke risk and provoke other thrombotic complications seen in COVID‐19. Early studies revealed that ischemic and hemorrhagic stroke is a complication in ~6% of COVID‐19 patients. In COVID‐19 patients, endothelial activation has been shown to be associated with elevated circulating markers of coagulation (eg, D‐dimers) 41 , 42 , 43 , 44 and inflammation (eg, TNF‐a, IL‐6, IL‐2, and MCP‐1). 45 Interestingly, COVID‐19 patients usually present with elevated platelet numbers, highly increased fibrinogen and slightly prolonged prothrombin and activated partial thromboplastin time. 46 , 47 These observations clearly suggest that in the brain, COVID‐19 infection of endothelial cells may result in cerebral endothelial dysfunction, inflammation, and heightened procoagulant state, culminating in the intensified microvascular stroke pathology often seen in COVID‐19. 35 , 48 While there is a growing movement to treat COVID‐19 patients with anticoagulants prophylactically to prevent thrombotic events, this approach does not hold for primary stroke prevention, and comes with increased bleeding risk. 49 Therefore, correcting the underlying mechanisms leading to intensified thrombosis is likely a more effective alternative, particular for seriously ill COVID‐19 patients. In present study, we have now shown that CBF, infarct size, and neurological outcomes in stroke are dramatically and significantly improved by hPMSC in a stem cell‐ACE2‐dependent/vascular MasR‐dependent fashion. While antithrombotic and anti‐inflammatory properties of hPMSCs‐derived ACE2 in the setting of brain vascular endothelium or SARS‐CoV‐2 have not yet been tested, this previously understudied effect of ACE2 and ACE2 products reveals an intensely important axis which may be exploited therapeutically to treat COVID‐19 using hPMSC which express, and are a source of, ACE2. Because COVID19‐mediated and experimental stroke models are both thrombotic phenomena exhibiting deficits in ACE2/MasR signaling, the successful demonstration that SARS‐CoV‐2‐induced ACE2 suppression mediates increased thrombotic risk and that stem cell ACE2/MasR activators may eliminate such risk could define highly novel, valuable, innovative, and safe approaches for clinically managing COVID‐19 stroke, and other thrombotic complications of COVID‐19 (pulmonary embolism).
Future studies will be necessary to investigate the potential protective mechanisms of hPMSC‐derived ACE‐2 in the setting of brain vascular endothelium of SARS‐CoV‐2 infection.
5. CONCLUSION
In summary, our study demonstrates that hPMSC administration provides powerful and acute protection against stroke injury when given immediately after reperfusion in the MCAO stroke model. This protection appears to be mediated through ACE‐2/MasR signaling pathway. Mechanistically, our study shows that ACE‐2 products (Ang‐1‐7 and/or Ang1‐9) maintain CBF in the post‐MCAO brain, which prevents development of ischemic reperfusion injury. These central findings of this study may have important relevance for the development of therapies for acute stroke treatment. While not yet tested, the observed suppression of ACE‐2 in COVID‐19 affected individuals, with the accompanying dysregulation of blood flow and coagulation, might also be responsive to this form of therapy. Thus, the rapid delivery of intraperitoneal hPMSCs and/or vesicles might have applicability in both acute ischemic stroke and vascular injury associated with SARS‐CoV‐2 infection.
CONFLICT OF INTEREST
A patent application by authors J.S.A., Y.W., and M.B. “Protective effect of intraperitoneal injection of human placenta stem cells in stroke” has been submitted to the LSUHSC‐Shreveport office of sponsored programs and technology transfer. R.E.K. declared expert testimony of medico‐legal cases. K.Y.S. declared research funding of COVID‐19 Intramural Grant (Co‐PI). 07/01/20‐12/31/21 Stem cell/ACE2 protects against cerebral thrombosis and inflammation in COVID‐19 models. All of the other authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
M.B.: conceptualization, methodology, data analysis and interpretation, collecting and assembly of data, writing—original draft; J.S.A.: conceptualization, methodology, resources, writing—review and editing, financial support; S.V.: methodology, collecting and assembly of data; L.A.W., J.W.Y.: data analysis and interpretation; Y.W.: resources; K.Y.S.: resources, writing—review and editing; O.C., R.E.K.: financial support.
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Health grants (NIGMS 5 P20 GM121307‐02 Center for Redox Biology and Cardiovascular Disease). J.S.A. was supported by the Department of Neurology, LSUHSC‐Shreveport. M.B. was supported by Intramural Malcolm Feist predoctoral fellowship (Center for Cardiovascular Diseases and Sciences‐LSUHSC‐Shreveport).
Barzegar M, Vital S, Stokes KY, et al. Human placenta mesenchymal stem cell protection in ischemic stroke is angiotensin converting enzyme‐2 and masR receptor‐dependent. Stem Cells. 2021;39(10):1335–1348. 10.1002/stem.3426
Funding information Malcolm Feist Predoctoral Fellowship‐CCDS LSUHSC‐Shreveport; National Institutes of Health grants, Grant/Award Number: P20 GM121307‐02; Department of Neurology, LSUHSC‐Shreveport
Contributor Information
Mansoureh Barzegar, Email: mansoureh.barzegar@lsuhs.edu.
Shantel Vital, Email: shantel.vital@lsuhs.edu.
Karen Y. Stokes, Email: karen.stokes@lsuhs.edu
Yuping Wang, Email: yuping.wang@lsuhs.edu.
Jungmi Winny Yun, Email: jyun527@gmail.com.
Luke A. White, Email: luke.white@lsuhs.edu
Oleg Chernyshev, Email: oleg.chernyshev@lsuhs.edu.
Roger E. Kelley, Email: roger.kelley@lsuhs.edu
Jonathan S. Alexander, Email: jonathan.alexander@lsuhs.edu.
DATA AVAILABILITY STATEMENT
This study did not generate any new unique reagents, data sets, or code. Further information and requests for resources and reagents should be directed to the corresponding author, Jonathan S. Alexander (jalexa@lsuhsc.edu).
REFERENCES
- 1. Kalladka D, Muir KW. Brain repair: cell therapy in stroke [in Eng]. Stem Cells Cloning. 2014;7:31‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Putaala J. Ischemic stroke in young adults [in Eng]. Continuum. 2020;26(2):386‐414. [DOI] [PubMed] [Google Scholar]
- 3. Putaala J, Yesilot N, Waje‐Andreassen U, et al. Demographic and geographic vascular risk factor differences in European young adults with ischemic stroke: the 15 cities young stroke study [in Eng]. Stroke. 2012;43(10):2624‐2630. [DOI] [PubMed] [Google Scholar]
- 4. von Sarnowski B, Putaala J, Grittner U, et al. Lifestyle risk factors for ischemic stroke and transient ischemic attack in young adults in the Stroke in Young Fabry Patients Study [in eng]. Stroke. 2013;44(1):119‐125. [DOI] [PubMed] [Google Scholar]
- 5. Kolikonda MK, Jandrasupalli KK, Lippmann S. Association of Coronavirus Disease 2019 and stroke: a rising concern [in Eng]. Neuroepidemiology. 2020;54(5):370‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Attwell D, Buchan AM, Charpak S, Lauritzen M, MacVicar BA, Newman EA. Glial and neuronal control of brain blood flow [in Eng]. Nature. 2010;468(7321):232‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mohammadi MT. Overproduction of nitric oxide intensifies brain infarction and cerebrovascular damage through reduction of claudin‐5 and ZO‐1 expression in striatum of ischemic brain [in Eng]. Pathol Res Pract. 2016;212(11):959‐964. [DOI] [PubMed] [Google Scholar]
- 8. Yilmaz G, Vital S, Yilmaz CE, Stokes KY, Alexander JS, Granger DN. Selectin‐mediated recruitment of bone marrow stromal cells in the postischemic cerebral microvasculature [in Eng]. Stroke. 2011;42(3):806‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Broughton BR, Lim R, Arumugam TV, et al. Post‐stroke inflammation and the potential efficacy of novel stem cell therapies: focus on amnion epithelial cells [in Eng]. Front Cell Neurosci. 2012;6:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhu MX, Wan WL, Li HS, et al. Early immune reconstitution after hematopoietic stem cell transplantation [in Chinese]. Beijing Da Xue Xue Bao Yi Xue Ban. 2016;48(3):515‐522. [PubMed] [Google Scholar]
- 11. Merino‐Gonzalez C, Zuniga FA, Escudero C, et al. Mesenchymal stem cell‐derived extracellular vesicles promote angiogenesis: potencial clinical application [in Eng]. Front Physiol. 2016;7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hofer HR, Tuan RS. Secreted trophic factors of mesenchymal stem cells support neurovascular and musculoskeletal therapies [in Eng]. Stem Cell Res Ther. 2016;7(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yilmaz G, Alexander JS, Erkuran Yilmaz C, Granger DN. Induction of neuro‐protective/regenerative genes in stem cells infiltrating post‐ischemic brain tissue [in Eng]. Exp Transl Stroke Med. 2010;2(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tang YH, Pennington LA, Scordino JW, Alexander JS, Lian T. Dynamics of early stem cell recruitment in skin flaps subjected to ischemia reperfusion injury [in Eng]. Pathophysiology. 2016;23(3):221‐228. [DOI] [PubMed] [Google Scholar]
- 15. Barzegar M, Wang Y, Eshaq RS, et al. Human placental mesenchymal stem cells improve stroke outcomes via extracellular vesicles‐mediated preservation of cerebral blood flow [in Eng]. EBioMedicine. 2020;63:103161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hao L, Zou Z, Tian H, et al. Stem cell‐based therapies for ischemic stroke [in Eng]. Biomed Res Int. 2014;2014:468748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dailey T, Metcalf C, Mosley YI, et al. An update on translating stem cell therapy for stroke from bench to bedside [in Eng]. J Clin Med. 2013;2(4):220‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mecca AP, Regenhardt RW, O'Connor TE, et al. Cerebroprotection by angiotensin‐(1‐7) in endothelin‐1‐induced ischaemic stroke [in Eng]. Exp Physiol. 2011;96(10):1084‐1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pena Silva RA, Heistad DD. Promising neuroprotective effects of the angiotensin‐(1‐7)‐angiotensin‐converting enzyme 2‐Mas axis in stroke [in Eng]. Exp Physiol. 2014;99(2):342‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu Z, Liu J, Xiao M, et al. Mesenchymal stem cell‐derived microvesicles alleviate pulmonary arterial hypertension by regulating renin‐angiotensin system [in Eng]. J Am Soc Hypertens. 2018;12(6):470‐478. [DOI] [PubMed] [Google Scholar]
- 21. Heitsch H, Brovkovych S, Malinski T, Wiemer G. Angiotensin‐(1‐7)‐stimulated nitric oxide and superoxide release from endothelial cells [in Eng]. Hypertension. 2001;37(1):72‐76. [DOI] [PubMed] [Google Scholar]
- 22. De Souza AM, Lopes AG, Pizzino CP, et al. Angiotensin II and angiotensin‐(1‐7) inhibit the inner cortex Na+‐ATPase activity through AT2 receptor [in Eng]. Regul Pept. 2004;120(1‐3):167‐175. [DOI] [PubMed] [Google Scholar]
- 23. Sun J, Luo Z, Wang G, et al. Notch ligand Jagged1 promotes mesenchymal stromal cell‐based cartilage repair [in Eng]. Exp Mol Med. 2018;50(9):126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bennion DM, Haltigan EA, Irwin AJ, et al. Activation of the neuroprotective angiotensin‐converting enzyme 2 in rat ischemic stroke [in Eng]. Hypertension. 2015;66(1):141‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nouraee C, Fisher M, Di Napoli M, et al. A brief review of edema‐adjusted infarct volume measurement techniques for rodent focal cerebral ischemia models with practical recommendations [in Eng]. J Vasc Interv Neurol. 2019;10(3):38‐45. [PMC free article] [PubMed] [Google Scholar]
- 26. Tang YH, Thompson RW, Nathan CA, Alexander JS, Lian T. Stem cells enhance reperfusion following ischemia: validation using laser speckle imaging in predicting tissue repair [in Eng]. Laryngoscope. 2018;128(6):E198‐e205. [DOI] [PubMed] [Google Scholar]
- 27. Bivalacqua TJ, Dalal A, Lambert DG, et al. Effects of candesartan and PD123319 on responses to angiotensin II in the anesthetized mouse [in Eng]. J Am Soc Nephrol. 1999;10(suppl 11):S98‐S100. [PubMed] [Google Scholar]
- 28. Lee S, Evans MA, Chu HX, et al. Effect of a selective mas receptor agonist in cerebral ischemia in vitro and in vivo [in Eng]. PLoS One. 2015;10(11):e0142087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sullivan R, Duncan K, Dailey T, Kaneko Y, Tajiri N, Borlongan CV. A possible new focus for stroke treatment – migrating stem cells [in Eng]. Expert Opin Biol Ther. 2015;15(7):949‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cunningham CJ, Redondo‐Castro E, Allan SM. The therapeutic potential of the mesenchymal stem cell secretome in ischaemic stroke [in Eng]. J Cereb Blood Flow Metab. 2018;38(8):1276‐1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dabrowska S, Del Fattore A, Karnas E, et al. Imaging of extracellular vesicles derived from human bone marrow mesenchymal stem cells using fluorescent and magnetic labels [in Eng]. Int J Nanomedicine. 2018;13:1653‐1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Busato A, Bonafede R, Bontempi P, et al. Labeling and magnetic resonance imaging of exosomes isolated from adipose stem cells [in Eng]. Curr Protoc Cell Biol. 2017;75:3.44.1‐3.44.15. [DOI] [PubMed] [Google Scholar]
- 33. Pena‐Silva RA, Heistad DD. Stages in discovery: angiotensin‐converting enzyme type 2 and stroke. Hypertension. 2015;66:15‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiang T, Yu JT, Zhu XC, et al. Angiotensin‐(1‐7) induces cerebral ischaemic tolerance by promoting brain angiogenesis in a Mas/eNOS‐dependent pathway [in Eng]. Br J Pharmacol. 2014;171(18):4222‐4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Panigada M, Bottino N, Tagliabue P, et al. Hypercoagulability of COVID‐19 patients in intensive care unit. A report of thromboelastography findings and other parameters of hemostasis [in eng]. J Thromb Haemost. 2020;18(7):1738‐1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ferrario CM, Trask AJ, Jessup JA. Advances in biochemical and functional roles of angiotensin‐converting enzyme 2 and angiotensin‐(1‐7) in regulation of cardiovascular function [in Eng]. Am J Physiol Heart Circ Physiol. 2005;289(6):H2281‐H2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rice GI, Thomas DA, Grant PJ, et al. Evaluation of angiotensin‐converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism [in Eng]. Biochem J. 2004;383(pt 1):45‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kucharewicz I, Pawlak R, Matys T, Pawlak D, Buczko W. Antithrombotic effect of captopril and losartan is mediated by angiotensin‐(1‐7) [in Eng]. Hypertension. 2002;40(5):774‐779. [DOI] [PubMed] [Google Scholar]
- 39. Fraga‐Silva RA, Pinheiro SV, Gonçalves AC, et al. The antithrombotic effect of angiotensin‐(1‐7) involves mas‐mediated NO release from platelets [in Eng]. Mol Med. 2008;14(1‐2):28‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS‐CoV‐2 infection [in Eng]. Eur J Intern Med. 2020;76:14‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Escher R, Breakey N, Lämmle B. Severe COVID‐19 infection associated with endothelial activation [in Eng]. Thromb Res. 2020;190:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus‐infected pneumonia in Wuhan, China [in Eng]. JAMA. 2020;323(11):1061‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China [in Eng]. Lancet. 2020;395(10223):497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li X, Wang L, Yan S, et al. Clinical characteristics of 25 death cases with COVID‐19: a retrospective review of medical records in a single medical center, Wuhan, China [in Eng]. Int J Infect Dis. 2020;94:128‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mehta P, McAuley DF, Brown M, et al. COVID‐19: consider cytokine storm syndromes and immunosuppression [in Eng]. Lancet. 2020;395(10229):1033‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ranucci M, Ballotta A, Di Dedda U, et al. The procoagulant pattern of patients with COVID‐19 acute respiratory distress syndrome [in Eng]. J Thromb Haemost. 2020;18:1747‐1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Klok FA, Kruip MJHA, van der Meer NJM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID‐19 [in Eng]. Thromb Res. 2020;191:145‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oxley TJ, Mocco J, Majidi S, et al. Large‐vessel stroke as a presenting feature of Covid‐19 in the young [in Eng]. N Engl J Med. 2020;382(20):e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Watson RA, Johnson DM, Dharia RN, et al. Anti‐coagulant and anti‐platelet therapy in the COVID‐19 patient: a best practices quality initiative across a large health system [in Eng]. Hosp Pract. 2020;48:169‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any new unique reagents, data sets, or code. Further information and requests for resources and reagents should be directed to the corresponding author, Jonathan S. Alexander (jalexa@lsuhsc.edu).