

Abstract
The epidemiological surveillance of SARS-CoV-2 by means of whole-genome sequencing has revealed the emergence and co-existence of multiple viral lineages or subtypes throughout the world. Moreover, it has been shown that several subtypes of this virus display particular phenotypes, such as increased transmissibility or reduced susceptibility to neutralizing antibodies, leading to the denomination of Variants of Interest (VOI) or Variants of Concern (VOC). Thus, subtyping of SARS-CoV-2 is a crucial step for the surveillance of this pathogen. Here, we present Covidex, an open-source, alignment-free machine learning subtyping tool. It is a shiny web app that allows an ultra-fast and accurate classification of SARS-CoV-2 genome sequences into the three most used nomenclature systems (GISAID, Nextstrain, Pango lineages). It also categorizes input sequences as VOI or VOC, according to current definitions.
The program is cross-platform compatible and it is available via Source-Forge https://sourceforge.net/projects/covidex or via the web application http://covidex.unlu.edu.ar.
Keywords: SARS-CoV-2, Subtyping, Machine learning, Web-application, VOC, VOI
1. Introduction
SARS-CoV-2 virus belongs to the species Severe acute respiratory syndrome-related coronavirus within the Betacoronavirus genus (Coronaviridae Study Group of the International Committee on Taxonomy of Viruses, 2020) and it is the etiological agent of the coronavirus disease 19 (COVID-19). Since the onset of the COVID-19 pandemic, scientific efforts have been centered globally on the characterization of SARS-CoV-2 genetic diversity. Typically, isolates are classified into clades (also termed subtypes or genotypes), which correspond to monophyletic groups of sequences on a phylogenetic tree.
Subtype classification is an important and complex problem in the field of virology (Gorbalenya and Siddell, 2021). For SARS-CoV-2, subtyping is a specially challenging issue because genome data is being generated at an unprecedented speed. In this sense, the high volume of sequences needs a naming system that can handle hundreds of thousands of viral genomes obtained in relative short periods of time. Additionally, different naming systems have been developed, most notably: GISAID (GISAID, 2020), Nextstrain (Clade Naming for SARS-CoV-2, 2021) and Pango lineages (Rambaut et al., 2020). Also, these classifications are updated periodically in order to reflect any changes in the relevant clusters under study. Remarkably, subtype classification supports the genetic surveillance of viral variants displaying particular phenotypic or epidemiological features, for example, higher transmissibility or decreased susceptibility to neutralizing antibodies, also known as Variants of Interest (VOI) or Variants of Concern (VOC) (WHO, 2021).
Several tools have been developed for viral sequence classification, including recently released applications for subtyping of SARS-CoV-2 sequences (Hadfield et al., 2018; Randhawa et al., 2020; Singer et al., 2020). Current SARS-CoV-2 classification software have at least one of the following limitations: i) they require the alignment of the input data against a set of reference sequences, which can be computationally expensive, particularly for long sequences such as the SARS-CoV-2 genome (>30 Kb); ii) the user must be familiar with command line usage; iii) they depend on the user to upload the data to an external server; iv) they require multiple steps for a correct installation; or v) they only classify sequences according to one of the multiple nomenclatures available. Alternatively, Covidex was developed as an open source alignment-free machine learning subtyping tool. It is an app that allows fast and accurate classification of viral genomes in all three (GISAID, Nextstrain and Pango) nomenclatures simultaneously. Covidex combines the analytical capacities of the R environment and the user-friendliness of the Shiny interface (Chang et al., 2020). It is available for free and without registration at sourceforge.net for macOS, Linux and Windows users with R and RStudio dependencies. Additionally, a web-based version is available that can be effortlessly deployed without any installation (covidex.unlu.edu.ar).
2. Tool description and performance
2.1. Classification algorithm
The overall classification algorithm is a modification of the method proposed by Solis-Reyes (Solis-Reyes et al., 2018) and it is divided in three phases. The first phase loads the user data in multi-fasta format and performs the k-mer counting operation using the k-mer package (Fig. 1) (Wilkinson, 2018). The dataset is splitted in subsets (according to the number of cores available) and processed in multiple threads parallelly in order to reduce computation time (Note: the latter is performed only if more than 2000 sequences are loaded). All outputs produced are then merged. Each k-mer count is normalized over the k-mer size (k = 6) and the sequence length. This k-mer size was selected in order to minimize the computation time, given the fact that k = 7 only produced slightly better results in terms of accuracy but with more than doubling the computation time (Supplementary Fig. 1). The second phase of the classification executes the call to the ranger package (Wright and Ziegler, 2017) predict function using three trained random forest models in sequential order (one for each nomenclature). From these, a probability score is calculated as the proportion of trees that supports the corresponding classification. The final phase generates a classification table with the probability for each classification. The proportion of N bases in the genome and the genome length are also informed (we recommend N < 1% and length > 29,500 in order to obtain the most accurate results). Additionally, any VOC or VOI detected following the Pango nomenclature is highlighted in the table. A report can be generated that includes the results table, date of analysis, model information and stats summarizing the proportions of VOC and VOI variants detected in the analysis. Fig. 2 shows an overview of the covidex app.
Fig. 1.

Workflow overview of Covidex. First, viral sequences are loaded in FASTA format. Next, normalized k-mer counts are obtained from these sequences. Three random forest models are then used to classify the query sequences and probability scores based on the number of trees that calls for each class are calculated. Finally, the classification results are presented and a report can be generated for download.
Fig. 2.

Overview of the Covidex app. The user is expected to load a sequence file and press RUN. A results table will be shown. Additionally, the user can download an automatic report.
2.2. Models description and tool performance
Covidex includes three classification models (one for each nomenclature) that have been developed for SARS-CoV-2 and that are widely used worldwide. To train Covidex models, approximately 65,000 SARS-CoV-2 sequences representing each clade from GISAID were downloaded from the database (Elbe and Buckland-Merrett, 2017). These sequences were classified with Pangolin and NextClade (Nextclade, 2021; Pangolin, 2021). For each model, a subset of training sequences and their corresponding subtypes were selected at random retaining 85% of the sequences in each class, provided the class had more than 5 and less than 600 sequences. If a subtype had more than 600 sequences, a maximum of 500 sequences were included in the training dataset in order to reduce the size of the final classification model. Once the random forest models were trained, a subsample of the excluded sequences of each class was used to test their performance. All three models performed well, with an accuracy of 0.9777, 0.9952 and 0.9656 for GISAID, Nextstrain and Pango models, respectively (Table 1). Additionally, multi-class AUC were even higher for GISAID and Pango models, but lower for the Nextstrain model. Correlation heatmaps, Metrics Tables, Precision-Recall curves and other statistics are available for each model (Supplementary File 1).
Table 1.
Classification models stats. Basic statistics for each classification model. Models were derived with SARS-CoV-2 sequences downloaded on 2021/11/27. Accuracy was calculated as sequences correctly labeled / number of sequences. Multi-class AUC is the mean AUC from all pairwise class comparisons.
| Model | Number of classes | Number of trees | Training dataset size | Testing dataset size | Accuracy | Multi-class AUC |
|---|---|---|---|---|---|---|
| GISAID | 10 | 1000 | 66,126 | 13,230 | 0.9777 | 0.9931 |
| Nextstrain | 22 | 1000 | 63,972 | 12,810 | 0.9952 | 0.9879 |
| Pango | 1437 | 500 | 65,467 | 12,346 | 0.9656 | 0.9926 |
Additionally, the time performance of Covidex was compared with two other subtyping tools available for SARS-CoV-2 lineage classification: Pangolin 2.0 and NextClade. We evaluated the performance of the tools by using a subset of 1000 randomly selected sequences. According to the results obtained, Covidex was the fastest tool, taking only 12 s to complete the task. In turn, Pangolin 2.0 and NextClade took 21 s and 52 s, respectively.
2.3. VOC and VOI variants detection
Covidex also generates a report that includes a summary of VOC and VOI variants detected. To evaluate the performance of the tool in this regard, 7 new testing datasets were obtained from the GISAID database on 2021-11-27, namely 5 for VOC variants (Alpha, Beta, Gamma, Delta and Omicron) and 2 for VOI (Lambda and Mu). In all cases, Covidex showed very high accuracy (Supplementary Fig. 2). This result underscores the versatility of Covidex as a fast and accurate tool for rapid VOC and VOI variants detection.
3. Conclusions
Ready-to-use software for analyzing sequences available across all platforms can certainly improve the efficiency of viral genomes subtyping. Covidex is freely accessible as a web application and it is also available to run locally on most systems with sufficient memory to run basic java-based applications. It combines accurate and fast machine learning models with an easy-to-use interface. It has been created to aid in real-time genome subtyping in the current COVID-19 pandemic. It classifies sequences in all three major nomenclatures in use, making it a first-choice software for this kind of analysis. The classification models are updated regularly in order to include any changes in the dynamic nomenclatures. Additionally, the computation time of Covidex is extremely low, particularly due to its multi-thread capability, making it feasible to classify large volumes of data (>100,000 sequences) in a personal computer.
Declaration of Competing Interest
The authors have no competing interests to declare.
Acknowledgements
We gratefully acknowledge all the Authors, the Originating laboratories responsible for obtaining the specimens, and the Submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based (Supplementary File 2). Icons used in Fig. 1 were modified from https://www.flaticon.com/authors/freepik.
We also thank Gabriel Tolosa and Alejandro Iglesias for assistance with the server at the Centro de Investigación, Docencia y Extensión en Tecnologías de la Información y la Comunicación (CIDETIC), Universidad Nacional de Luján, Argentina.
This work was funded by Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), grant No (PICT 2016-1327) and Departamento de Ciencias Básicas, Universidad Nacional de Luján (CDD-CB 143/20).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.meegid.2022.105261.
Appendix A. Supplementary data
The following are the supplementary data related to this article.
Supplementary Fig. 1.
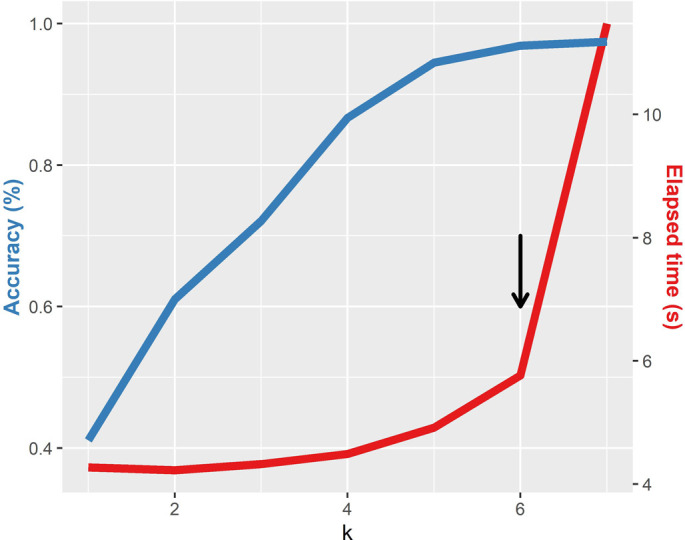
Accuracy score and running time for the random forest algorithm, at different values of k, for a set of 8000 whole SARS-CoV-2 genomes. The black arrow shows the chosen k (highest accuracy with an overall low time).
Supplementary Fig. 2.

VOC and VOI variants detection. Covidex performance on detecting variants of relevance was analyzed. For each VOC and VOI category a sample dataset was created by downloading from GISAID database (with high coverage and complete filters on). Sequences: number of sequences in the dataset; Accuracy: percentage of correctly labeled variants.
Supplementary material 1: This file contains performance statistics of all three classification models.
Supplementary material 2: GISAID sequences authors acknowledgement table.
References
- Chang W., Cheng J., Allaire J.J., Xie Y., McPherson J. 2020. shiny: Web Application Framework for R. [Google Scholar]
- Clade Naming for SARS-CoV-2 . 2021. Year-letter Genetic Clade Naming for SARS-CoV-2 on Nextstrain.org. (URL (accessed 3.22.20)) [Google Scholar]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health: data, disease and diplomacy. Global Chall. 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GISAID . Clade and Lineage Nomenclature. 2020. Clade and lineage nomenclature.https://www.gisaid.org/references/statements-clarifications/clade-and-lineage-nomenclature-aids-in-genomic-epidemiology-of-active-hcov-19-viruses/ (accessed 3.22.21) [Google Scholar]
- Gorbalenya A.E., Siddell S.G. Recognizing species as a new focus of virus research. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34:4121–4123. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nextclade 2021. https://clades.nextstrain.org (accessed 12.16.21)
- Pangolin Cov-lineages. 2021. https://cov-lineages.org/resources/pangolin.html (accessed 12.16.21)
- Rambaut A., Holmes E.C., O’Toole Á., Hill V., McCrone J.T., Ruis C., du Plessis L., Pybus O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020;5:1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randhawa G.S., Soltysiak M.P.M., El Roz H., de Souza C.P.E., Hill K.A., Kari L. Machine learning using intrinsic genomic signatures for rapid classification of novel pathogens: COVID-19 case study. PLoS One. 2020;15 doi: 10.1371/journal.pone.0232391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer J., Gifford R., Cotten M., Robertson D. CoV-GLUE: a web application for tracking SARS-CoV-2 genomic variation (preprint) Life Sci. 2020 doi: 10.20944/preprints202006.0225.v1. [DOI] [Google Scholar]
- Solis-Reyes S., Avino M., Poon A., Kari L. An open-source k-mer based machine learning tool for fast and accurate subtyping of HIV-1 genomes. PLoS One. 2018;13 doi: 10.1371/journal.pone.0206409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Tracking SARS-CoV-2 Variants. 2021. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed 11.25.21)
- Wilkinson S. 2018. kmer: An R Package for Fast Alignment-free Clustering of Biological Sequences. [DOI] [Google Scholar]
- Wright M.N., Ziegler A. ranger: a fast implementation of random forests for high dimensional data in C++ and R. J. Stat. Softw. 2017;77:1–17. doi: 10.18637/jss.v077.i01. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1: This file contains performance statistics of all three classification models.
Supplementary material 2: GISAID sequences authors acknowledgement table.