

Abstract
Poor nutrition, lack of exercise, and genetic predisposition all contribute to the growing epidemic of obesity. Overweight/obesity create an environment of chronic inflammation that leads to negative physiological and neurological outcomes, such as diabetes, cardiovascular disease, and anxiety/depression. While the whole body contributes to metabolic homeostasis, the neuroimmune system has recently emerged as a key regulator of metabolism. Microglia, the resident immune cells of the brain, respond both directly and indirectly to dietary fat, and the environment in which microglia develop contributes to their responsiveness later in life. Thus, high maternal weight during pregnancy may have consequences for microglial function in offspring. Here, we discuss the most recent findings on microglia signaling in overweight/obesity with a focus on perinatal programming.
Keywords: Microglia, obesity, development, circuit refinement, priming
The obesity epidemic
Excess weight gain leading to a diagnosis of overweight or obesity has become increasingly prevalent, with almost 40% of the worldwide population diagnostically overweight or obese [1]. Research has demonstrated a strong genetic contribution to obesity risk, but other factors, such as the availability of high-calorie low-nutrition foods, lack of exercise, and stress, all play a significant role in the increasing obesity epidemic [2,3]. Further, the COVID-19 pandemic has led to a disheartening rise in overweight/obesity, particularly in children and juveniles [4]. Overweight/obesity is associated with multiple comorbidities, ranging from metabolic to neurological. Dementia, Alzheimer’s disease (AD), and anxiety/depression are all correlated with obesity, though often the mechanisms or clinical significance of such correlations is unclear [5–9]. Regardless, the staggering rise of obesity, in conjunction with the known and suspected comorbidities, renders investigations into obesity as a critical area for study.
Obesity creates an environment of chronic inflammation, and microglia, the resident immune cells of the brain, both respond to and propagate inflammation. Microglia are essential for brain development and have been implicated in numerous disease pathologies [10]. Recent studies have also highlighted the relevance of microglial ‘priming’ (see Glossary) or the two-hit hypothesis, in which an initial insult (e.g. stress or high-fat diet exposure during early life) primes microglia to over-respond to future insults (e.g. bacterial infection) [11–14]. In 2014, an estimated 38.9 million pregnant women worldwide were overweight or obese [15], and almost 20% of children are considered obese [16]; thus, a substantial proportion of the population are at risk for these recently defined microglial susceptibilities.
Is there an ethologically relevant model of obesity?
Studies investigating the impact of overweight/obesity on physiology span a wide range of model systems and encompass human clinical data. Rodent models of increased dietary saturated fat content are frequently used to model overweight/obesity; 10% kcal from fat is generally used for ‘low-fat diet’, while 45% and 60% are often used for ‘high-fat diet (HFD)’ and ‘very high-fat diet (VHFD)’ (or ‘diet-induced obesity (DIO)’) respectively. When modeling overweight/obesity, use of the very high-fat diet can expedite studies because mice will gain more weight quickly, but this very high fat content may not be as translatable to a human high-fat diet [17]. Unfortunately, rodent dietary compositions and high-fat diet paradigms (e.g., the source of the fat and length of time the animal is on high-fat diet) are not standard. This is, in part, due to the fact that ‘control’ diets are also not standard, with some researchers using standard chow and others using purified control diets [18,19]. Additionally, some institutes use both standard chow and breeding chow, which differ substantially in their fat content (standard chow often containing 13%kcal from fat, and breeding chow up to 24% kcal from fat) and other nutrient composition. Thus, when studying overweight/obesity, researchers should strive to be transparent about their methods by including details of dietary compositions used, including for control diets.
It takes a village: metabolic homeostasis is a whole-body effort
Evolutionarily, humans are programmed to seek out nutrient-dense foods, particularly those rich in fats, to ensure ample energy sources. Upon consumption of an energy-dense meal, excess fats are stored in adipocytes in adipose tissue and excess carbohydrates are converted to glycogen stores primarily in liver and skeletal muscle tissue [20]. During a fast (such as overnight while one is sleeping), fats are mobilized for energy while glycogen stores are broken down to restore decreasing blood glucose levels to a homeostatic state [21]. Dysregulated blood glucose levels are a hallmark of metabolic disease and can result in severe complications throughout the body, such as cardiovascular disease.
In the case of consistent excess nutrient consumption, blood glucose levels will remain elevated and stimulate the secretion of insulin from the endocrine pancreas [21]. Consistently elevated insulin levels can result in insulin resistance – a metabolic condition associated with major depressive disorder [22] where peripheral tissues become less responsive to insulin and unable to clear elevated circulating glucose from the blood. Excess nutrient consumption additionally results in increasing levels of adipokine secretion (adipose-derived cytokines) [23,24]. Adipokines are pro-inflammatory molecules that act locally and travel through the vascular system, contributing to chronic, whole-body inflammation. A variety of adipokines have been described, but seminal work linking obesity and inflammation first identified tumor necrosis factor-α (TNF-α) as an obesity-induced adipokine that influenced glucose homeostasis [25]. More recent work has identified adipose tissue-resident macrophages as the primary source of adipokine secretion [23,24].
The hypothalamus, and in particular the arcuate nucleus of the hypothalamus (ARC), plays a pivotal role in controlling food intake via nutrient sensing and integration of peripheral hormones such as ghrelin and leptin [26]. Ghrelin, which is secreted by the stomach, informs the brain of diminished nutrient status via signaling on hypothalamic neuropeptide Y/agouti-related peptide (AgRP) and proopiomelanocortin (POMC) neurons [27]. Leptin is produced from adipocytes and acts on the same neuronal populations, but instead informs the brain of sufficient nutrient status, thus resulting in diminished energy intake and increased energy expenditure [26]. In response to increased caloric intake (e.g. high-fat diet, a significant contributor to overweight/obesity), saturated fatty acids (SFAs) create an inflammatory response in the hypothalamus [28]. The inflammatory response to diet is classically thought to occur over days and weeks, and this timeline is true for peripheral inflammation. However, inflammatory responses in the hypothalamus have been visualized as early as 24-hours post dietary fat increase [28]. In persistent high-fat diet studies, bone marrow-derived myeloid cells can infiltrate the hypothalamus and further propagate the inflammatory response [29]. Further studies are warranted to dissect the contribution of acute versus chronic inflammatory responses to high-fat diet-induced pathologies. Of note, how lipids enter the brain is highly complex. While SFAs are thought to cross the blood brain barrier, their entry into the brain is likely tightly regulated. Circumventricular organs – regions of highly fenestrated capillaries where polypeptide hypothalamic hormones exit the brain without disrupting the blood brain barrier – are rich in nutrient sensing ependymal cells called tanycytes, and interesting work has suggested a key role for tanycytes, particularly in the hypothalamus, in mediating lipid homeostasis [30–32]. Different brain regions likely have different energetic needs and routes of acquisition for circulating factors, including fatty acids, and the interactions with and transport of lipids across the blood brain barrier has been recently reviewed [33,34], though this is an area where future study will be informative.
An emerging pattern: the innate immune system and obesity
Recent studies have highlighted the importance of the innate immune system, in particular toll-like receptors (TLRs), as a molecular link between obesity and inflammation. Microglia constitutively express TLR1–13, and microglial TLR signaling has been implicated in a number of disease pathologies [35,36]. TLRs are pattern-recognition receptors that classically initiate a pro-inflammatory cascade in response to infectious pathogens (e.g. bacterial, viral, fungal) [37]. TLR activity has been thoroughly characterized in innate immune cells, including macrophages and monocytes, but increasing evidence has demonstrated that TLRs are expressed and functionally relevant in a variety of other cell types. Among the TLRs, TLR2 and TLR4 are the most well-characterized for their roles in obesity and metabolic homeostasis (Figure 1). TLR2 is increased in muscle and white adipose tissue in a mouse VHFD model of obesity, and contributes to insulin resistance, particularly SFA-induced insulin resistance, in skeletal muscle [38,39]. In obese human patients, TLR4 expression is elevated in muscle tissue and correlated with insulin resistance severity [40]. In mouse models of obesity, Tlr4 expression is increased in adipose tissue as well as in the hypothalamus [41,42]. Bone marrow transplantation from TLR4-deficient mice demonstrated that TLR4 signaling in hematopoietic-derived cells promotes insulin resistance in a mouse model of high-fat diet [43]. Reduction of HFD-induced (14 weeks) Tlr4 over-expression in the ARC resulted in weight loss and improved glucose homeostasis in rats [44]. Both TLR2 and TLR4 are cell-surface receptors that canonically respond to microbial components such as the bacterial endotoxin lipopolysaccharide (LPS). In experimental models of TLR activation, LPS is often derived from Escherichia coli. Interestingly, recent studies have demonstrated that the source of LPS has profound effects on inflammatory responses and metabolic homeostasis [45]. Until recently, LPS was not thought to enter the brain, and thus neuroinflammatory responses to LPS administration were thought to be dependent on peripheral responses [46]. Interestingly, plasma LPS has been shown to be increased in response to high-fat diet, potentially through changes to the gut microbiome, and both palmitate and lauric acid, a fatty-acid component of LPS, are sufficient to initiate TLR signaling in vitro [47–51]. Given that LPS has been shown to enter the rat brain under healthy conditions [46], and that the origin of LPS can have strikingly different effects on neuroinflammatory and metabolic outcomes [45], more work will need to be done to dissect the origin and contribution of microbiota-derived LPS in HFD. At this point, studies involving exogenous LPS administration represent a tool to study TLR activation, rather than a model to study LPS-dependent effects of dietary fat intake. Studies have also suggested that non-immune cell TLR expression may play a role in obesity. TLR2 is upregulated with age or high-fat diet in POMC neurons, and mice lacking TLR2 in non-immune cells showed an increased susceptibility to high-fat diet-induced weight gain, suggesting that neuronal TLR2 is protective against age- or high-fat diet-dependent metabolic challenge [52]. Thus, there is a clear role for the innate immune system, and in particular TLRs, in the pathology of obesity and associated metabolic comorbidities, though much work is still needed.
Figure 1. TLR2 and TLR4 react to dietary fat content throughout the body.
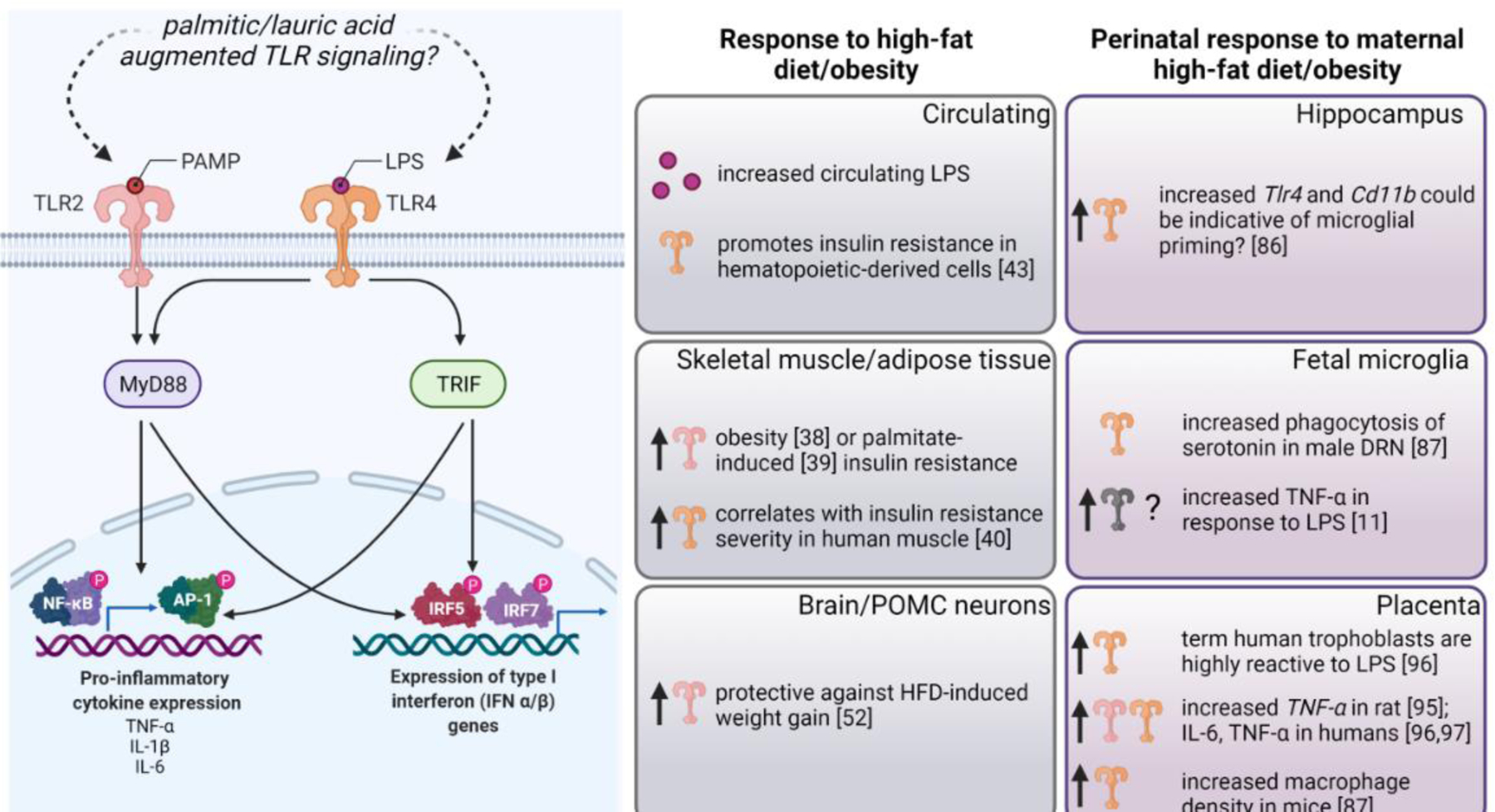
Left Panel: PAMPs (pathogen-associated molecular patterns) such as LPS canonically activate TLR2 and TLR4, respectively. Recent literature suggests that saturated fatty acids, such as palmitic or lauric acid, augment TLR2/4 signaling. TLR2 signaling initiates through the adaptor protein MyD88 (Myeloid differentiation primary response 88) and results in activation of the transcription factors NF-KB (nuclear factor-kappaB) and AP-1 (activation protein-1) which promote transcription of pro-inflammatory genes such as TNF-α, IL-1β, and IL-6. TLR4 signaling can initiate MyD88-dependent inflammatory signaling or TRIF (TIR-domain-containing adapter-inducing interferon-β)-dependent signaling, resulting in increased expression of type I interferon genes. Center panels: In response to high-fat diet/obesity, circulating LPS levels increase, hematopoietic-derived cells promote insulin-resistance in a TLR4-dependent manner. TLR2 is upregulated in skeletal muscle and adipose tissue in response to obesity or palmitate-induced insulin resistance. TLR4 is upregulated in skeletal muscle and adipose tissue and TLR4 levels correlate with insulin resistance severity in skeletal muscle. Increased TLR2 in POMC neurons in response to high-fat diet protects against weight gain. Right panels: In response to maternal high-fat diet/obesity, Tlr4 and Cd11b are increased in the neonatal hippocampus, potentially indicative of microglial priming. Male maternal HFD offspring microglia phagocytose excess serotonin in the DRN as early as e14.5. Isolated fetal microglia from obese dams have exaggerated TNF-α production in response to LPS, though the signaling upstream of this (i.e. which TLR may be over-reactive) is unknown. The placenta shows a robust immune response to obesity/high-fat diet in humans and rodents, and both TLR2 and TLR4 likely play a role in this response. Created with BioRender.
Microglia, the resident immune cells of the brain, are increasingly recognized as being highly influenced by dietary fat content. In murine studies, adult hypothalamic microglia increase production of inflammation related genes such as Tnf-α, interleukin-6 (Il-6), and Il-1β in response to increased fat content [53]. Considering the whole-body changes that occur in response to high-fat diet, this was for some time thought to be a response to increased body weight and the associated peripherally derived pro-inflammatory signals. However, hypothalamic microglia number and cell size are increased in response to high-fat diet in as little as three-days – well before high-fat diet-induced weight gain is apparent [28,54]. Increased microglia size and density were paralleled by increased levels of pro-inflammatory genes such as Il-6 and Tnf-α [28]. Interestingly, pro-inflammatory gene expression returns to baseline after 1-week of high-fat diet feeding, but becomes elevated again after 1-month of high-fat diet feeding [28]. Thus, inflammation in the hypothalamus, similar to peripheral tissues, appears to be biphasic in response to high-fat diet/obesity. Importantly, microglial heterogeneity is widely accepted and has been recently reviewed [55]. In addition to spatial heterogeneity, microglia also demonstrate sex-specific heterogeneity [56,57]. We have focused on the hypothalamus here because of its longstanding role as a central regulator of metabolic homeostasis [58]. However, it is likely that microglia in different brain regions respond differently to HFD in sex and spatiotemporal specific ways [59,60].
Dietary fat is not alone in influencing microglial responses in adult animals. Indeed, lack of dietary fiber [61] or excess dietary sugar [62] drive increased microglial activity in mouse hippocampus and hypothalamus, respectively. A binge sucrose paradigm that aimed to mimic sucrose content in a contemporary Western diet demonstrated increased microglial density and decreased microglial ramification in rat cortex, amygdala, hypothalamus, and hippocampus, suggesting that despite known regional heterogeneity, microglia throughout the brain are susceptible to diet [63]. While beyond the scope of this review, the microbiome, which is heavily influenced by diet [64], is also increasingly recognized as a modulator of microglial function (see recent review [65]), and microbiota represent an additional source of fatty acids that may influence microglial function [66].
Neuro-immunometabolism is a burgeoning field, and recent literature has demonstrated that microglial energetics impact cellular function [67]. LPS – likely through Tlr4 – induces aerobic glycolysis in rodent microglia, triggering the release of interleukin-1 beta (IL-1β) [68]. Microglial energetics have also been shown to be impacted by high-fat diet [54,69]. In particular, high-fat diet increases expression of microglial mitochondrial Uncoupling Protein 2 (UCP2), which is sufficient to promote inflammation and obesity in mice [54]. This process appears to be plastic as blockade of UCP2 after neuroinflammation was established was still able to rescue microglial inflammatory responses and metabolic dysfunction. Increased Ucp2 was associated with reduced mitochondrial size, likely due to mitochondrial fission via dynamin-related protein 1 (DRP1), which was increased in response to high-fat diet [54]. In rats, high-fat diet interrupted the circadian rhythmicity of clock and immune-related genes, with high-fat diet fed rats showing a bioenergetic shift toward increased energy production in microglia during the inactive period [69]. Studies investigating the impact of diet on neuro-immunometabolism are scarce but given the aforementioned role for Tlr4 and the knowledge that Tlr4 is influenced by dietary changes, it is possible that microglial metabolism may be shifted in response to high-fat diet, potentially in a Tlr4-dependent manner.
A critical window of microglial plasticity?
The introduction of a high-fat diet postnatally in rodents appears to have regional, temporal, and sex-specific consequences on microglia. In response to HFD (but not VHFD/very low-carbohydrate diet), microglia density and TNF-α expression specifically within the ARC increases in conjunction with circulating leptin and Il-1β in males [70]. Blockade of this expansion with the antimitotic drug arabinofuranosyl cytidine (AraC) is sufficient to normalize circulating leptin and Il-1β levels [70]. Male mice transitioned to a high-fat high-sucrose (western style) diet at weaning have substantially fewer ramified hippocampal microglia (with a corresponding increase in the number of microglia with an increased soma size and thick, short branches – a morphology highly correlated with ‘reactive’ microglia) [71]. Given that ramified microglia play a pivotal role in synaptic pruning during development [72,73], the shift in microglial morphology away from a highly ramified morphology may have functional and/or behavioral consequences. In contrast, introducing adult mice to the same diet did not influence microglial morphology [71]. However, both juvenile and adult mice introduced to the western style diet had increased white adipose tissue (WAT) accumulation, and WAT mass was significantly correlated with ionized calcium binding adaptor molecule 1 (Iba1; a marker for microglia) immunoreactivity [71]. Together, this suggests that microglia are more plastic during the adolescent period than in adulthood.
Recent studies have suggested that aging also represents a critical window where microglia may be more susceptible to high-fat diet [74,75]. Short term (3 days) high-fat diet exposure was sufficient to increase IL-1β levels in the hippocampus and amygdala of aged (24 months) rats and diminish long-term memory [75]. Further, short term high-fat diet exposure increased microglial number in the amygdala of aged rats, but not the hippocampus, in comparison to young (3 months) high-fat diet rats. However, it is still unclear if this is truly a compounding effect of high-fat diet and aging. The young high-fat diet animals tended to have fewer microglia than young or aged control animals, and the difference in microglial number between the aged control and aged high-fat diet groups was marginal [74]. Interestingly, short term high-fat diet had a substantial effect on phagocytosis in amygdalar, but not hippocampal, microglia in vitro. In both brain regions, microglia from aged rats phagocytosed more fluorescent beads than microglia from young rats; short term high-fat diet almost completely abolished the aging-related increase in phagocytosis only in amygdalar microglia [74]. Whether this represents a detrimental or neuroprotective effect of short term high-fat diet remains unknown.
Mother knows best: maternal high-fat diet impacts microglia in a spatiotemporal, sex-dependent manner
The epidemic of obesity does not spare expectant mothers. In the United States, over 50% of women fall into the category of overweight (or obese) at their first prenatal visit [76]. Maternal high-fat diet, one contributor to high maternal weight, is associated with a range of detrimental outcomes for offspring, including metabolic imbalances and psychiatric disorders such as autism spectrum disorders, anxiety, and depression [77–80]. In human cohorts, dissecting the contribution of maternal dietary fat content and/or maternal weight from other factors represents a substantial challenge. Maternal age, gestational diabetes, autoimmunity, and infection during pregnancy – all factors that can influence the immune system – are often interrelated confounds [81]. Further, non-immune factors (e.g. maternal education or socioeconomic status) are linked in parallel with neurological outcomes in offspring. Despite these confounds, longitudinal studies have demonstrated that maternal obesity is associated with offspring sex-biased cognitive and social impairments [82]. Future clinical studies focused on quantifying maternal nutrition (to dissect the contribution of maternal dietary intake from maternal obesity) and offspring outcomes will be important [83].
Microglia, unlike other yolk sac-derived macrophages that are renewed throughout their life span from hematopoietic precursors, are unique in that the pool of microglia that develops during embryogenesis and early postnatal development represent the microglia population throughout life [84,85]. Microglia are therefore subject to persistent influence from alterations to the maternal environment. Despite this, few studies have interrogated the role for maternal overnutrition in fetal or neonatal microglial development or function. One of the first descriptions of this showed that neonatal rat hippocampal microglia are basally activated (increased gene expression of Cd11b and Tlr4) in response to a maternal very high-saturated- or very high trans-fat diet (4 weeks before mating) [86]. This study also found that male maternal very high-saturated- or very high-trans-fat offspring spent less time in the open arms of an elevated plus maze [86], often interpreted as increased anxiety-like behavior. A seminal study demonstrated that maternal VHFD (12–14 weeks pre-mating) primes fetal mouse microglia to overproduce inflammatory cytokines, including TNF-α, in response to immune challenge (LPS), particularly in males [11]. Interestingly, this priming was also apparent in placental macrophages from obese dams, and the placenta is rapidly emerging as a tissue of interest in maternal diet-dependent brain changes (See Box 1). More recently, maternal high-fat diet (6 weeks pre-mating) has been demonstrated to increase Iba1 density in fetal male and female offspring placenta and brain tissue as early as embryonic day 14.5 (e14.5) in mice [87]. In male fetal brain tissue, increased Iba1 density was associated with increased microglial volume and increased phagocytosis of serotonin in the dorsal raphe nucleus (DRN), a key reward center, leading to lasting decreased serotonin bioavailability in the brain. How increased Iba1 density in response to maternal high-fat diet may impact the fetal female brain is yet unknown. In tandem, this study demonstrated robust inflammatory responses from male and female human placental tissue (11 weeks post conception) and female fetal brain tissue in response to maternal placenta (decidua) triglyceride accumulation, a proxy for maternal fat intake [87].
BOX 1: The placenta as an emerging contributor to microglial state?
The environment of chronic inflammation created by maternal high-fat diet/obesity impacts the developing fetus as well as the placenta [92], the organ that develops during early gestation and surrounds the fetus to both restrict and facilitate macromolecule and nutrient transport. Recent studies have demonstrated that (i) maternal high-fat diet alters placental morphology and gene expression in a sex-specific manner in mice, rats, and humans [87,93], and (ii) the placenta acts as a crucial interface between the maternal environment and the developing fetal brain [94]. In a mouse model of maternal HFD, Tlr4 is increased in placental tissue as early as e14.5 [87]. Similarly, Tlr2 and Tlr4 are increased in fetal placenta at e20.5 in rat models of maternal HFD [95]. In humans, TLR4 is increased in term placental trophoblast and macrovascular endothelial cells from obese women [96]. Term trophoblasts are highly responsive to LPS challenge [96], suggesting a functional relevance behind obesity-dependent increases in placental TLR4. Further, placental macrophages accumulate in placentae from obese mothers in conjunction with upregulation of inflammatory markers such as interleukin-6 (Il-6) and tumor necrosis factor-α (TNF-α) [96,97]. Of note, placental macrophages are of fetal, and not maternal, origin, and are posited to share a common origin with microglia in the fetal yolk sac [98–100]. Thus, the placenta is uniquely poised to translate maternal insults, such as maternal overnutrition, to microglia in the developing brain.
More studies have focused on the effect of maternal diet on juvenile and adult microglia. During the juvenile period, microglia play a critical role throughout the brain in synapse and receptor refinement [12,88]. In postnatal day 20 (P20) rats, LPS significantly increases IL-1β protein levels in the hippocampus of maternal very high-saturated fat diet offspring of both sexes [86]. In P30 mice, male and female maternal VHFD (4 weeks pre-mating) offspring had decreased mature lysosomes in microglia and increased microglial-synapse contact in the corpus collosum, despite no changes in gross microglial number or morphology [89]. This was associated with decreased expression of myelin-related transcripts and altered myelin organization in the hippocampus, a projection region of the corpus collosum, only in male offspring [89]. In contrast to other studies [86], no changes were observed in male or female offspring time spent in the open arm of an elevated plus maze [89], pointing to possible species-specific differences. VHFD mouse offspring did, however, show decreased preference for a novel social stimulus, a phenotype that appears to be driven by male offspring [89]. Other studies have demonstrated baseline increases in Iba1 immunoreactivity in the adult hippocampus of male and female maternal very high saturated-fat diet rat offspring [86] and female-specific increases in adult expression of CD68 in maternal HFD offspring (4 weeks pre-mating) [90]. Interestingly, male HFD offspring who were maintained on a HFD at weaning also demonstrated increased CD68 as adults, suggesting different windows of susceptibility in males and females. In adolescent maternal VHFD (6 weeks pre-mating) offspring (6 weeks-old), microglia in female mouse amygdala showed basally higher Iba1 gene expression and immunoreactivity as compared to males [91]. Increased Iba1 in female offspring could be prevented by maternal dietary intervention (changing dams to a control diet at birth). TNF-α was also increased in brain lysates from maternal VHFD female offspring only (preventable by maternal dietary intervention at birth), and female maternal VHFD offspring showed deficits in social behavior [91]. Social behavior changes are also increased in children born to obese women, and social behavior deficits have been seen in other mouse studies of maternal VHFD [82].
An interesting line of study has suggested that dietary intervention, such as reverting dams from a high-fat to low-fat diet at birth, can prevent inflammatory phenotypes and associated social behavior deficits [91]. This is notable because while it is unlikely that women who consume a high-fat diet while pregnant will completely alter their diet postpartum, it does suggest that targeted clinical interventions during neonatal life may have a significant impact on behavioral outcomes. In support of this, another recent study demonstrated that switching maternal high-fat diet offspring to a high-fiber diet (or increasing the fiber in the diet of obese dams) could reduce the expression of inflammatory markers in hippocampus and prefrontal cortex of offspring and rescue social behavior deficits [82]. In sum, the reaction of microglia to maternal diet appears to be dependent on a number of factors, such as brain region and offspring sex.
Concluding Remarks and Future Perspectives
An important consideration when discussing the role of overweight/obesity in any facet of research is the underlying cause(s) of the excess weight. In humans, every overweight individual is unique in their genetic predisposition and environment. In contrast, in rodents, by far the most prevalent organism in obesity research, the genetics and environment can be carefully controlled. The benefit of rodent research is that the impact of a single factor, and in the case of obesity this is often a change in dietary fat, can be interrogated. However, these studies often fail to parse out the impact of increased weight versus the manipulation that caused the weight gain. The merit of rodent models is not in question, but rather we implore that caution should be used when extrapolating rodent studies to humans, as a singular influence is rarely the cause of overweight/obesity in humans.
Microglial activity during critical developmental windows is increasingly recognized as playing a key role in brain development and function. Microglia are particularly susceptible to environmental perturbations during development, and this is particularly relevant in the context of the rapidly expanding obesity epidemic. Recent research has begun to focus on how the innate immune system, and microglial signaling in particular, are impacted by maternal overweight/obesity. These studies have led to seminal discoveries demonstrating that maternal overnutrition results in microglial priming. However, microglial priming in response to maternal overnutrition appears to be region, sex, environment, and age-dependent, leading to several as of yet unanswered questions in this research area (see Outstanding Questions).
Outstanding Questions Box.
Key components of brain function are determined in utero. How does maternal diet influence microglial activity in utero?
What mechanisms underlie brain region-specific microglial responses to overnutrition?
How does fetal sex influence microglial response to maternal overnutrition?
Do alterations in placental form or function influence microglial activity?
How can rodent models more accurately reflect the complexities of human obese populations (i.e. consider relationships between available nutrition, socioeconomic status, and environmental stressors that could have synergistic effects)?
Highlights.
Obesity is a growing epidemic, and the impacts of maternal and pediatric obesity are only recently gaining attention
Toll-like receptors play a critical role in direct and indirect microglial responses to overnutrition
Microglia are sensitive to maternal overweight/obesity during critical developmental windows
Microglial priming in response to maternal overweight/obesity is brain region, age, and sex-biased
Glossary
- Adipokine
adipose-tissue derived cytokine, also called adipocytokine. Adipokines often act on immune cells can have pro-inflammatory, anti-inflammatory, or metabolic downstream effects
- Arcuate Nucleus of the Hypothalamus (ARC)
A central nervous system nucleus (collection of neuronal cell bodies) primarily controlling food intake, often in response to levels of the adipokine leptin
- Cd11b
cluster of differentiation molecule 11b, also known as ITGAM (Integrin alpha M), Mac-1 (macrophage-1 antigen), or CR3 (complement receptor 3). A canonical marker of macrophages/monocytes often used to aid in experimentally isolating/identifying these cell types
- CD68
cluster of differentiation molecule 68, primarily localizes to lysosomes and is often used as a marker for actively phagocytic microglia
- C-reactive protein (CRP)
a liver-derived marker for inflammation in circulating blood. CRP activates the complement system, and is increased in response to increasing levels of IL-6
- Diet-induced obesity (DIO)
a (primarily rodent) model of high-fat diet that results in significant weight gain. Often, a diet of 60% kcal from fat (versus standard chow containing 10–15%) is provided ad libitum for an extended period of time (e.g. 10+ weeks) to induce obesity
- Dorsal raphe nucleus (DRN)
one of the raphe nuclei located in the midbrain comprised of serotonergic, GABAergic, dopaminergic, and glutamatergic neuronal cell bodies. The home of the majority of serotonergic neurons
- Dynamin-related protein 1 (DRP1)
also known as dynamin-1-like protein. A GTPase that plays an essential role in promoting mitochondrial fission
- High-Fat Diet (HFD)
A purified rodent diet usually consisting of 45% kcal from fat. This diet was developed to mimic a ‘fast-food diet’ in humans
- Interleukin-1 beta (IL-1β)
a pro-inflammatory cytokine secreted by immune cells in response to pathogen – in particular IL-1β is a downstream target gene of TLRs
- Interleukin-6 (IL6)
a cytokine that can have both pro- and anti-inflammatory actions
- Ionized calcium binding adaptor molecule 1 (Iba1)
also known as allograft inflammatory factor 1 (Aif1), a macrophage marker often used to identify microglia as its expression is specific to microglia in the CNS
- Lipopolysaccharide (LPS)
An integral component of the outer membrane of gram-negative bacteria often used to mimic bacterial infection in experimental settings
- Neuropeptide Y/agouti-related peptide (AgRP)
Neuropeptide Y (NPY) and agouti-related peptide (or protein; AgRP) are co-expressed in neuronal cell bodies in the ventromedial arcuate nucleus of the hypothalamus. AgRP and NPY increase appetite/decrease metabolism
- Priming
in the context of macrophages/microglia, priming refers to a primary incendiary event that does not necessarily affect basal macrophage/microglial function, but does result in macrophage/microglial over-reaction to subsequent insult
- Proopiomelanocortin (POMC) neurons
a neuronal population in the arcuate nucleus of the hypothalamus whose activity reduces food intake
- Saturated fatty acids (SFAs)
a type of fat where the fatty acid chain consists of only single bonds. Animal fat and highly processed foods are generally high in saturated fat, and high-saturated fat intake may increase risk for metabolic or cardiovascular disease
- Toll-like receptors (TLRs)
TLRs are single-pass membrane receptors expressed on a broad population of immune cells that respond to common pathogenic peptide patterns (often called PAMPs, or pathogen-associated molecular patterns)
- Tumor necrosis factor-α (TNF-α)
a pro-inflammatory cytokine produced in macrophages downstream of TLR signaling
- Uncoupling Protein 2 (UCP2)
UCP2 is widely expressed and reduces mitochondrial membrane potential/ROS (radical oxygen species) production
- Very high-fat diet (VHFD)
A purified rodent diet usually consisting of 60% kcal from fat. Long-term VHFD feeding is obesogenic
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Obesity and overweight [Online]. Available: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. [Accessed: 17-Nov-2021]
- 2.McPherson R (2007) Genetic contributors to obesity. Can J Cardiol 23, 23A–27A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swinburn BA et al. (2011) The global obesity pandemic: shaped by global drivers and local environments. Lancet 378, 804–814 [DOI] [PubMed] [Google Scholar]
- 4.Lange SJ (2021) Longitudinal Trends in Body Mass Index Before and During the COVID-19 Pandemic Among Persons Aged 2–19 Years — United States, 2018–2020. MMWR Morb Mortal Wkly Rep 70, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mrak RE (2009) Alzheimer-type neuropathological changes in morbidly obese elderly individuals. Clin Neuropathol 28, 40–45 [DOI] [PubMed] [Google Scholar]
- 6.Pedditzi E et al. (2016) The risk of overweight/obesity in mid-life and late life for the development of dementia: a systematic review and meta-analysis of longitudinal studies. Age Ageing 45, 14–21 [DOI] [PubMed] [Google Scholar]
- 7.Anstey KJ et al. (2011) Body mass index in midlife and late-life as a risk factor for dementia: a meta-analysis of prospective studies. Obes Rev 12, e426–437 [DOI] [PubMed] [Google Scholar]
- 8.Simon GE et al. (2006) ASSOCIATION BETWEEN OBESITY AND PSYCHIATRIC DISORDERS IN THE US ADULT POPULATION. Arch Gen Psychiatry 63, 824–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blasco BV et al. (2020) Obesity and Depression: Its Prevalence and Influence as a Prognostic Factor: A Systematic Review. Psychiatry Investig 17, 715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bachiller S et al. (2018) Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Frontiers in Cellular Neuroscience 12, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edlow AG et al. (2018) Placental Macrophages: A Window Into Fetal Microglial Function in Maternal Obesity. International Journal of Developmental Neuroscience DOI: 10.1016/j.ijdevneu.2018.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao P et al. (2021) Early-life inflammation promotes depressive symptoms in adolescence via microglial engulfment of dendritic spines. Neuron 0, [DOI] [PubMed] [Google Scholar]
- 13.Li J-W et al. (2018) Microglial priming in Alzheimer’s disease. Ann Transl Med 6, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Block CL et al. (2020) Prenatal Environmental Stressors Impair Postnatal Microglia Function and Adult Behavior in Males,bioRxiv doi: 10.1101/2020.10.15.336669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C et al. (2018) Estimated global overweight and obesity burden in pregnant women based on panel data model. PLoS One 13, e0202183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogden CL et al. (2020) Trends in Obesity Prevalence by Race and Hispanic Origin—1999–2000 to 2017–2018. JAMA 324, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Speakman JR (2019) Use of high-fat diets to study rodent obesity as a model of human obesity. Int J Obes 43, 1491–1492 [DOI] [PubMed] [Google Scholar]
- 18.Warden CH and Fisler JS (2008) Comparisons of diets used in animal models of high fat feeding. Cell Metab 7, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pellizzon MA and Ricci MR (2020) Choice of Laboratory Rodent Diet May Confound Data Interpretation and Reproducibility. Current Developments in Nutrition 4, nzaa031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gesta S et al. (2007) Developmental origin of fat: tracking obesity to its source. Cell 131, 242–256 [DOI] [PubMed] [Google Scholar]
- 21.Aronoff SL et al. (2004) Glucose Metabolism and Regulation: Beyond Insulin and Glucagon. Diabetes Spectrum 17, 183–190 [Google Scholar]
- 22.Watson KT et al. (2021) Incident Major Depressive Disorder Predicted by Three Measures of Insulin Resistance: A Dutch Cohort Study. AJP DOI: 10.1176/appi.ajp.2021.20101479 [DOI] [PubMed] [Google Scholar]
- 23.Xu H et al. (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112, 1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weisberg SP et al. (2003) Obesity is associated with macrophage accumulation in adipose tissue DOI: 10.1172/JCI19246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hotamisligil GS et al. (1993) Adipose Expression of Tumor Necrosis Factor-α: Direct Role in Obesity-Linked Insulin Resistance. Science at < 10.1126/science.7678183> [DOI] [PubMed] [Google Scholar]
- 26.Kwon O et al. (2016) Leptin signalling pathways in hypothalamic neurons. Cell. Mol. Life Sci 73, 1457–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen HY et al. (2004) Orexigenic Action of Peripheral Ghrelin Is Mediated by Neuropeptide Y and Agouti-Related Protein. Endocrinology 145, 2607–2612 [DOI] [PubMed] [Google Scholar]
- 28.Thaler JP et al. (2012) Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 122, 153–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valdearcos M et al. (2017) Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab 26, 185–197.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prevot V et al. (2018) The Versatile Tanycyte: A Hypothalamic Integrator of Reproduction and Energy Metabolism. Endocrine Reviews 39, 333–368 [DOI] [PubMed] [Google Scholar]
- 31.Hofmann K et al. (2017) Tanycytes and a differential fatty acid metabolism in the hypothalamus. Glia 65, 231–249 [DOI] [PubMed] [Google Scholar]
- 32.Geller S et al. (2019) Tanycytes Regulate Lipid Homeostasis by Sensing Free Fatty Acids and Signaling to Key Hypothalamic Neuronal Populations via FGF21 Secretion. Cell Metab 30, 833–844.e7 [DOI] [PubMed] [Google Scholar]
- 33.Rhea EM and Banks WA (2021) Interactions of Lipids, Lipoproteins, and Apolipoproteins with the Blood-Brain Barrier. Pharm Res 38, 1469–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pifferi F et al. (2021) Lipid Transport and Metabolism at the Blood-Brain Interface: Implications in Health and Disease. Frontiers in Physiology 12, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olson JK and Miller SD (2004) Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 173, 3916–24 [DOI] [PubMed] [Google Scholar]
- 36.Fiebich BL et al. (2018) Role of Microglia TLRs in Neurodegeneration. Frontiers in Cellular Neuroscience 12, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Medzhitov R (2001) Toll-like receptors and innate immunity. Nat Rev Immunol 1, 135–145 [DOI] [PubMed] [Google Scholar]
- 38.Caricilli AM et al. (2008) Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J Endocrinol 199, 399–406 [DOI] [PubMed] [Google Scholar]
- 39.Senn JJ (2006) Toll-like Receptor-2 Is Essential for the Development of Palmitate-induced Insulin Resistance in Myotubes *. Journal of Biological Chemistry 281, 26865–26875 [DOI] [PubMed] [Google Scholar]
- 40.Reyna SM et al. (2008) Elevated Toll-Like Receptor 4 Expression and Signaling in Muscle From Insulin-Resistant Subjects. Diabetes 57, 2595–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi H et al. (2006) TLR4 links innate immunity and fatty acid–induced insulin resistance. J Clin Invest 116, 3015–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milanski M et al. (2009) Saturated Fatty Acids Produce an Inflammatory Response Predominantly through the Activation of TLR4 Signaling in Hypothalamus: Implications for the Pathogenesis of Obesity. Journal of Neuroscience 29, 359–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saberi M et al. (2009) Hematopoietic Cell-Specific Deletion of Toll-like Receptor 4 Ameliorates Hepatic and Adipose Tissue Insulin Resistance in High-Fat-Fed Mice. Cell Metabolism 10, 419–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Y et al. (2017) Knockdown of Tlr4 in the Arcuate Nucleus Improves Obesity Related Metabolic Disorders. Sci Rep 7, 7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anhê FF et al. (2021) Metabolic endotoxemia is dictated by the type of lipopolysaccharide. Cell Reports 36, 109691. [DOI] [PubMed] [Google Scholar]
- 46.Vargas-Caraveo A et al. (2017) Lipopolysaccharide enters the rat brain by a lipoprotein-mediated transport mechanism in physiological conditions. Sci Rep 7, 13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JY et al. (2001) Saturated Fatty Acids, but Not Unsaturated Fatty Acids, Induce the Expression of Cyclooxygenase-2 Mediated through Toll-like Receptor 4 *. Journal of Biological Chemistry 276, 16683–16689 [DOI] [PubMed] [Google Scholar]
- 48.Cani PD et al. (2007) Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 56, 1761–1772 [DOI] [PubMed] [Google Scholar]
- 49.Huang S et al. (2012) Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res 53, 2002–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Z et al. (2012) Saturated fatty acids activate microglia via Toll-like receptor 4/NF-κB signalling. Br J Nutr 107, 229–241 [DOI] [PubMed] [Google Scholar]
- 51.Little JP et al. (2012) The saturated fatty acid palmitate induces human monocytic cell toxicity toward neuronal cells: exploring a possible link between obesity-related metabolic impairments and neuroinflammation. J Alzheimers Dis 30 Suppl 2, S179–183 [DOI] [PubMed] [Google Scholar]
- 52.Shechter R et al. (2013) Hypothalamic neuronal toll-like receptor 2 protects against age-induced obesity. Sci Rep 3, 1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valdearcos M et al. (2014) Microglia Dictate the Impact of Saturated Fat Consumption on Hypothalamic Inflammation and Neuronal Function. Cell Reports 9, 2124–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim JD et al. (2019) Microglial UCP2 Mediates Inflammation and Obesity Induced by High-Fat Feeding. Cell Metabolism 30, 952–962.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan Y-L et al. (2020) Microglial regional heterogeneity and its role in the brain. Mol Psychiatry 25, 351–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bordt EA et al. (2019) Microglia and sexual differentiation of the developing brain: A focus on ontogeny and intrinsic factors. Glia DOI: 10.1002/glia.23753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.VanRyzin JW et al. (2020) Microglia and sexual differentiation of the developing brain: A focus on extrinsic factors. Glia 68, 1100–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roh E et al. (2016) Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp Mol Med 48, e216–e216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Troutman TD et al. (2021) Exploiting dynamic enhancer landscapes to decode macrophage and microglia phenotypes in health and disease. Molecular Cell 81, 3888–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Folick A et al. (2021) Microglial Lipid Biology in the Hypothalamic Regulation of Metabolic Homeostasis. Front Endocrinol (Lausanne) 12, 668396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shi H et al. (2021) A fiber-deprived diet causes cognitive impairment and hippocampal microglia-mediated synaptic loss through the gut microbiota and metabolites. Microbiome 9, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao Y et al. (2017) Dietary sugars, not lipids, drive hypothalamic inflammation. Molecular Metabolism 6, 897–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patkar OL et al. (2021) A binge high sucrose diet provokes systemic and cerebral inflammation in rats without inducing obesity. Sci Rep 11, 11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leeming ER et al. (2019) Effect of Diet on the Gut Microbiota: Rethinking Intervention Duration. Nutrients 11, 2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith CJ (2021) Emerging roles for microglia and microbiota in the development of social circuits. Brain, Behavior, & Immunity - Health 16, 100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Colombo AV et al. (2021) Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife 10, e59826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bernier L-P et al. (2020) Immunometabolism in the Brain: How Metabolism Shapes Microglial Function. Trends in Neurosciences 43, 854–869 [DOI] [PubMed] [Google Scholar]
- 68.York EM et al. (2021) Neuroinflammatory inhibition of synaptic long-term potentiation requires immunometabolic reprogramming of microglia. Glia 69, 567–578 [DOI] [PubMed] [Google Scholar]
- 69.Milanova IV et al. (2019) Diet-Induced Obesity Disturbs Microglial Immunometabolism in a Time-of-Day Manner. Frontiers in Endocrinology 10, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.André C et al. (2017) Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes 66, 908–919 [DOI] [PubMed] [Google Scholar]
- 71.Reichelt AC et al. (2021) Age-dependent and region-specific alteration of parvalbumin neurons, perineuronal nets and microglia in the mouse prefrontal cortex and hippocampus following obesogenic diet consumption. Sci Rep 11, 5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kettenmann H et al. (2013) Microglia: New Roles for the Synaptic Stripper. Neuron 77, 10–18 [DOI] [PubMed] [Google Scholar]
- 73.Béchade C et al. (2013) Microglial control of neuronal activity. Frontiers in Cellular Neuroscience 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spencer SJ et al. (2019) High-fat diet worsens the impact of aging on microglial function and morphology in a region-specific manner. Neurobiol Aging 74, 121–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spencer SJ et al. (2017) High-fat diet and aging interact to produce neuroinflammation and impair hippocampal- and amygdalar-dependent memory. Neurobiology of Aging 58, 88–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Deputy N et al. (2019) Prevalence and Trends in Prepregnancy Normal Weight — 48 States, New York City, and District of Columbia, 2011–2015. MMWR Morb Mortal Wkly Rep 66, 1402–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vickers MH et al. (2000) Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am J Physiol Endocrinol Metab 279, E83–7 [DOI] [PubMed] [Google Scholar]
- 78.Edlow AG (2017) Maternal obesity and neurodevelopmental and psychiatric disorders in offspring. Prenat Diagn 37, 95–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kott J and Brummelte S (2019) Trick or treat? Evaluating contributing factors and sex-differences for developmental effects of maternal depression and its treatment. Horm Behav 111, 31–45 [DOI] [PubMed] [Google Scholar]
- 80.Krakowiak P et al. (2012) Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 129, e1121–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bordeleau M et al. (2021) From Maternal Diet to Neurodevelopmental Disorders: A Story of Neuroinflammation. Front Cell Neurosci 14, 612705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu X et al. (2021) High-fiber diet mitigates maternal obesity-induced cognitive and social dysfunction in the offspring via gut-brain axis. Cell Metabolism 33, 923–938.e6 [DOI] [PubMed] [Google Scholar]
- 83.Cortés-Albornoz MC et al. (2021) Maternal Nutrition and Neurodevelopment: A Scoping Review. Nutrients 13, 3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ajami B et al. (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10, 1538–1543 [DOI] [PubMed] [Google Scholar]
- 85.Ginhoux F et al. (2010) Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 330, 841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bilbo SD and Tsang V (2010) Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. The FASEB Journal 24, 2104–2115 [DOI] [PubMed] [Google Scholar]
- 87.Ceasrine AM et al. (2021) Maternal diet disrupts the placenta-brain axis in a sex-specific manner. bioRxiv doi: 10.1101/2021.11.12.468408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kopec AM et al. (2018) Microglial dopamine receptor elimination defines sex-specific nucleus accumbens development and social behavior in adolescent rats. Nature Communications 9, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bordeleau M et al. (2021) Maternal high-fat diet modifies myelin organization, microglial interactions, and results in social memory and sensorimotor gating deficits in adolescent mouse offspring. Brain, Behavior, & Immunity - Health 15, 100281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Contu L et al. (2019) Pre- and Post-natal High Fat Feeding Differentially Affects the Structure and Integrity of the Neurovascular Unit of 16-Month Old Male and Female Mice. Frontiers in Neuroscience 13, 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kang SS et al. (2014) Dietary intervention rescues maternal obesity induced behavior deficits and neuroinflammation in offspring [DOI] [PMC free article] [PubMed]
- 92.Roberts KA et al. (2011) Placental structure and inflammation in pregnancies associated with obesity. Placenta 32, 247–54 [DOI] [PubMed] [Google Scholar]
- 93.Song L et al. (2017) Prenatal high-fat diet alters placental morphology, nutrient transporter expression, and mtorc1 signaling in rat. Obesity (Silver Spring) 25, 909–919 [DOI] [PubMed] [Google Scholar]
- 94.Goeden N et al. (2016) Maternal Inflammation Disrupts Fetal Neurodevelopment via Increased Placental Output of Serotonin to the Fetal Brain. J. Neurosci 36, 6041–6049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thompson JA et al. (2015) The contribution of Toll-like receptors to placental inflammation in diet-induced maternal obesity. Placenta 36, 1204–1206 [DOI] [PubMed] [Google Scholar]
- 96.Yang X et al. (2016) Causal relationship between obesity-related traits and TLR4-driven responses at the maternal-fetal interface. Diabetologia 59, 2459–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Challier JC et al. (2008) Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 29, 274–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim JS et al. (2008) Involvement of Hofbauer cells and maternal T cells in villitis of unknown aetiology. Histopathology 52, 457–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim MJ et al. (2009) Villitis of unknown etiology is associated with a distinct pattern of chemokine up-regulation in the feto-maternal and placental compartments: implications for conjoint maternal allograft rejection and maternal anti-fetal graft-versus-host disease. J Immunol 182, 3919–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ceasrine AM et al. (2021) Single cell profiling of Hofbauer cells and fetal brain microglia reveals shared programs and functions. bioRxiv doi: 10.1101/2021.12.03.471177 [DOI] [Google Scholar]