

Introduction
Voltage gated calcium channels (VGCCs) in sensory neurons play an essential role in neurotransmitter release at the spinal dorsal horn presynaptic terminal, and are therefore necessary for all forms of somatosensation, including pain. Because of this, VGCC remain a viable therapeutic target for the treatment of pain. However, virtually everything we know about the biophysical and pharmacological properties of VGCCs in sensory neurons was obtained through the study of rodent sensory neurons, or heterologous expression systems. While heterologous expression systems have played an essential role in drug development, it is now clear that the biophysical[30; 33] and pharmacological[2; 12; 19] properties of ion channels in heterologous expression systems may different significantly from system to system as well as from channels present in native tissues. More recent data have documented potentially important species differences as well.[31; 41; 42] To address these issues, we have characterized HVA current subtypes in acutely dissociated human and rat DRG neurons.
Materials and Methods
Human subjects
Data were collected from a total of 21 organ donors. The L4 and L5 DRG were recovered by the Center for Organ Recovery and Education (CORE) staff with consent from next of kin as described in Zhang et al. 2017[42]. DRG were placed in ice cold collection media comprised of 129.5 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1 mM CaCl2, and 30 mM HEPES, and had been filter sterilized after the pH had been adjusted to 7.35 with 5N NaOH. They were transported to the laboratory on ice and processed immediately. All procedures were approved by the University of Pittsburgh Committee for Oversight of Research and Clinical Training Involving Decedents.
Animals
Data were collected on a total of 10 adult (200–350g) male Sprague-Dawley rats. At least 3 rats were used for each endpoint. Females were not used simply because this was a convenience sample of rats that were available in the facility. Rats were pair housed in a temperature and humidity controlled, Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) accredited animal housing facility on a 12:12 light:dark schedule (lights on at 7AM). Food and water were available ad libitum. All procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with National Institutes of Health guidelines for the use of laboratory animals in research.
Isolation and plating of human DRG neurons
DRG were processed as described in Moy et al., 2020. Briefly, following mechanical and enzymatic digestion, cells were spun down for 4 min at 450 G. The pellet was resuspended in L-15 basal media containing (in a 500 mL solution) 60 mg imidazole, 15 mg aspartic acid, 15 mg glutamic acid, 15 mg cystine, 5 mg ß-alanine, 10 mg myo-inositol, 10 mg Choline-Cl, 5 mg p-aminobenzoic acid, 25 mg fumaric acid, 2 mg vitamin B12, and 5 mg of lipoic acid. Resuspended cells were placed on a Percoll gradient and spun at 199 G for 10 min. The pellet was then resuspended in Complete Media, which contained Basal Media, as well as 10% FBS, 10 ng/mL nerve growth factor (NGF 2.5S), 0.3 mg/mL glutamine, 0.225 mg/mL glucose, 2.55 mg/mL ascorbic acid, 0.12 mg/mL glutathione, and 0.2% (w/v) NaHCO3. Resuspended neurons were plated on 12 mm glass coverslips coated with poly-L-lysine. After plating, cells were placed in incubator at 37°C with 5% CO2 for 4–6 h prior to flooding with complete media. Although initial experiments used cells up to 80 h after plating, we excluded cells in culture >48 h from data analysis to minimize contamination from the voltage-gated sodium channel-derived outward current as described below.
Isolation and plating of rat DRG
Sensory neurons from L4 and L5 were obtained from adult rats, enzymatically and mechanically digested and plated as described in Lu et al., 2006. Cells were plated on 5mm coverslips coated with laminin and poly-L-ornithine. After plating, cells were stored for 2h in a CO2 incubator prior to flooding with L15 Complete Media. Rat sensory neurons were used within 12h of plating.
Whole cell patch clamp
Whole cell patch clamp recordings were conducted using a HEKA EPC10 using Patchmaster (v2×90.2) software (Harvard Bioscience, Inc, Holliston, MA). Data were acquired at 10–20 kHz. Borosilicate glass electrodes (World Precision Instruments, Sarasota, FL) pulled with a Sutter P-97 (Sutter Instruments, Novato CA) to a resistance of 1–2 MΩ when filled with internal solution (see below). Amplifier circuitry was used for capacitance and series resistance compensation. Cells were held at −60 mV. A p/4 leak subtraction protocol was employed from a holding potential of −80 mV for all protocols with the exception of the IV protocols, in which a holding potential of −100 or −50 mV was used to include or exclude LVA currents, respectively.
A series of voltage-clamp protocols were used to characterize the biophysical properties of voltage-gated Ca2+ currents in DRG neurons. The first of these was an I-V protocol which included a 505 ms pre-pulse to −80 mV, followed by 50 ms test pulses to potentials ranging between −60 and +55 mV in 5 mV steps, followed by a return step to −80 mV, as used previously [17]. This protocol was run with an inter-sweep interval (ISI) 5 s. The second was an I-V protocol used to estimate current decay during a prolonged voltage-step, referred to here as open-state inactivation. The protocol was the same as the first I-V protocol, except the test pulse was 500 ms in duration, and the protocol was only run from −60 to +35 mV. The third protocol was used to estimate steady-state inactivation, based upon previous studies [22]. This protocol consisted of a 500 ms conditioning step ranging from −90 to +10 mV, in 10 mV increments, followed by a 10 ms step back to −90 mV (to enable open channels to close), which was followed by a 50 ms test pulse to the potential corresponding to the peak inward current (generally between −10 and +10 mV). The final voltage step was to −80 mV. This protocol was run with an ISI of 10 s. The fourth protocol was a two-pulse protocol used to estimate the extent of G-protein βγ subunit-dependent inhibition of VGCC. The protocol consisted of an 80 ms conditioning step to either −70 or +80 mV, followed by a 10 ms step to −90 mV, followed by a 30 ms voltage step to the peak inward current. This test step was followed by a return to −70 mV.[22] This protocol was run with an ISI of 5 s. The fifth protocol was used to evoke tail currents. The protocol consisted of a 100 ms conditioning step to −90 mV, followed by a 50 ms voltage step to the potential associated with peak inward current, followed by a series of voltage steps ranging between −80 and −50 mV in 10 mV steps. This protocol was run with an ISI of 5 seconds. Finally, a subpopulation of neurons was stimulated with a voltage-protocol representing an action potential waveform, similar to protocols previously conducted by others.[3; 25] To generate this voltage-clamp protocol, a subpopulation of neurons (8 cells from 1 donor) were studied in current clamp with physiological internal and external solutions (see below). Action potentials were evoked with a 5 ms current injection. The waveform used for the voltage clamp protocol had properties (duration, overshoot, afterhyperpolarization amplitude, etc.) that were most similar to those of the mean of the population of neurons studied. This protocol was used to stimulate neurons 10 times at 5 Hz (to be able to estimate use dependent changes in the evoked currents).
Solutions
To isolate Ca2+ currents, we used an extracellular solution containing: 100 mM Choline-Cl, 3 mM KCl, 10 mM HEPES, 10 mM glucose, 5 mM BaCl2·2H2O, 0.6 mM MgCl2, adjusted to a pH 7.4 with Tris-Base and to 325 mOsm with sucrose. The electrode solution was composed of 100 mM CsCl, 5mM NaCl, 40 mM TEA-Cl, 1 mM CaCl2, 2 mM MgCl2, 11 mM EGTA, 10 mM HEPES, 2 mM ATP-Mg, 1 mM GTP adjusted to a pH 7.4 with Tris-Base and 310 mOSm with sucrose.
For current clamp experiments, cells were superfused with bath solution containing in mM: 130 NaCl, 3 KCl, 2.5 CaCl2, 0.6 MgCl2, 10 HEPES, and 10 glucose, pH 7.4 (osmolality 325 mOsm). The electrode solution contained (in mM): K-methanesulfonate 110, KCl 30, NaCl 5, CaCl2 1, MgCl2 2, HEPES 10, EGTA 11, Mg-ATP 2, Li-GTP 1.
Drugs and chemicals
The L-type HVA current blocker nifedipine (Sigma-Aldrich, St. Louis, MO) was diluted in DMSO as a 100 mM stock solution and stored at −20°C until use. ω-agatoxin IVA and ω-conotoxin GVIA (Alomone Labs, Jerusalem, Israel), which block N-type and P/Q-type HVA currents respectively, were diluted in diH2O as a 500 μM stock solution and stored at −20°C until use. CdCl2 (Thermo Fisher Scientific, Waltham, MA) was diluted in diH2O to a stock solution of 100 mM and stored at RT until use. DAMGO (Sigma-Aldrich, St. Louis MO) was diluted in diH2O to a stock solution of 1 mM and stored at −20°C until use. On the day of use, stock solutions were diluted in BaCl2 bath to a final concentration, indicated in parentheses: nifedipine (1 μm), ω-agatoxin IVA (200 nM), ω-conotoxin GVIA (200 nM), CdCl2 (100 μM), DAMGO (1 μM).
Drug application
For all pharmacological experiments, bath containing VGCC blockers and/or DAMGO was superfused into the recording chamber via a gravity system at a rate of 1 ml/min. Neurons were considered responders to VGCC blockers or DAMGO if there was a decrease in peak inward current and/or an increase in the slope ΔI/ΔTime of currents evoked at 0.2 Hz prior to blocker/DAMGO application. The full effect of VGCC blocker or DAMGO was considered to be achieved when Imax values stabilized over three pulses. Cells in which there was no detectable change in the slope and/or peak inward current following blocker/DAMGO application were classified as non-responders. Because of the slow rates of washout for both ω-agatoxin IVA and ω-conotoxin GVIA, drugs were added sequentially as described in Lu et al., 2010 and the order of drug application for blockers was randomized to minimize the influence of the order in which blockers were added to the bath.
Data analysis
Inclusion/exclusion criteria. Neurons were excluded 1) if uncompensated voltage error was > 6 mV, 2) if outward current amplitude exceeded inward current amplitude (see below), and 3) if current density was <13 pA/pF. Seven human and 4 rat neurons were rejected for failing the first criterion, 70 human and 1 rat neurons studied < 48 hrs in culture were rejected for the second criterion, and 19 human and 0 rat neurons were rejected for the third criterion. Furthermore, if neurons contained low voltage activated (LVA) currents (i.e., currents activated at potentials < −30 mV), the data generated were not included in the analysis of HVA currents.
The biophysical properties of voltage-gated Ca2+ currents were analyzed with a combination of Fitmaster (V2×73.5, Harvard Bioscience) and SigmaPlot (v14.0, SYSTAT, Chicago, IL). Peak inward current was used for the I-V plots, and tail currents evoked following the first I-V protocol were used for G-V plots. G-V plots were fitted to a Boltzmann equation to extract the V1/2, slope, and Gmax. Pooled data were normalized to Gmax. Steady-inactivation data were fitted to a Boltzmann equation modified to account for non-inactivated current.
Kinetic properties including current activation, inactivation, and deactivation, were determined with a single exponential fitted to the rising phase (activation), falling phase (inactivation), and tail currents (deactivation). Activation time constants (tau) were estimated from exponential fits of currents evoked with the first I-V protocol. Inactivation tau were estimated from exponential fits of currents evoked with the second I-V. Deactivation tau were estimated from exponential fits of currents evoked with the tail current protocol. Voltage dependence of these kinetic properties were determined by an exponential fit of the tau values plotted as a function of the voltage to derive the tau of tau. This tau of tau value indicates the voltage range over which the time constant of activation, inactivation or deactivation has changed by 63%, where a smaller voltage ranged (smaller tau) reflects a greater voltage-dependent change in the time constant.
VGCC blocker and DAMGO data were plotted as a fraction of the total current. The fraction of blocker and/or DAMGO sensitive current were determined as the difference between the current evoked before and after the blocker and/or DAMGO relative to the total current. The total current evoked with the AP waveform was determined by digitally subtracting the current evoked in the presence of CdCl2 from the total current. The integral of the current was used to determine the charge and was measured during the rising and falling phases were determined based upon the peak of the AP. Blockers or DAMGO sensitive charge was determined relative to total charge. In experiments measuring Ca2+ influx sensitive to VGCC blockers, drugs were added sequentially or, in the case of both nifedipine and conotoxin, were added individually prior to application of DAMGO. Data generated using the two-pulse protocol were analyzed to determine the percent potentiation driven by the pre-pulse and were calculated as the percent change from P1. Data were analyzed using two-tailed t-tests or one-, two-, or three-way ANOVAs for parametric data and Mann-Whitney and Kruskal-Wallis tests for non-parametric data. Chi-square analysis was used for proportion data. Correlation data were analyzed using a Pearson’s two-tailed test. Significance was assigned if p < 0.05. When p < 0.05 for the ANOVA, data were analyzed with a Holm-Sidak post-hoc test to determine differences between groups.
Results
A total of 195 neurons were studied from 21 human donors (17 male, 4 female), ranging in age from 21–77 years (46 average age) and 93 neurons from 12 male rats, ranging in age from 3 to 6 months.
Characteristics of HVA currents in rat and human DRG neurons
The same voltage-clamp protocol was used to evoke Ba2+ currents from rat and human DRG neurons (Figure 1A). Currents in most neurons from both species had a high threshold for activation, demonstrated little inactivation over the 50 ms step duration, had peak inward current between −10 and +5 mV, and inactivated and deactivated relatively slowly (resulting in large tail currents). While the peak inward currents in human DRG neurons (−6.01 ± 0.40 nA, n = 81) were larger than those in rat DRG neurons (−4.09 ± 0.35 nA, n = 70, p < 0.01), the current density (peak current normalized to membrane capacitance) was larger in rat DRG neurons (Figure 1B, p < 0.01).
Figure 1.
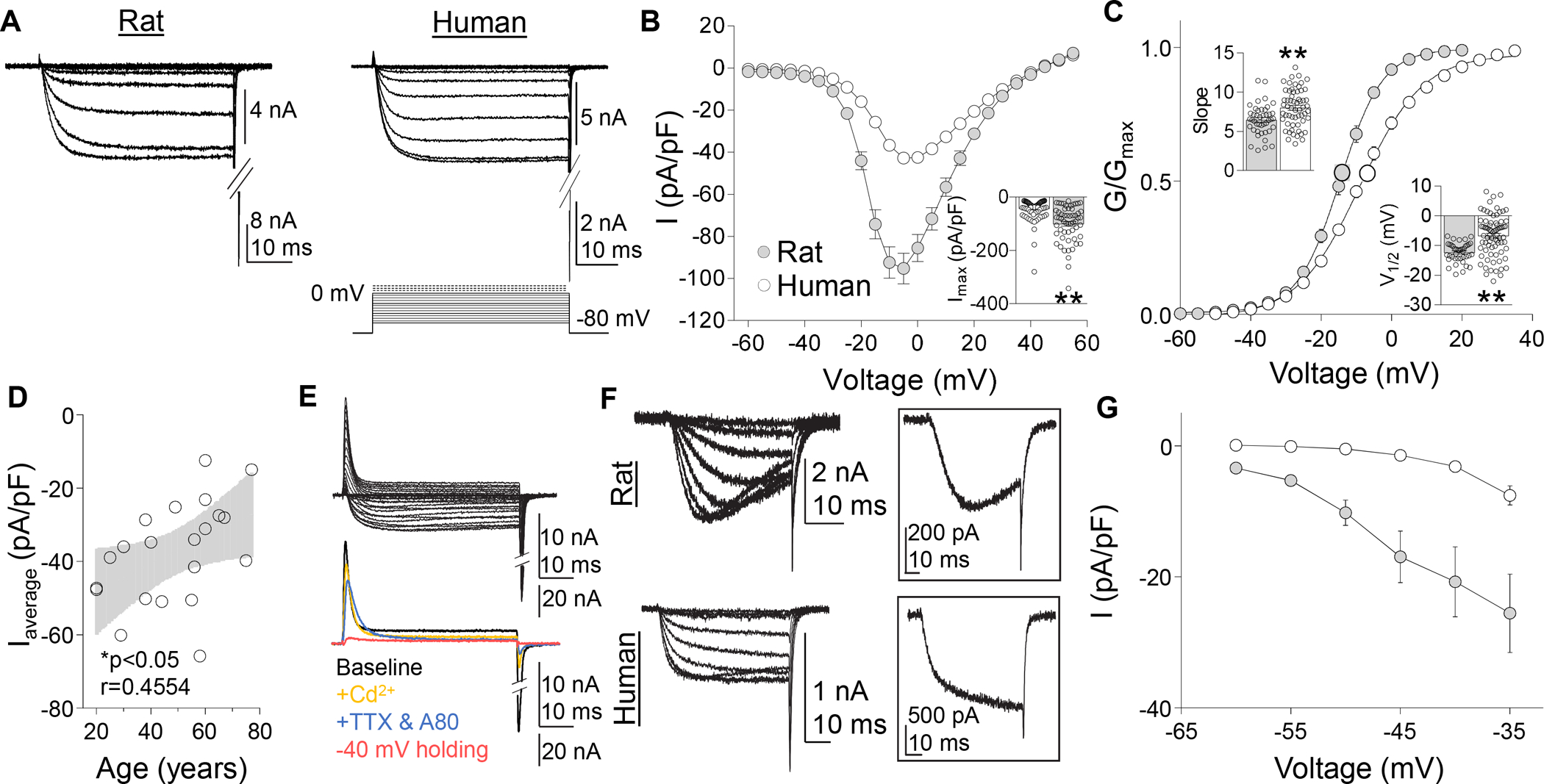
Voltage gated Ca2+ currents in rat and human DRG neurons. (A) A series of depolarizing voltage steps were used to evoke HVA currents were evoked in rat and human DRG neurons. (B) Pooled HVA I-V data from human and rat DRG neurons. Inset: Variability in peak inward HVA current evoked in human and rat DRG neurons (n = 87 human, 57 rat), where each point represents a single neuron. (C) Pooled G-V data from the same neurons plotted in (B). G-V data for each neuron was used to estimate Gmax, the voltage-dependence of activation (slope), and the voltage (V1/2) at which conductance was half of Gmax. Insets: Variability in slope and V1/2. (D) There was a negative correlation between the mean peak current density (Imax pA/pF) per donor (n = 22) and donor age. Black line is a linear fit of the data with grey area the 95% confidence interval. (E) With time in culture, there was an increase in a voltage-dependent transient outward current (top traces), that was not Ca2+ dependent, but was abolished by blockade of TTX-sensitive and -insensitive voltage gated sodium currents with 300 nM TTX, 300 nM A803467, and changing the holding potential is −40 mV (bottom traces). (F) LVA currents (activation threshold < −35 mV) were detected in a subpopulation of both human and rat DRG neurons. LVA currents detected in rat DRG neurons were slowly activating, slowing inactivating, and slowly deactivating. No open-state inactivation was detected in human LVA currents. Inset: LVA current evoked at −35 mV in rat (top trace) and human (bottom trace) DRG neurons. (G) Pooled I-V data for LVA currents detected in human (n = 18) and rat (n = 18) DRG neurons. Currents were evoked following a 500 ms pre-pulse to −100 mV. *p < 0.05, **p < 0.01. Data are mean ± SEM.
Of note, while data from several recent studies suggests that properties of putative nociceptors are enriched in human DRG neurons with a small cell body diameter, human DRG neurons selected for study were not chosen based on the cell body size. Indeed, the membrane capacitance of the neurons studied ranged from 55 to 687 pF. Nevertheless, to determine whether there was an association between cell body size and HVA current density, we analyzed HVA currents as a function of membrane capacitance (as a more sensitive measure of cell body size). Results of this analysis indicated that while there was a significant correlation (p < 0.01) between peak inward current and cell body capacitance (i.e., there were larger currents in larger neurons), there was no association between current density and cell body size (p > 0.05). This contrasts with the positive relationship between cell body size and HVA current density we have previously reported in rat DRG neurons (where the current density increases as the cell body size increases).[17] To further explore this relationship in human DRG neurons, we compared the current density in the 20% of neurons with the smallest cell body size to that observed in the 20% of neurons with the largest cell body size. Again, there was no significant difference between these groups with respect to HVA current density.
Tail currents recorded in response to membrane repolarization at the end of the 50 ms test pulse were used to generate G-V curves. Only neurons which did not contain LVA current (activating <−35 mV) were included in this analysis. Although HVA currents in human DRG neurons were larger than those in rat DRG neurons, the G-V curve for human neurons was right-shifted compared to that of rat, suggesting that differences in G-V and current density were not due to clamp control. Rather, the difference in the G-V appeared to be largely due to the voltage-dependence of activation (slope of the G-V) of HVA currents which was less in human neurons than that of rodent sensory neurons (8.0 ± 0.31mV per e-fold increase in G, n = 59 vs 6.5 ± 0.30 mV per e-fold increase in G, n = 43, p < 0.01, two-tailed t-test, Figure 1C, inset). The V1/2 of HVA current activation in human sensory neurons was also significantly more depolarized than that of HVA current in rat sensory neurons (−6.7 ± 0.79 mV, n = 73 vs. −12.4 ± 0.51, n = 40, p < 0.01, two-tailed t-test, Figure 1C, inset).
Because of evidence from rodent sensory neurons indicating that HVA current decreases with age,[14] we assessed if the age affected HVA current density, as human neurons studied were from donors considerably older than the rats used for this analysis. This was done by averaging the peak inward HVA current density of each neuron studied from a donor and then plotting the average per donor as a function of donor age. Consistent with previous rodent data, there was a significant negative correlation between current density and age (p < 0.05, Pearson’s two-tailed correlation test Figure 1D).
To confirm that we were isolating only HVA currents in both rat and human neurons, we added 100 μM Cd2+ to eliminate all HVA currents in human and rat. In acutely dissociated human DRG neurons, inward currents were completely blocked by Cd2+ (Figure 1E, inset). This was also true for neurons in culture for > 48h, but a large outward current emerged with time in culture (Figure 1E). Initial experiments suggested that this outward current was Ca2+-dependent, which we assumed would other be carried by either Cl− or K+ channels. Subsequent analysis ruled out either channel subtype, however, and confirmed that the outward current was Ca2+-independent. This left voltage-gated sodium channels as the only remaining likely possibility based on the composition of the external and internal bath solutions used. This was subsequently confirmed by the observation that the outward current was abolished by a combination of 300 nM tetrodotoxin (TTX, to block TTX sensitive channels), 1 μM A803467 (to block NaV1.8) and a holding potential of −40 mV (to block NaV1.9). Thus, we suggest that the outward current was carried by a combination of TTX-sensitive and -insensitive voltage gated sodium channels. Due to concerns that outward current would contaminate VGCC data, all neurons in this study were in culture for < 48h. Additionally, cells showing a large (peak outward current > peak inward current) outward current, including those within the 48h window, were eliminated from analysis (70 of 195 neurons).
LVA currents, those activating at potentials < −35 mV, were detected in DRG neurons from both rat and human (Figure 1F). Interestingly, in contrast to the transient LVA currents detected in rodent sensory neurons, the LVA currents in human sensory neurons exhibited little open-state inactivation (Figure 1F inset). LVA currents in the rodent also tended to activate at lower potentials than those in human sensory neurons (Figure 1G) and were present at a higher density. To minimize the potential impact of LVA currents on the properties of HVA currents in human and rodent sensory neurons, neurons with a current > 48 pA at −35 mV were excluded from further analysis. This was done for 18 human DRG neurons and 18 rat DRG neurons.
Steady-state inactivation of HVA currents in rat and human DRG neurons
Typical currents evoked with a steady-state inactivation protocol are shown for rat and human in Figure 2A. No inactivation (<10% current inactivated) was observed in 6/60 rat neurons and 0 of 83 human neurons (p < 0.01, Fisher Exact test). In a small fraction of neurons (12 of 60 rat, 0 of 83 human neurons; p < 0.01, Fisher Exact test) currents appeared to inactivate in two phases with a clear plateau between each inactivation phase. In the remainder of neurons studied (42 of 60 neurons in rat, 83 of 83 neurons in human), steady-state inactivation was present, but resulted in the inactivation of only a fraction of the total inward current (Figure 2B). Of the current that did inactivate, the V1/2 was more depolarized in human, which appeared to be due to a difference, at least in part, in slope (Figure 2B, inset). There was also a trend towards a difference in the fraction of inactivated current in rat and human neurons (p = 0.051, Figure 2B, inset).
Figure 2.
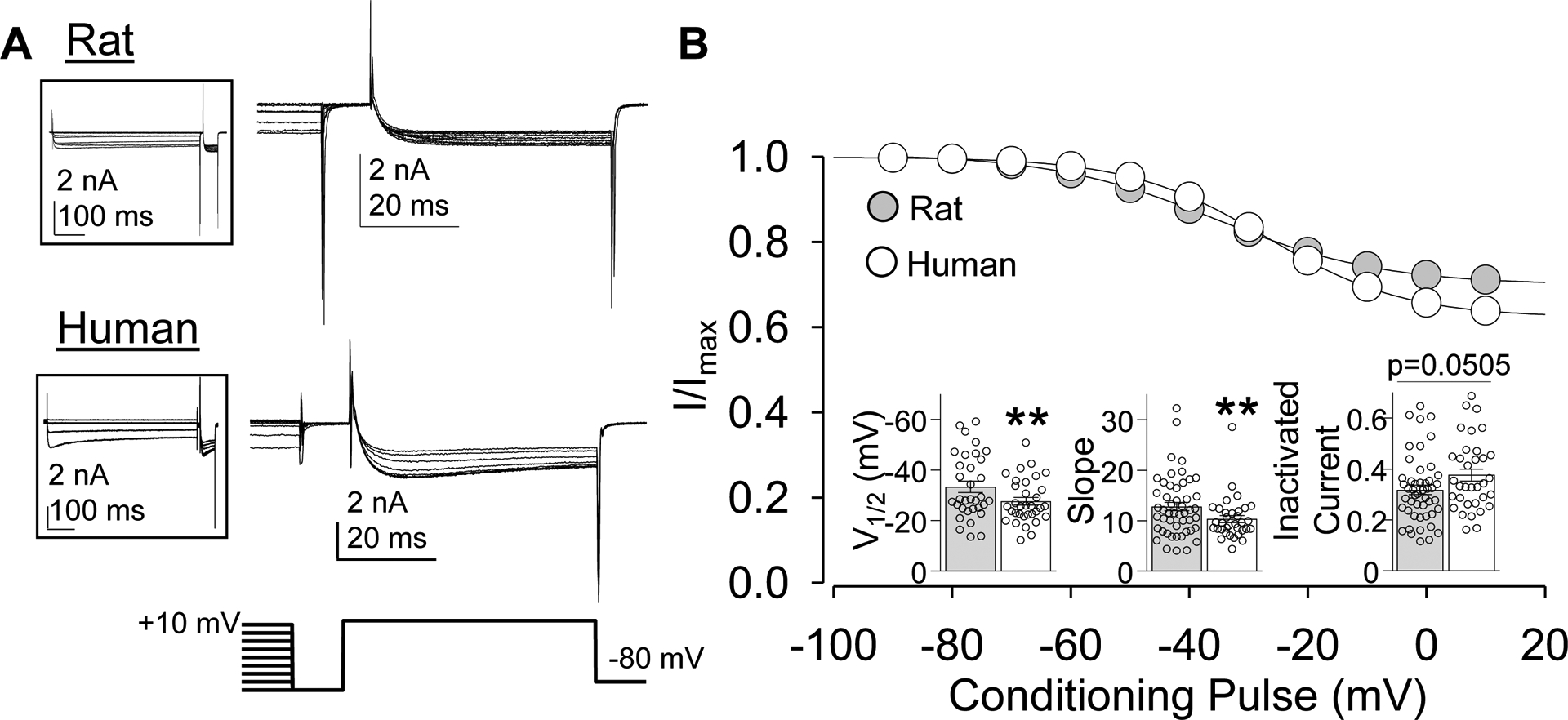
Steady-state inactivation in HVA currents in rat and human DRG neurons. (A) A three-pulse protocol (shown beneath current traces) was used to assess steady-state inactivation of HVA currents. Most of the current evoked during the conditioning pre-pulse has been eliminated for clarity. Full current sweep is shown in insets. (B) Steady-state inactivation data were fitted with a modified Boltzmann equation enabling an estimate of the fraction of total current subject to steady state inactivation, in addition to the V1/2 of inactivation and the slope of the inactivation curve. Data for each neuron was normalized to the fitted maximal current value (Imax). Data from rat (n = 49) and human (n = 34) were pooled and plotted. Insets: Variability in V1/2 slope and fraction of inactivated current. Data are mean ± SEM. **p < 0.01.
Kinetic properties of HVA currents in rat and human DRG neurons
While rat HVA currents were relatively homogeneous with respect to the rate of activation, there was heterogeneity in the activation rate of human HVA currents with a subpopulation in which currents activated relatively quickly (time constant of activation, tau < 3ms at 0 mV) and another in which the currents activated more slowly (tau > 3 ms). These current types were referred to as fast and slow (Figure 3A). Pooled data supported this impression, where rat currents generally activated more quickly than those in human (Figure 3B). The HVA current from rat DRG neurons was also relatively homogeneous with respect to the small but consistent voltage-dependent decrease in the tau of activation. In contrast, there were at least three subpopulations of human DRG neurons that could be defined by both the tau of activation (Fast and Slow), and whether the tau of activation was voltage dependent. That is, the tau of activation decreased in a voltage-dependent manner in the majority (19 of 19 and 9 of 17) neurons with fast (Fast) and slow (Slow) HVA currents. However, in a subpopulation of neurons with slow HVA currents, there was little evidence of a voltage-dependent change in tau (Flat) (Figure 3B). The voltage-dependent change in the tau of HVA current activation (tau of tau’s) was significantly different between Fast and Slow currents in human neurons (p < 0.05, one-way ANOVA followed by Holm-Sidak post hoc test, Figure 3C), indicating that the voltage-dependent change in the tau of activation was greater in Fast HVA currents than in Slow HVA currents in human DRG neurons.
Figure 3.
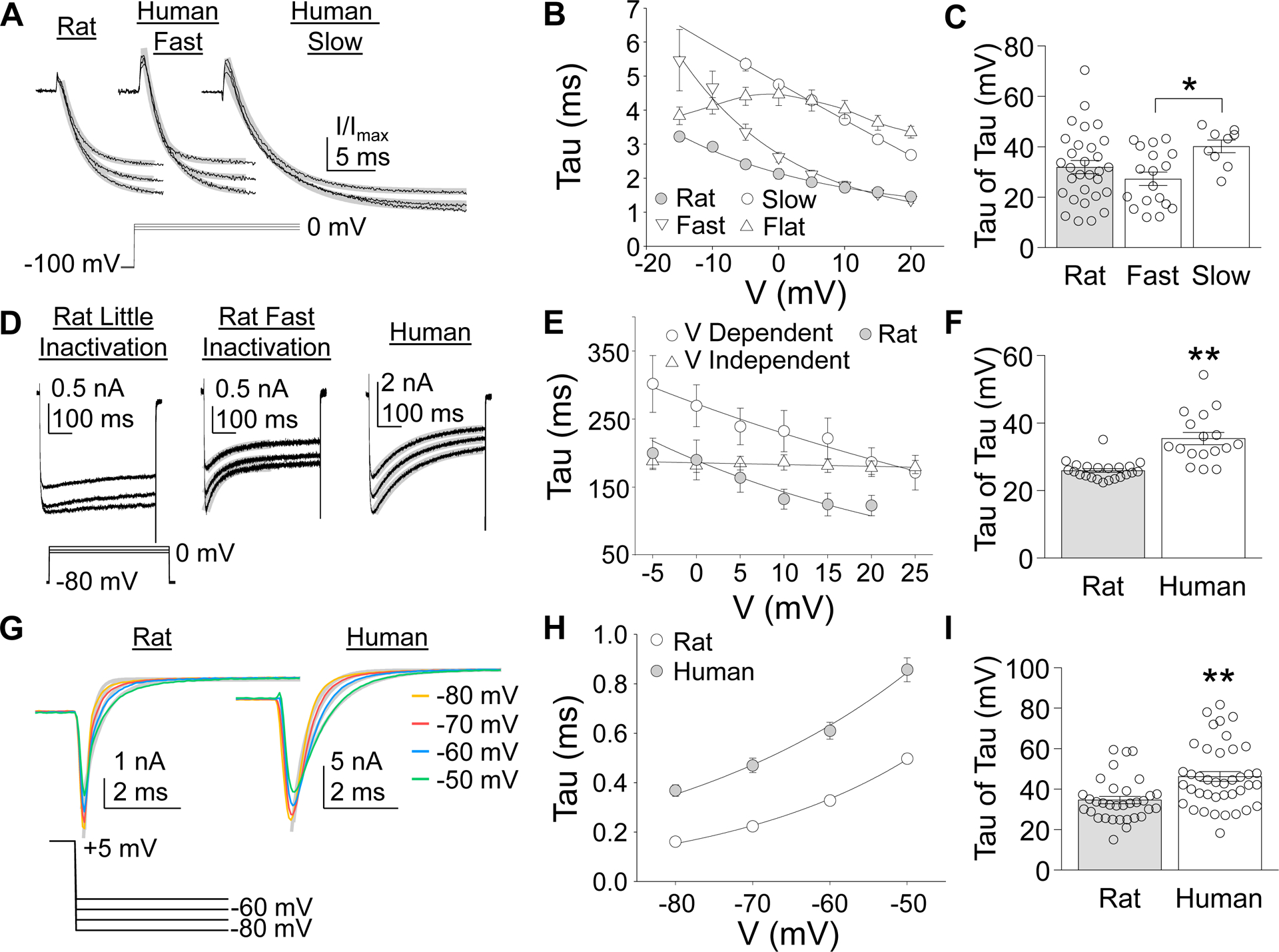
Kinetic Properties of HVA currents in rat and human DRG neurons. (A) The initial increase in current associated with a depolarizing voltage step was fitted with a single exponential to estimate the time constant (tau) of current activation. There was little variability between rat neurons with respect to the rates of HVA current activation. However, there were at least two types of HVA current in human that could be distinguished based on the rate of current activation across a range of voltage steps. (B) The tau of HVA current activation was plotted against the voltage at which the current was activated. The voltage-dependent decrease in the tau of activation of HVA current from rat DRG neurons were well fitted with a single exponential. The voltage-dependent decrease in the tau of activation of Fast and Slow HVA currents observed in human DRG neurons, illustrated in (A) could also be fitted with a single exponential. However, there was a third subpopulation of human DRG neurons in which the rate of HVA current activation demonstrated little voltage-dependence (Flat). (C) Variability in the voltage dependence of current HVA current activation in rat and the two subpopulations of human neurons in which it was possible to estimate the voltage dependence of the time constant of current activation (tau of the tau). (D) The presence of open-state inactivation of HVA currents was estimated with 500 ms depolarizing voltage steps. There was a subpopulation of rat DRG neurons in which HVA currents demonstrated little inactivation (left traces) and a second subpopulation in which an initial phase of fast inactivation was detected (middle traces). HVA currents in human DRG neurons demonstrated a consistent rate of inactivation across the entire voltage step (right traces). (E) The time constant (tau) of HVA current inactivation in all rat neurons in which fast inactivation was detected decreased with increasing voltage. In contrast, there were two subpopulations of HVA currents in human neurons, one in which there was a small but consistent voltage-dependent decrease (V-Dependent) in the tau of inactivation, and a second in which there was no voltage-dependent (V-Independent) change in the tau pf inactivation. (F) Variability in the voltage-dependent change in the tau of HVA current inactivation. (G) A tail current protocol was used to determine the voltage-dependence of HVA current deactivation. HVA current deactivation was relatively consistent in both rat and human DRG neurons. (H) While the time constant of HVA current deactivation in human DRG neurons was consistently larger than that in rat DRG neurons, currents in both species demonstrated a voltage-dependent increase in the tau of deactivation. (I) Variability in the voltage-dependent change in the tau of HVA current deactivation. * is p < 0.05, ** is p < 0.01.
In contrast to the activation data, there was greater heterogeneity in the open-state inactivation data in rat, resulting in two neuronal subpopulations, one with little inactivation and another showing a rapidly inactivating component (26 of 58 neurons, Figure 3D). Interestingly while slow inactivation was detected in all human neurons, there appeared to be two subpopulations of neurons based on whether the time constant of slow inactivation was voltage-dependent. That is, in one subpopulation of human DRG neurons (17 of 59), there was a small but consistent decrease in the tau of inactivation with increasing voltage (V Dependent, Figure 3E). No voltage-dependent change in the tau of inactivation was detected in the other subpopulations of neurons (42 of 59, V Independent, Figure 3E). In contrast, HVA currents in all rat neurons in which open state inactivation was detected, demonstrated a voltage-dependent decrease in the tau of inactivation (Figure 3E). There was a significant difference between HVA currents in rat and human DRG neurons with respect to the voltage-dependence of the tau of inactivation (p < 0.01, Student’s t-test, Figure 3F), which was greater HVA current evoked in DRG neurons from rats than humans.
Consistent with activation and inactivation, HVA current deactivation rates appeared to be faster in DRG neurons from rat compared to human (Figure 3G). Pooled data supported this impression confirming that the tau of deactivation was faster at all potentials assessed (Figure 3H). Furthermore, while the tau of deactivation was voltage-dependent in all HVA currents in both rat and human DRG neurons, there was a significant difference between rat and human with respect to the voltage-dependence of the tau of deactivation (p < 0.01, Student’s T test, Figure 3I), which was greater HVA current evoked in DRG neurons from rats than humans.
Constitutive inhibition of HVA currents in rat and human DRG neurons
Pre-pulse potentiation was observed in rat and human DRG neurons prior to the application of the GPCR agonist DAMGO (Figure 4A). Variability in the degree of potentiation was relatively small in most rat neurons compared to human. Furthermore, there appeared to be a subpopulation of human neurons in which the percent increase in current associated with the pre-pulse (% Potentiation) was relatively large (> 2 SDs from the mean). Because of the presence of this subpopulation, potentiation of HVA currents was, on average, significantly greater in neurons from human compared to rat (p < 0.01, Student’s T test, Figure 4B).
Figure 4.
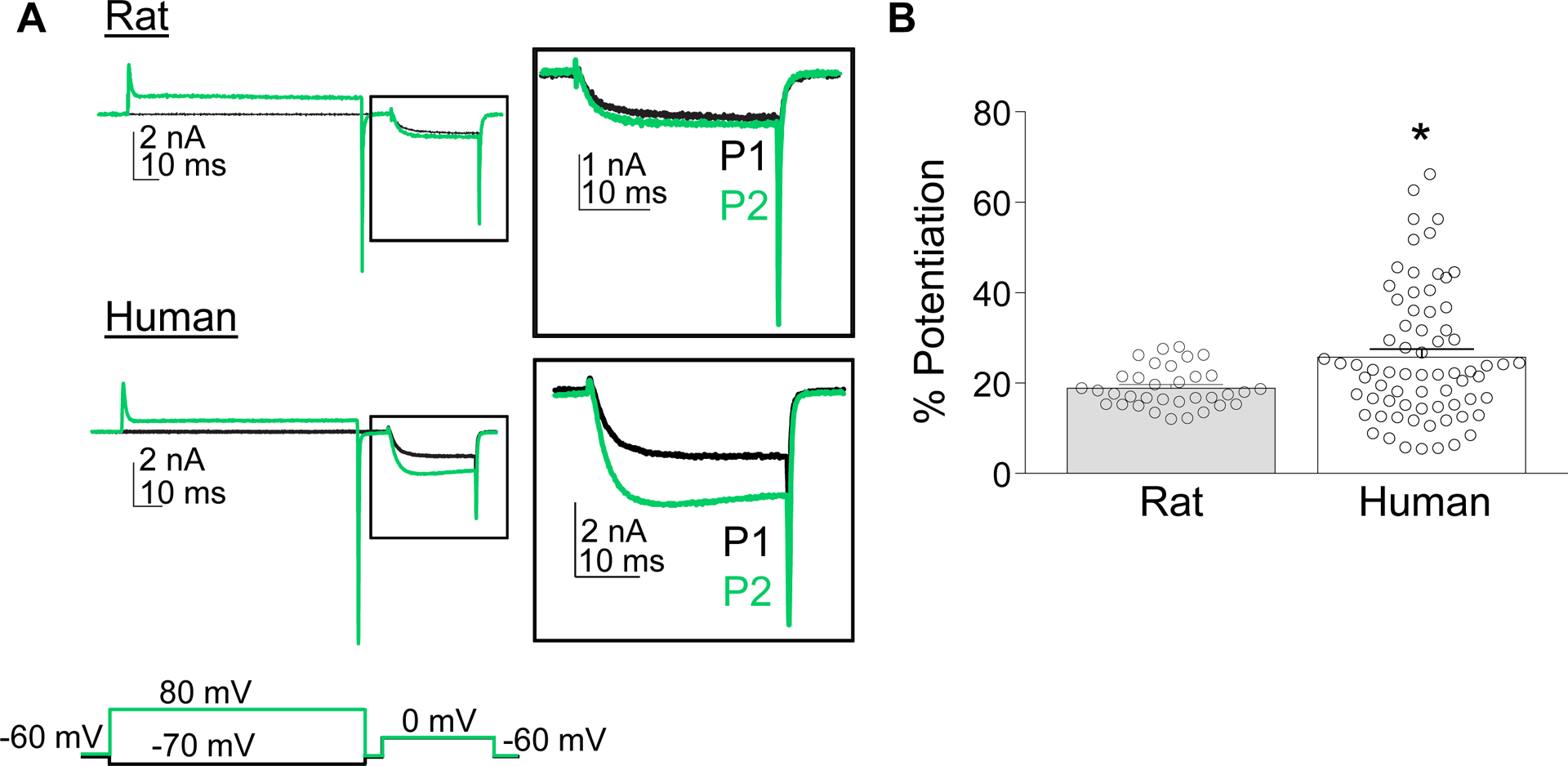
Pre-pulse potentiation in rat and human neurons. (A) Pre-pulse potentiation was assessed by comparing the current evoked at 0 mV following a 500 ms conditioning voltage step to either −70 mV (black trace) or +80 mV (green trace). Inset: Magnified view of the current evoked during the test pulse. (B) Variability in the pre-pulse potentiation (% Potentiation) of HVA currents in rat and human DRG neurons. Data are mean ± SEM. * is p < 0.05.
HVA current subtypes in rat and human DRG neurons
There was heterogeneity in both rat and human neurons with respect to the amount of current blocked by nifedipine, ω-conotoxin GVIA and ω-agatoxin IVA, with the greatest source of heterogeneity being the proportion of nifedipine or conotoxin sensitive current (Figure 5A). Nevertheless, there were significant species-dependent differences in the proportion of Ca2+ currents sensitive to the HVA blockers, with significantly more nifedipine sensitive currents in human neurons and significantly more conotoxin sensitive current in rat neurons (two-way ANOVA, Holm-Sidak post-hoc, Figure 5B). There were no species differences in the proportion of current sensitive to ω-agatoxin IVA or the current resistant to the three HVA blockers used (Figure 5B).
Figure 5.
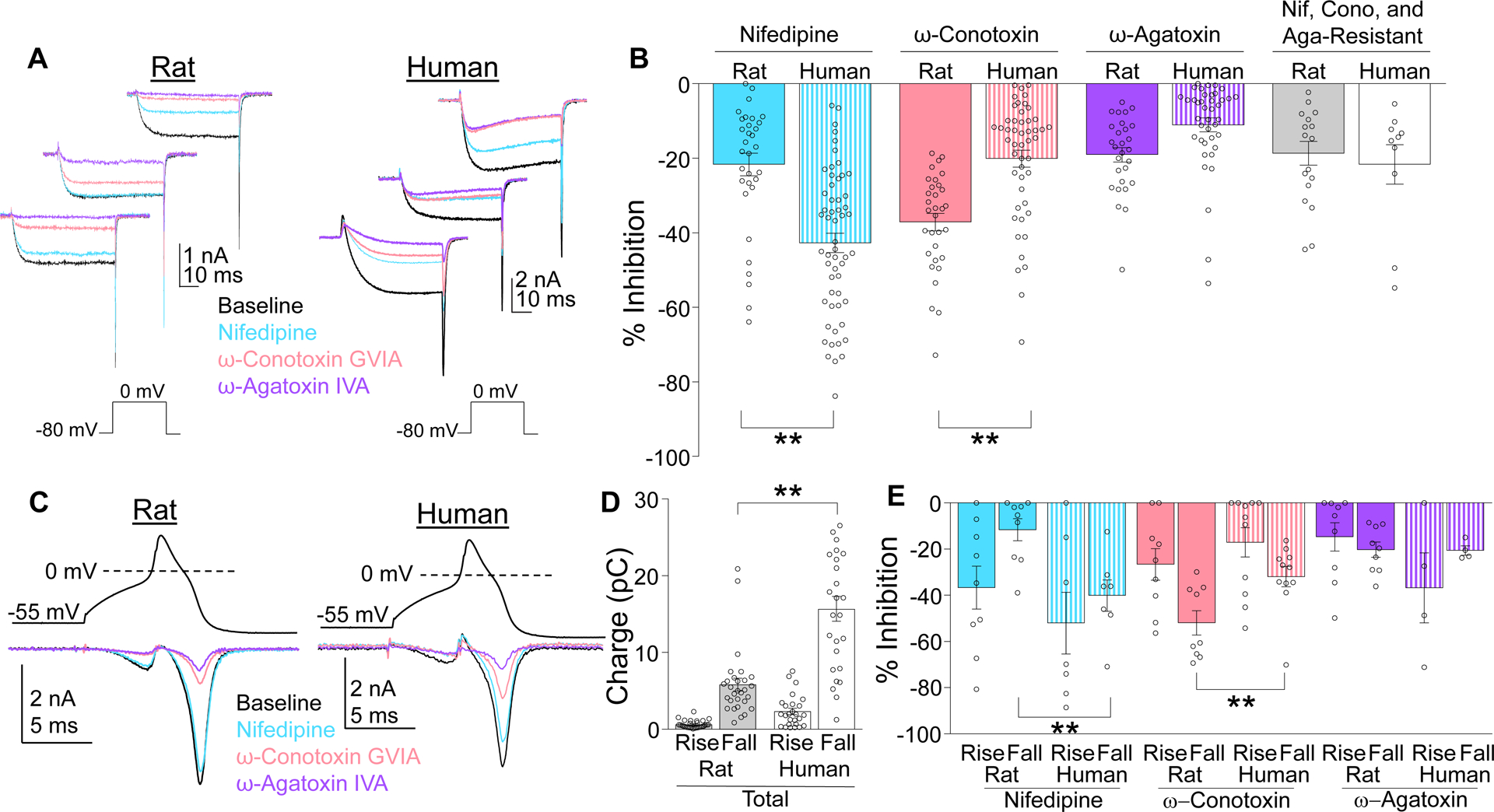
Subtypes of HVA currents differ in rat and human. (A) HVA currents were evoked in rat and human neurons before (Baseline) and after serial application of HVA channel blockers nifedipine (1 μM), ω-conotoxin GVIA (200 nM), and ω-agatoxin IVA (200 nM). Blockers were applied in random order. Each family of traces is from a different neuron. (B) In both species there was heterogeneity with respect to the percent of total HVA current (% Inhibition) blocked by each compound. Pooled data revealed a species difference with respect to the relative contribution of nifedipine- and ω-conotoxin GVIA-sensitive current. There was a significant interaction between species and blocker (two-way ANOVA, p < 0.01). The results of post-hoc comparisons (Holm-Sidak test) are indicated. (C) HVA current evoked with a typical action potential used as the voltage-clamp protocol was associated with Ca2+ influx during both the rising (Rise) and falling (Fall) phase of the waveform. The application of HVA channel blockers enabled determination of the channel subtypes contributing to the action potential-induced Ca2+ influx. (D) There was a species difference in Ca2+ influx (charge) evoked with the action potential waveform. (E) There was a species difference in the relative contribution of HVA current subtypes to the Ca2+ influx evoked with the action potential waveform, although this was only manifest in the influx associated with the falling phase of the action potential. There was again a significant interaction between species and blocker (p < 0.01 two-way ANOVA), with post hoc comparisons (Holm-Sidak test) indicated. Data are mean ± SEM. ** is p < 0.01.
Although there were clear species differences in HVA currents evoked by a square voltage step, as a more physiologically-relevant voltage protocol, we assessed the relative contributions of these HVA subtypes evoked by an action potential (AP) waveform, as has been employed previously.[3] Data were collected by sequentially adding each HVA blocker as in Figure 5A. Because a p/4 protocol was not used with the AP waveform, Cd2+ was added at the end of recording to enable subtraction of capacitive and leak currents. Consistent with larger current amplitudes in human neurons, larger currents were evoked in human neurons with the AP waveform (Figure 5C, black traces), as total Ba2+ influx was significantly larger in human (11.48 ± 1.49 pC) than rat (7.36 ± 0.90 pC, p < 0.05) neurons, a difference largely due to greater Ba2+ influx during the falling phase of the AP (Figure 5D). Similar to results generated with a square pulse, the relative contribution of HVA current subtypes to total charge influx evoked with the AP waveform was primarily blocked by nifedipine and conotoxin (Figure 5E). Interestingly, nifedipine-sensitive current appeared to contribute to Ba2+ influx during both the rising and falling phase of the AP waveform in human, but not rat neurons. Finally, to assess the presence of a difference in the relative contribution of nifedipine- and conotoxin-sensitive current to the Ba2+ influx evoked with a square pulse or an AP waveform, proportion of block data were analyzed with a three-way ANOVA (species × stimulus type × channel blocker). Consistent with results of the analysis of data generated with each stimulus independently, there was a significant interaction between species and drug. However, the absence of a significant interaction between all three factors suggests that the apparent differences between the proportion of Ba2+ influx sensitive to an HVA blocker is stimulus independent.
As noted above, there was no evidence of an association between cell body size and HVA current density in human DRG neurons. However, previous evidence suggests that N-type currents are enriched in putative nociceptive rat DRG neurons,[18; 29] consistent with the results of the present study. We therefore determined if there was a relationship between cell size and the proportion of current blocked by L-type/N-type channel blockers. We ran a two-way ANOVA on % inhibition by either L-type (nifedipine) or N-type (conotoxin) channel blockers in the smallest 20% and largest 20% of neurons and found that while there was a significant effect of drug (p<0.05), there was no significant difference by size or interaction.
μ-opioid receptor-induced suppression of HVA currents in human and rat DRG neurons
Given evidence of species differences in the relative contribution of HVA current subtypes and of HVA subtype specific modulation by G-protein coupled receptors,[5] we assessed the impact of the μ-opioid receptor (MOR), a canonical inhibitory GPCR, on HVA currents in rat and human DRG neurons. In line with previously published data from our lab[21] and others,[4] the selective MOR agonist, DAMGO (1 μM) blocked ~50% of the Ba2+ current elicited by a square pulse in 58.8% of rat and 72% of human neurons (Figure 6A,B). HVA currents evoked during both the rising and falling phase of the AP waveform were blocked by DAMGO in neurons from both rat and human. (Figure 6C). When only neurons considered responders based on data collected in the square pulse were included, there were neurons in which there was no detectable suppression of the current evoked with the AP waveform. Nevertheless, on average, the percentage of current blocked during the rising and falling phases were similar between rat and human neurons (Figure 6D).
Figure 6.
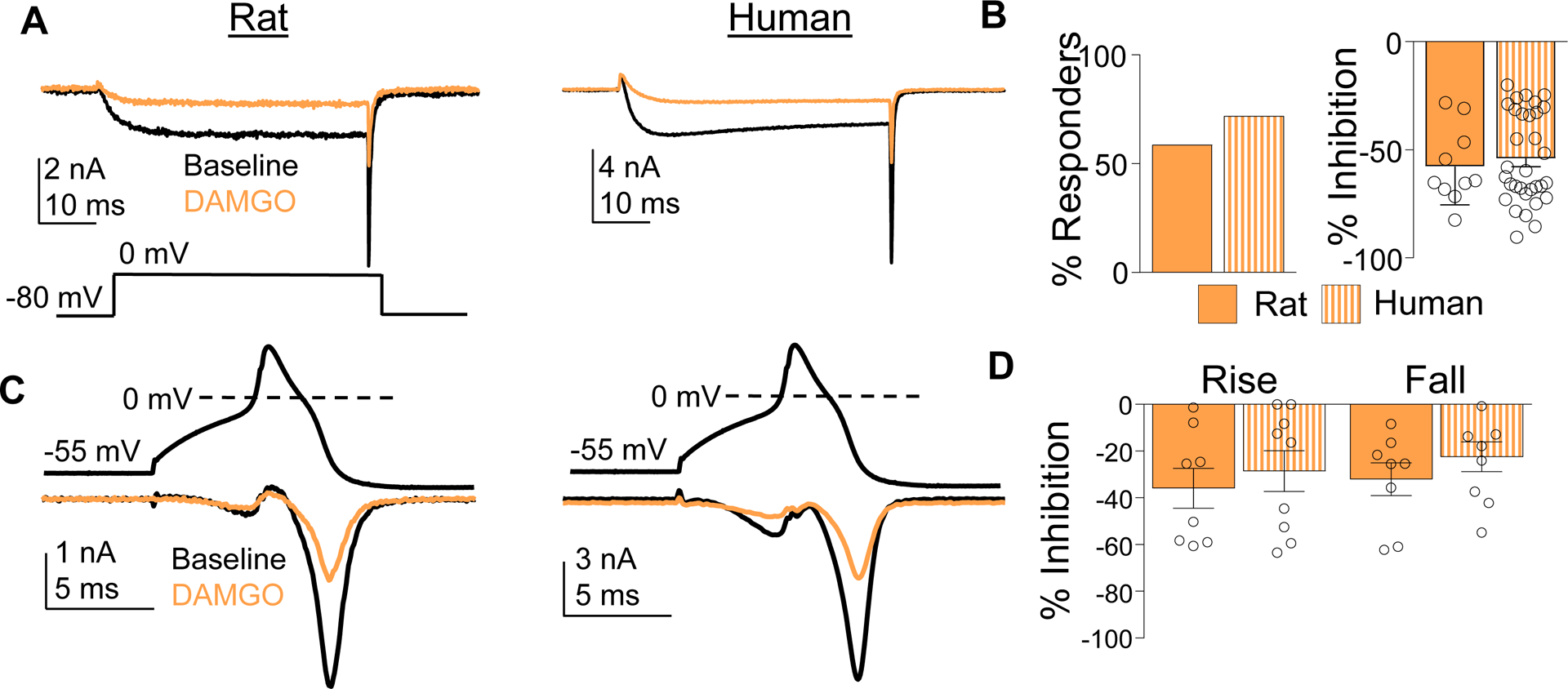
Inhibition of HVA currents with μ-opioid receptor agonist DAMGO. (A) HVA current was evoked in rat and human DRG neurons before (Baseline, black trace) and after (DAMGO, orange trace) application of DAMGO (1 μM). (B) The percentage of neurons responsive to DAMGO (% Responders) and the magnitude of the HVA current inhibition (% Inhibition), was comparable in rat and human DRG neurons. (C) HVA current evoked during the rising and falling phase of an action potential waveform was inhibited by DAMGO in DAMGO responsive rat and human DRG neurons. (D) Pooled data indicate that the percent of HVA current blocked by DAMGO during the rising and falling phase of the action potential was comparable (p > 0.05, two-way ANOVA). Data are mean ± SEM.
Based on previous data suggesting that the Gβγ subunit displaces the VGCC β subunit from the α subunit, resulting in a depolarizing shift in the voltage dependence of activation,[24] we used the pre-pulse potentiation protocol to quantify the extent of the DAMGO-induced inhibition of HVA current that could be reversed with a depolarizing pre-pulse. Only neurons considered responsive to DAMGO were included in this analysis. DAMGO-induced suppression of HVA current was reversed with a depolarizing pre-pulse in both rat and human DRG neurons (Figure 7A). There was heterogeneity in rat and human with respect to the number of neurons that responded to DAMGO in which current suppression was reversed with pre-pulse potentiation (Figure 7A), which was detected in 85.2% of human and 66.7% of rat (p > 0.05). Of those neurons that pre-pulse potentiation was detected, there was no species-dependent difference in the magnitude of the potentiation (Figure 7B). These results are consistent with a Gβγ-dependent suppression of HVA current in the majority of both rat and human DRG neurons.
Figure 7.
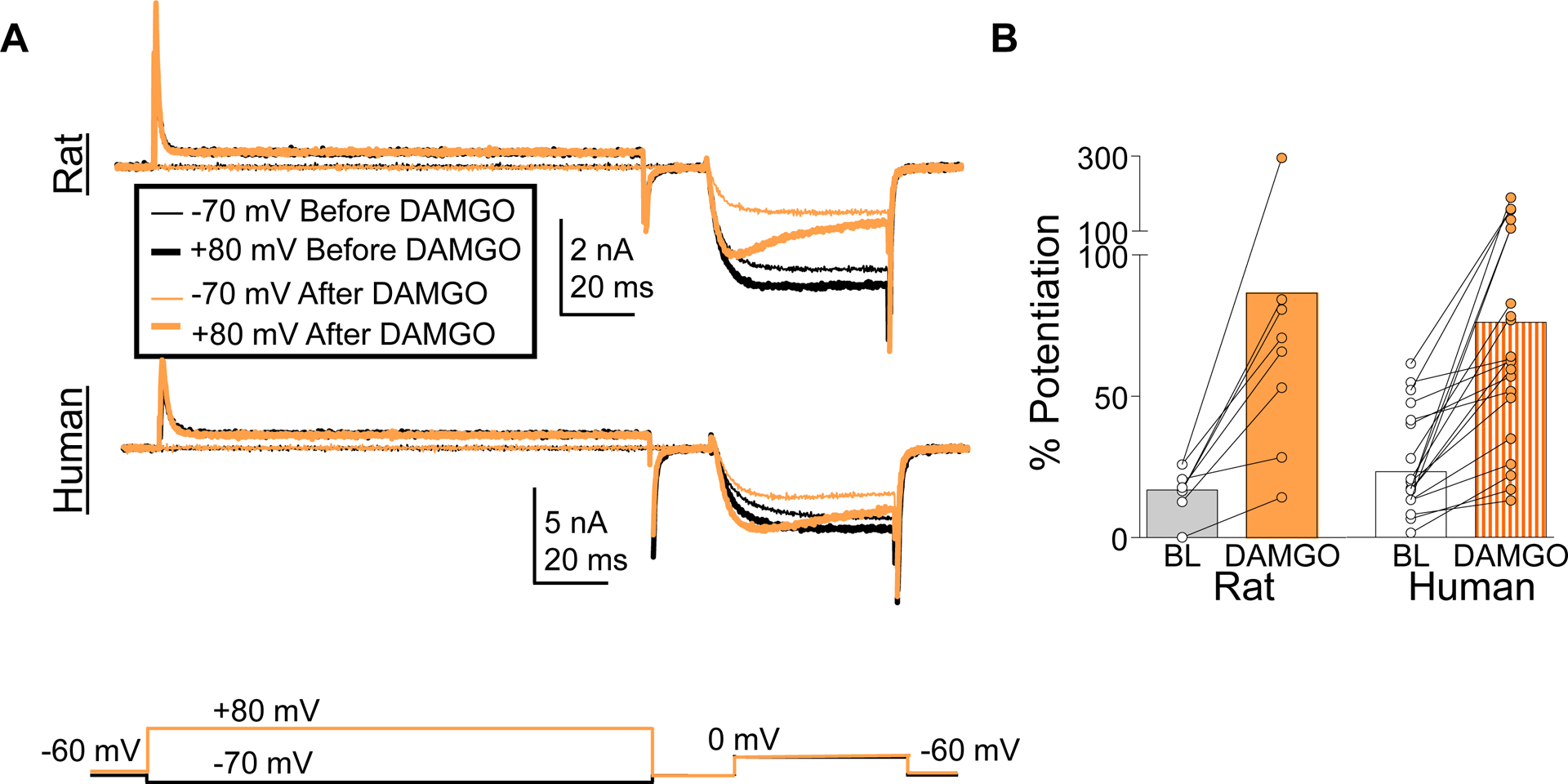
DAMGO-induced inhibition of HVA current in rat and human DRG neurons was reversed with a pre-pulse potentiation protocol. (A) HVA currents evoked with the protocol described in Figure 4 before and after application of DAMGO (1 μM). (B) Pooled data indicate that there was no difference between rat and human DRG neurons with respect to the increase in current associated with pre-pulse potentiation (% Potentiation). That is, there was a main effect of DAMGO (p < 0.01), but not species difference (p > 0.05) or a significant interaction between DAMGO and species (p > 0.05, two-way ANOVA).
The impact of DAMGO on N- and L-type currents
Rapid Gi-protein-dependent suppression of HVA currents is generally thought to be due to a change in N-type channel gating.[35] However, our data suggest that other HVA subtypes may also be involved, since DAMGO blocked 54% of the current in human neurons, while N-type currents comprised only ~20% of the total HVA current in human neurons. Therefore, we assessed the impact of nifedipine and ω-conotoxin GVIA on the current suppressed by DAMGO (Figure 8A). In the rat, ω-conotoxin-GVIA reduced the percent responders and there was as trend toward a reduction in the magnitude of inhibition, whereas in the human neurons nifedipine had a dominant effect on the actions of DAMGO, at least with respect to the % of neurons responsive to DAMGO (Figure 8B).
Figure 8.
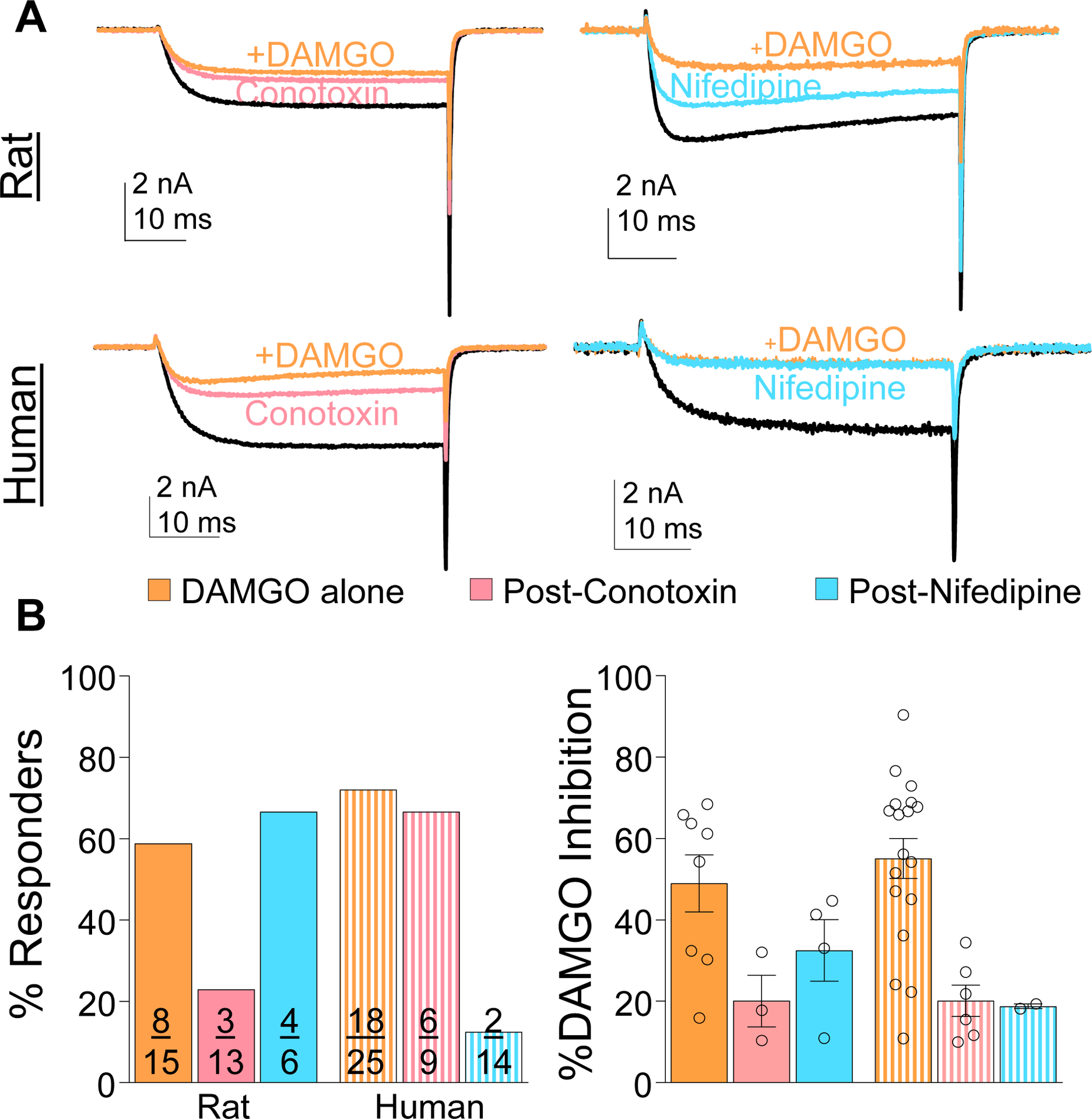
HVA current subtypes inhibited by DAMGO in rat and human DRG neurons. (A) HVA currents were evoked in rat and human DRG neurons before and after application of ω-conotoxin GVIA (200 nM, left traces) or nifedipine (10 μM, right traces), and then again after the application of DAMGO (1 μM). (B) There was a species difference in the percentage of neurons responsive to DAMGO when applied alone or in the presence of an HVA channel blocker (p < 0.05, chi-square left panel), but the impact of HVA channel blocker on the magnitude of the current inhibited by DAMGO (% DAMGO Inhibition) was not significant (right panel).
Discussion
The aim of this study was to compare properties of HVA currents in rat and human DRG neurons. HVA currents isolated in both rat and human DRG neurons were similar in their biophysical and pharmacological properties. However, we identified three major differences between rat and human HVA currents: (1) human HVA current density was significantly smaller than that of rat, (2) a subpopulation of human neurons displayed significantly more constitutive inhibition relieved by a depolarizing pre-pulse, and (3) the proportion of HVA current subtypes was significantly different with a greater proportion of nifedipine sensitive current and smaller proportion of ω-conotoxin IVA sensitive current in humans compared to rat. A smaller species-dependent difference was the heterogeneity in kinetic properties of HVA currents in human DRG neurons not observed in rat. Finally, although the focus of this study was HVA currents, there were potentially interesting species-dependent differences in the magnitude and biophysical properties of LVA currents.
While we suggest that the differences observed were species dependent, other factors may also have contributed, including the age, time in culture, length of time between death and DRG recovery, and cause of death. Age is important to consider because of evidence in rodent neurons that HVA current density decreases with age[14] and the fact that the median age for human donors was older than the rat by any comparative aging scale. Indeed, consistent with the age effect previously observed in rats, current density in humans DRG neurons decreased with increased age (Figure 1D). Nevertheless, even when data from only the youngest human donors was included in the HVA current density analysis, the species difference persisted (data not shown). Similarly, evidence against a time-in-culture effect is that statistically significant differences in current density and kinetics persisted when only acutely dissociated human DRG neurons were analyzed (data not shown). The impact of time after death is difficult to account for, as there appears to be a species difference in ability of sensory neurons to survive under hypoxic conditions: human neurons survive longer following cross clamp than rodent sensory neurons.[42] Cause of death is also difficult to address experimentally. Nevertheless, we detected no influence of variables that could be used to differentiate the donor populations including cause of death, on HVA properties. This provides at least indirect evidence that cause of death is unlikely to have played a large role in the species differences in HVA currents. Thus, we suggest differences observed between rat and human DRG neurons with respect to HVA currents are largely species-dependent.
There are at least two implications of lower HVA current density in human DRG neurons. First, Ca2+ influx at any level of activity will be smaller. Previous data from rat suggest that while the activity-dependent increase in intracellular Ca2+ is entirely dependent on the activation of VGCC, the Ca2+ influx through these channels is only responsible for the initiation of the evoked transient. Other Ca2+-regulatory machinery (e.g., that underlying Ca2+-induced Ca2+ release from intracellular stores, and the plasma membrane Ca2+-ATPase) are largely responsible to determining the magnitude and duration of the evoked transient at even moderate activity levels for relatively short burst durations.[8; 9; 17] As a result, the magnitude and duration of activity-dependent increases in intracellular Ca2+ are largely independent of VGCCs at even moderate activity levels for relatively short burst durations.[10; 17; 39] This dissociation is even more apparent with inflammation, where the magnitude and duration of activity-dependent increases of intracellular Ca2+ are increased despite decreased HVA current density.[18; 27; 28] The relatively small current evoked by action potential discharge in human DRG neurons[21] suggests other Ca2+ regulatory processes do not compensate for low HVA current density. Thus, the first implication of the lower HVA current density in human DRG neurons is that activity-dependent increases in intracellular Ca2+ will be smaller and more spatially-restricted than in rat neurons, suggesting that cellular processes dependent on Ca2+ (e.g. kinases and phosphatases) would have to respond to smaller increases in intracellular Ca2+ and/or be closer to the point of influx. That said, as discussed below, the slower kinetics of the HVA currents observed in human DRG neurons (Figure 3), may also help compensate for the lower HVA current density when Ca2+ influx is driven by more physiologically relevant stimuli such as the action potential waveform. Second, lower HVA current density and more localized, activity-dependent increases in Ca2+ may impact activity-dependent changes in gene expression.[36] In light of compelling data linking neural activity, Ca2+, and changes in gene expression in rodent DRG neurons,[7] initial injury-induced changes in gene expression in sensory neurons[15; 38] are thought to be influenced by activity-dependent increases in intracellular Ca2+.[6] However, if activity fails to drive global increases in intracellular Ca2+ in human DRG neurons, then this form of plasticity would not be as robust in human DRG neurons and/or other mechanisms may instead link neuronal activity and gene expression. From an experimental perspective, the tight association between VGCC activity and the magnitude and duration of Ca2+ transients in human DRG neurons suggests that Ca2+ imaging may provide a more accurate measure of activity in human DRG neurons than rat, even if transients are smaller and shorter duration.[10]
While this study was not designed to identify subpopulations of sensory neurons, there were no unique characteristics of the human DRG neurons with large pre-pulse potentiation of HVA current. This population of neurons (12 neurons with potentiation > 1 standard deviation above the mean) had similar cell body diameter, HVA current density, and proportions of L- and N-type currents (although only 7 neurons received a subtype selective channel blocker). Nevertheless, we ruled out three potential functional consequences of the large constitutive influence on channel gating. First, constitutive suppression of HVA currents did not influence current density in human DRG neurons as current density was still lower when potentiated currents were used to estimate peak current density. Similarly, if a peak inward HVA current was potentiated by a depolarizing pre-pulse, this might counter other mechanisms associated with decreased HVA current, thereby maintaining Ca2+ influx during a burst of action potentials. However, we observed a negative correlation between pre-pulse potentiation and the degree of change in current influx in response to 10 stimuli at 10 Hz (Pearson’s, p < 0.05). Furthermore, and potentially most importantly, there was no association between the magnitude of the pre-pulse potentiation and either the magnitude of the DAMGO-induced suppression of HVA current, or the percentage of neurons responsive to DAMGO. Therefore, if pre-pulse potentiation reflects long-term GPCR signaling, the underlying mechanisms are distinct from those underlying short-term GPCR-dependent suppression of HVA currents. Additional experiments will be needed to elucidate the mechanisms and functional implications of constitutive suppression of HVA currents in human DRG neurons.
Given the therapeutic efficacy of ziconotide, an N-type VGCC blocker presumed to block transmitter release from central terminals of nociceptive afferents in the spinal dorsal horn,[34] we were surprised that L-type currents comprised the majority of human HVA current. There are at least two likely explanations for differences in the relative density of HVA current subtypes. One is species differences in HVA channel subtype trafficking within the neuron (e.g. in the cell body versus axon terminals). The second possibility is species differences in HVA channel subtype transcription, translation and/or protein turnover. If the difference in HVA subtype density is due to trafficking, then results demonstrating that N-type channels underlie Ca2+ influx in presynaptic terminals in rodents are likely also true in humans.[40] However, two observations favor the second possibility: 1) Despite the presence of N-type channels in human DRG neurons, DAMGO appeared to act preferentially through L-type channels, suggesting these, and not N-type channels,[11] may be a target for presynaptic suppression of transmitter release; 2) genes encoding L-type channels (CACNA1(C/D/F) are expressed at relatively higher levels in human DRG than that encoding N-type channels (CACNA1B), while the opposite is true in mouse.[26] If the differences in HVA current subtypes are not just restricted to the cell soma, then the relatively higher density of L-type channels in human DRG neurons would explain the limited therapeutic efficacy of drugs such as gabapentin that appear to preferentially reduce N-type currents.[37]
While not as dramatic as current density differences, pre-pulse potentiation, and HVA current subtypes, there were interesting implications associated with slower kinetics of activation, inactivation, and deactivation observed in human HVA currents compared to rat. For example, slower rates of activation and deactivation delay Ca2+ influx via HVA channels during an action potential, with influx corresponding to the falling phase and afterhyperpolarization of the action potential. This shift in timing maximizes the driving force on Ca2+ ions and, as noted above, may compensate for lower HVA current density observed when current is evoked with a square pulse (e.g., Figure 1). Ca2+ influx data generated with an AP waveform (Figure 5D) are consistent with this suggestion. Similarly, the heterogeneity observed in the voltage-dependence of activation and deactivation of HVA current in human neurons may suggest species differences in expressed splice variants of HVA genes. For example, splice variants for genes encoding L- and N-type channels can influence voltage-dependence of activation[1; 16; 25] and the kinetics of channel activation.[23]
Finally, while the focus of this study was on HVA currents, it was difficult to ignore what appeared to be marked differences between rodent and human DRG neurons with respect to the amplitude and kinetics of LVA currents. That is, the LVA current density in human DRG neurons was smaller than in rat neurons, and the current detected demonstrated little open-state inactivation. Because rodent data suggest LVA currents play important roles in neuronal excitability and pain associated with tissue injury,[13; 20; 32] further investigation of LVA currents in human sensory neurons is warranted. Taken together, these data add to a growing list of differences between rodent and human neurons and once again underscore the need to conduct research using human neurons to successfully develop effective therapeutics.
Supplementary Material
Acknowledgments:
We thank members of the Gold lab and the Pittsburgh Center for Pain Research for their feedback in writing this manuscript. We especially thank the Center for Organ Recovery for tissue recovery. Most importantly, we thank the next of kin of tissue donors for their generous consent to the use of the donor’s tissue for research purposes. This work was supported by the National Institutes of Health (R01NS083347 and R01DK107966 (MSG), F32NS103231 and T32NS073548 (JEH) and a grant from the Bloch Foundation (MSG) and from Grünenthal GmbH. RJ, TC, and WS are, or were employees of Grünenthal GmbH. While MSG has received research grants from Grünenthal, none of the authors have a conflict of interest with the material in this manuscript.
Abbreviations:
- HVA
High voltage activating
- LVA
Low voltage activating
- VGCC
Voltage gated Ca2+ current
- MOR
μ opioid receptor
- DAMGO
[D-Ala2, N-MePhe4, Gly-ol]-enkephalin
- GPCR
G-protein coupled receptor
- DRG
Dorsal root ganglion
- AP
Action potential
References
- [1].Allen SE, Darnell RB, Lipscombe D. The neuronal splicing factor Nova controls alternative splicing in N-type and P-type CaV2 calcium channels. Channels (Austin) 2010;4(6):483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Birnir B, Korpi ER. The impact of sub-cellular location and intracellular neuronal proteins on properties of GABA(A) receptors. Curr Pharm Des 2007;13(31):3169–3177. [DOI] [PubMed] [Google Scholar]
- [3].Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 2002;22(23):10277–10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bourinet E, Soong TW, Stea A, Snutch TP. Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proceedings of the National Academy of Sciences 1996;93(4):1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. The Journal of physiology 2016;594(19):5369–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fields RD, Eshete F, Dudek S, Ozsarac N, Stevens B. Regulation of gene expression by action potentials: dependence on complexity in cellular information processing. Novartis Found Symp 2001;239:160–172; discussion 172–166, 234–140. [DOI] [PubMed] [Google Scholar]
- [7].Fields RD, Lee PR, Cohen JE. Temporal integration of intracellular Ca2+ signaling networks in regulating gene expression by action potentials. Cell calcium 2005;37(5):433–442. [DOI] [PubMed] [Google Scholar]
- [8].Fuchs A, Rigaud M, Sarantopoulos CD, Filip P, Hogan QH. Contribution of Calcium Channel Subtypes to the Intracellular Calcium Signal in Sensory Neurons: The Effect of Injury. Anesthesiology: The Journal of the American Society of Anesthesiologists 2007;107(1):117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gemes G, Bangaru ML, Wu HE, Tang Q, Weihrauch D, Koopmeiners AS, Cruikshank JM, Kwok WM, Hogan QH. Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. The Journal of neuroscience : the official journal of the Society for Neuroscience 2011;31(10):3536–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hartung JE, Gold MS. GCaMP as an indirect measure of electrical activity in rat trigeminal ganglion neurons. Cell calcium 2020;89:102225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Heinke B, Gingl E, Sandkühler J. Multiple Targets of μ-Opioid Receptor-Mediated Presynaptic Inhibition at Primary Afferent Aδ- and C-Fibers. The Journal of Neuroscience 2011;31(4):1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, Kort M, Carroll W, Marron B, Atkinson R, Thomas J, Liu D, Krambis M, Liu Y, McGaraughty S, Chu K, Roeloffs R, Zhong C, Mikusa JP, Hernandez G, Gauvin D, Wade C, Zhu C, Pai M, Scanio M, Shi L, Drizin I, Gregg R, Matulenko M, Hakeem A, Gross M, Johnson M, Marsh K, Wagoner PK, Sullivan JP, Faltynek CR, Krafte DS. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proceedings of the National Academy of Sciences of the United States of America 2007;104(20):8520–8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Joksimovic SL, Joksimovic SM, Tesic V, García-Caballero A, Feseha S, Zamponi GW, Jevtovic-Todorovic V, Todorovic SM. Selective inhibition of CaV3.2 channels reverses hyperexcitability of peripheral nociceptors and alleviates postsurgical pain. Science Signaling 2018;11(545):eaao4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kostyuk P, Pronchuk N, Savchenko A, Verkhratsky A. Calcium currents in aged rat dorsal root ganglion neurones. The Journal of physiology 1993;461:467–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kupari J, Usoskin D, Lou D, Parisien M, Hu Y, Fatt M, Lönnerberg P, Spångberg M, Eriksson B, Barkas N. Single cell transcriptomics of primate sensory neurons identifies cell types associated with human chronic pain. bioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lipscombe D, Andrade A. Calcium Channel CaVα₁ Splice Isoforms - Tissue Specificity and Drug Action. Curr Mol Pharmacol 2015;8(1):22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. The Journal of physiology 2006;577(Pt 1):169–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lu SG, Zhang XL, Luo ZD, Gold MS. Persistent inflammation alters the density and distribution of voltage-activated calcium channels in subpopulations of rat cutaneous DRG neurons. Pain 2010;151(3):633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].McCool BA, Harpold MM, Stauderman KA, Brust PF, Lovinger DM. Relative contributions of G protein, channel, and receptor to voltage-dependent inhibition of neuronal N-type and P/Q-type calcium channels in HEK 293 cell lines. Neurosci Lett 1997;239(2–3):89–92. [DOI] [PubMed] [Google Scholar]
- [20].Messinger RB, Naik AK, Jagodic MM, Nelson MT, Lee WY, Choe WJ, Orestes P, Latham JR, Todorovic SM, Jevtovic-Todorovic V. In vivo silencing of the CaV3. 2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. PAIN® 2009;145(1–2):184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Moy JK, Hartung JE, Duque MG, Friedman R, Nagarajan V, Loeza-Alcocer E, Koerber HR, Christoph T, Schroder W, Gold MS. Distribution of functional opioid receptors in human dorsal root ganglion neurons. Pain 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Patil PG, Brody DL, Yue DT. Preferential Closed-State Inactivation of Neuronal Calcium Channels. Neuron 1998;20(5):1027–1038. [DOI] [PubMed] [Google Scholar]
- [23].Pinggera A, Lieb A, Benedetti B, Lampert M, Monteleone S, Liedl KR, Tuluc P, Striessnig J. CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol Psychiatry 2015;77(9):816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Proft J, Weiss N. G Protein Regulation of Neuronal Calcium Channels: Back to the Future. Molecular Pharmacology 2015;87(6):890–906. [DOI] [PubMed] [Google Scholar]
- [25].Raingo J, Castiglioni AJ, Lipscombe D. Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat Neurosci 2007;10(3):285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ray P, Torck A, Quigley L, Wangzhou A, Neiman M, Rao C, Lam T, Kim JY, Kim TH, Zhang MQ, Dussor G, Price TJ. Comparative transcriptome profiling of the human and mouse dorsal root ganglia: an RNA-seq-based resource for pain and sensory neuroscience research. Pain 2018;159(7):1325–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Scheff NN, Gold MS. Trafficking of Na+/Ca2+ Exchanger to the Site of Persistent Inflammation in Nociceptive Afferents. The Journal of Neuroscience 2015;35(22):8423–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Scheff NN, Lu S-G, Gold MS. Contribution of endoplasmic reticulum Ca2+ regulatory mechanisms to the inflammation-induced increase in the evoked Ca2+ transient in rat cutaneous dorsal root ganglion neurons. Cell calcium 2013;54(1):46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Scroggs RS, Fox AP. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. The Journal of physiology 1992;445:639–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Seebohm G, Lerche C, Busch AE, Bachmann A. Dependence of I(Ks) biophysical properties on the expression system. Pflugers Arch 2001;442(6):891–895. [DOI] [PubMed] [Google Scholar]
- [31].Sheahan TD, Valtcheva MV, McIlvried LA, Pullen MY, Baranger DAA, Gereau RWt. Metabotropic Glutamate Receptor 2/3 (mGluR2/3) Activation Suppresses TRPV1 Sensitization in Mouse, But Not Human, Sensory Neurons. eNeuro 2018;5(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Snutch TP, Zamponi GW. Recent advances in the development of T-type calcium channel blockers for pain intervention. British Journal of Pharmacology 2018;175(12):2375–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tscherter A, David F, Ivanova T, Deleuze C, Renger JJ, Uebele VN, Shin HS, Bal T, Leresche N, Lambert RC. Minimal alterations in T-type calcium channel gating markedly modify physiological firing dynamics. The Journal of physiology 2011;589(Pt 7):1707–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang YX, Pettus M, Gao D, Phillips C, Scott Bowersox S. Effects of intrathecal administration of ziconotide, a selective neuronal N-type calcium channel blocker, on mechanical allodynia and heat hyperalgesia in a rat model of postoperative pain. Pain 2000;84(2–3):151–158. [DOI] [PubMed] [Google Scholar]
- [35].Weiss N, Zamponi GW. Opioid Receptor Regulation of Neuronal Voltage-Gated Calcium Channels. Cellular and Molecular Neurobiology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proceedings of the National Academy of Sciences 2001;98(20):11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wiffen PJ, Derry S, Bell RF, Rice ASC, Tölle TR, Phillips T, Moore RA. Gabapentin for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2017(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yasko JR, Moss IL, Mains RE. Transcriptional Profiling of Non-injured Nociceptors After Spinal Cord Injury Reveals Diverse Molecular Changes. Frontiers in Molecular Neuroscience 2019;12(284). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yilmaz E, Watkins SC, Gold MS. Paclitaxel-induced increase in mitochondrial volume mediates dysregulation of intracellular Ca(2+) in putative nociceptive glabrous skin neurons from the rat. Cell calcium 2017;62:16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol Rev 2015;67(4):821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang X, Hartung JE, Friedman RL, Koerber HR, Belfer I, Gold MS. Nicotine Evoked Currents in Human Primary Sensory Neurons. The Journal of Pain 2019;20(7):810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang X, Priest BT, Belfer I, Gold MS. Voltage-gated Na+ currents in human dorsal root ganglion neurons. eLife 2017;6:e23235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.