

Abstract
The T cell receptor (TCR) endows T cells with antigen specificity and is central to nearly all aspects of T cell function. Each naïve T cell has a unique TCR sequence that is stably maintained during cell division. In this way, the TCR serves as a molecular barcode that tracks processes such as migration, differentiation, and proliferation of T cells. Recent technological advances have enabled sequencing of the TCR from single cells alongside deep molecular phenotypes on an unprecedented scale. In this Review, we discuss strengths and limitations of TCR sequences as molecular barcodes, and their application to study immune responses following PD-1 blockade in cancer. Additionally, we consider applications of TCR data beyond use as a barcode.
Single cell TCR seq – Bringing high-throughput single-cell precision to T cell immunology
T cells are a crucial component of the adaptive immune response, endowed with the ability to respond to harmful or threatening stimuli (e.g. pathogens, cancer) in an antigen-specific manner. Naïve T cells traffic through the spleen, lymph nodes (LNs), and blood [1] in search of antigen presenting cells (APCs) to communicate these threats and initiate T cell activation. T cell activation is a tightly regulated process which requires an antigen-specific signal (signal 1) provided through T cell receptor (TCR) interactions with peptide bound to Major Histocompatibility Complexes (pMHC) on the APC, and a costimulatory signal (signal 2) delivered by the CD28 costimulatory receptor on the T cell binding to B7 ligands on the APC. Inflammatory signals (signal 3) promote full effector differentiation [2, 3]. Effector differentiation is accompanied by extensive proliferation, significant transcriptional and epigenetic reprogramming, and licensing to produce effector molecules (e.g., secretion of inflammatory cytokines, cytotoxicity) to combat the perceived threat. In acute antigen settings, memory T cell populations develop, which are poised to better protect the host against antigen reencounter. If antigen is not cleared, T cell exhaustion (see Glossary) develops, characterized by a progressive loss of effector functions and adoption of a dysfunctional state programmed by a fixed epigenetic landscape [4, 5]. The crucial role of antigenic stimulation (either acute or chronic) renders the TCR a gateway to T cell fate and function. Additionally, the affinity of the TCR for pMHC can delineate T cell phenotype, such as the propensity to become an effector or memory T cell [6, 7]. Consequently, all stages of the T cell life cycle - from generation in the thymus, to activation in the periphery, to the development of a memory or exhausted state - starts with the TCR binding pMHC.
Elegant studies in mice have shown the tremendous plasticity of individual naïve T cells. Fate mapping studies, including transfers of single naïve CD4+ or CD8+ T cells into new mice followed by infection, have demonstrated the ability of a single T cell clone to give rise to diverse cell states [8–12]. In humans, such adoptive transfer studies are challenging. One alternative approach is in vivo labeling with deuterium to mark proliferating cells [13]. Using this approach, one study reported that the human CD8+ T cell memory pool following vaccination with the live yellow fever virus vaccine originates from CD8+ T cells that extensively divided during the first two weeks of infection, and these memory T cells were maintained as quiescent populations with a doubling time of over 450 days [13]. While this study provides proof-of-concept for the feasibility of long term lineage tracing in humans, the use of labeling approaches such as deuterium is limited, and there is a significant need to develop additional approaches to bring single cell precision to human studies on a larger scale.
Recent methodological advancements in technologies have dramatically changed the experimental and computational landscape for understanding individual T cells through high-throughput sequencing of the TCR (single cell (sc)TCR seq) with paired gene expression (scRNA seq). This technology provides the capacity to interrogate clonal T cell dynamics on an unprecedented scale in mice and humans, and a new tool kit to relate TCR information to biological phenotypes. With this explosion in new data, a current focus is to identify the best methodologies to interpret these data and develop computational pipelines to harness this type of information. In this Review, we focus on recent advances on the use of the TCR as a molecular barcode to quantify clonal expansion and diversity, as well as to identify cells of shared clonal lineage between tissues (Figure 1). We illustrate the value of these type of data in providing insights into anti-tumor immune responses and mechanisms of protection following Programmed Death-1 (PD-1) immunotherapy in cancer (Figure 2). We also consider challenges associated with this method (Box 1), including i) ambiguities in antigen-specificity and the polyspecific nature of TCR binding to pMHC (Box 2), ii) the presence of both antigen-specific T cells and bystander T cells in tissue sites of interest (Box 3), and iii) statistical inference issues related to sample size (Box 4). Lastly, we discuss ways to leverage the TCR sequence as more than a barcode, including how features within the TCR sequence influence T cell fate and function.
Figure 1 (Key Figure): The T cell receptor (TCR) sequence as a molecular barcode and beyond.

The TCR sequence can be used as a barcode to track clonal expansion, diversity, and migration, as well as a set of features to inform T cell fate and function. The pre-immune repertoire of T cells is highly diverse at the TCR level, in which virtually all naïve T cells have a unique TCR sequence [14, 15]. With T cell activation comes clonal expansion, where an individual T cell will divide numerous times, and the TCR is heritably maintained during cell division [3]. In this way, sister clones can be tracked within or between tissues based on sharing of the TCR sequence [27–29, 36, 38, 39, 50, 51]. Additionally, the TCR also acts as a set of features that influence T cell fate and function following antigen encounter [68, 69, 71]. In the figure, T cells (which can refer to either CD4+ or CD8+ T cells) with the same color TCR are considered clones based on matching sequences. Abbreviations used: TCR = T cell receptor; LN = lymph node; Tconv = conventional CD4+ T cell (non-Treg), Treg = CD4+ regulatory T cell; pMHCI = peptide/Major Histocompatibility Class I complex. Created with BioRender.com.
Figure 2: Use of T cell receptor (TCR) sequencing to inform mechanisms of PD-1-based immunotherapy.
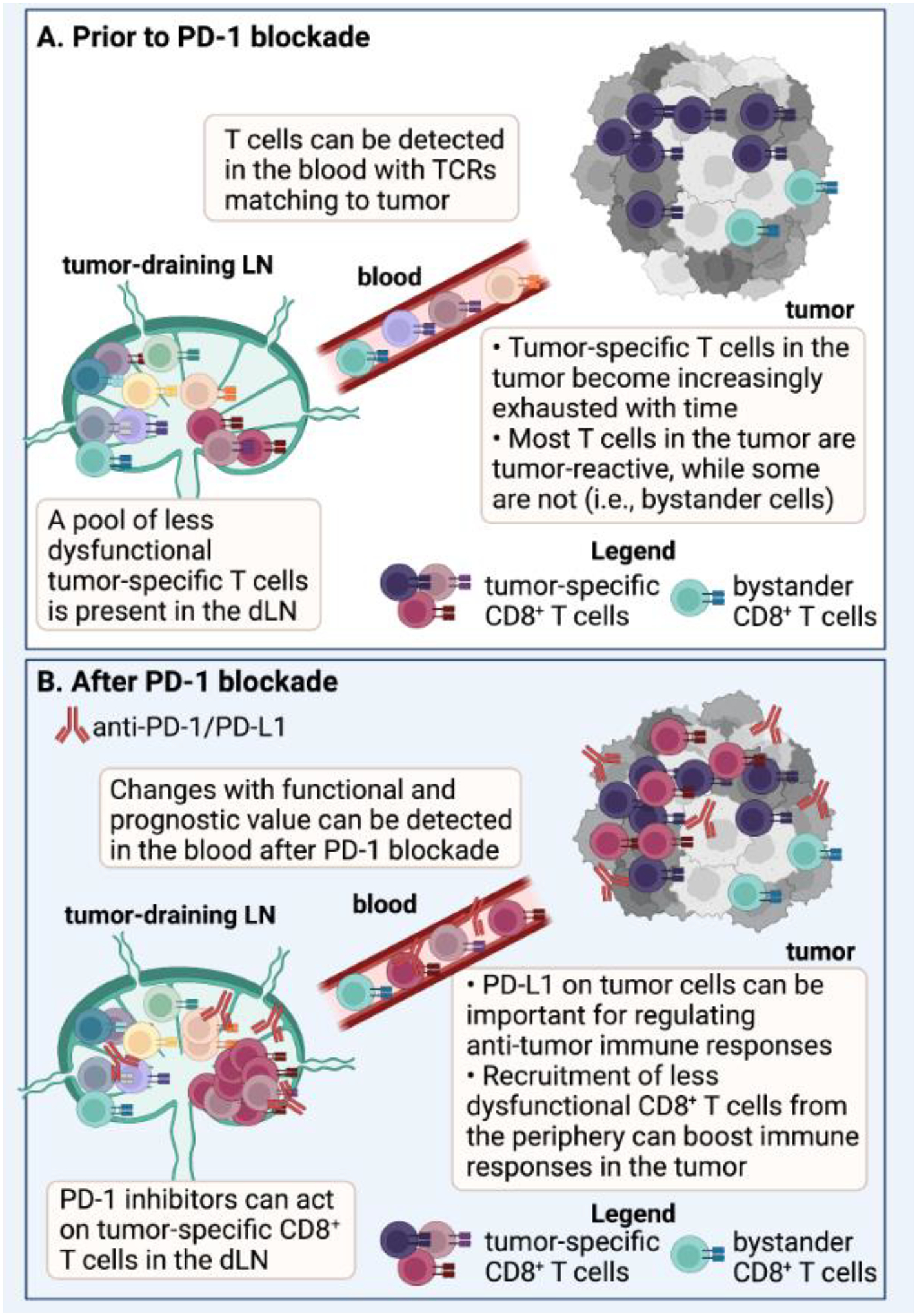
(A) The TCR sequence has been used as a molecular barcode to track T cells with shared TCRs between the tumor-draining lymph node (LN), blood, and tumors in mice [27, 63]. Similarly, the TCR has been used as a barcode to track T cells with matching TCRs between blood and tumor in humans [27–29, 35]. The tumor-draining LN contains a reservoir of less dysfunctional T cells, while the T cells found in the tumor often display a more exhausted or dysfunctional phenotype [31, 56, 63]. In addition to tumor-specific T cell clones, bystander T cells can also be present in the tumor that are not specific for canonical tumor antigens [107], but may still contribute functionally to the anti-tumor response [108, 109]. (B) Work has shown that Programmed Death 1 (PD-1) pathway inhibitors can act on T cells in the tumor-draining LN in mice [61, 62], and immunological changes that have functional and prognostic significance can be detected in the peripheral blood of humans [40, 67, 131–133]. An important mechanism of protection following PD-1 pathway blockade is recruitment of new, potentially less dysfunctional T cell clones from the periphery into the tumor [30, 36, 38–40, 67]. However, studies in mice and humans suggest that PD-1 inhibitors also likely act on T cells in the tumor as well [56–59]. Abbreviations in Figure: dLN = draining lymph node; TCR = T cell receptor, PD-1 = Programmed Death 1. Created with BioRender.com.
Box 1. Limitations of the barcode method.
One of the most significant limitations of TCR seq is conducting analyses across multiple individuals since the vast majority of the TCR repertoire is private. For example, autoimmunity studies have remarked on matching TCRs expanded across lesions and time points, but such matching largely occurs only within an individual [44, 45, 47]. Hence, although many individuals were afflicted with the same autoimmune disease presumably mediated by similar autoantigens, barcode-based approaches have thus far failed to identify TCRs that are pathogenic across patients.
Another significant limitation of TCR seq is the difficulty of inferring cognate antigen(s) from the TCR sequence. There have been efforts to experimentally link TCR sequences to antigen-specificity. One approach is to clone TCRs and express them in new cells (i.e., T cells from healthy donors or Jurkat cells) using lentiviral transduction, followed by testing the ability of these cells to bind and/or respond to antigens [35, 64]. Alternatively, TILs have been expanded in vitro using CD40-activated B cells and then screened for reactivity to different neoantigens [88]. Another approach is to combine tetramer reagents [89, 90] or dextramer reagentsI with TCR sequencing to simultaneously detect the TCR sequence, transcriptome, and antigen-binding capabilities with single cell resolution. This can be done by either FACS-based sorting on T cells that can bind tetramer or dextramer before performing single cell sequencing (using fluorescently labeled reagents) or by detecting the tetramer or dextramer computationally after sequencing (using DNA-barcoded reagents.) The latter approach may be useful for tracking both the expansion and distribution of individual TCR clones while accounting for antigen specificity, and has the advantage that ex vivo manipulations of T cells (i.e., in vitro expansion, cloning of TCRs, etc.) are not required. However, using tetramer/dextramer reagents also comes with limitations, including that the antigens of interest must be known, and these reagents preferentially bind high affinity TCRs [15, 22]. Consequently, developing approaches to infer antigen specificity directly from the TCR sequence would be extremely useful, and there are significant ongoing efforts aimed at creating methods to do so, including; i) clustering TCRs into antigen specificity groups [91–94], which may relate similar TCR sequences to the ability to bind a shared antigen, ii) T-scan, a high throughput platform for identifying antigens recognized by T cells that uses lentiviral delivery of antigens into APCs for processing onto MHC [95], and iii) high-throughput screening with iterative TCR affinity-based selection of peptide libraries presented by yeast cells [96]. This latter approach revealed similarity among the family of peptides recognized by a TCR, which was termed the “TCR fingerprint” [97], and might be used to help circumvent the issue that a single TCR can bind multiple different pMHC complexes.
Box 2. Predicting antigen specificity from the TCR sequence.
Moving forward, new, and better algorithms to predict antigen specificity from the TCR sequence will be extremely useful. A key complication to the goal of constructing antigen-specific TCR groups might be that TCRs in general are polyspecific rather than monospecific for pMHC complexes [98–100], meaning they are capable of recognizing multiple pMHC ligands [100]. Considering the diversity of potential antigens (and the complexity of MHC), developing models to predict cognate pMHCs of a given TCR is challenging. Recent efforts have reframed this problem to predict whether a TCR will recognize a given peptide, and have shown some success with [101] and without [102, 103] modeling MHC restriction. Several challenges in this endeavor, however, remain unsolved. First, public databases [104, 105] are helpful resources of TCR and pMHC pairs that bind, but it is difficult to systematically identify and validate TCR and pMHC pairs that definitively do not bind. These negative examples are equally important in training algorithms to distinguish the rules of TCR-pMHC recognition. Second, the amount of training data for these algorithms is limited relative to the staggering diversity of sequence interactions involved. Lastly, TCR fingerprinting reports feature recurrent observations of “off-target” ligands: several pMHCs that are perplexingly dissimilar from the family of recognized pMHCs [96, 106]. While some physiochemical interaction rules may be discernable from amino acid sequences, many interactions may arise from unknown variability in the 3D conformation of these molecules. Continued efforts to collect training data, with attention to HLA haplotype diversity and TCR cross-reactivity, may eventually lead to accurate predictions of TCR recognition.
Better methods also are needed to assess functionality of T cells on a clonal basis. Knowing the antigen-specificity of a T cell is a first step to inferring whether a T cell of interest is functionally relevant to a given immune response. Just because a T cell is antigen-specific does not necessarily mean it is functional (i.e., is capable of producing effector cytokines, killing cells). Similarly, just because a T cell is not antigen-specific (i.e., bystander cells) does not necessarily mean that it cannot contribute to an immune response of interest (Box 3).
Box 3. “Bystander” CD8+ T cells in anti-tumor immune responses.
There is evidence that T cells that are not specific to tumor antigens, termed “bystander” T cells, can infiltrate tumors [107]. Using the TCR as a barcode to identify T cells in peripheral sites based solely on matching TCR sequences to T cells in the tumor may enable detection of either tumor antigen-specific or bystander T cells since both can be present in tumors [107]. Despite the notion that “bystander” T cells do not recognize canonical tumor antigens, some data suggest that these cells can functionally contribute to the anti-tumor immune response. For instance, in cancers associated with viral infection, such as hepatocellular carcinoma, Hepatitis B virus, and human papillomavirus (HPV)+ head and neck cancer, virus-specific CD8+ T cells infiltrate the tumor [108] and can be associated with a favorable prognosis in patients [109]. Moreover, CD8+ T cells with viral specificities are also found among the tumor infiltrate in cancers with no viral etiology. For example, using mass cytometry and pMHC tetramer staining to test a large panel of tumor-specific antigens, tumor-associated antigens, and irrelevant antigens in lung cancer, one study found bystander CD8+ T cells to be more abundant than tumor-specific CD8+ T cells [107]. At the protein level, bystander and tumor-specific CD8+ TILs shared largely overlapping phenotypes, but CD39 expression was exclusive to tumor-specific CD8+ TILs [107]. Conversely, another study showed that, at the RNA level, neoantigen and tumor associated antigen-specific melanoma-infiltrating CD8+ T cells occupied the same exhausted clusters, while bystander CD8+ TILs were segregated to different, more cytotoxic areas of the transcriptional space [35]. Consistent with these results, in melanoma, neoantigen-specific CD8+ T cells could be detected more easily among TILs with the highest gene signatures of exhaustion [110]. In CD8+ TILs in melanoma, the degree of TCR sharing across tissues can recapitulate the T cell phenotype: the most cytotoxic CD8+ TILs can exhibit the most highly shared clonotypes, while the most exhausted CD8+ TILs can be exclusively found in the tumor [27, 28]. If indeed bystander CD8+ TILs are the most cytotoxic [35], it is possible that the clonotypes that are most shared will be bystander, suggesting that circulating tumor-directed CD8+ T cells might be enriched for sister clones of bystander cells. Nevertheless, the potential role of bystander CD8+ TILs for the anti-tumor immune response should not be discounted. While CD39+ CD8+ TILs (e.g. likely tumor-specific) have been associated with favorable prognosis in human lung cancer [111], it is possible that bystander CD8+ TILs might also actively contribute to tumor rejection [112]. A better understanding of how these bystander or non-tumor antigen-specific T cells influence anti-tumor immunity is needed to determine the functional relevance of these populations.
Box 4. Diversity as a means to extract the relevance of TCRs.
TCR repertoire diversity has been a classic metric of comparison, with clonal expansion indicated by a reduction in diversity [113]. Many diversity-based metrics have been applied to bulk TCR seq data including Shannon entropy, Abundance-based Coverage Estimator (ACE), and Chao index [114], and often correspond with phenotypes of interest in T cells; notably, tumor mutation load [115], cancer progression [116], and disease activity in RA [117]. With scTCR data, considering how robust these metrics are to sample size is important. While bulk TCR seq routinely collects 105-106 TCRs per sample, sample sizes in single-cell studies are variable and typically orders of magnitude lower (102-103 TCRs) than bulk [118]. The distribution of clonotype sizes is heavily skewed, featuring a long tail of unexpanded clones that represent a minimal fraction of the true underlying repertoire, even with exhaustive sequencing assays [119–121]. Many diversity estimators are exquisitely sensitive to sample size, generating different results in simulated samples of the same repertoire [114]. Strategies proposed to address this issue include: i) down-sampling the larger sample for comparison to match the smaller [122], ii) creating diversity metric reference distributions for different sample sizes to enable statistical testing and reporting of confidence intervals around diversity estimates [123, 124], and iii) characterizing the diversity of each sample instead by a rarefaction curve, which quantifies how quickly the observed number of clonotypes grows with sample size [114]. The impact of sample size on the metric used should be closely examined before inferring relationships between TCR diversity and phenotypes of interest.
An important underlying question is whether diversity metrics will continue to be one of the most useful approaches to elucidate the role of the TCR repertoire in health and disease. While diversity metrics have been useful, a considerable amount of information is lost when TCR seq data are reduced to a single feature. Deep learning studies with TCR seq data [125] demonstrate that there is flexibility in deriving information from the TCR repertoire, and that convolutional neural networks may successfully extract antigen-relevant motifs from TCR sequences. Ultimately, clustering TCRs into antigen specificity groups [91–94] may be the path forward for comparing private TCR repertoires, analogous to the way that clustering transcriptional profiles in scRNAseq data has enabled cell type-based comparisons between individuals. Association studies between TCR clusters and somatic mutations in cancer, for example, has led to the exciting identification of a novel immunogenic cancer antigen, Heat Shock Transcription Factor X-linked 1 [93]. Emerging strategies that examine the co-occurrence patterns of TCR sequences have shown early success in identifying disease-related TCRs across individualsII [126]. An important challenge to address in these approaches is disentangling genetic influences on the TCR repertoire [85, 87, 127] from antigenic causes of TCR co-occurrence.
Methods for tracking T cells of interest
The diversity of the pre-immune repertoire of naïve T cells is extremely high in higher order mammals (i.e., humans, mice, non-human primates), with virtually all naïve T cells having a different TCR sequence [14, 15]. Based on the number of V, D, and J gene segments in humans, there are theoretically 1014-1019 possible different TCR sequences, though the observed number tends to be closer to 108, likely due to constraints during T cell development (i.e., failure to undergo positive selection, negative selection, etc.) [16–18]. When a naïve T cell becomes activated, the process of effector differentiation is coupled with proliferation [3]. In acute lymphocytic choriomeningitis virus (LCMV) infection in mice, it is thought that each individual CD8+ T cell can divide up to 15–20 times, expanding the number of antigen-specific T cells by up to 50,000 fold compared to the preimmune repertoire [19, 20]. However, this likely represents the upper limit of clonal expansion possible under optimal conditions. In autoimmunity and cancer, clonal expansion likely does not reach this extent, due at least in part, to suboptimal T cell activation conditions (which include low-affinity antigens, low levels of inflammation, etc.). Regardless of the degree of clonal expansion, T cells of a single clonotype are numerically rare in the periphery and exist amongst the rest of the diverse T cell repertoire that is not involved in the immune response of interest. The ability to distinguish T cell clones of interest from the rest of the T cell repertoire is fundamental to determining mechanisms associated with T cell-driven immune responses.
Antigen-directed vs. TCR-directed methods for detecting T cells of interest
There are a number of methods to identify and track T cells of interest [21]. Historically, antigen-specific T cells have been identified based on their ability to bind and/or recognize specific antigens (referred to as antigen-directed methods). Tetramers are the gold standard for detecting T cells based on their ability to bind a specific antigen bound to MHC. Tetramers are soluble pMHC molecules complexed to a streptavidin core that is labeled with either a fluorescent marker or a DNA barcode that can be tracked. While tetramers have been extremely useful for identifying antigen-specific T cell populations in diverse disease settings, they have a number of limitations, including: i) the antigen must be known, limiting studies to TCRs that recognize well-characterized antigens, ii) studies of the breadth of the response are limited to the number of antigens that are known and can be tracked at one time, iii) tetramers are often inefficient at detecting TCRs with low affinity for pMHC, which may be a significant fraction of responding T cells in cancer and autoimmunity, and iv) tetramers can only be used in individuals with MHC haplotypes for which there are tetramer reagents available, limiting the patient population in which these T cells can be tracked [15, 22]. Another antigen-directed method is the use of antigen stimulation in vitro followed by sorting of activated T cells, evaluating either the activation marker expression or the expansion levels [23–25]. While this can be a useful approach for determining if antigen-specific T cells are present in a sample, it is difficult to use these cells for downstream functional assays, or to characterize the phenotype and function of these cells at steady state prior to in vitro stimulation. Considering these limitations, developing alternative methods to identify antigen-specific T cell populations that do not rely on knowing each individual antigen could broaden the scope of the T cell response under investigation.
The TCR sequence provides an alternative to antigen-directed methods for identifying T cells that are relevant to immune responses of interest. The TCR sequence is generated through V(D)J recombination during T cell development in the thymus [26]. This process essentially involves the random recombination of V, D, and J gene segments to generate unique sequences for the beta chain (V, D, and J) and alpha chain (V, J) of each TCR [26]. Because these somatically recombined DNA sequences are conserved during mitosis of mature T cells, expanded T cell clones harbor identical TCR sequences. When TCR sequencing assays recover identical TCR sequences in a given individual, clonal expansion is generally inferred. The alternative inference — that identical but independent V(D)J recombination events were sampled from the same individual — is highly unlikely given the enormous diversity of the pre-immune repertoire. Consequently, the TCR sequence can serve as a molecular barcode to track T cells of a shared origin (e.g., clonally-related sister cells) through biological processes such as T cell migration and differentiation within and across tissues (Figure 1). In sequencing assays that link TCRs to unique molecular identifiers (UMIs), the TCR can be used to quantify the extent to which a T cell clone has clonally expanded. TCR analyses also provide a useful approach for enriching circulating T cells that are relevant to a response (as opposed to using surface marker expression); indeed, this approach does not rely on the assumption that antigen-specific T cells will express any given marker [27, 28]. The simultaneous analysis of TCR sequences, gene expression (using scRNA seq), and protein expression (using “cellular indexing of transcriptomes and epitopes by sequencing” or CITE seq) allows for dissection of the transcriptional and phenotypic states of T cells without altering their function, enabling deep profiling of T cell populations.
Using the TCR as a barcode in cancer
The TCR has been used as a barcode to identify CD8+ T cells in peripheral blood that share TCRs with CD8+ tumor-infiltrating lymphocytes (TILs) in tumors (referred to as “tumor matching”), as shown in mice with MC38 colon adenocarcinoma [27], as well as in patients with advanced melanoma and non-small cell lung (NSCL) cancer [27–30]. The TCR has also been valuable for studying the dynamics of individual CD8+ T cell clones, including clone-by-clone comparisons of transcriptional states across tissues and inferring lineage relationships between populations of CD8+ T cells in mouse (including MC38, B16-F10, CT26, and a genetically engineered mouse model (GEMM) of lung adenocarcinoma caused by oncogenic Kras and loss of p53) and human tumors (including melanoma, NSCL cancer, colorectal cancer (CRC)) [27–38]. A recent study in humans used paired scRNA seq and scTCR seq to analyze CD4+ T cells and CD8+ T cells across 21 cancer types (including melanoma and breast, gastric, pancreatic, kidney, and esophageal cancers) from 316 patients; the study identified two major developmental trajectories that contributed to T cell exhaustion across multiple tumor types, based on transcriptional information [32]. Additionally, scTCR seq has been useful to inform mechanisms of response following PD-1-based immunotherapy in cancer patients [30, 36–41], as later discussed. While these studies have shown that scTCR seq data can be promising for focused analyses on T cell populations of interest, there are limitations to this method (Box 1, Box 2, Box 3). One limitation is that some baseline level of clonal expansion may be needed for the TCR sequence to be useful, due to the detection limits for identifying rare or unexpanded populations by single cell sequencing [42]. Because of the importance of clonal expansion, the barcode method may have more utility in immunologically “hot” tumors (i.e. high mutational burden, high CD8+ T cell infiltration, high inflammation) than in immunologically “cold” tumors (i.e. low mutational burden, low CD8+ T cell inflammation, low inflammation).
Using the TCR as a barcode in autoimmunity
Leveraging the TCR as a molecular barcode in immune compartments associated with autoimmune disease can identify expanded clones contributing to ongoing inflammation. For example, scTCRseq in the affected joints of Psoriatic Arthritis patients revealed predominantly CD8+ clonal expansions, providing the strongest evidence to date that psoriatic arthritis may be CD8+ T cell-mediated [43]. Several studies that have sequenced TCRs in the affected joints and peripheral blood of Rheumatoid Arthritis (RA) patients have remarked that a given individual will often demonstrate clonal expansion of the same TCR across multiple joints [44–47]. Longitudinal studies of TCRs infiltrating the central nervous system (CNS) in multiple sclerosis (MS) [48, 49] have similarly revealed that the same clone can be active over time within a given individual. Comparison of profiles of clonally expanded T cells obtained from the cerebral spinal fluid (CSF) of patients with multiple sclerosis (MS) and healthy donors revealed that clonally expanded T cells from the CSF of patients with MS had heightened expression of genes related to T cell activation and cytotoxicity [50]. Additionally, in a mouse model of MS (experimental autoimmune encephalomyelitis; EAE), combined scRNA seq and TCR seq identified a stem-like pathogenic precursor CD4+ T cell population in the intestine (TCF-1+ SlamF6+ IL-17+) that gave rise to a more pathogenic subset (GM-CSF+ IFNγ+ CXCR6+ Th17 CD4+ T cells), capable of migrating to the CNS, and highlighting the utility of the TCR as a barcode to track pathogenic autoreactive CD4+ T cells between different tissues [51]. However, as in cancer where the TCR as a barcode may be more relevant for immunologically “hot” than “cold” tumors, the TCR may be more useful for some types and/or stages of autoimmunity than others. For example, in RA and MS, antigen persists throughout disease, while in Type 1 Diabetes, the antigen (insulin-producing beta cells) decreases over time, can be completely eliminated, and the pathogenic T cell population may largely contract with the loss of the antigen-expressing cells. Frozen pancreas tissue from 45 patients with Type 1 Diabetes (ranging from 1 week to greater than 50 years post-diagnosis) was analyzed for the presence of autoreactive CD8+ T cells using pMHC tetramers; over time, the presence of autoantigen-specific CD8+ T cells declined, and these cells became progressively more difficult to detect with increasing time post-diabetes onset [52]. Consequently, using the TCR as a barcode may be more informative in settings in which pathogenic T cells are likely present in the target autoimmune organ, which may also vary with type and/or stage of autoimmune disease.
Using TCR analyses to inform on mechanisms of anti-tumor immunity following PD-1 immunotherapy
Harnessing the power of the immune system to target and destroy cancer cells using immunotherapy has transformed cancer care. Inhibitors of the PD-1 pathway have shown particular promise, and are now approved by the U.S. Food and Drug Administration for use in over 20 types of advanced stage cancers [53–55]. However, most patients do not have durable tumor regression, and some cancer indications are highly intractable to PD-1 blockade (i.e., pancreatic cancer, prostate cancer, glioblastoma) [54]. Consequently, there are intense efforts to understand the immunological basis for improved anti-tumor immunity following PD-1 blockade to facilitate the development of alternative approaches in patients unlikely to respond. In this section, we discuss how TCR analyses have provided insight into which T cells are essential for driving effective clinical responses following anti-PD-1 antibody cancer therapy. Considerations for monitoring responses to PD-1 inhibitors in patients are discussed in Box 5.
Box 5. Clinician’s Corner Box – Clinical Applications of TCR Sequencing.
High-throughput TCR sequencing has enabled characterization of the repertoire and dynamics of T cell responses with immunotherapy. Recent studies have demonstrated the potential utility of analyzing the clonal composition of blood T cells to predict clinical responses to checkpoint blockadeIII, IV [40, 67]. A correlation has been observed between highly expanded circulating clones and response to therapy in metastatic melanoma patients receiving anti-PD-1 antibody with or without anti-CTLA-4 antibodyV [40]. Of note, expanded clones at baseline and on-treatment were correlated with a 6-month positive clinical response, highlighting the potential importance of both the pre-existing immune response and early on-treatment dynamics.
Longitudinally monitoring relevant circulating T cell clones also may inform clinical responses and progression. In a study of three metastatic NSCL patients receiving anti-PD-1 antibody, the presence of “ubiquitous” CD8+ T cell clones – clonotypes also found in all tissue compartments sampled, including blood, tumor, adjacent normal tissue, and LNs – correlated with systemic tumor control when traced over a three year periodIV. Conversely, the persistence of experimentally-verified, tumor-reactive CD8+ T cell clones with transcriptional features of exhaustion in the blood correlated with disease progression in a study of 14 melanoma patients receiving anti-PD-1 antibody [35]. Another study in patients with resectable NSCL cancer undergoing neoadjuvant PD-1 blockade found that following definitive surgical resection, the frequency of neoantigen-specific TCRs substantially decreased in the blood [64]. The reduction in these TCRs may suggest that tumor antigen is required to sustain these T cells, since exhausted CD8+ T cells (in mouse chronic LCMV infection) require continual antigen stimulation for their survival and maintenance [128, 129]. Additional work is needed to determine if T cell death is the major mechanism contributing to decreased T cells after surgical tumor resection. While these studies suggest the potential clinical utility of clonal analyses, translation into the clinical setting remains in its nascency, and it remains to be seen whether clonal analyses can be feasibly implemented for clinical testing. Nonetheless, TCR sequencing studies have provided key insights into how T cell repertoires change with immunotherapies and have provided a basis for improving clinical management in patients with immunotherapy-resistant tumors.
One potential application of coupling TCR sequencing with high-resolution transcriptional profiling is as an alternative approach towards predictive biomarker discovery. Using paired TCRseq with RNAseq, two recent studies utilized a program called COMET [130] to identify candidate cell surface markers shared by blood T cells with TCR sequences matching tumor-infiltrating lymphocytes from patients with metastatic melanoma [27, 28]. One primary advantage of focusing on cell surface markers is the ease-of-use in clinical settings, as flow cytometry-based assays are already established. Further studies to validate and define the clinical relevance of these markers are ongoing.
Anatomical site of action of PD-1 inhibitors – Tumor vs. LN
Early studies examining the role of interactions between the PD-1 receptor and its ligand Programmed Death Ligand-1 (PD-L1) in anti-tumor immunity focused on the direct interaction between tumor cell expression of PD-L1 and T cell PD-1 [53, 54, 56]. In mouse models: i) Transgenic expression of PD-L1 in mastocytoma tumor cells (P815) limited T cell cytotoxic function [57]; ii) PD-L1 expression on tumor cells (MC38 colon adenocarcinoma) was sufficient to suppress the anti-tumor CD8+ T cell response, and selectively deleting PD-L1 on tumor cells could re-sensitize tumors to CD8+ T cell-mediated killing [58]; and iii) PD-L1 expressed by the tumor cell (methylcholanthrene-induced sarcomas) was crucial for promoting immune escape [59]. Collectively, these studies supported the notion that PD-1/PD-L1 signals in the tumor microenvironment (TME) could play an important role in suppressing anti-tumor immunity (Figure 2).
One outstanding question from these studies was whether effective anti-tumor immune responses were driven by reinvigorated CD8+ T cells within the tumor bed, or by newly recruited tumor-specific T cells. Newer work suggests that the migration of tumor-specific CD8+ T cells from outside of the tumor is a key driver of effective anti-tumor responses with PD-1 blockade [53, 56, 60–62] (Figure 2). In mouse tumor models, reservoirs of tumor-specific CD8+ T cells were identified in tumor-draining LN in lung adenocarcinoma (oncogenic Kras, loss of p53 tumor model) and mesothelioma (AE17 tumor model) [31, 62, 63], and the clonal repertoire within tumor-draining LNs was reflective of the intratumoral repertoire [63]. ScTCR seq from patients with NSCL cancer similarly confirmed a reservoir of neoantigen-specific CD8+ T cells within tumor-draining LNs [64]. In mice, treatment-induced expansion of tumor-specific CD8+ T cells (using bulk TCRseq) was more pronounced in the draining LN (dLN) than in B16 melanoma tumors [65], and surgical removal of the tumor-dLN diminished the therapeutic efficacy of PD-1 pathway blockade (in MC38 colon adenocarcinoma and CT26 colon carcinoma) [61]. Additionally, blocking T cell migration from the tumor-dLN to the tumor using the S1P agonist FTY720 abolished such anti-tumor effects following PD-1 blockade [62, 66]; this suggested that T cell trafficking from the tumor-dLN to the tumor was important for productive anti-tumor immune responses following PD-1 blockade. Together, these findings support a model whereby the anti-tumor effects of PD-1 blockade derive, at least partially, from T cells recruited beyond the tumor, also highlighting the importance of the tumor-dLN in this process (Figure 2).
Protective effects of PD-1 blockade - Novel vs. pre-existing clones in the tumor?
Clonotype characterization at the single-cell level provided further evidence for the essential role of CD8+ T cells recruited from the periphery in response to anti-PD-1 antibody therapy (Figure 2). Paired scRNA seq and scTCR seq on site-matched tumors before and at ~9 weeks following anti-PD-1 antibody therapy from patients with non-melanoma skin cancers (11 basal cell carcinoma, 4 cutaneous squamous cell carcinoma) revealed that ~68% of clonally expanded tumor-infiltrating T cells were undetectable in the tumor prior to therapy [38]. Furthermore, clonotype sequences present in the tumor prior to PD-1 blockade showed minimal to no expansion post-treatment. The recruitment of “novel” clones from the blood to the tumor following PD-1 blockade was termed clonal replacement [38]. Another study examining pre- and post-PD-1 blockade tumors from patients with breast cancer also observed that ~40% of expanded clones were novel just nine days after receiving anti-PD-1 antibody [37]. Additional studies showed that the presence of clonal replacement correlated with improved clinical anti-tumor response [36, 39, 40, 67]. However, work in patients with NSCL cancer has led to the proposal of an additional mechanism for protective immune responses following PD-1 blockade, termed clonal revival [30]. With clonal revival, new, less dysfunctional CD8+ T cell clones populate the tumor (from the periphery, local expansion within the tumor, or both), replenishing the more terminally exhausted pre-existing pool of CD8+ TILs. However, these clones can either have novel TCRs to the tumor following PD-1 blockade (like clonal replacement), or TCRs matching to pre-existing clones in the tumor [30]. Collectively, while all of these studies support a model where the anti-tumor immune response boosted by PD-1 blockade is largely generated beyond the tumor [30, 36–40, 67] (Figure 2), whether most protective responses following PD-1 blockade are due to truly novel clones that were absent from the tumor before blockade or to both novel and pre-existing clones, remains to be determined. Of note, timing of sampling post-PD-1 blockade may influence the frequency of the clonally expanded TIL population that is novel vs. pre-existing. Additionally, the inability to broadly and comprehensively sample the entire repertoire of TCRs within a given tumor may be a confounding factor in classifying T cell clones as being “novel” or “pre-existing”, because if not enough T cells were sequenced, clones that were pre-existing may be misclassified as novel. The addition of bulk TCRseq may be useful to increase sampling depth. Moreover, further work is needed to understand the functional differences between newly recruited T cells and T cells residing in the tumor; the limited data are transcriptional [36–39]. Thus, the answers to these questions will be important for determining how to optimally harness peripheral T cell populations for therapeutic gain in PD-1-based immunotherapies.
Beyond a barcode – How does the TCR sequence impact T cell fate?
The TCR sequence is more than a clonal identity barcode. The amino acids of the TCR direct the reactivity, and in many contexts, the cellular phenotype, of the T cell. This concept was demonstrated in patients with CRC or breast cancer, where statistical classifiers could identify TILs (unsorted, which included both CD4+ and CD8+ subsets) based on TCR amino acid physiochemical features such as hydrophobicity and electrostatic charge [68]. This classifier distinguished TILs from T cells in adjacent normal tissue with 93% accuracy in a testing data set (data held out from the initial training set). This idea was extended to broadly characterize “cancer-associated” TCRs based on physicochemical features with a deep convolutional neural network called DeepCat [69]. While the prior study developed a specific classifier for breast cancer and another specific classifier for CRC [68], DeepCat demonstrated that tumor-infiltrating TCRs had convergent features across as many as 32 cancer types [69]. Predictions made by DeepCat achieved an AUC >0.95 for multiple early-stage, treatment-naïve cancers, demonstrating the possibility of TCR repertoire-based biomarkers for cancer detection. Because nonspecific factors such as chronic inflammation can characterize some late-stage cancers, it will be important to similarly test future TCR biomarkers with respect to early-stage disease [69]. It should be noted, however, that the cancer type-specific classifiers developed in the aforementioned study of statistical classifiers [68] achieved better accuracy than achieved using DeepCat [69], suggesting that some infiltration-promoting TCR features might be cancer type-specific. This speculation raises the possibility that patient-specific classifiers might achieve the best longitudinal accuracy in tracking tumor-directed TCRs. Broadly-trained classifiers such as DeepCat, however, are poised to uncover general biology across patients and cancer types. Given the success of studies such as DeepCat in cancer [69], a question is whether physicochemical property-based TCR analyses would shed light on T cells that drive autoimmunity. While one study has shown that self-reactive CD4+ and CD8+ T cells were enriched for hydrophobicity at two positions of the TCR sequence [70], there has not been an attempt as comprehensive as DeepCat for predicting convergent features of pathogenic TCRs across diverse autoimmune diseases. Extending these approaches to try to elucidate the features of the TCR that broadly promote autoreactivity are thus warranted.
While these approaches demonstrated the predictive value of TCR amino acids in terms of tumor infiltration, the functional phenotype of these cells remained unknown. Focusing on T cell phenotypes, an orthogonal study from our group developed a TCR scoring system that ultimately described which CD4+ T cells in the tumor microenvironment were more likely to express a regulatory T cell (Treg) phenotype over a conventional effector CD4+ T cell phenotype (Tconv) based on features of the TCR sequence [71]. Training data for this scoring system used human circulating Tregs, which were expected to largely consist of clones that became Tregs in the thymus [72]. After developing the scoring system based on data from individuals without cancer, TILs were scored from basal and squamous cell carcinoma patients; T cell clones containing Tregs harbored significantly higher-scoring TCRs than other T cells. When the TCR was used as a barcode to mark clonally-related cells, some T cell clones consisted of both Treg and Tconv cells [71]. These clones, likely consisting of peripherally-induced Tregs and Tregs that have lost their suppressive phenotype (exTregs), also demonstrated significantly more Treg-like TCRs than clones that maintained the conventional effector phenotype [71]. While the extent to which peripherally-induced Tregs contributed to this result remains to be determined, the identification of TCR sequence features that could promote suppressive phenotypes in the tumor is an important step toward understanding the T cell population involved in the anti-tumor response. These Treg-promoting TCR sequence features are also significantly enriched in TCRs known to be autoreactive [71], suggesting that such TCR scoring might be used to screen for autoimmune pathogenic TCRs, most of which have been elusive to date.
As evidenced by these three studies [68, 69, 71], studying TCR sequences as a set of features through the lens of physicochemical properties (e.g. hydrophobicity, volume, charge of the T cells) [68, 69, 71] is a promising approach to translate otherwise incomparable private TCR sequences into quantities that are easily analyzable across individuals and biological contexts. Numerous attempts to identify tumor-infiltrating TCRs have failed to uncover cancer-specific TCR sequences across patients [73–78]. Moreover, TCR sequences seen in multiple patients have been reported in the repertoires of healthy donors, suggesting that exact TCR sequence matches between individuals may reflect public clonotypes rather than shared antigens [73, 75, 77, 78]. Physiochemical properties, however, enable the ability to draw similarities between TCRs that do not precisely match in terms of sequence. Analyses that couple the TCR as a set of features with the existing molecular barcode approach might hold great power to unlock information regarding clonal and functional dynamics of the T cell response.
Concluding Remarks
Single-cell sequencing of the TCR has the potential to dramatically change the experimental and computational landscapes of both preclinical and clinical immunology, providing clonal single-cell precision to T cell responses in settings that otherwise would be intractable to traditional fate mapping approaches. This is truly an unprecedented time for human immunology with the use of these approaches to understand T cell responses in health and disease. However, several outstanding questions remain (Outstanding Questions Box). The TCR sequence has clearly demonstrated its utility in identifying T cell populations of interest to a given immune response. However, a key outstanding question is assessing the functional relevance of T cells on a clonal basis. While pairing deep molecular profiling and surface protein information with the TCR sequence provides an opportunity to thoroughly characterize T cells based on clonotype, defining T cell states based on transcriptional data alone is not trivial [79]. The addition of other parameters such as defining open chromatin regions to profile the epigenetic landscape of a cell (such as by using single cell assay Assay for Transposase-Accessible Chromatin with high-throughput sequencing or ATAC seq) could be useful to better annotate cell states. However, ultimately developing methods that can couple functional information with these sequencing-based parameters would be extremely useful.
Outstanding Questions.
How do we determine which clonotypes are relevant to an immune response of interest in humans and mice? Linking the TCR sequence to the identity of the antigen the T cell can bind constitutes a first step for distinguishing TCRs that are “relevant”. Paired gene expression and surface protein information can be useful for coupling transcriptional/phenotypic features with specific TCR sequences to infer functional relevance to responses. However, further work is needed to determine if additional functional metrics can be ascertained from T cells along with the TCR sequence to more definitively classify T cells as being functionally relevant.
How does clonotype information relate to antigen-specificity? Will the ability to infer antigen-specificity from TCR sequences be confounded by the flexibility in terms of pMHC complexes that bind T cells, or will it be possible to confidently predict antigen-specificity from the TCR sequence?
To what extent does MHC haplotype shape the ability of TCRs to bind pMHC, and will that influence how TCR data can be leveraged? Is there so much diversity within the T cell repertoire that the ability to make comparisons between individuals is limited?
To what extent is a certain degree of clonal expansion a prerequisite for using the TCR as a molecular barcode? Will the TCR have limited utility as a barcode in settings where there is not a certain degree of clonal expansion?
How does the sequence of the TCR directly influence T cell fate and function beyond simply acting as a unique identifying barcode to track clonally expanded populations?
Can clonal analyses serve as a clinically useful predictive biomarker, particularly for cancer patients receiving immunotherapy? Will the ability of TCR data to serve as a ‘useful biomarker’ differ between immunologically “hot” and “cold” tumors? Can longitudinal tracking of TCR sequences within a single patient be a useful metric for monitoring changes in the immune response over time?
While paired scRNA seq and TCR seq are currently being widely employed to uncover mechanisms associated with response or resistance to PD-1 cancer immunotherapy, there is also significant interest in using this approach to study immune-related adverse events (irAEs). IrAEs are pathogenic events that occur following immunotherapy in cancer patients, and are well documented following immune checkpoint blockade (e.g. anti-CTLA-4 or PD-1 antibody) [80]. IrAEs are extremely diverse, impacting a wide range of organ systems, and varying in severity from mild to life threatening [80]. The idea of using the TCR as a molecular barcode to track pathogenic T cell responses in settings such as irAEs is appealing, since theoretically, the full complement of antigens involved in the response need not be known necessarily. However, further work is needed to determine whether the technical limitations of using this method will impact utility in these settings as well as in other specific disease contexts. TCR data have been useful in settings such as immunologically hot cancers (e.g. advanced melanoma, NSCL cancer) where there are highly expanded CD8+ T cell clones and the tumor is well infiltrated by CD8+ T cells. In settings where there may be less clonal expansion and/or low levels of T cell infiltration in tissues of interest at the time of analysis, it is unclear how reliable the TCR will be as a molecular barcode to identify relevant clones. Finally, in some pathogenic immune reactions (i.e., systemic autoimmunity, graft vs. host disease, some irAEs), aberrant immune activation is systemic, targeting one or more antigens that are diffuse throughout the body, in contrast to organ-specific pathogenic reactions. Additional work is needed to determine the utility of the TCR as a barcode in different immune contexts, at steady state as well as in patients receiving immunomodulatory agents (e.g. immunosuppression).
Another major limitation for cancer, autoimmunity, and irAEs is that the repertoires of antigens driving responses are largely unknown, in contrast to infectious diseases [81–83]. Consequently, alternative strategies may be required to identify TCRs relevant to the disease-specific immune response. Many autoimmune diseases feature risk polymorphisms in HLA genes, which constrain the TCR repertoire during thymic selection [84–86]. Recent innovations have leveraged the relationship between HLA and TCR variation to uncover which TCR repertoire features are associated with genetic risk for autoimmune disease [87]. Moving forward, combining methods to better link TCR sequences detected within sites of interest, antigen prediction, and MHC information may be useful to provide a clearer picture of the populations driving protective or pathogenic immune responses. Finally, the TCR has been the focus of intense study for decades; however, active areas of investigation include how the TCR sequence shapes T cell fate and function, as well as what the contribution of antigen and genetic variation in MHC to TCR recognition and T cell function are. Future studies aimed at deciphering how the TCR sequence itself influences T cell fate decisions will be essential for understanding how the complex interplay among the TCR, peptide and MHC dictates T cell responses.
Highlights.
The TCR can act as a molecular barcode in mice and humans to track T cells with clonal-relatedness through processes such as migration, differentiation, and proliferation.
The TCR sequence provides a means to enrich T cells of interest in diverse types of cancer and some types of autoimmunity.
Using the TCR as a barcode in cancer has been useful for identifying mechanisms of PD-1 blockade-based immunotherapy and features associated with clinical responses in diverse cancer types.
Using the TCR sequence to identify T cell populations of interest is currently limited by our inability to confidently infer antigen-specificity from the TCR sequence.
Beyond acting as a molecular-identity barcode, the TCR sequence consists of a set of features that can inform T cell fate and function.
Acknowledgements
We apologize to our colleagues whose work was not cited due to space constraints. This work was supported in part by T32GM007753 from the National Institute of General Medical Sciences (which supports K.A.L.), the National Cancer Institute T32 CA233414 (which supports B.Y.L.); the Yale SPORE in Skin Cancer P50 CA121974 (to H.M.K); grants from the National Institutes of Health P01 AI073748, U24 AI11867, R01 AI22220, UM 1HG009390, P01 AI039671, P50 CA121974, R01 CA227473 (to D.A.H.); grants from the National Institutes of Health P01 AI039671, P01 AI56299, P01 CA236749 and P01 AI108545 (to A.H.S); and 1P01AI148102-01A1, 1U01HG012009-01, and 2R01AR063759-06A1 (to S.R.). A. I. D. receives funding from the Parker Institute for Cancer Immunotherapy, Cook, McGlynn and Amoroso Funds, Merck, Novartis, Genentech/Roche, Seagen, Incyte and BMS.
Glossary
- Abundance-based Coverage Estimator (ACE)
estimator of population diversity
- AUC
measure of the entire two-dimensional area underneath a Receiver Operating Characteristic (ROC) curve, generated from the sensitivity and specificity of a classifier over a range of test thresholds; single measure of the classifier’s ability to distinguish positive and negative classes. More accurate classifiers have higher AUC values (up to 1.0)
- Bystander T cells
In tumors, T cells that do not bind tumor-associated antigens
- Chao index
estimator of population diversity
- Clonal replacement
recruitment of less dysfunctional peripheral T cell clonotypes to the tumor that were not detectable in the tumor prior to PD-1 blockade
- Clonal revival
replenishment of highly dysfunctional T cell clones in the tumor with new, less dysfunctional T cells. The source of the latter can be the periphery, local expansion within the tumor, or both; these T cells can either be novel clonotypes or clonotypes pre-existing in the tumor
- Clonotype
paired alpha and beta TCR nucleotide sequence of a T cell
- Clone
set of T cells that are clonal progeny due to shared clonotype and shared host
- Convolutional neural network
algorithm that optimizes a series of mathematical transformations (layers, consisting of neurons) on input data to generate a prediction. Each neuron processes only the part of the previous layer that lies in its receptive field (i.e. a subsequence of the TCR)
- Dextramer
similar to a tetramer; contains a peptide bound to an MHC molecule; labeled to allow detection by flow cytometry or sequencing
- Graft vs. host disease
when following transplant, cells from the graft (e.g., bone marrow or stem cells) attack the host
- FTY720
drug blocking the binding of Sphingosine-1-phosphate receptor 1 to sphingosine-1-phosphate
- MHC haplotype
combination of MHC alleles found in a single individual
- Monospecific
a T cell recognizing only a single peptide/MHC complex
- Neoadjuvant
treatment given prior to definitive, curative treatment (i.e., surgery or radiation)
- Neoantigens
Antigens generated by somatic mutations in tumor cells, allowing the expression by tumor cells and not non-tumor cells
- PD-1 blockade
type of cancer immunotherapy; antibodies against either PD-1 or the ligand PD-L1 are administered to block PD-1:PD-L1 signaling
- Physicochemical properties
intrinsic physical and chemical characteristics of a substance (e.g. hydrophobicity or electrostatic charge.)
- Polyspecific
T cell recognizing multiple peptide/MHC complexes
- Private TCR sequences
unique to a single individual
- Public clonotypes
T cells sharing the same TCR sequences found in multiple individuals
- Rarefaction curve
relationship (X-Y plot) between the number of unique observations (i.e. TCR sequences, Y axis) and the number of samples (X axis)
- Shannon entropy
estimator of population diversity
- T cell exhaustion
differentiation state caused by chronic exposure to antigen and inflammation, and defined by progressive loss of function
- T-scan
high throughput platform for identifying antigens recognized by T cells that uses lentiviral delivery of antigens into APCs for processing unto MHC
- TCR fingerprint
family of peptides recognized by a single TCR;can be represented by a matrix of amino acid weights along the peptide sequence
- Treg
subset of CD4+ T cells expressing the transcription factor FoxP3; suppressive role in immune responses
- UMI
index added to sequencing libraries before any PCR amplification step. In droplet-based scRNA seq (e.g. 10X), the UMI is added to each cell during the initial processing step to separately index each individual cell’s transcriptome
- V, D, and J gene segments
brought together via recombination to generate the TCR or BCR
- V(D)J recombination
Somatic gene rearrangement during T and B cell development generating TCR and BCR repertoires, respectively
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Masopust D and Schenkel JM (2013) The integration of T cell migration, differentiation and function. Nature reviews. Immunology 13, 309–320. 10.1038/nri3442 [DOI] [PubMed] [Google Scholar]
- 2.Curtsinger JM and Mescher MF (2010) Inflammatory cytokines as a third signal for T cell activation. Current opinion in immunology 22, 333–340. 10.1016/j.coi.2010.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith-Garvin JE et al. (2009) T cell activation. Annual review of immunology 27, 591–619. 10.1146/annurev.immunol.021908.132706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLane LM et al. (2019) CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annual review of immunology 37, 457–495. 10.1146/annurev-immunol-041015-055318 [DOI] [PubMed] [Google Scholar]
- 5.Wherry EJ and Kurachi M (2015) Molecular and cellular insights into T cell exhaustion. Nature reviews. Immunology 15, 486–499. 10.1038/nri3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang JT et al. (2014) Molecular regulation of effector and memory T cell differentiation. Nature immunology 15, 1104–1115. 10.1038/ni.3031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daniels MA and Teixeiro E (2015) TCR Signaling in T Cell Memory. Front Immunol 6, 617. 10.3389/fimmu.2015.00617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerlach C et al. (2013) Heterogeneous differentiation patterns of individual CD8+ T cells. Science 340, 635–639. 10.1126/science.1235487 [DOI] [PubMed] [Google Scholar]
- 9.Buchholz VR et al. (2013) Disparate individual fates compose robust CD8+ T cell immunity. Science 340, 630–635. 10.1126/science.1235454 [DOI] [PubMed] [Google Scholar]
- 10.Tubo NJ et al. (2016) Most microbe-specific naive CD4(+) T cells produce memory cells during infection. Science 351, 511–514. 10.1126/science.aad0483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tubo NJ et al. (2013) Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell 153, 785–796. 10.1016/j.cell.2013.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plumlee CR et al. (2013) Environmental cues dictate the fate of individual CD8+ T cells responding to infection. Immunity 39, 347–356. 10.1016/j.immuni.2013.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akondy RS et al. (2017) Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552, 362–367. 10.1038/nature24633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sewell AK (2012) Why must T cells be cross-reactive? Nature reviews. Immunology 12, 669–677. 10.1038/nri3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jenkins MK et al. (2010) On the composition of the preimmune repertoire of T cells specific for Peptide-major histocompatibility complex ligands. Annual review of immunology 28, 275–294. 10.1146/annurev-immunol-030409-101253 [DOI] [PubMed] [Google Scholar]
- 16.Arstila TP et al. (1999) A direct estimate of the human alphabeta T cell receptor diversity. Science 286, 958–961. 10.1126/science.286.5441.958 [DOI] [PubMed] [Google Scholar]
- 17.Robins HS et al. (2009) Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 114, 4099–4107. 10.1182/blood-2009-04-217604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein L et al. (2014) Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nature reviews. Immunology 14, 377–391. 10.1038/nri3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murali-Krishna K et al. (1998) Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8, 177–187. 10.1016/s1074-7613(00)80470-7 [DOI] [PubMed] [Google Scholar]
- 20.Butz EA and Bevan MJ (1998) Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity 8, 167–175. 10.1016/s1074-7613(00)80469-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joglekar AV and Li G (2021) T cell antigen discovery. Nat Methods 18, 873–880. 10.1038/s41592-020-0867-z [DOI] [PubMed] [Google Scholar]
- 22.Martinez RJ and Evavold BD (2015) Lower Affinity T Cells are Critical Components and Active Participants of the Immune Response. Front Immunol 6, 468. 10.3389/fimmu.2015.00468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawakami Y et al. (1994) Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proceedings of the National Academy of Sciences of the United States of America 91, 6458–6462. 10.1073/pnas.91.14.6458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Bruggen P et al. (1991) A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254, 1643–1647. 10.1126/science.1840703 [DOI] [PubMed] [Google Scholar]
- 25.Reiss S et al. (2017) Comparative analysis of activation induced marker (AIM) assays for sensitive identification of antigen-specific CD4 T cells. PloS one 12, e0186998. 10.1371/journal.pone.0186998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schatz DG and Ji Y (2011) Recombination centres and the orchestration of V(D)J recombination. Nature reviews. Immunology 11, 251–263. 10.1038/nri2941 [DOI] [PubMed] [Google Scholar]
- 27.Pauken KE et al. (2021) Single-cell analyses identify circulating anti-tumor CD8 T cells and markers for their enrichment. J Exp Med 218. 10.1084/jem.20200920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lucca LE et al. (2021) Circulating clonally expanded T cells reflect functions of tumor-infiltrating T cells. J Exp Med 218. 10.1084/jem.20200921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gueguen P et al. (2021) Contribution of resident and circulating precursors to tumor-infiltrating CD8(+) T cell populations in lung cancer. Sci Immunol 6. 10.1126/sciimmunol.abd5778 [DOI] [PubMed] [Google Scholar]
- 30.Liu B et al. (2021) Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nature Cancer. 10.1038/s43018-021-00292-8 [DOI] [PubMed] [Google Scholar]
- 31.Schenkel JM et al. (2021) Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1(+) CD8(+) T cells in tumor-draining lymph nodes. Immunity. 10.1016/j.immuni.2021.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng L et al. (2021) Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474. 10.1126/science.abe6474 [DOI] [PubMed] [Google Scholar]
- 33.Zhang L et al. (2018) Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272. 10.1038/s41586-018-0694-x [DOI] [PubMed] [Google Scholar]
- 34.Bhatt D et al. (2021) STARTRAC analyses of scRNAseq data from tumor models reveal T cell dynamics and therapeutic targets. J Exp Med 218. 10.1084/jem.20201329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oliveira G et al. (2021) Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature 596, 119–125. 10.1038/s41586-021-03704-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu TD et al. (2020) Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278. 10.1038/s41586-020-2056-8 [DOI] [PubMed] [Google Scholar]
- 37.Bassez A et al. (2021) A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nature medicine 27, 820–832. 10.1038/s41591-021-01323-8 [DOI] [PubMed] [Google Scholar]
- 38.Yost KE et al. (2019) Clonal replacement of tumor-specific T cells following PD-1 blockade. Nature medicine 25, 1251–1259. 10.1038/s41591-019-0522-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J et al. (2020) Compartmental Analysis of T-cell Clonal Dynamics as a Function of Pathologic Response to Neoadjuvant PD-1 Blockade in Resectable Non-Small Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 26, 1327–1337. 10.1158/1078-0432.CCR-19-2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fairfax BP et al. (2020) Peripheral CD8(+) T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nature medicine 26, 193–199. 10.1038/s41591-019-0734-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watson RA et al. (2021) Immune checkpoint blockade sensitivity and progression-free survival associates with baseline CD8(+) T cell clone size and cytotoxicity. Sci Immunol 6, eabj8825. 10.1126/sciimmunol.abj8825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiffelle J et al. (2020) T-cell repertoire analysis and metrics of diversity and clonality. Curr Opin Biotechnol 65, 284–295. 10.1016/j.copbio.2020.07.010 [DOI] [PubMed] [Google Scholar]
- 43.Penkava F et al. (2020) Single-cell sequencing reveals clonal expansions of pro-inflammatory synovial CD8 T cells expressing tissue-homing receptors in psoriatic arthritis. Nat Commun 11, 4767. 10.1038/s41467-020-18513-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Striebich CC et al. (1998) Selective accumulation of related CD4+ T cell clones in the synovial fluid of patients with rheumatoid arthritis. J Immunol 161, 4428–4436 [PubMed] [Google Scholar]
- 45.Klarenbeek PL et al. (2012) Inflamed target tissue provides a specific niche for highly expanded T-cell clones in early human autoimmune disease. Ann Rheum Dis 71, 1088–1093. 10.1136/annrheumdis-2011-200612 [DOI] [PubMed] [Google Scholar]
- 46.Chini L et al. (2002) Evidence of clonotypic pattern of T-cell repertoire in synovial fluid of children with juvenile rheumatoid arthritis at the onset of the disease. Scand J Immunol 56, 512–517. 10.1046/j.1365-3083.2002.01153.x [DOI] [PubMed] [Google Scholar]
- 47.Chang MH et al. (2021) Arthritis flares mediated by tissue-resident memory T cells in the joint. Cell Rep 37, 109902. 10.1016/j.celrep.2021.109902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacobsen M et al. (2002) Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain 125, 538–550. 10.1093/brain/awf059 [DOI] [PubMed] [Google Scholar]
- 49.Skulina C et al. (2004) Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proceedings of the National Academy of Sciences of the United States of America 101, 2428–2433. 10.1073/pnas.0308689100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pappalardo JL et al. (2020) Transcriptomic and clonal characterization of T cells in the human central nervous system. Sci Immunol 5. 10.1126/sciimmunol.abb8786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schnell A et al. (2021) Stem-like intestinal Th17 cells give rise to pathogenic effector T cells during autoimmunity. Cell 184, 6281–6298 e6223. 10.1016/j.cell.2021.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coppieters KT et al. (2012) Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med 209, 51–60. 10.1084/jem.20111187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pauken KE et al. (2021) Emerging concepts in PD-1 checkpoint biology. Semin Immunol, 101480. 10.1016/j.smim.2021.101480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ribas A and Wolchok JD (2018) Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. 10.1126/science.aar4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vaddepally RK et al. (2020) Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers (Basel) 12. 10.3390/cancers12030738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Topalian SL et al. (2020) Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 367. 10.1126/science.aax0182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iwai Y et al. (2002) Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proceedings of the National Academy of Sciences of the United States of America 99, 12293–12297. 10.1073/pnas.192461099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Juneja VR et al. (2017) PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med 214, 895–904. 10.1084/jem.20160801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noguchi T et al. (2017) Temporally Distinct PD-L1 Expression by Tumor and Host Cells Contributes to Immune Escape. Cancer Immunol Res 5, 106–117. 10.1158/2326-6066.CIR-16-0391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spitzer MH et al. (2017) Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 168, 487–502 e415. 10.1016/j.cell.2016.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fransen MF et al. (2018) Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight 3. 10.1172/jci.insight.124507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dammeijer F et al. (2020) The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer cell. 10.1016/j.ccell.2020.09.001 [DOI] [PubMed] [Google Scholar]
- 63.Connolly KA et al. (2021) A reservoir of stem-like CD8(+) T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci Immunol 6, eabg7836. 10.1126/sciimmunol.abg7836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caushi JX et al. (2021) Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 596, 126–132. 10.1038/s41586-021-03752-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aoki H et al. (2018) TCR Repertoire Analysis Reveals Mobilization of Novel CD8(+) T Cell Clones Into the Cancer-Immunity Cycle Following Anti-CD4 Antibody Administration. Front Immunol 9, 3185. 10.3389/fimmu.2018.03185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spranger S et al. (2014) Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer 2, 3. 10.1186/2051-1426-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Valpione S et al. (2020) Immune-awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat Cancer 1, 210–221. 10.1038/s43018-019-0022-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ostmeyer J et al. (2019) Biophysicochemical Motifs in T-cell Receptor Sequences Distinguish Repertoires from Tumor-Infiltrating Lymphocyte and Adjacent Healthy Tissue. Cancer research 79, 1671–1680. 10.1158/0008-5472.CAN-18-2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beshnova D et al. (2020) De novo prediction of cancer-associated T cell receptors for noninvasive cancer detection. Science translational medicine 12. 10.1126/scitranslmed.aaz3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stadinski BD et al. (2016) Hydrophobic CDR3 residues promote the development of self-reactive T cells. Nature immunology 17, 946–955. 10.1038/ni.3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lagattuta KA, Kang JB, Nathan A, Pauken KE, Jonsson AH, Rao DA, Sharpe AH, Ishigaki K and Raychaudhuri S (In press) Repertoire analyses reveal TCR sequence features that influence T cell fate. Nature immunology, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thornton AM et al. (2010) Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol 184, 3433–3441. 10.4049/jimmunol.0904028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Munson DJ et al. (2016) Identification of shared TCR sequences from T cells in human breast cancer using emulsion RT-PCR. Proceedings of the National Academy of Sciences of the United States of America 113, 8272–8277. 10.1073/pnas.1606994113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakanishi K et al. (2016) Characterization of the T-cell receptor beta chain repertoire in tumor-infiltrating lymphocytes. Cancer Med 5, 2513–2521. 10.1002/cam4.828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beausang JF et al. (2017) T cell receptor sequencing of early-stage breast cancer tumors identifies altered clonal structure of the T cell repertoire. Proceedings of the National Academy of Sciences of the United States of America 114, E10409–E10417. 10.1073/pnas.1713863114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sherwood AM et al. (2013) Tumor-infiltrating lymphocytes in colorectal tumors display a diversity of T cell receptor sequences that differ from the T cells in adjacent mucosal tissue. Cancer Immunol Immunother 62, 1453–1461. 10.1007/s00262-013-1446-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levy E et al. (2016) Immune DNA signature of T-cell infiltration in breast tumor exomes. Sci Rep 6, 30064. 10.1038/srep30064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang T et al. (2017) The Different T-cell Receptor Repertoires in Breast Cancer Tumors, Draining Lymph Nodes, and Adjacent Tissues. Cancer Immunol Res 5, 148–156. 10.1158/2326-6066.CIR-16-0107 [DOI] [PubMed] [Google Scholar]
- 79.Lahnemann D et al. (2020) Eleven grand challenges in single-cell data science. Genome Biol 21, 31. 10.1186/s13059-020-1926-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martins F et al. (2019) Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol 16, 563–580. 10.1038/s41571-019-0218-0 [DOI] [PubMed] [Google Scholar]
- 81.Robinson WH et al. (2002) Autoantigen microarrays for multiplex characterization of autoantibody responses. Nature medicine 8, 295–301. 10.1038/nm0302-295 [DOI] [PubMed] [Google Scholar]
- 82.Northrup L et al. (2016) Combining antigen and immunomodulators: Emerging trends in antigen-specific immunotherapy for autoimmunity. Adv Drug Deliv Rev 98, 86–98. 10.1016/j.addr.2015.10.020 [DOI] [PubMed] [Google Scholar]
- 83.Haen SP et al. (2020) Towards new horizons: characterization, classification and implications of the tumour antigenic repertoire. Nat Rev Clin Oncol 17, 595–610. 10.1038/s41571-020-0387-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klein L et al. (2009) Antigen presentation in the thymus for positive selection and central tolerance induction. Nature reviews. Immunology 9, 833–844. 10.1038/nri2669 [DOI] [PubMed] [Google Scholar]
- 85.Sharon E et al. (2016) Genetic variation in MHC proteins is associated with T cell receptor expression biases. Nat Genet 48, 995–1002. 10.1038/ng.3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Melenhorst JJ et al. (2008) Contribution of TCR-beta locus and HLA to the shape of the mature human Vbeta repertoire. J Immunol 180, 6484–6489. 10.4049/jimmunol.180.10.6484 [DOI] [PubMed] [Google Scholar]
- 87.Ishigaki K, Lagattuta K, Luo Y, James E, Buckner J and Raychaudhuri S (In press) HLA autoimmune risk alleles restrict the hypervariable region of T cell receptors. Nature Genetics, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arnaud M et al. (2021) Sensitive identification of neoantigens and cognate TCRs in human solid tumors. Nat Biotechnol. 10.1038/s41587-021-01072-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang SQ et al. (2018) High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat Biotechnol. 10.1038/nbt.4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma KY et al. (2021) High-throughput and high-dimensional single-cell analysis of antigen-specific CD8(+) T cells. Nature immunology 22, 1590–1598. 10.1038/s41590-021-01073-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dash P et al. (2017) Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93. 10.1038/nature22383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang H et al. (2020) Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat Biotechnol. 10.1038/s41587-020-0505-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang H et al. (2020) Investigation of Antigen-Specific T-Cell Receptor Clusters in Human Cancers. Clinical cancer research : an official journal of the American Association for Cancer Research 26, 1359–1371. 10.1158/1078-0432.CCR-19-3249 [DOI] [PubMed] [Google Scholar]
- 94.Pogorelyy MV et al. (2019) Detecting T cell receptors involved in immune responses from single repertoire snapshots. PLoS Biol 17, e3000314. 10.1371/journal.pbio.3000314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kula T et al. (2019) T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell 178, 1016–1028 e1013. 10.1016/j.cell.2019.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Birnbaum ME et al. (2014) Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157, 1073–1087. 10.1016/j.cell.2014.03.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bentzen AK et al. (2018) T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide-MHC complexes. Nat Biotechnol. 10.1038/nbt.4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wooldridge L et al. (2012) A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem 287, 1168–1177. 10.1074/jbc.M111.289488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hiemstra HS et al. (1999) Quantitative determination of TCR cross-reactivity using peptide libraries and protein databases. European journal of immunology 29, 2385–2391. [DOI] [PubMed] [Google Scholar]
- 100.Wucherpfennig KW et al. (2007) Polyspecificity of T cell and B cell receptor recognition. Semin Immunol 19, 216–224. 10.1016/j.smim.2007.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lu T et al. (2021) Deep learning-based prediction of the T cell receptor–antigen binding specificity. Nature Machine Intelligence 3, 864–875. 10.1038/s42256-021-00383-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moris P et al. (2021) Current challenges for unseen-epitope TCR interaction prediction and a new perspective derived from image classification. Brief Bioinform 22. 10.1093/bib/bbaa318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Springer I et al. (2020) Prediction of Specific TCR-Peptide Binding From Large Dictionaries of TCR-Peptide Pairs. Front Immunol 11, 1803. 10.3389/fimmu.2020.01803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shugay M et al. (2018) VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res 46, D419–D427. 10.1093/nar/gkx760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tickotsky N et al. (2017) McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 33, 2924–2929. 10.1093/bioinformatics/btx286 [DOI] [PubMed] [Google Scholar]
- 106.Karapetyan AR et al. (2019) TCR Fingerprinting and Off-Target Peptide Identification. Front Immunol 10, 2501. 10.3389/fimmu.2019.02501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Simoni Y et al. (2018) Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579. 10.1038/s41586-018-0130-2 [DOI] [PubMed] [Google Scholar]
- 108.Eberhardt CS et al. (2021) Functional HPV-specific PD-1(+) stem-like CD8 T cells in head and neck cancer. Nature 597, 279–284. 10.1038/s41586-021-03862-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cheng Y et al. (2021) Non-terminally exhausted tumor-resident memory HBV-specific T cell responses correlate with relapse-free survival in hepatocellular carcinoma. Immunity 54, 1825–1840 e1827. 10.1016/j.immuni.2021.06.013 [DOI] [PubMed] [Google Scholar]
- 110.Li H et al. (2019) Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 176, 775–789 e718. 10.1016/j.cell.2018.11.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yeong J et al. (2021) Intratumoral CD39(+)CD8(+) T Cells Predict Response to Programmed Cell Death Protein-1 or Programmed Death Ligand-1 Blockade in Patients With NSCLC. J Thorac Oncol 16, 1349–1358. 10.1016/j.jtho.2021.04.016 [DOI] [PubMed] [Google Scholar]
- 112.Maurice NJ et al. (2021) The Ugly Duckling Turned to Swan: A Change in Perception of Bystander-Activated Memory CD8 T Cells. J Immunol 206, 455–462. 10.4049/jimmunol.2000937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nikolich-Zugich J et al. (2004) The many important facets of T-cell repertoire diversity. Nature reviews. Immunology 4, 123–132. 10.1038/nri1292 [DOI] [PubMed] [Google Scholar]
- 114.Laydon DJ et al. (2015) Estimating T-cell repertoire diversity: limitations of classical estimators and a new approach. Philos Trans R Soc Lond B Biol Sci 370. 10.1098/rstb.2014.0291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li B et al. (2016) Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat Genet 48, 725–732. 10.1038/ng.3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Valpione S et al. (2021) The T cell receptor repertoire of tumor infiltrating T cells is predictive and prognostic for cancer survival. Nat Commun 12, 4098. 10.1038/s41467-021-24343-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jiang X et al. (2020) Comprehensive TCR repertoire analysis of CD4(+) T-cell subsets in rheumatoid arthritis. J Autoimmun 109, 102432. 10.1016/j.jaut.2020.102432 [DOI] [PubMed] [Google Scholar]
- 118.Pai JA and Satpathy AT (2021) High-throughput and single-cell T cell receptor sequencing technologies. Nat Methods 18, 881–892. 10.1038/s41592-021-01201-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Warren RL et al. (2011) Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res 21, 790–797. 10.1101/gr.115428.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Robins HS et al. (2010) Overlap and effective size of the human CD8+ T cell receptor repertoire. Science translational medicine 2, 47ra64. 10.1126/scitranslmed.3001442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Elhanati Y et al. (2018) Predicting the spectrum of TCR repertoire sharing with a data-driven model of recombination. Immunological reviews 284, 167–179. 10.1111/imr.12665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Venturi V et al. (2007) Methods for comparing the diversity of samples of the T cell receptor repertoire. J Immunol Methods 321, 182–195. 10.1016/j.jim.2007.01.019 [DOI] [PubMed] [Google Scholar]
- 123.Miho E et al. (2018) Computational Strategies for Dissecting the High-Dimensional Complexity of Adaptive Immune Repertoires. Front Immunol 9, 224. 10.3389/fimmu.2018.00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gupta NT et al. (2015) Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358. 10.1093/bioinformatics/btv359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sidhom JW et al. (2021) DeepTCR is a deep learning framework for revealing sequence concepts within T-cell repertoires. Nat Commun 12, 1605. 10.1038/s41467-021-21879-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pogorelyy MV et al. (2018) Method for identification of condition-associated public antigen receptor sequences. Elife 7. 10.7554/eLife.33050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Akolkar PN et al. (1993) Influence of HLA genes on T cell receptor V segment frequencies and expression levels in peripheral blood lymphocytes. J Immunol 150, 2761–2773 [PubMed] [Google Scholar]
- 128.Shin H et al. (2007) Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J Exp Med 204, 941–949. 10.1084/jem.20061937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wherry EJ et al. (2004) Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proceedings of the National Academy of Sciences of the United States of America 101, 16004–16009. 10.1073/pnas.0407192101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Delaney C et al. (2019) Combinatorial prediction of marker panels from single-cell transcriptomic data. Mol Syst Biol 15, e9005. 10.15252/msb.20199005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Huang AC et al. (2017) T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65. 10.1038/nature22079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huang AC et al. (2019) A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nature medicine 25, 454–461. 10.1038/s41591-019-0357-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kamphorst AO et al. (2017) Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proceedings of the National Academy of Sciences of the United States of America 114, 4993–4998. 10.1073/pnas.1705327114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Resources
- I. https://www.immudex.com/resources/educational-material/single-cell-levelidentification-of-antigen-specific-t-cells-with-dcode-dextramer/
- II. https://www.biorxiv.org/content/10.1101/375162v1 .
- III. https://www.biorxiv.org/content/10.1101/2020.11.15.383786v2.full.pdf .
- IV. https://www.biorxiv.org/content/10.1101/2021.09.27.461389v1.full.pdf .
- V. https://www.biorxiv.org/content/10.1101/2020.11.15.383786v2.full.pdf .