

SUMMARY
The innate immune response is critical for recognizing and controlling infections through the release of cytokines and chemokines. However, severe pathology during some infections, including SARS-CoV-2, is driven by hyperactive cytokine release, or cytokine storm. The innate sensors that activate production of pro-inflammatory cytokines and chemokines during COVID-19 remain poorly characterized. Here we show that both TLR2 and MYD88 expression were associated with COVID-19 disease severity. Mechanistically, TLR2 and MyD88 were required for β-coronavirus–induced inflammatory responses, and TLR2-dependent signaling induced the production of pro-inflammatory cytokines during coronavirus infection independent of viral entry. TLR2 sensed the SARS-CoV-2 envelope protein as its ligand. Additionally, blocking TLR2 signaling in vivo provided protection against the pathogenesis of SARS-CoV-2 infection. Overall, our study provides a critical understanding of the molecular mechanism of β-coronavirus sensing and inflammatory cytokine production, which opens new avenues for therapeutic strategies to counteract the ongoing COVID-19 pandemic.
Keywords: COVID-19, MHV, SARS-CoV-2, β-Coronavirus, inflammatory cytokines, cytokine storm, TNF-α, IFN-γ, TLR2, envelope protein, NF-κB, ERK, inflammation
INTRODUCTION
β-Coronaviruses belong to the family Coronaviridae and are positive-stranded enveloped RNA viruses. Several members of the β-coronavirus genus have emerged to cause zoonotic epidemic infections this century, including severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) and Middle East respiratory syndrome (MERS) coronavirus (MERS-CoV)1. The currently circulating SARS-CoV-2, which is responsible for the ongoing pandemic of coronavirus disease 2019 (COVID-19), is also a member of the β-coronavirus genus2.
β-coronavirus infection leads to robust pro-inflammatory cytokine release, including TNF-α, IL-6, IL-1β, and IFN-γ3–6. While these cytokines are a critical component of the innate immune response and aid in clearing viral infections, the dysregulated release of pro-inflammatory cytokines can lead to a cytokine storm, which causes severe damage to host tissues and organs by inducing inflammatory cell death3,6,7. COVID-19 has caused more than 2.8 million deaths globally to date, and increased levels of pro-inflammatory cytokines are directly associated with the pathogenesis8. A lack of fundamental understanding of the underlying pathways driving this dysregulated cytokine release has hampered the development of targeted therapeutic strategies. A recent study discovered the molecular mechanisms linking cytokine storm to mortality, showing that TNF-α and IFN-γ are the key factors among the cytokines that are increased in patients with COVID-19. These two specific cytokines act synergistically to activate robust inflammatory cell death (PANoptosis) thereby causing tissue and organ damage and lethality6. However, the host factors that control the expression of inflammatory cytokines including TNF-α and IFN-γ during coronavirus infection are largely unknown.
The host innate immune system can recognize pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs) during infection to induce inflammatory responses to eliminate pathogens9. The five PRR families identified to date include Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), C-type lectin receptors (CLRs), and the absent in melanoma 2 (AIM2)-like receptors (ALRs). PAMPs can include moieties such as cell wall components of pathogens, i.e. lipopolysaccharide (LPS) and lipoproteins, glycans, conserved proteins such as flagellin, or pathogen nucleic acids including RNA and DNA; these moieties are generally conserved among pathogen species but are distinct from host components10. Several PRRs have been reported to be involved in sensing β-coronavirus infection, including MDA511, TLR712,13, and NLRP35. MDA5 contributes to mouse hepatitis virus (MHV)-induced type I interferon (IFN) expression, but plays an inhibitory role in inflammatory cytokine expression in vivo11. TLR7 is required for MERS-CoV–triggered type I IFN induction in vivo and for MHV-induced type I IFN activation in plasmacytoid dendritic cells12,13, whereas the role of TLR7 in β-coronavirus–induced inflammatory cytokine release has not been examined. NLRP3 activation leads to the release of the pro-inflammatory cytokines IL-1β and IL-18 in response to β-coronavirus infection5. Myd88, a TLR adaptor protein, is required for the production of other inflammatory cytokines such as TNF-α and IL-6 following β-coronavirus infection14. However, the sensors upstream of Myd88 that control the activation of the inflammatory signaling pathway remain unclear during coronavirus infection.
In this study, we screened several innate sensors upstream of Myd88 to determine the receptors required for the production of pro-inflammatory cytokines following β-coronavirus infection and to understand how these critical cytokines are produced, particularly given the pathogenic role of TNF-α and IFN-γ in cytokine storm-induced mortality in patients with COVID-196. We found that TLR2 can sense the envelope protein from SARS-CoV-2 and was required for inflammatory cytokine release during β-coronavirus infection. Blocking TLR2 also protected against SARS-CoV-2–induced lethality in vivo. Our findings suggest an essential role of TLR2 in the disease development of COVID-19 and identify potential strategies for therapeutic intervention against this deadly pandemic disease.
RESULTS
MYD88 and TLRs are associated with the severity of COVID-19
Myd88 is important for pro-inflammatory cytokine production during MHV and SARS-CoV-1 infection14,15. To determine whether MYD88 or another TLR adaptor TRIF play a role in SARS-CoV-2–induced inflammatory responses and pathogenesis, we first re-analyzed a publicly available dataset16 for MYD88 and TRIF expression in patients with differing severities of COVID-19 (Figures 1A and 1B). We found that MYD88 expression showed a positive correlation with the pathogenesis of COVID-19 (Figure 1A), suggesting that MYD88 is associated with SARS-CoV-2 infection in humans. By contrast, TRIF was significantly elevated in only patients with critical COVID-19 (Figure 1B). MYD88 is a key adaptor shared by most TLRs. So far, 10 TLRs have been identified in humans, named from TLR1 to TLR1017. With the exception of TLR3, which signals exclusively through TRIF, all other TLRs utilize MYD88 to trigger inflammatory cytokine production17. To investigate whether any TLRs were positively correlated with COVID-19 severity, as MYD88 was, we re-analyzed the dataset16 for TLR expression in patients with differing severities of COVID-19 (Figures 1C–1J). We found that the expression pattern of TLR2 was increased with severity of COVID-19, similar to the phenomenon observed with MYD88 (Figure 1D). Also, the expression of TLR1, TLR4, TLR5, TLR8, and TLR9 was significantly elevated in patients with severe and critical COVID-19 (Figures 1C, 1F, 1G, 1I, and 1J). By contrast, expression of TLR3 did not show any correlation with the disease development of COVID-19, and the expression of TLR7 was increased in only patients with moderate COVID-19. All these data suggest an association of MYD88 and certain TLRs (TLR1, TLR2, TLR4, TLR5, TLR8, and TLR9) with disease progression in patients with COVID-19.
Figure 1. MYD88 and TLRs are associated with the severity of COVID-19.

(A–J) Absolute RNA counts of MYD88 (A), TRIF (B), TLR1 (C), TLR2 (D), TLR3 (E), TLR4 (F), TLR5 (G), TLR7 (H), TLR8 (I), and TLR9 (J) in patients with mild-to-moderate (n = 11), severe (n = 10), and critical (n = 11) COVID-19, and 13 healthy controls16. (K) Immunoblot analysis of phospho-ERK (pERK), total ERK (tERK), pIκB, and tIκB in WT, Myd88–/–, and Trif–/– bone marrow-derived macrophages (BMDMs) after infection with MHV at a MOI of 0.1 for the indicated time. Actin was used as the internal control. (L) Real-time PCR analysis of Tnf expression in WT, Myd88–/–, and Trif–/– BMDMs after infection with MHV at a MOI of 0.1 for the indicated time, presented relative to levels of the host gene Gapdh. Significant differences compared to the healthy group are denoted as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; ns: not significant (one-way ANOVA). Exact P values are presented in Supplementary Table 1. Data are shown as mean ± SEM (A–J, L). Data are representative of three independent experiments (K and L).
To confirm the role of Myd88 in β-coronavirus–induced inflammatory responses, we infected mouse bone marrow-derived macrophages (BMDMs) with MHV, the prototype of this genus which mimics many of the key aspects of human coronavirus biology and can be investigated under BSL-2 conditions18. Inflammatory cytokine expression largely depends on the ERK and NF-κB signaling pathways19,20, and the NF-κB pathway is activated in patients with COVID-1916. In line with this, we found activation of NF-κB by MHV infection in BMDMs, as indicated by the phosphorylation of IκB (Figure 1K). Meanwhile, ERK was also activated following MHV infection (Figure 1K). Consistent with previous reports that Myd88 is required for β-coronavirus–induced inflammatory responses14,15, loss of Myd88 abolished the activation of both ERK and NF-κB after MHV infection (Figure 1K). In contrast, TRIF was dispensable for MHV-induced ERK and NF-κB activation (Figure 1K). In agreement with the activation of signaling pathways, transcription of inflammatory cytokine genes Tnf, Il1b, and Il6 was induced following MHV infection in WT and Trif–/– BMDMs but not in Myd88–/– BMDMs (Figures 1L, Extended Data Fig. 1A and 1B). Given that the type I interferons (IFNs) are critical for the development of COVID-1916, we also assessed the role of the innate sensors required for the induction of type I IFNs following β-coronavirus infection using Mda5–/– and Mavs–/– BMDMs. Consistent with published reports11,21, MDA5 was required for β-coronavirus–induced type I IFN expression (Extended Data Fig. 2A and 2B). Collectively, these data indicate that MYD88 and some TLRs are correlated with the disease development of COVID-19, and that MYD88 is essential for β-coronavirus–induced inflammatory cytokine expression while MDA5 is required for type I IFN expression.
TLR2 is required for inflammatory responses to β-coronavirus
TLR2, 3, 4, 7, 8, 9, and 10 have been reported to recognize viral infection under certain conditions22,23. TLR3 is a TRIF-dependent innate sensor23, and TLR10 has been shown to be nonfunctional in mice24. Additionally, TLR8 functions controversially in mice, compared to its role in humans25. To determine which TLR(s) are responsible for sensing β-coronavirus infection and signaling through Myd88, we infected BMDMs deficient in TLR2, 4, 7, or 9 with MHV and monitored the transcription of inflammatory cytokine genes. We found that loss of TLR4, 7, or 9 had no effect on MHV-induced expression of Il1b, Il6, or Tnf in BMDMs, while TLR2 deficiency abolished transcription of each of these genes (Figures 2A–2C). This suggests that TLR2 is the innate sensor that triggers β-coronavirus–induced inflammatory cytokine expression. To further confirm the role of TLR2 during β-coronavirus infection, we examined the activation of inflammatory signaling pathways in Tlr2–/– BMDMs following MHV infection. MHV-induced activation of ERK and NF-κB was abrogated in Tlr2–/– BMDMs (Figure 2D). By contrast, TLR7 deficiency had no effect on MHV-induced activation of ERK and NF-κB (Figure 2D). Additionally, secretion of IL-6 and TNF-α was abolished in Tlr2–/– BMDMs infected with MHV, while loss of TLR7 did not affect the release of these two cytokines (Extended Data Fig. 3A and 3B). Release of inflammatory chemokines including CXCL10 (IP-10), CCL3 (MIP-1α), CXCL1 (KC), RANTES, MCP-1, and G-CSF was also reduced or completely eliminated in Tlr2–/– BMDMs following MHV infection at 3 and 6 h post-infection, whereas loss of TLR7 had minimal effect on the expression of these chemokines (Extended Data Fig. 3C–3H). These findings suggest that TLR2 specifically is required for the expression of these inflammatory cytokines and chemokines following MHV infection.
Figure 2. TLR2 is required for the inflammatory response during β-coronavirus infection.

(A–C) Real-time PCR analysis of the expression of Il1b (A), Il6 (B), and Tnf (C) in WT, Tlr2–/–, Tlr4–/–, Tlr7–/–, and Tlr9–/– bone marrow-derived macrophages (BMDMs) after infection with MHV at a MOI of 0.1 for the indicated time, presented relative to levels of the host gene Gapdh. (D) Immunoblot analysis of phospho-ERK (pERK), total ERK (tERK), pIκB, and tIκB in WT, Tlr2–/–, and Tlr7–/– BMDMs after infection with MHV at a MOI of 0.1 for the indicated time. GAPDH was used as the internal control. (E–L) TNF-α (E), IFN-γ (F), IL-1α (G), IL-6 (H), CXCL10 (I), MCP-1 (J), G-CSF (K), and CCL3 (L) release from human peripheral blood mononuclear cells (PBMCs) infected with SARS-CoV-2 at a MOI of 0.5 for 20 h with or without the TLR2 inhibitor (oxPAPC; TLR2 in) or TLR4 inhibitor (CLI-095; TLR4 in). Significant differences compared to the media control infection group are denoted as *P < 0.05 and ***P < 0.001; ns: not significant (one-way ANOVA) (E–L). Exact P values are presented in Supplementary Table 1. Data are representative of three independent experiments (A–D) or two independent experiments (E–L). Data are shown as mean ± SEM (n = 3 biological replicates) (A–C and E–L).
Studies on serum cytokines in patients with COVID-19 have indicated that many cytokines and chemokines, such as IL-6, TNF-α, IFN-γ, IL-1α, IL-1ra, G-CSF, MCP-1, CCL3, and CXCL10, are elevated in response to SARS-CoV-2 infection and are positively correlated with disease severity4,16,26. To investigate the role of TLR2 in SARS-CoV-2–induced cytokine and chemokine expression, we infected human peripheral blood mononuclear cells (PBMCs) with SARS-CoV-2 in the presence of a TLR2 inhibitor. First, we identified an optimal TLR2 inhibitor by confirming the effectiveness and specificity of two different inhibitors on TLR2 signaling in BMDMs stimulated with a TLR2 ligand, Pam3CSK4 (Pam3). We found that oxPAPC was more potent in inhibiting Pam3-induced inflammatory signaling than C29 was (Extended Data Fig. 4A). Additionally, oxPAPC had no effect on activation of ERK and NF-κB signaling induced by a TLR7 ligand, R848 (Extended Data Fig. 4B), suggesting that oxPAPC specifically inhibits TLR2-dependent inflammatory responses. In human PBMCs, the inhibition of TLR2 signaling by oxPAPC treatment significantly reduced SARS-CoV-2–induced secretion of TNF-α, IFN-γ, IL-1α, IL-6, CXCL10, MCP-1, G-CSF, and CCL3 (Figures 2E–2L). By contrast, the TLR4-specific inhibitor had minimal effects on the release of these cytokines and chemokines in response to SARS-CoV-2 infection (Figures 2E–2L).
Taken together, these results indicate that TLR2 can sense β-coronavirus infection and is responsible for β-coronavirus–induced inflammatory cytokine production.
TLR2 can sense the envelope protein of β-coronaviruses
TLR2 is a cell surface innate immune sensor with the ability to recognize diverse ligands from viruses, fungi, bacteria, and parasites27. TLR2 has been reported to sense several viral proteins following infection including the dUTPase of Epstein-Barr virus, glycoprotein B of cytomegalovirus, and capsid of hepatitis B virus27. We therefore hypothesized that TLR2 recognizes viral proteins from β-coronaviruses as well. To investigate whether viral proteins independent of functioning virions could activate TLR2, we used heat-inactivated MHV (MHV-HI)28. MHV-HI activated ERK and NF-κB signaling pathways in WT BMDMs (Figure 3A). In contrast, this activation was reduced or eliminated in Tlr2–/– and Myd88–/– BMDMs (Figure 3A). These data suggest that activation of the TLR2 signaling pathway by β-coronaviruses does not require viral replication. It has been reported that chloroquine, when added before infection, can inhibit coronavirus entry29. We also observed that the cell fusion during live MHV infection was suppressed by chloroquine treatment, further suggesting that chloroquine inhibits viral entry (Extended Data Fig. 5A). To test whether the activation of the TLR2 signaling pathway requires the internalization of viral proteins from β-coronaviruses, we treated BMDMs with 10 μM chloroquine for 30 min before adding the MHV-HI. We observed that chloroquine treatment did not inhibit the activation of MHV-HI–induced TLR2-dependent inflammatory signaling pathways (Figure 3B), suggesting that the ligands sensed by TLR2 in β-coronaviruses are viral surface proteins. Based on these findings, we hypothesized that even though SARS-CoV-2 cannot infect murine cells due to the receptor specificity30, it would still be able to activate the inflammatory signaling pathways in BMDMs. Indeed, SARS-CoV-2–stimulated BMDMs showed activation of the ERK and NF-κB signaling pathways (Figure 3C). This may support the recent finding that SARS-CoV-2 inoculation induced lung inflammation in WT C57BL/6 mice31. To further confirm the ability of SARS-CoV-2 proteins to activate inflammatory signaling without infection, we used heat-inactivated SARS-CoV-2 to stimulate the BMDMs. We found that heat-inactivated SARS-CoV-2 stimulated inflammatory signaling pathways, which were substantially reduced in TLR2- and Myd88-deficient BMDMs (Figure 3D). Collectively, these data suggest that TLR2 can sense the surface proteins of β-coronaviruses before entry.
Figure 3. TLR2 can sense the envelope protein of β-coronaviruses.

(A) Immunoblot analysis of phospho-ERK (pERK), total ERK (tERK), pIκB, and tIκB in WT, Tlr2–/–, and Myd88–/– bone marrow-derived macrophages (BMDMs) after stimulation with heat-inactivated MHV (MHV-HI) at a MOI of 0.1 for the indicated time. GAPDH was used as the internal control. (B) Immunoblot analysis of pERK, tERK, pIκB, and tIκB in WT, Tlr2–/–, and Tlr7–/– BMDMs after stimulation with MHV-HI for the indicated time. Chloroquine (CQ) at a final concentration of 10 μM was added 30 min before the stimulation. Actin was used as the internal control. (C) Immunoblot analysis of pERK, tERK, pIκB, and tIκB in WT BMDMs after stimulation with SARS-CoV-2 for the indicated time. Actin was used as the internal control. (D) Immunoblot analysis of pERK, tERK, pIκB, and tIκB in WT, Tlr2–/–, and Myd88–/– BMDMs after stimulation with heat-inactivated SARS-CoV-2 (SARS2-HI) for the indicated time. Actin was used as the internal control. (E) Immunoblot analysis of pERK, tERK, pIκB, and tIκB in WT, Tlr2–/–, and Myd88–/– BMDMs after stimulation with 1 μg/ml of the envelope (E) protein of SARS-CoV-2 for the indicated time. Actin was used as the internal control. (F–I) Real-time PCR analysis of the expression of TNF (F), IFNG (G), IL6 (H), and IL1B (I) in human peripheral blood mononuclear cells (PBMCs) after stimulation with 1 μg/ml of the E or spike (S) protein from SARS-CoV-2 or Pam3CSK4 (Pam3) for 4 h, presented relative to levels of the host gene GAPDH. (J) Immunoprecipitates and total lysates from 293T cells after incubation of purified E protein from SARS-CoV-2 with overexpressed human TLR2, TLR3 or mouse TLR2. Student’s t test was used for statistical analysis between E and Pam3 treated groups; ns: not significant. Data are representative of three independent experiments (A–J). ns, not significant (two-sided student’s t test) (F–I). Data are shown as mean ± SEM (n = 2 biological replicates plus 2 technical replicates) (F–I).
There are three viral proteins on the surface of β-coronaviruses, namely spike (S) protein, envelope (E) protein, and membrane (M) protein32. It was previously reported that a strain of SARS-CoV-1 lacking the E protein was not able to activate the NF-κB pathway, thus significantly reducing the production of inflammatory cytokines after infection in mice33. To determine whether the E protein can be sensed by TLR2 to activate inflammatory signaling pathways, we stimulated BMDMs with purified E protein from SARS-CoV-2. We found that the E protein activated ERK and NF-κB signaling pathways in WT BMDMs, and this activation was greatly attenuated in both Tlr2–/– and Myd88–/– BMDMs (Figure 3E). In contrast, we did not observe any activation after stimulating BMDMs with the purified S protein from SARS-CoV-2 (Extended Data Fig. 6A). To further confirm the effect of the E protein on the activation of inflammatory signaling, we evaluated the expression of inflammatory cytokines in response to different doses of E protein. We found that the E protein could induce robust inflammatory cytokine expression at the concentration of 5 ng/ml, although the effect was not as strong as the traditional TLR2 ligand Pam3 at this concentration (Extended Data Fig. 6B and 6C). When a higher concentration of E protein was added, its ability to induce inflammatory cytokine expression was comparable to that of Pam3 (Extended Data Fig. 6B and 6C). Additionally, transcription of Il1b, Il6, and Tnf were strongly upregulated in WT BMDMs stimulated with the E protein but not with the S protein, and the loss of TLR2 attenuated the expression of Il1b, Il6, and Tnf induced by the E protein (Extended Data Fig. 6D–6F).
ERK and NF-κB signaling are also required for transcriptional upregulation of Nlrp334, one of the prerequisite steps in NLRP3 inflammasome activation. To further confirm the effect of the E protein on activating the inflammatory signaling pathways in BMDMs, we evaluated Nlrp3 upregulation and ATP-induced NLRP3 inflammasome activation after priming with the E protein. We observed Nlrp3 upregulation and caspase-1 cleavage in WT BMDMs in response to priming with the E protein, while S protein priming failed to induce the Nlrp3 upregulation and inflammasome activation (Extended Data Fig. 6G and 6H). The Nlrp3 upregulation and inflammasome activation were reduced in both Tlr2–/– and Myd88–/– BMDMs (Extended Data Fig. 6G and 6H). Consistently, pyroptosis activation and IL-18 release following E protein plus ATP treatment were reduced in both Tlr2–/– and Myd88–/– BMDMs, compared to those in WT BMDMs (Extended Data Fig. 6H and 6I). Together, these data indicate that the E protein of SARS-CoV-2 can activate TLR2-dependent signaling pathways.
To further examine the role of the E protein in driving inflammatory signaling in human cells, we stimulated human PBMCs with the E or S protein of SARS-CoV-2. We found that the E protein could induce expression of TNF, IFNG, IL6 and IL1B similar to that induced by Pam3 in human PBMCs, whereas the S protein could not induce the expression of these inflammatory cytokine genes (Figures 3F–3I). These data suggest that the E protein of β-coronaviruses can act as a PAMP in human cells. To verify whether TLR2 can directly interact with the E protein of SARS-CoV-2, we conducted co-immunoprecipitations with TLR2 and the E protein. We found that both human and mouse TLR2 could interact with the E protein of SARS-CoV-2 (Figure 3J), further supporting the conclusion that the E protein of SARS-CoV-2 can induce inflammatory responses in both human and mouse cells.
Taken together, these data indicate that TLR2 can sense the E protein of β-coronaviruses to initiate inflammatory signaling pathways and cytokine production.
SARS-CoV-2 E protein induces TLR2-driven lung inflammation
Given that SARS-CoV-2 infection induces severe lung inflammation, we next determined whether the E protein could trigger lung inflammation in vivo. It has been reported that Pam3 can induce lung inflammation after intratracheal instillation35. Consistent with this inflammation, we detected large amounts of CD45+ cells accumulating in the lungs of WT mice administered Pam3 (Figure 4A). Administration of the E protein from SARS-CoV-2 also triggered the recruitment of large numbers of inflammatory cells in the lungs of WT mice but not in lungs of Tlr2–/– mice (Figure 4A). Also, there was no increase of CD45+ cells in the lungs of mice treated with the S protein compared to those administrated PBS (Extended Data Fig. 7A), which is consistent with our in vitro findings that the S protein does not induce inflammatory signaling. Since overt inflammatory cytokine release can lead to tissue damage, we assessed whether administration of the E protein resulted in lung damage. We found that administration of Pam3 triggered cell death in the mouse lungs (Figure 4A). Similarly, E protein administration also led to comparable cell death in the mouse lungs (Figure 4A), suggesting that the E protein of SARS-CoV-2 has the potential to induce tissue damage during infection. By contrast, no cell death was detected in the lungs of mice treated with the S protein (Extended Data Fig. 7A). Furthermore, we evaluated the amounts of inflammatory cytokines and chemokines in the serum of the mice. We found that levels of IL-6 were elevated in WT mice after stimulating with the E protein of SARS-CoV-2 (Figure 4B). By contrast, the amount of IL-6 in Tlr2–/– mice administered the E protein or WT and Tlr2–/– mice administrated the S protein was close to the basal level, suggesting there was no activation of IL-6 production in these mice (Figure 4B). Similarly, CXCL10 and G-CSF levels were increased in WT mice stimulated with the E protein but not in Tlr2–/– mice administered the E protein or mice administrated the S protein (Figure 4B). To further confirm the inflammatory effect of the E protein in the lungs of mice, we collected the bronchoalveolar lavage fluid (BALF) at 6 h after intratracheal instillation. We found that the S protein did not induce secretion of any of the cytokines tested in the BALF of both WT and Tlr2–/– mice, while the E protein significantly increased the amounts of IL-6, TNF-α, CXCL1, GM-CSF and CCL3 in the BALF of WT mice (Figure 4C). Taken together, these data indicate that the E protein of SARS-CoV-2 can induce TLR2-dependent inflammation in the lungs of mice.
Figure 4. The envelope protein of SARS-CoV-2 can induce TLR2-dependent inflammation and damage in the lungs.

(A) CD45 immuno-staining and TUNEL staining of lung samples obtained from mice 24 h after being intratracheally instilled with PBS (n= 3 biological replicates), the envelope (E) from SARS-CoV-2 (n = 4 biological replicates), or Pam3CSK4 (Pam3) (n = 4 biological replicates). Red arrows indicate TUNEL-positive cells. Scale bar, 100 μm (black) or 25 μm (red). (B) Levels of IL-6, CXCL10, and G-CSF in the serum of mice 24 h after being intratracheally instilled with PBS or the E or S protein from SARS-CoV-2. (C) Levels of IL-6, TNF-α, CXCL1, GM-CSF, and CCL3 in the bronchoalveolar lavage fluid (BALF) of mice 6 h after being intratracheally instilled with PBS or the E or S protein from SARS-CoV-2. Significant differences compared to WT group treated with E protein are denoted as *P < 0.05, **P < 0.01, *** P < 0.001 and ****P < 0.0001 (one-way ANOVA) (B and C). Exact P values are presented in Supplementary Table 1. Data are representative of two independent experiments (A–C). Data are shown as mean ± SEM (n = 3–5 biological replicates) (B and C).
TLR2 inhibitor protects against SARS-CoV-2 pathology in vivo
Given the critical role of TLR2 in SARS-CoV-2 infection-induced inflammatory cytokine release in vitro, we assessed whether blocking TLR2 signaling could affect the mortality of mice after infection with SARS-CoV-2. Due to the receptor specificity of SARS-CoV-2, we used mice expressing human ACE2 introduced under the control of the human cytokeratin 18 (K18), known as K18-hACE2 transgenic mice. Two doses of the TLR2 inhibitor were administrated during the infection. Treating the mice with the TLR2 inhibitor significantly reduced the release of IL-6, CXCL10, and MCP-1 and led to a non-significant decrease in the amounts of TNF-α, IFN-γ, and G-CSF released in the BALF at day 2 post-infection (Figure 5A). Additionally, TLR2 inhibitor treatment led to significantly increased survival of the SARS-CoV-2–infected mice, compared to the control mice injected with PBS (Figure 5B). In line with this, the body weight of mice treated with the TLR2 inhibitor decreased more slowly and recovered faster compared with the body weight change in the control group (Figure 5C). Collectively, these data suggest that TLR2 senses SARS-CoV-2 infection in vivo and that blocking TLR2 signaling could provide protection against SARS-CoV-2 infection.
Figure 5. Blocking TLR2 signaling protects against SARS-CoV-2 infection in vivo.

(A) Levels of TNF-α, IFN-γ, IL-6, G-CSF, CXCL10, and MCP-1 in the bronchoalveolar lavage fluid (BALF) of mice 2 days after SARS-CoV-2 infection. (B) Pooled survival of 8- to 10-week-old K18-hACE2 transgenic mice after infection with SARS-CoV-2 (2 × 104 pfu/mouse). (C) Pooled body weight change of infected mice in (B). Significant differences compared to the group treated with oxPAPC are denoted as *P < 0.05, **P < 0.01, and ***P < 0.001 (two-sided student’s t test (A), log-rank test (B), or two-way ANOVA (C)). Exact P values are presented in Supplementary Table 1. Data are shown as mean ± SEM (n = 3–4 biological replicates) (A) or (n = 14–16 biological replicates) (B and C).
Discussion
Inflammatory signaling leading to increases in cytokine secretion and cytokine storm are common phenotypes following β-coronavirus–induced zoonotic infection3,7. Recently, it has been shown that the inflammatory cytokines TNF-α and IFN-γ specifically play a critical role in the pathogenesis of COVID-196. Although it is well known that β-coronavirus infections can trigger robust inflammatory cytokine release, the innate sensors that are required for inflammatory cytokine expression in this context have remained unclear. In this study, we provided the first genetic evidence that TLR2 can sense β-coronavirus infection via recognition of the envelope protein to induce release of inflammatory cytokines including TNF-α and IFN-γ. We also showed that the activation of TLR2 signaling in response to β-coronavirus infection was independent of viral entry and replication.
TLR2 is involved in sensing different kinds of pathogens including bacteria, viruses, fungi, and parasites27. The ligand diversity of TLR2 could be explained by its ability to form heterodimers with TLR1 or TLR627. Also, TLR10 can interact with TLR2 to sense some ligands under certain conditions36. The residual NF-κB and ERK signaling activation in Tlr2–/– BMDMs stimulated with the E protein of SARS-CoV-2 could be due to recognition of the E protein by TLR1 or TLR6 to activate the inflammatory response. It is also possible that some other innate sensors may recognize the E protein. More studies are required to confirm whether this occurs.
The E protein is a structural protein of coronaviruses which has ion channel activity37. It can form protein-lipid pores in the membrane to allow ion transport, which could provide the activation signal for NLRP3 inflammasome assembly37. Therefore, the E protein may be able to provide both the priming and activation signal for the NLRP3 inflammasome. It is likely that priming and then transfecting with the E protein would activate the NLRP3 inflammasome and lead to the processing of the pyroptosis executioner gasdermin D that we observed. The phenomenon is similar to lipopolysaccharide (LPS), the cell wall component of gram-negative bacteria, which can trigger inflammatory responses and also activate pyroptosis. Therefore, the E protein of β-coronaviruses could be a virulence factor like LPS that can induce an endotoxic-like shock on its own in vivo. However, to test this hypothesis requires large amounts of purified E proteins.
The effect of cytokine storm on the development of COVID-19 has been extensively discussed since SARS-CoV-2 emerged, and increases in serum levels of pro-inflammatory cytokines have been linked to disease pathogenesis and mortality8. Based on this evidence, several clinical trials are ongoing to block various cytokines, including IL-6 and TNF-α38. However, blocking IL-6 has had limited clinical success, and more recent work suggests that TNF-α and IFN-γ are the two key cytokines to target to inhibit disease pathogenesis6. Therefore, identifying the sensor and ligand upstream of TNF-α and IFN-γ production in response to SARS-CoV-2 infection may provide critical information for clinical trials and direct the use of drugs to target specific pathways. Our findings identify more targets that could be pursued for therapeutic strategies to prevent or treat COVID-19. TLR2 inhibitors or antibodies could be useful to suppress the production of inflammatory cytokines and chemokines, thereby preventing the onset of cytokine storm. Additionally, targeting TLR2 would have fewer side effects than directly targeting MYD88, since MYD88 is also the adaptor for other TLRs and IL1R and IL18R39; inhibiting MYD88 would lead to the suppression of other critical cellular responses. In our study here, inhibiting TLR2 signaling reduced the release of TNF-α and IFN-γ, but this reduction was not significant at the time point analyzed in the lungs of mice during SARS-CoV-2 infection. One possible explanation is that some other innate sensors could also regulate the expression of TNF-α and IFN-γ in vivo, allowing their expression to potentially be restored or not significantly changed after TLR2 inhibitor administration. Further studies are required to clarify this. Additionally, to treat COVID-19, doses and timing to block TLR2 signaling should be seriously investigated, since optimal pro-inflammatory cytokine levels are generally protective to the host during infections40.
Beyond the potential of blocking TLR2, antibodies targeting the E protein of SARS-CoV-2 could possess dual roles, neutralizing the viral particles and simultaneously preventing the sensing by TLR2 and reducing inflammatory cytokine production. However, the timing of these interventions in the inflammatory signaling pathways should be carefully evaluated. Broad and long-lasting blockade of cytokine production may lead to delayed viral clearance and tissue repair. Detailed studies are required to analyze the effects of cytokine inhibition in different infection phases on the development of COVID-19.
Overall, our study identified the key inflammatory signaling pathways activated during β-coronavirus infection, which greatly improves our understanding of the molecular mechanism of β-coronavirus–induced cytokine production. These findings provide critical insights into strategies that can be pursued to control the development of the ongoing COVID-19 pandemic.
METHODS
Mice
Nlrp3−/− 41, Trif–/– 42, Tlr2–/– 43, Tlr4–/– 44, Tlr7–/– 45, Tlr9−/− 46, Myd88–/– 47, Mda5–/– 48, and Mavs–/– 49,50 mice have been described previously. For SARS-CoV-2 infections, K18-ACE-2 transgenic mice were purchased from The Jackson Laboratory (Stock Number 034860). All mice were bred at the Animal Resources Center at St. Jude Children’s Research Hospital and were backcrossed to the C57BL/6 background. Mice were kept with a 12:12 light:dark cycle. Humidity was maintained between 30 and 70% and the temperature ranged from 20 to 23.3 °C. Age- and sex-matched 6- to 9-week old male and female mice were used for intratracheal instillation and 8-to 10-week old male mice were used for in vivo SARS-CoV-2 infection studies. Non-infectious animal studies were conducted under protocols approved by the St. Jude Children’s Research Hospital committee on the Use and Care of Animals. SARS-CoV-2 infections were performed at the University of Tennessee Health Science Center under ABSL3 conditions in accordance with the approval of the Institutional Animal Care and Use Committee of University of Tennessee Health Science Center (Protocol #20–0132).
Bone marrow-derived macrophages (BMDMs)
Primary BMDMs were cultured for 6 days in IMDM (Thermo Fisher Scientific, 12440–053) supplemented with 10% FBS (Biowest, S1620), 30% L929-conditioned medium, 1% non-essential amino acids (Thermo Fisher Scientific, 11140–050), and 1% penicillin and streptomycin (Thermo Fisher Scientific, 15070–063). Then BMDMs were seeded into 12-well plates at a density of 1 million cells per well and incubated overnight before use.
Murine hepatitis virus (MHV) propagation
The murine hepatitis virus (A59 strain) was amplified in 17Cl-1 cells as previously described51. Briefly, 17Cl-1 cells were inoculated with MHV-A59 at a multiplicity of infection of 0.1. At 48 h post-infection, the whole flask with media and cells was frozen at –80°C and then thawed at 37°C. After repeating the freezing-thawing cycle twice, the supernatant was collected and centrifuged at 2000 × g for 10 min to remove the cell debris. Then the virus was purified by ultracentrifugation at 30,000 rpm for 1h, after which the pellets were resuspended with fresh media. Virus titer was measured by plaque assay in 17Cl-1 cells.
Isolation of peripheral blood mononuclear cells (PBMCs)
Whole blood was obtained from anonymous healthy donors who provided informed consent at St. Jude Children’s Research Hospital following IRB-approved protocols. PBMCs were harvested from the blood by density gradient using Percoll (GE Healthcare, 17–5445-01). PBMCs were maintained in RPMI 1640 supplemented with 10% FBS.
SARS-CoV-2 culture
The SARS-CoV-2 isolate USA-WA1/2020 was obtained from BEI Resources (NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281) and propagated in Vero-E6 cells (ATCC, VERO C1008) at an MOI of 0.1 in Minimal Essential Medium (MEM; Corning, 17–305-CV) supplemented with 5% heat-inactivated FBS (Gibco), 1% L-glutamine (Corning, 25–005-Cl) and 5 mM penicillin/streptomycin (Gibco, 30–001-Cl). Viral titer was measured by plaque assay as described previously for alphaviruses52. All experiments with live SARS-CoV-2 were conducted in a biosafety level 3 laboratory.
Cell stimulation/infection
For chemical ligand-induced inflammatory signaling responses, 1 × 106 BMDMs seeded in 12-well plates were pretreated with oxPAPC (Invivogen, tlrl-oxp1) at the final concentration of 30 μg/ml or C29 (MedChem Express, HY-100461) at the final concentration of 50 μM for 1 h. Then the cells were stimulated with 1 μg/ml Pam3CSK4 (Pam3) or 1 μg/ml R848 for the indicated time. To check the NLRP3 inflammasome activation after viral protein priming, 1 × 106 BMDMs seeded in 12-well plates were stimulated with the envelope (E) (Abclonal, RP01263) or spike (S) (Abclonal, RP01283LQ) protein of SARS-CoV-2 at the final concentration of 1 μg/ml for 4 h and then treated for 45 min with 5 mM ATP (Roche, 10127531001).
For MHV infection, 1 × 106 BMDMs seeded in 12-well plates were infected at a MOI of 0.1 in DMEM plain media (Sigma, D6171). After 2 h of incubation, cells were supplemented with 10% FBS. Samples were collected as the indicated timepoints. For heat-inactivated MHV stimulation, MHV was inactivated at 65°C for 30 min, and then inactivated MHV at a volume equivalent to MOI of 0.1 of live MHV was added into BMDMs for the indicated time.
For SARS-CoV-2 infection of BMDMs, 1 × 106 BMDMs were seeded into 12-well plates and infected at a MOI of 0.5 for the indicated time. Cells were lysed with RIPA lysis buffer. For PBMC infection, 1 × 106 PBMCs from 3 healthy donors were seeded into 24-well plates and infected at a MOI of 0.5. Supernatants from mock-treated and infected PBMCs were collected after infection for 20 h. To inhibit TLR2 activity, oxPAPC was used at a final concentration of 30 μg/ml together with SARS-CoV-2. To inhibit TLR4 activity, CLI-095 was used at a final concentration of 1 μg/ml together with SARS-CoV-2. The heat-inactivated SARS-CoV-2 was obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, Heat Inactivated, NR-52286.
Cell fusion analysis
BMDMs (106 cells/well) seeded into 12-well plates were treated with 10 μM chloroquine (CQ) for 30 min. Then the cells were infected with MHV at a MOI of 0.1. At 8 h post-infection, DNA stain, the NUCLEAR-ID Red (Enzo, ENZ-52406), were added to the cells after diluting 5000-fold. Images were analyzed using IncuCyte S3 software.
Co-immunoprecipitation assay
Plasmids expressing Flag-tagged human TLR2, TLR3 or mouse TLR2 were transfected into 293T (ATCC, CRL-3216) cells for 48 h, then cells were lysed in NP-40 lysis buffer (0.1% NP-40, 150 mM NaCl, 50 mM HEPES), and 20 min later cell lysates were centrifuged at 13000 rpm for 10 min. Supernatant was collected and incubated with 5 μg of purified E protein of SARS-CoV-2 and 1.5 μg of the indicated primary antibody on a rocking platform at 4°C. After overnight incubation, protein A/G PLUS-Agarose (Santa Cruz Biotechnology) beads were added. Two hours later, the beads were collected by centrifugation after washing with lysis buffer four times. Finally, samples were harvested after boiling in 2× SDS loading buffer at 100°C for 5 min.
SARS-CoV-2 infection in mice
8- to 10-week-old K18-ACE-2 transgenic male mice were administered intraperitoneally 200 μl of DPBS alone or DPBS containing oxPAPC at a dose of 2 mg/kg body weight. At 1 h post-administration, mice were anesthetized with 5% isoflurane and then inoculated intranasally with SARS-CoV-2 in 50 μl DPBS containing around 2 × 104 PFU. At 3 days post-infection, the mice were administered another dose of DPBS or DPBS plus oxPAPC. Mice were weighed daily and monitored over a period of 14 days for survival. For BALF collection, mice were sacrificed at day 2 post-infection.
Intratracheal instillation and histopathology
Age- and sex-matched 6- to 9-week old male and female mice were used, and each mouse was administrated with 25 μg of the SARS-CoV-2 envelope (abclonal, RP01263) or spike (abclonal, RP01283LQ) protein (LPS contamination is less than 0.1 EU/μg of protein, tested by LAL method) via intratracheal instillation. PBS and Pam3CSK4 were used as negative and positive controls, respectively. At 24 h post-administration, serum was collected, and lungs were fixed in 10% formalin, and then embedded in paraffin by standard procedures. For immunohistochemistry, formalin-fixed paraffin-embedded lungs were cut into 4 μM sections. CD45 (BD Pharmingen™, 553076, 1:500) and TUNEL (Promega, PRG7130) staining was performed according to the manufacturer’s instructions and examined by a pathologist blinded to the experimental groups. BALF collection was performed at 6 h post-instillation.
Gene expression analysis from patients with COVID-19
Nanostring nCounter data was kindly shared by Hadjadj et al. for healthy patients and patients with moderate, severe, and critical COVID-1916. Expression of MYD88, TRIF, TLR1, TLR2, TLR3, TLR4, TLR5, TLR7, TLR8 and TLR9 were generated based on average expression of selected genes using Morpheus.
Immunoblot analysis
For caspase-1 analysis, BMDMs were lysed together with the supernatant using 50 μl caspase lysis buffer (1× protease inhibitors, 1× phosphatase inhibitors, 10% NP-40, and 25 mM DTT). Then 125 μl 4× SDS loading buffer was added for boiling the samples. For analysis of all other proteins, the supernatants of stimulated cells were discarded, and then cells were washed once with PBS, after which RIPA lysis buffer was added to lyse the cells.
Electrophoresis was conducted to separate proteins in 10%−12% polyacrylamide gels. Then proteins were transferred onto PVDF membranes and blocked with 5% skim milk for 1 h at room temperature. All the primary antibodies were incubated overnight at 4°C, while secondary antibodies with HRP were incubated for 1 h at room temperature. Images were developed via a GE Amersham Imager 600. And final images were analyzed via Fiji for MacOS X (version 2.0.0-rc-67/1.52c).
The antibodies used were: anti-caspase-1 (AdipoGen, AG-20B-0042, 1:2000), anti-GSDMD (abcam, ab209845, 1:1000), anti-pERK (Cell Signaling Technology [CST], #9101, 1:1000), anti-pIκB (CST, #2859, 1:1000), anti-ERK (CST, #9102, 1:1000), anti-IκB (CST, #9242, 1:1000), anti-β-actin (Proteintech, 66009–1-IG, 1:5000), anti-GAPDH (CST, #5174, 1:1000) and HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, anti-rabbit [111–035-047], 1:5000; anti-mouse [315–035-047], 1:5000).
Real-time (RT) PCR analysis
RNA extraction was conducted with TRIzol (Thermo Fisher Scientific, 15596026). cDNA synthesis was done by using the First Strand cDNA Synthesis Kit (Applied Biosystems, 4368814) according to the manufacturer’s instructions. Real-time PCR was performed with 2× SYBR Green (Applied Biosystems, 4368706) on an ABI 7500 fast RT PCR machine. Primers used were as follows: mouse—Gapdh: 5′-CGT CCC GTA GAC AAA ATG GT-3′, 5′-TTG ATG GCA ACA ATC TCC AC-3′; Il1b: 5′-GAT CCA CAC TCT CCA GCT GCA-3′, 5′-CAA CCA ACA AGT GAT ATT CTC CAT G-3′; Il6: 5’-GAC AAA GCC AGA GTC CTT CAG AGA G-3′, 5′-CTA GGT TTG CCG AGT AGA TCT C-3′; Tnf: 5’-CAT CTT CTC AAA ATT CGA GTG ACA A-3′, 5′-TGG GAG TAG ACA AGG TAC AAC CC-3′; Nlrp3: 5’-TCA GAT TGC TGT GTG TGG GAC TGA-3′, 5′- AGC TCA GAA CCA ATG CGA GAT CCT-3′; Ifna: 5’-GCT AGG CHY TRT GCT TTC CT-3′, 5′-CAC AGR GGC TGT GTT TCT TC-3′; Ifnb: 5’-GCC TTT GCC ATC CAA GAG ATG C-3′, 5′-ACA CTG TCT GCT GGT GGA GTT C-3′.
Human—GAPDH: 5′-CGG AGT CAA CGG ATT TGG TCG TAT-3′, 5′-AGC CTT CTC CAT GGT GGT GAA GAC-3′; IL6: 5’-GCC TTC GGT CCA GTT GCC TT-3′, 5′-GCA GAA TGA GAT GAG TTG TC-3′; TNF: 5’-ATG ACT TCC AAG CTG GCC GT-3′, 5′-TCC TTG GCA AAA CTG CAC CT-3′; IL1B: 5’-CCA CAG ACC TTC CAG GAG AAT G-3′, 5′-GTG CAG TTC AGT GAT CGT ACA GG-3′; IFNG: 5’- GAG TGT GGA GAC CAT CAA GGA AG-3′, 5′- TGC TTT GCG TTG GAC ATT CAA GTC-3′.
Cytokine analysis
For in vivo analyses and studies using BMDMs, cytokines were measured by multiplex ELISA (Millipore, MCYTOMAG-70K) or IL-18 ELISA (Invitrogen, BMS618–3) according to the manufacturer’s instructions. For studies using PBMCs, the release of pro-inflammatory cytokines in the supernatant was measured by multiplex ELISA (Millipore, HCYTMAG-60K-PX29) according to the manufacturer’s instructions. For multiplex ELISAs, all the antibodies were prepared by the manufacturer.
Statistical analysis
GraphPad Prism 7.0 software was used for data analysis. Data are represented as mean ± SEM. Statistical significance was determined by unpaired two-tailed Student’s t test for two groups comparisons, one-way ANOVA with Dunnett’s multiple comparisons test for comparisons of more than two groups, two-way ANOVA for comparisons of more than two groups with two or more time points, or log-rank test for survival experiments. P values less than 0.05 were considered statistically significant where *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
DATA AVAILABILITY STATEMENT
The datasets generated and analyzed during the current study are contained within the manuscript and accompanying supplemental figures. Nanostring nCounter data was kindly shared by Hadjadj et al. for healthy patients and patients with moderate, severe, and critical COVID-19.
Extended Data
Extended Data Fig. 1. Myd88 is required for MHV-induced inflammatory responses.

(A and B) Real-time PCR analysis of the expression of Il1b (A) and Il6 (B) in WT, Myd88–/–, and Trif–/– bone marrowderived macrophages (BMDMs) after infection with MHV at a MOI of 0.1 for the indicated time, presented relative to levels of the host gene Gapdh. Data are representative of three independent experiments. Data are shown as mean ± SEM (n = 3) (A and B).
Extended Data Fig. 2. MDA5 is required for MHV-induced type I interferon expression.
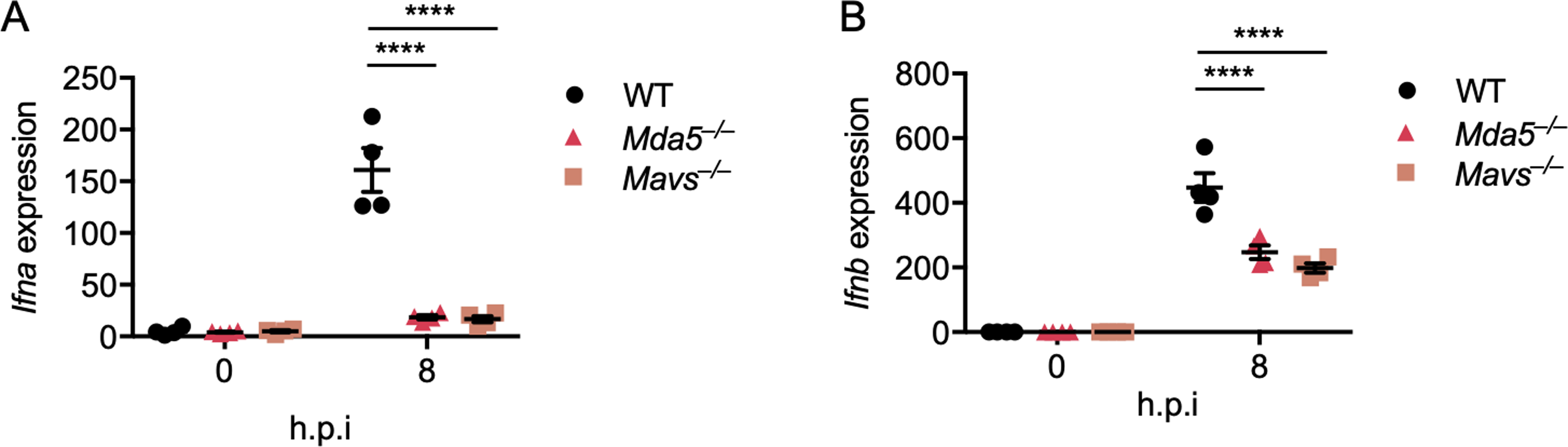
(A and B) Real-time PCR analysis of the expression of Ifna (A) and Ifnb (B) in WT, Mda5–/–, and Mavs–/– bone marrowderived macrophages (BMDMs) after infection with MHV at a MOI of 0.1 for the indicated time, presented relative to levels of the host gene Gapdh. Significant differences compared to the WT group infected with MHV are denoted as ****P < 0.0001 (two-way ANOVA) (A and B). Data are representative of three independent experiments. Data are shown as mean ± SEM (n = 3) (A–B).
Extended Data Fig. 3. TLR2 is essential to Inflammatory cytokine expression during MHV infection.

(A–H) Release of IL-6 (A), TNF-α (B), CXCL10 (C), CCL3 (D), CXCL1 (E), RANTES (F), MCP-1 (G), and G-CSF (H) from bone marrow-derived macrophages (BMDMs) infected with MHV at a MOI of 0.1 for the indicated time. Data are representative of three independent experiments. Data are shown as mean ± SEM (n = 3) (A–H).
Extended Data Fig. 4. oxPAPC is a potent TLR2 inhibitor.

(A) Immunoblot analysis of phospho-ERK (pERK), total ERK (tERK), pIκB, and tIκB in WT and Tlr2–/– bone marrow-derived macrophages (BMDMs) after stimulation with 1 μg/ml of Pam3CSK4 (Pam3) for the indicated time with or without the TLR2 inhibitor oxPAPC or C29. (B) Immunoblot analysis of pERK, tERK, pIκB, and tIκB in WT and Tlr7–/– BMDMs after stimulation with 1 μg/ml of R848 for the indicated time with or without the TLR2 inhibitor oxPAPC or C29. Data are representative of two independent experiments.
Extended Data Fig. 5. Chloroquine can inhibit MHV-induced cell fusion.

(A) Cell fusion of wild type (WT) bone marrow-derived macrophages (BMDMs) after infection with MHV at a MOI of 0.1 for 8 h in the absence or presence of 10 μM chloroquine (CQ). The upper panel was obtained via IncuCyte after staining with NUCLEAR-ID Red, and the lower panel is the corresponding phase channel. The yellow outline indicates the fused cells. Scale bar, 30 μm. Data are representative of three independent experiments.
Extended Data Fig. 6. TLR2 can sense the envelope protein but not spike protein of SARS-CoV-2.

(A) Immunoblot analysis of phospho-ERK (pERK), total ERK (tERK), pIκB, and tIκB in WT, Tlr2–/–, and Myd88–/– BMDMs after stimulation with 1 μg/ml of the spike (S) protein from SARS-CoV-2 for the indicated time. Actin was used as the internal control. (B and C) Real-time PCR analysis of the expression of Il6 (B) and Tnf (C) in WT BMDMs after stimulating with the envelope (E) protein of SARS-CoV-2 or Pam3CSK4 (Pam3) for 2 h, presented relative to levels of the host gene Gapdh. (D–G) Real-time PCR analysis of the expression of Il1b (D), Il6 (E), Tnf (F), and Nlrp3 (G) in WT, Tlr2–/–, and Myd88–/– BMDMs after stimulation with 1 μg/ml of the E or S protein from SARSCoV-2 for 4 h, presented relative to levels of the host gene Gapdh. (H) Immunoblot analysis of pro- (p45) and cleaved caspase-1 (p20; CASP1) and pro (p55) and cleaved gasdermin D (p30; GSDMD) in BMDMs primed with 1 μg/ml of the E or S protein from SARS-CoV-2 for 4 h and then stimulated with ATP for 45 min. Actin was used as the internal control. (I) Release of IL-18 from BMDMs after the treatment in (H). Significant differences compared to the WT group stimulated with E protein are denoted as ****P < 0.0001 (one-way ANOVA) (D–G, I). Data are representative of two (B and C) or three (A, D–I) independent experiments. Data are shown as mean ± SEM (n = 3) (B–G and I).
Extended Data Fig. 7. The spike protein of SARS-CoV-2 cannot induce inflammation and damage in mouse lungs.

(A) CD45 immuno-staining and TUNEL staining of lung samples obtained from mice 24 h after being intratracheally instilled with the spike (S) protein from SARS-CoV-2. Data are representative of two independent experiments. Scale bar, 100 μm (black) or 25 μm (red).
Supplementary Material
ACKNOWLEDGEMENTS
We thank all the members of the Kanneganti laboratory for their comments and suggestions during the development of this manuscript. We thank A. Burton (St. Jude Children’s Research Hospital) for her technical support. We also thank R. Tweedell, PhD, for scientific editing and writing support and Surekha Surendrathanan for technical assistance. Work from our laboratory is supported by the US National Institutes of Health (AI101935, AI124346, AR056296, and CA253095 to T.-D.K.) and the American Lebanese Syrian Associated Charities (to T.-D.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The following reagents were deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281, and Heat Inactivated, NR-52286.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing interest.
REFERENCES
- 1.MacLachlan NJ & Dubovi EJ Chapter 24 Coronaviridae. in Fenner’s Veterinary Virology 435–461 (Academic Press, 2017). doi: 10.1016/B978-0-12-800946-8.00024-6. [DOI] [Google Scholar]
- 2.Hu B, Guo H, Zhou P. & Shi Z-L Characteristics of SARS-CoV-2 and COVID-19. Nature Reviews Microbiology 1–14 (2020) doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Channappanavar R. & Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol 39, 529–539 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lucas C. et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng M. et al. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem (2020) doi: 10.1074/jbc.RA120.015036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karki R. et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell (2020) doi: 10.1016/j.cell.2020.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jose RJ & Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. The Lancet Respiratory Medicine 8, e46–e47 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Del Valle DM et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nature Medicine 26, 1636–1643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amarante-Mendes GP et al. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Briard B, Place DE & Kanneganti T-D DNA Sensing in the Innate Immune Response. Physiology (Bethesda) 35, 112–124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zalinger ZB, Elliott R, Rose KM & Weiss SR MDA5 Is Critical to Host Defense during Infection with Murine Coronavirus. Journal of Virology 89, 12330–12340 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cervantes-Barragan L. et al. Control of coronavirus infection through plasmacytoid dendritic-cell–derived type I interferon. Blood 109, 1131–1137 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Channappanavar R. et al. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Invest 130, 3625–3639 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheahan T. et al. MyD88 Is Required for Protection from Lethal Infection with a Mouse-Adapted SARS-CoV. PLOS Pathogens 4, e1000240 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou H, Zhao J. & Perlman S. Autocrine Interferon Priming in Macrophages but Not Dendritic Cells Results in Enhanced Cytokine and Chemokine Production after Coronavirus Infection. mBio 1, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hadjadj J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Zayat SR, Sibaii H. & Mannaa FA Toll-like receptors activation, signaling, and targeting: an overview. Bull Natl Res Cent 43, 187 (2019). [Google Scholar]
- 18.Ghosh S. et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell (2020) doi: 10.1016/j.cell.2020.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carter AB, Monick MM & Hunninghake GW Both Erk and p38 Kinases Are Necessary for Cytokine Gene Transcription. Am J Respir Cell Mol Biol 20, 751–758 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Liu T, Zhang L, Joo D. & Sun S-C NF-κB signaling in inflammation. Signal Transduction and Targeted Therapy 2, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang D, Geng T, Harrison AG & Wang P. Differential roles of RIG-I-like receptors in SARS-CoV-2 infection. bioRxiv 2021.02.10.430677 (2021) doi: 10.1101/2021.02.10.430677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SMY et al. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc Natl Acad Sci U S A 111, 3793–3798 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lester SN & Li K. Toll-Like Receptors in Antiviral Innate Immunity. J Mol Biol 426, 1246–1264 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasan U. et al. Human TLR10 Is a Functional Receptor, Expressed by B Cells and Plasmacytoid Dendritic Cells, Which Activates Gene Transcription through MyD88. The Journal of Immunology 174, 2942–2950 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Barrat FJ TLR8: No gain, no pain. J Exp Med 215, 2964–2966 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oliveira-Nascimento L, Massari P. & Wetzler LM The Role of TLR2 in Infection and Immunity. Front Immunol 3, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kampf G, Voss A. & Scheithauer S. Inactivation of coronaviruses by heat. J Hosp Infect (2020) doi: 10.1016/j.jhin.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burkard C. et al. Coronavirus Cell Entry Occurs through the Endo-/Lysosomal Pathway in a Proteolysis-Dependent Manner. PLOS Pathogens 10, e1004502 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoffmann M. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181, 271–280.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oladunni FS et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nature Communications 11, 6122 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siu YL et al. The M, E, and N Structural Proteins of the Severe Acute Respiratory Syndrome Coronavirus Are Required for Efficient Assembly, Trafficking, and Release of Virus-Like Particles. Journal of Virology 82, 11318–11330 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeDiego ML et al. Inhibition of NF-κB-Mediated Inflammation in Severe Acute Respiratory Syndrome Coronavirus-Infected Mice Increases Survival. Journal of Virology 88, 913–924 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao W, Ma L, Cai C. & Gong X. Caffeine Inhibits NLRP3 Inflammasome Activation by Suppressing MAPK/NF-κB and A2aR Signaling in LPS-Induced THP-1 Macrophages. Int J Biol Sci 15, 1571–1581 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cabanski M. et al. Genome-wide transcriptional profiling of mononuclear phagocytes recruited to mouse lungs in response to alveolar challenge with the TLR2 agonist Pam3CSK4. Am J Physiol Lung Cell Mol Physiol 297, L608–618 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Guan Y. et al. Human TLRs 10 and 1 Share Common Mechanisms of Innate Immune Sensing but Not Signaling. J.I 184, 5094–5103 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Nieto-Torres JL et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 485, 330–339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lythgoe MP & Middleton P. Ongoing Clinical Trials for the Management of the COVID-19 Pandemic. Trends in Pharmacological Sciences 41, 363–382 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loiarro M, Ruggiero V. & Sette C. Targeting TLR/IL-1R Signalling in Human Diseases. Mediators of Inflammation vol. 2010 e674363 https://www.hindawi.com/journals/mi/2010/674363/ (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Slaats J, ten Oever J, Veerdonk, van de FL & Netea MG IL-1β/IL-6/CRP and IL-18/ferritin: Distinct Inflammatory Programs in Infections. PLOS Pathogens 12, e1005973 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanneganti TD et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem 281, 36560–8 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto M. et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–3 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Takeuchi O. et al. Differential Roles of TLR2 and TLR4 in Recognition of Gram-Negative and Gram-Positive Bacterial Cell Wall Components. Immunity 11, 443–451 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Hoshino K. et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162, 3749–3752 (1999). [PubMed] [Google Scholar]
- 45.Hemmi H. et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88–dependent signaling pathway. Nature Immunology 3, 196–200 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Hemmi H. et al. A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Adachi O. et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9, 143–150 (1998). [DOI] [PubMed] [Google Scholar]
- 48.Gitlin L. et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A 103, 8459–8464 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suthar MS et al. IPS-1 Is Essential for the Control of West Nile Virus Infection and Immunity. PLOS Pathogens 6, e1000757 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar H. et al. Essential role of IPS-1 in innate immune responses against RNA viruses. Journal of Experimental Medicine 203, 1795–1803 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schickli JH, Zelus BD, Wentworth DE, Sawicki SG & Holmes KV The murine coronavirus mouse hepatitis virus strain A59 from persistently infected murine cells exhibits an extended host range. J. Virol 71, 9499–9507 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee J. et al. Emergence and Magnitude of ML336 Resistance in Venezuelan Equine Encephalitis Virus Depend on the Microenvironment. Journal of Virology 94, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are contained within the manuscript and accompanying supplemental figures. Nanostring nCounter data was kindly shared by Hadjadj et al. for healthy patients and patients with moderate, severe, and critical COVID-19.