

Abstract
Objective:
Rett Syndrome, CDKL5-Deficiency Disorder, FOXG1 Disorder, and MECP2 Duplication Disorder are Developmental Encephalopathies with shared and distinct features. Though historically linked, no direct comparison has been performed. The first head-to-head comparison of clinical features in these conditions is presented.
Methods:
Comprehensive clinical information was collected from 793 individuals enrolled in the Rett Syndrome and Related Disorders Natural History Study. Clinical features including clinical severity, regression, and seizures were cross-sectionally compared between diagnoses to test the hypothesis that these are 4 distinct disorders.
Results:
Distinct patterns of clinical severity, seizure onset age, and regression were present. Individuals with CDKL5-Deficency Disorder were the most severely affected and had the youngest age of seizure onset (2 months) whereas children with MECP2-duplication syndrome had the oldest median age of seizure onset (64 months) and lowest severity scores. Rett syndrome and FOGX1 were intermediate in both features. Smaller head circumference correlates with increased severity in all disorders and earlier age of seizure onset in MECP2-duplication syndrome. Developmental regression occurred in all Rett syndrome participants (median 18 months) but only 23–34% of the other disorders. Seizure incidence prior to the baseline visit was highest for CDKL5-Deficency Disorder (96.2%) and lowest for Rett syndrome (47.5%). Other clinical features including seizure types and frequency differed amongst groups.
Interpretation:
While these Developmental Encephalopathies share many clinical features, clear differences in severity, regression, and seizures warrant considering them as unique disorders. These results will aid in the development of disease specific severity scales, precise therapeutics, and future clinical trials.
Introduction
Developmental Encephalopathies (DE) is an emerging term for a group of severe, genetically-based disorders with onset of progressive postnatal neurological symptoms including epilepsy, developmental impairments, autonomic dysfunction, mood disorders, motor impairments, and potential systemic symptoms.1 A number of named disorders fall within this rubric. Rett syndrome (RTT, MIM 312750), has a number of features, such as relatively normal initial motor-cognitive development, regression, loss of hand function, poor or no spoken language, gait abnormalities, intellectual impairment, microcephaly, and stereotypic hand movements that can be seen in many of these disorders.2 As RTT was the earliest described and is more prevalent than most of these rare conditions, terms such as “RTT-like” emerged in the literature to describe genetic disorders with overlapping features, such as postnatal onset microcephaly and abnormalities in spoken language and hand use. Recently, the DE nomenclature is becoming more prevalent in the literature as a more encompassing term, but imprecise terms such as “RTT-like” remain. For example, a search of Pubmed for “developmental encephalopathy” yields 17 results starting in 2015 and “Rett-like” yields 72 since 2006. The persistence of this imprecise nomenclature is likely due to the lack of direct comparisons to investigate the commonalities and unique features of these disorders. In this study, we compare the features, epilepsy, head size/growth, and severity, in four disorders representative of the DEs.
RTT affects ~1 in 10,000 live female births and classic RTT is caused by mutations in the X-linked Methyl-CpG binding protein 2 (MECP2) in over 95% of cases.3,4 Variants of RTT, including the early seizure variant and congenital variant, were subsequently described when groups of individuals presented with some, but not all, clinical features of typical RTT. Many of these individuals were found to have mutations in different genetic loci, CDKL5 (MIM 300203) or FOXG1 (MIM 164871, 613454) respectively.5 Furthermore, although loss of function mutations in MECP2 are associated with RTT, duplication of the MECP2 locus causes a distinct but related neurodevelopmental disorder, MECP2 Duplication Disorder (MDD, MIM 300260) that primarily affects males.6
Common features amongst people with mutations in CDKL5, FOXG1, MECP2 Duplication, and RTT led to these disorders being termed variant forms of RTT or RTT-like. However, further characterization has led to the recognition that while similarities exist, significant developmental, clinical, and genetic differences warrant their consideration as distinct disorders. For example, MDD displays more severe autistic features, later severe/refractory seizures, and recurrent respiratory infections.6–8 CDKL5 deficiency disorder (CDD) is characterized by early-onset seizures, global developmental delay, cortical visual impairment, and severely impaired gross motor function.9,10 FOXG1 disorder, previously known as the congenital RTT variant, is characterized by postnatal microcephaly typically associated with corpus callosum abnormalities, and marked dyskinetic movements.11,12 Thus while shared features exist between these disorders, the unique features indicate each should be considered as distinct entities: a critically important concept as the therapeutic approach to each is likely to be equally distinct.
No direct comparison between all of these disorders has been performed. Since 2014, a National Institutes of Health sponsored Natural History Study has been following individuals with these disorders. Here, we present a direct comparison of these four related disorders using cross-sectional data collected at the baseline study visit on overall clinical severity, regression, and seizures. This information is critical to the development of future clinical trials and could potentially help with earlier diagnosis, assessment, and treatment of these disorders.
Methods
Data Sources:
Participants were recruited at one of 15 clinical sites that are part of the multicenter NIH funded Natural History Study of Rett Syndrome and Related Disorders (clinicaltrials.gov NCT02738281). The protocol for the study received prior approval by the appropriate Institutional Review Boards of Baylor College of Medicine, Children’s Hospital of Philadelphia, Cleveland Clinic, Gillette Children’s Hospital, and Vanderbilt University. The remaining sites (Boston Children’s Hospital, Cincinnati Children’s Hospital, Greenwood Genetic Center, Oakland children’s Hospital, Rush Medical Center, UCSD, University of Colorado-Denver, and Washington University) relied on the single-IRB agreement provided by the University of Alabama at Birmingham. Informed consent was obtained for each subject. Assent to participate was waived as these individuals were incapable of providing such. All data analyzed were extracted from the baseline visit for each participant enrolled in the Natural History Study between December 2015 and July 2018. Participants were evaluated by a neurologist or geneticist to confirm the diagnosis of RTT or other phenotypes; prior genetic testing for alterations in MECP2 (mutations or duplications), CDKL5, and FOXG1 was reviewed. Based on the prior classification system2, participants were then classified as having typical/classic RTT (T-RTT), atypical RTT (A-RTT), MECP2 mutations without Rett syndrome (N-RTT), MDD, CDD, FOXG1 disorder, FOXG1 duplication, or other (mutations in other genes).2 For this study, data extraction included age of enrollment, age of regression, age of seizure onset, frequency of seizures, and seizure characteristics/semiology. Demographics, associated features, co-morbidities, and somatic measurements were also extracted. Disease severity at the baseline visit was quantified using the RTT Clinical Severity Scale3 (supplementary table 4) and qualitatively assessed using the RTT Clinical Global Impression-Severity Scale.13 These two scales were performed on all of the participants in the natural history study, allowing a direct comparison between groups. We choose these collective measures to give an overall assessment of the similarities and differences in these syndromes.
Data Definitions and Categorization:
Data were extracted from the Rett Consortium database, hosted by the Rare Diseases Clinical Research Network (RDCRN) through the Data Management and Coordinating Center at the University of South Florida. Clinical severity scale, age of regression, and age at seizure onset were used as primary comparisons across disorders. Other measures examined were seizure frequency, seizure semiology, head circumference, age of enrollment, and various demographic data. These measures were chosen as they are core features for all four conditions.
Participants were analyzed in groups by clinical diagnosis. Participants with FOXG1 duplication (n=3), atypical congenital variant (n=9), atypical delayed onset variant (n=2), atypical early seizure variant (n=1), and atypical preserved speech variant (n=10), mutations in other genes (n= 3), or no available diagnosis (n=2) were excluded from our analysis due to small sample sizes. Based on their genotypes, five subjects in our sample were listed in a diagnostic category different then their genetic results (confirmed by testing report). These participants’ diagnoses were relabeled before proceeding with data analysis. These subjects were: 1 participant with CDKL5 mutation (c.470 T> C) listed as classic Rett syndrome; 2 participants with FOXG1 c.460dupG listed as FOXG1 duplications instead of FOXG1 Disorder; 1 participant with a CDKL5 mutation listed as atypical early seizure variant; and 1 participant with a FOXG1 mutation listed as atypical preserved speech variant. The physicians that made these initial diagnoses likely did so by clinical criteria and not genetic diagnosis.
The clinical severity scale (CSS) used was previously published by Schanen et al.14 and has been applied in both clinical studies3,15 and a clinical trial.16,17 Details of the clinical severity scale can be found in Schanen et al. and the scale is presented in supplementary table 4.14 All the investigators at each site were trained on the CSS at site initiation and most investigators had previous training and experience with this scale as part of previous industry sponsored trials.16,17
Age of regression was defined based on parent-reported age at regression. Regression was defined in the NHS as the age which parents reported a loss of a skill (in years and months; n= 590) or date of regression (dd/mm/yyyy; n= 13) after questioning by a clinician. We excluded one participant with typical RTT, one participant with A-RTT, and one participant with N-RTT MECP2 mutations from the analysis of age of regression and age of seizure onset as no date of birth was recorded. Two additional A-RTT participants were noted to not have documented regression, which is needed for the diagnosis of A-RTT. On review of physician interviews both patients had evidence of regression, however they were excluded from the age of regression analysis as this was not clearly documented.
Age of seizure onset was identified through either the physician-entered age (in years and months) in the development log or by calculating the age from the first reported seizure start date (dd/mm/yyyy) in the seizure log. In the seizure log, “1” was entered if there was no recorded start day (n= 59 RTT, 3 A-RTT, 4 N-RTT, 7 MDD, 0 CDD, 2 FOXG1) or start month and day (n=78 RTT, 5 A-RTT, 4 N-RTT, 7 MDD, 0 CDD, 1 FOXG1). We chose to use physician-entered data from the development log if there was a discrepancy between the two ages (n=133) as this was the clinician actively asking the family to remember when the first seizure occurred. Early-onset seizures were defined as a seizure onset age less than or equal to five months. Seizure severity was defined in an ordinal fashion based on seizure frequency using the bounds defined in the natural history database including: absent, less than monthly, less than weekly to monthly, weekly, more than weekly, and daily. Seizure semiology was classified based on investigator impression from a list of seizure types which included: absence, atonic, complex partial, generalized, infantile spasm, myoclonic, tonic-clonic, partial, non-epileptic Rett spell, and not a seizure. Clinicians used their judgement to map parent reported description of seizures to one of these pre-defined seizure types.
In order to prevent floor effects of using percentiles, head circumference (HC) measurements obtained in the study were converted to Z scores using the LMS method18 according to:
where A is the HC (in cm), and L, M, and S are data extracted from the normal HC curves generated from the study by Rollins et al.19 Using this method, the raw HC measurement and percentile were converted to Z-scores. The accuracy of this method was tested on a handful of subjects by inputting the age and HC measurement into an on-line Z-score calculator (https://simulconsult.com/resources/measurement.html; data not shown). We then compared HC Z-scores to the clinical variables described above.
Statistical Analysis:
Classical descriptive statistics were generated using median and interquartile range for continuous variables and frequency and percentage for categorical variables. To measure the differences between groups, the chi square or Fischer’s exact tests were used for categorical variables. The Kruskal-Wallis test was used for continuous variables; for post-hoc pairwise comparisons a Bonferroni correction was used with an adjusted p-value for significance of 0.001667 (two tailed alpha=0.05, therefore the corrected p-value= 0.025/number of comparisons; 15 comparisons total).
Kaplan-Meier survival analysis was used to estimate the time to epilepsy onset and the lifetime prevalence of epilepsy, as well as the time to regression onset and lifetime prevalence of regression. Event time was defined as the age at onset of seizures or regression. If the event had not occurred at the age of the baseline study visit, the data was censored for that participant. The log-rank test was used to compare the survival curves.
Linear regression and Spearman’s rank correlation were used to describe the relationship between HC z-score and age of seizure onset, HC z-score and clinical severity, clinical severity (minus the epilepsy sub-score) and seizure onset age, and seizure onset age and regression age. All statistical analyses were performed using STATA.
Results
The Rett and Rett-Related National History Study enrolled 793 participants as of July 1, 2018. Our final analysis included 96.2% (763/793) of these total participants (Table 1). This included 535 participants with T-RTT (529 female, 3 male, 3 gender not available), 39 participants with A-RTT (38 female, 1 male), 44 participants with N-RTT (27 female, 16 male, 1 gender not available), 53 participants with CDD (43 female, 10 male), 43 participants with FOXG1 disorder (22 female, 20 male, 1 gender not available), and 49 participants with MDD (5 female, 44 male).
Table 1:
Participant Demographics
| Total Number of Participants | Gender (n) | Age at Baseline Visit (Years) | |||||
|---|---|---|---|---|---|---|---|
| Female | Male | Not Reported | Median | IQR | P-value | ||
| T-RTT | 535 | 529 | 3 | 3 | 13.5 | 7.2 – 20.9 | -- |
| A-RTT | 39 | 38 | 1 | 0 | 9.8 | 4.3 – 21.5 | 0.037028 |
| N-RTT | 44 | 27 | 16 | 1 | 6 | 2.8–10.2 | <0.000001 |
| CDD | 53 | 43 | 10 | 0 | 5.7 | 3.1 – 9.9 | <0.000001 |
| FOXG1 | 43 | 22 | 20 | 1 | 5.1 | 2.4 – 8.8 | <0.000001 |
| MDD | 49 | 5 | 44 | 0 | 7.4 | 4.1 – 12.1 | 0.000002 |
At the baseline study visit, the age of participants with T-RTT (median 13.5 years, IQR 7.2– 20.9 years) was significantly greater than the age of participants with N-RTT (median 6, IQR 2.8–10.1, p<1×10−6), CDD (median 5.7 years, IQR 3.1–9.9 years, p<1×10−6), FOXG1 disorder (median 5.1 years, IQR 2.4–8.8 years, p<1×10−6), and MDD (median 7.4 years, IQR 4.1– 12.1 years, p=0.000002). The age of participants with T-RTT did not differ significantly from the participants with A-RTT (median 9.8, IQR 4.3–23.5, p=0.37028).
Clinical Severity:
Scores on the Clinical Severity Scale (Figure 1A) varied by diagnosis and were lowest (less affected) in those with N-RTT (median score 12; IQR 7–23), followed by A-RTT (15; IQR 10–22) and MDD (16; IQR 11–21.5), then T-RTT (24; IQR 19–31), FOXG1 (28; IQR 22–31), and finally CDD (29; IQR 25–33). The scores for participants with T-RTT were significantly higher (more affected) than those with A-RTT (p<1×10−6), N-RTT (p<1×10−6), and MDD (p<1×10−6). Scores for participants with T-RTT, CDD, and FOXG1 did not vary significantly. Scores for participants with A-RTT, N-RTT, and MDD did not vary significantly. Overall, A-RTT, MDD, and N-RTT all had lower scores on the clinical severity scale and were all significantly different from the three with higher scores (T-RTT, FOXG1, CDD).
Figure 1: Clinical Severity for the Different DEs.
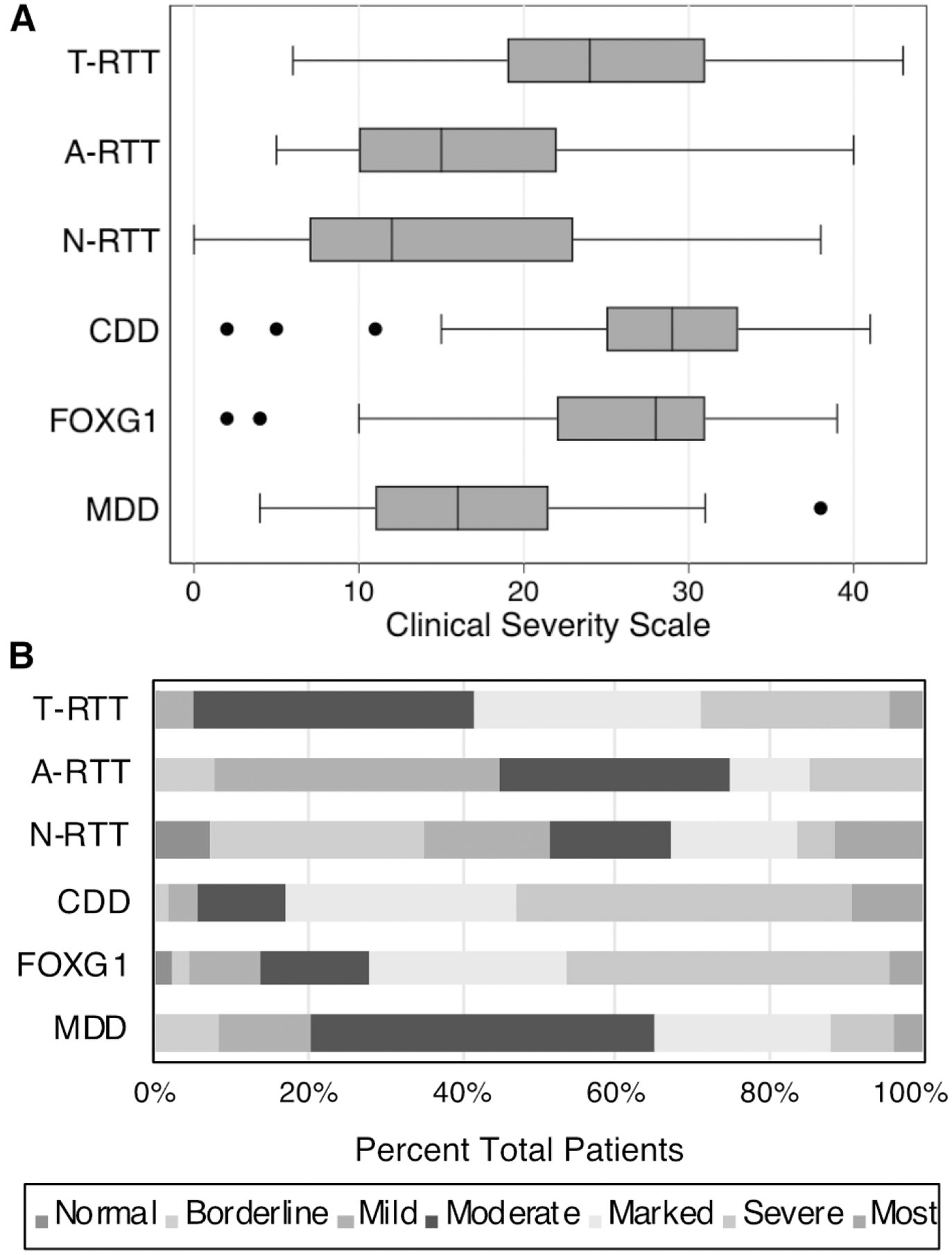
(A) Boxplot of Clinical Severity Scale by diagnosis. Higher scores indicate greater severity. (B) Clinical Global Impression-Severity Scale. Degree of impairment is indicated in the key (normal, borderline impaired, mildly impaired, moderately impaired, markedly impaired, severely impaired, the most impaired).
These findings were mirrored in the Clinical Global Impression Severity Scale (Figure 1B). 58.2% of participants with T-RTT had clinical global impressions of “markedly impaired”, “severely impaired”, or “the most impaired” versus 83.0% of participants with CDD (χ2= 12.5, p<0.001), 72.1% of participants with FOXG1 disorder (χ2=3.2, p=0.07), 34.7% of participants with MDD (χ2= 10.04, p=0.002), 32.5% of participants with N-RTT (χ2=10.6, p=0.001), and 25% of participants with A-RTT (χ2= 15.6, p<0.001).
Regression:
At the baseline visit, regression was reported for 100% (534/534) of participants with T-RTT, 100% (36/36) of participants with A-RTT, 30.2% (13/34) of participants with N-RTT, 34.0% (18/53) of participants with CDD, 35.8% (15/42) of participants with FOXG1 disorder, and 22.5% (11/49) of participants with MDD (Table 2). The median age of regression was 18 months (IQR 15–24 months) for participants with T-RTT and was 23 months (IQR 18–33) for participants with A-RTT. While the majority of participants with the other disorders did not regress, the median age of regression was lower than T-RTT for participants with CDD (6.5 months, IQR 6–12 months, p=0.000014). The median age of regression for participants with N-RTT (24, IQR 12–44, p=0.149637), FOXG1 disorder (12 months, IQR 4–18 months, p=0.002782), and MDD (36 months, IQR 12–72, p=0.012849) did not differ significantly from participants with T-RTT, however there was a trend towards earlier regression for participants with FOXG1 and later regression for participants with MDD and N-RTT.
Table 2:
Regression
| Regression prior to baseline visit | Age of Regression (Months) | ||||
|---|---|---|---|---|---|
| Freq. (n) | % | Median | IQR | P value | |
| T-RTT (N= 534) | 534 | 100% | 18 | 15–24 | -- |
| A-RTT (N=36) | 36 | 100% | 23 | 18–33 | 0.013468 |
| N-RTT (N=43) | 13 | 30.2% | 24 | 12–42 | 0.149637 |
| CDD (N=53) | 18 | 34.0 % | 6.5 | 6–12 | 0.000014 |
| FOXG1 (N=42) | 15 | 35.7 % | 12 | 4–18 | 0.002782 |
| MDD (N= 49) | 11 | 22.5 % | 36 | 12–72 | 0.012849 |
Age of regression when estimated over the lifetime of the group using a Kaplan-Meier survival curve (Figure 2) demonstrated that diagnosis was significantly associated with overall age of regression (log-rank test, χ2=300.23, p<0.0001). The cumulative proportion of participants with reported regression approached 100% for T-RTT, 100% for A-RTT, 39.36 for N-RTT, 36.8% for CDD, 35.6% for FOXG1 disorder, and 32.3% for MDD. 50% of T-RTT participants experienced regression within the first 18 months of life. 50% of A-RTT participants experienced regression within the first 22 months of life.
Figure 2: Cumulative incidence of regression.
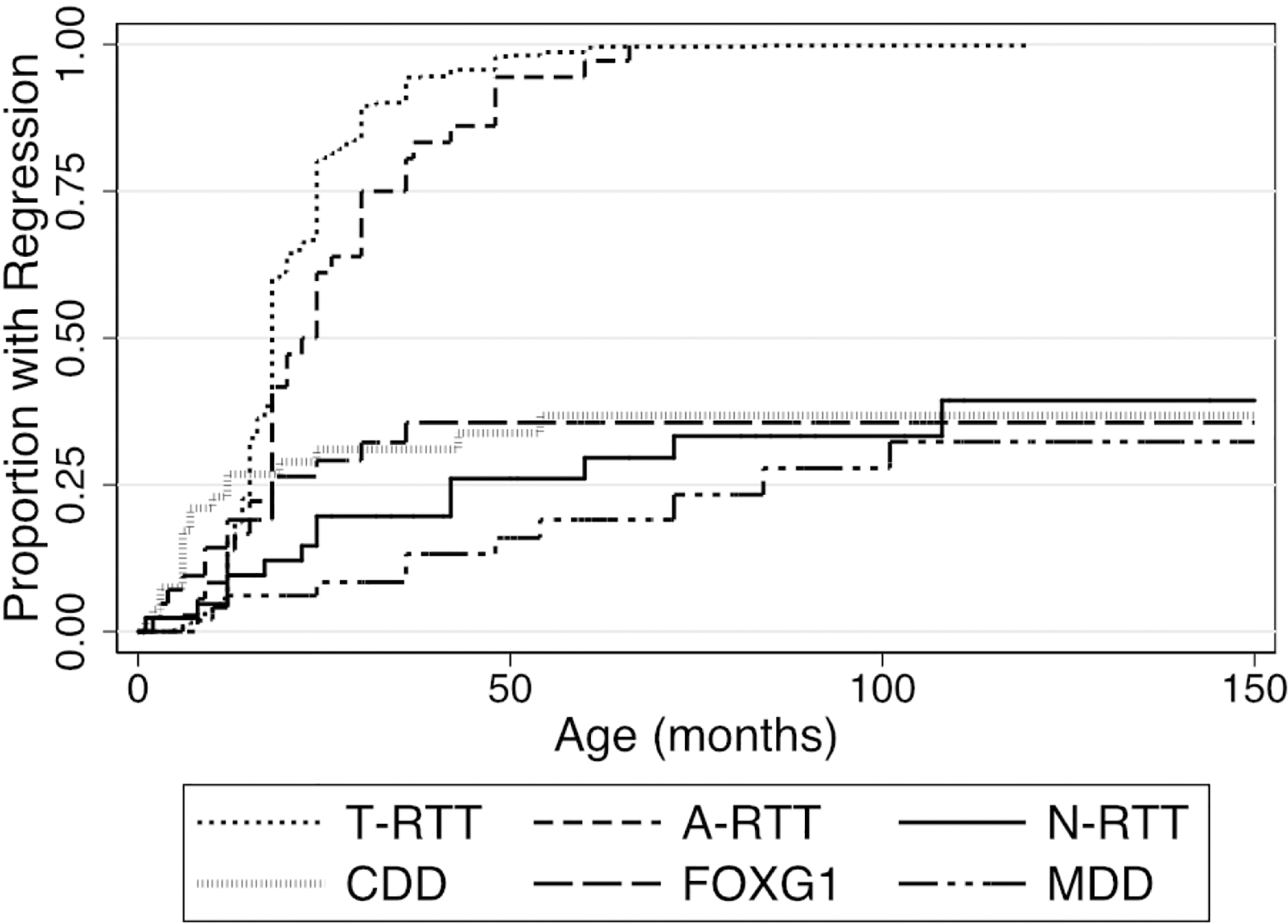
The Y axis represents one minus the cumulative survival score. Event time was defined as the age at onset of regression, or as the age at the baseline study visit if censored.
Seizures:
Seizures were reported at any time prior to the baseline study visit for 47.5% (254/535) of T-RTT participants, 41.0% (16/39) of participants with A-RTT, 29.5% (13/44) of participants with N-RTT, 98.1% (52/53) of participants with CDD, 69.8% (30/43) of participants with FOXG1 disorder, and 44.9% (22/49) of participants with MDD (Table 3). The age of seizure onset was significantly later for T-RTT (median 47.5 months, IQR 27–77 months) than CDD (median 2 months, IQR 1–3 months, p<1×10−6) or FOXG1 disorder (median 11.5 months, IQR 7–18 months, p<1×10−6). The age of seizure onset for T-RTT did not vary significantly from A-RTT (median 39.5 months, IQR 13–56 months, p=0.13), N-RTT (median 24 months, IQR 11–47 months, p=0.028), or MDD (median 64 months, IQR 24–120 months, p=0.20). In this population, 0% of T-RTT, 0% of A-RTT, 0% of N-RTT, 88.5% (46/52) of CDD, 9.5% (4/42) of FOXG1 disorder, and 0% of MDD participants had early onset seizures.
Table 3:
Seizures
| Seizures prior to baseline visit | Early Onset Seizures | Age of Seizure Onset (Months) | |||||
|---|---|---|---|---|---|---|---|
| Freq.(n) | % | Freq.(n) | % | Median | IQR | P value | |
| T-RTT | 254 | 47.5 % | 0 | 0 % | 47.5 | 22–77 | -- |
| A-RTT | 16 | 41.0 % | 0 | 0% | 39.5 | 13– 56 | 0.129 |
| N-RTT | 13 | 29.5 % | 0 | 0% | 24 | 11–47 | 0.028 |
| CDD | 52 | 98.1 % | 46 | 88.5 % | 2 | 1–3 | <1×10−6 |
| FOXG1 | 30 | 69.8 % | 4 | 9.5 % | 11.5 | 7–18 | <1×10−6 |
| MDD | 22 | 44.9 % | 0 | 0 % | 64 | 24–120 | 0.203 |
Estimates of total lifetime prevalence of seizures were higher when calculated using the Kaplan-Meier survival curve (Figure 3A), but still differed significantly by diagnosis (log-rank test, χ2= 683.34, p<0.0001). The cumulative proportion of participants with seizures approached 75% for T-RTT, 50.4% for A-RTT, 40.4% for N-RTT, and 96% for CDD. For FOXG1 disorder and MDD, the cumulative proportion of participants with seizures was respectively 73.3% and 84.3% before the last participant, however the oldest participant in both groups developed seizures, which increased the cumulative proportion to 100% in the Kaplan-Meier analysis. Based on the survival analysis, we found that seizures appeared earlier in CDD and FOXG1 disorder than T-RTT. 50% of participants had seizure onset within 173 months (14.4 years) in T-RTT, 169 months for A-RTT (14.1 years), 2 months in CDD, 14 months in FOXG1 disorder, and 120 months (10 years) in MDD.
Figure 3: Seizures.
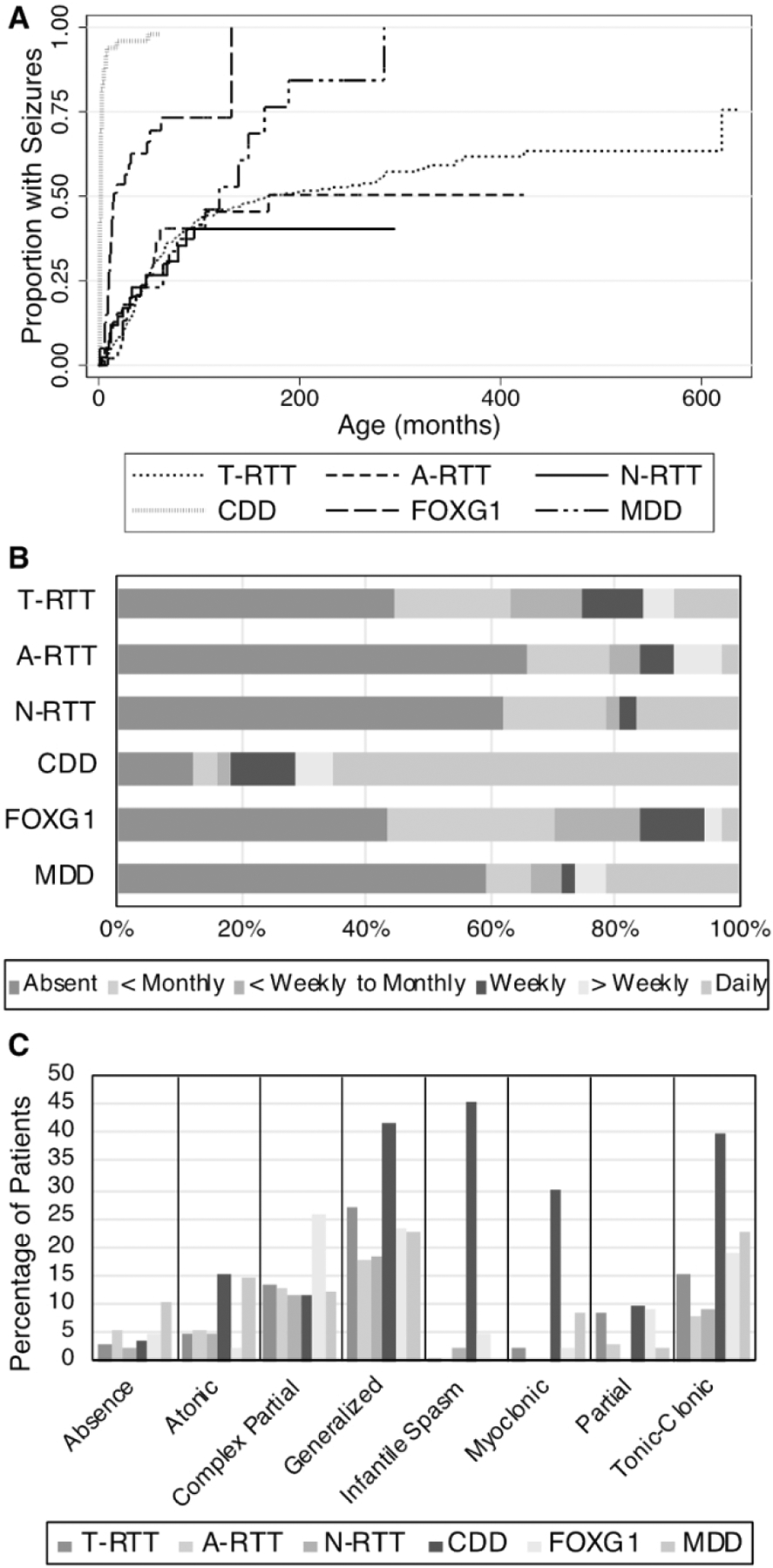
(A) Cumulative incidence of seizures. The Y axis represents one minus the cumulative survival score. Event time was defined as the age of seizure onset, or as the age at the baseline visit if censored. (B) Reported seizure frequency. (C) Investigator impression of reported seizures.
In the year prior to the baseline study visit, 55.6% of T-RTT, 34.2% of A-RTT, 38.1% of N-RTT, 87.8% of CDD, 56.8% of FOXG1 disorder, and 40.5% of MDD participants had seizures (Figure 3B). Average seizure frequencies can be found in Supplementary Table 1. The majority of people (65.3%) with CDD had daily seizures. The majority of participants with N-RTT and MDD did not have seizures in the past year, but if seizures were reported they tended to occur frequently: 43.8% of N-RTT and 64% of MDD individuals with seizures reported had daily seizures. In contrast, people with FOXG1 disorder tended to have seizures less often, with 71% having an average seizure frequency of less than weekly or monthly.
Additionally, seizure semiology was captured at the baseline visit based on investigator impression (Figure 3C, Supplementary Table 2). In T-RTT, the most commonly reported seizure types were generalized seizures (26.9%), tonic-clonic seizures (15.1%), and complex partial seizures (13.6%). Participants with A-RTT most often presented with generalized seizures (17.9%) or complex partial seizures (12.8%). In participants with N-RTT, generalized seizures (18.2%), complex partial seizures (11.4%), and tonic-clonic seizures (9.1%) were the most commonly reported semiologies. Notably, participants with CDD presented with epileptic spasms (45.3 %) more often than those with other phenotypes (0.4% for T-RTT, 0% for A-RTT, 2.3% for N-RTT, 4.7% for FOXG1, 0% for MDD). The next most commonly reported seizure semiologies for CDD included generalized seizures (41.5%), tonic-clonic seizures (39.6%), and myoclonic seizures (30.2%). Participants with FOXG1 disorder most often presented with complex partial seizure (25.6%), generalized seizures (23.3%), or tonic-clonic seizures (18.6%). In MDD, generalized seizures (22.5%), tonic-clonic seizures (22.5%), and atonic seizures (14.3%) were the most commonly reported semiologies.
After comparing the three major clinical outcomes of the DEs (regression, clinical severity, and epilepsy) we aimed to determine if any clinical variables correlated with the outcome measures. HC Z-score, age of regression, age of seizure onset, and severity scores were all compared using a linear regression analysis. The comparisons resulted in notable relationships.
In all DEs analyzed, lower HC Z-Scores were associated with higher scores on the Clinical Severity Scale (Table 4, Figure 4B). The median HC Z-score was −1.8 (IQR −2.6, −0.9) for T-RTT, −0.4 (IQR −1.6, 0.1) for A-RTT, −0.6 (IQR −1.9, 0.8) for N-RTT, −1.0 (IQR −1.8, −0.5) for CDD, −3.1 (IQR −3.8, −1.9) for FOXG1, and −0.0 (IQR −1.5, 1.4) for MDD (Figure 4A).
Table 4:
Association of HC Z-Score and Clinical Severity Scale
| N | Linear Regression | Spearman’s Rank | |||||
|---|---|---|---|---|---|---|---|
| Model p-value | R2 | Coefficient Independent | 95% CI | Correlation | p-value | ||
| T-RTT | 472 | <0.0001 | 0.284 | −2.98 | −3.41, −2.55 | −0.535 | <0.0001 |
| A-RTT | 35 | 0.0002 | 0.349 | −3.171 | −4.706, −1.646 | −0.495 | 0.0025 |
| N-RTT | 38 | 0.0007 | 0.275 | −2.854 | −4.420, −1.288 | −0.602 | 0.0001 |
| CDD | 44 | 0.0071 | 0.160 | −2.771 | −4.745, −0.796 | −0.447 | 0.0024 |
| FOXG1 | 35 | 0.0001 | 0.386 | −4.403 | −6.371, −2.434 | −0.370 | 0.0287 |
| MDD | 40 | 0.0067 | 0.179 | −1.986 | −3.390, −0.683 | −0.456 | 0.0031 |
Figure 4: Association of HC Z-Score and Clinical Severity.
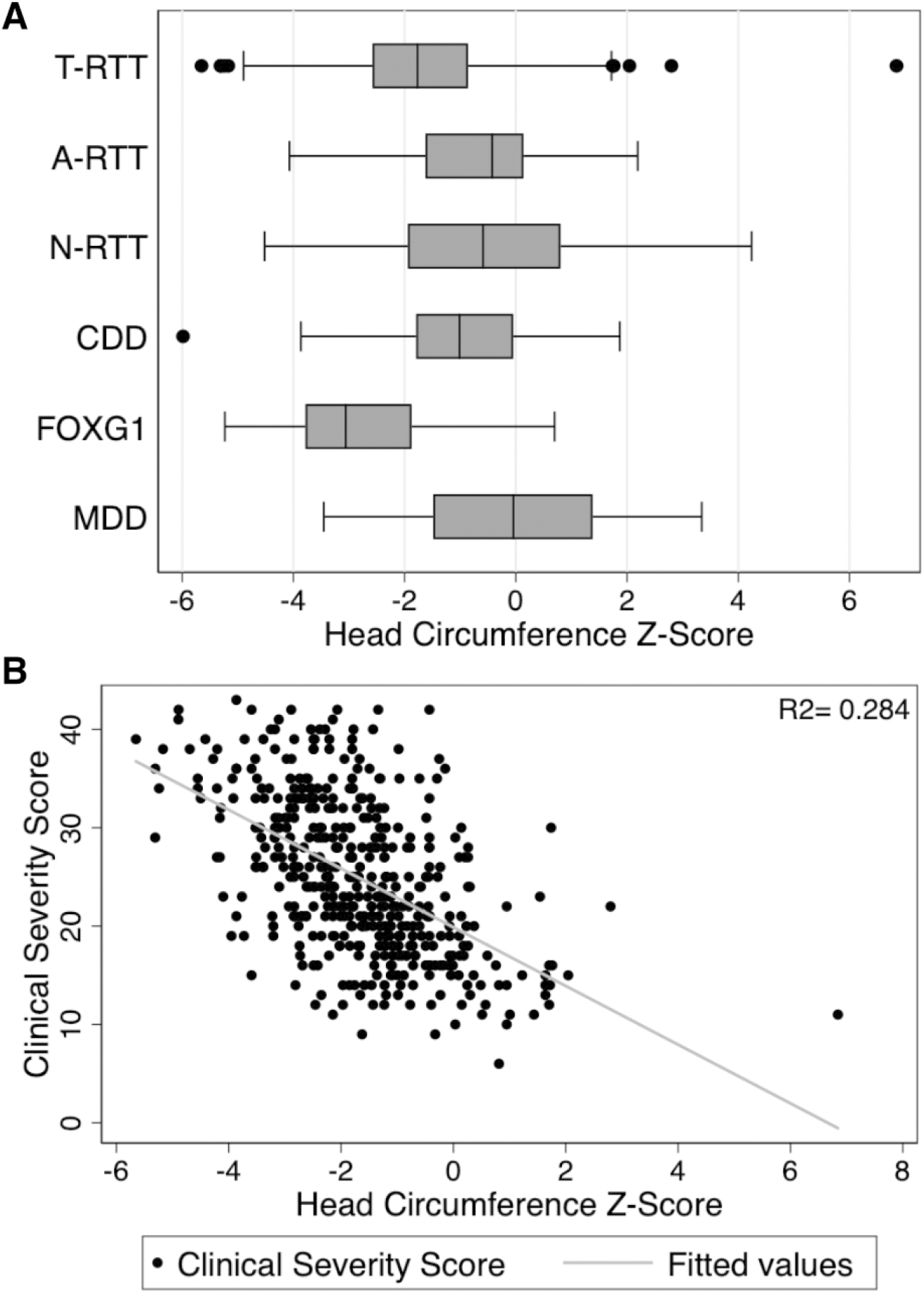
(A) Boxplot of HC Z-score by diagnosis (B) Clinical Severity Scale vs HC Z-score for T-RTT, with linear regression plotted in gray. Similar associations between HC and clinical severity were also observed for the other DEs (see Table 4).
For MDD, there was a significant association between HC Z-score and age of seizure onset (months, correlation coefficient= 21.307, p=0.0097). This association was not seen in T-RTT, A-RTT, N-RTT, CDD, of FOXG1 disorder. Additionally, no significant correlations existed between seizure onset age and Clinical Severity Scale or regression age for any of the groups in this study (Supplementary Table 3).
Discussion
RTT, CDD, FOXG1, and MDD are prototypical examples of the developmental encephalopathies. Each disorder is characterized by a lack of overt features at birth with unique progressive, developmentally timed onset of cognitive and motor disabilities, propensities to develop seizures, and other abnormalities. Due to the overlapping nature of these conditions, they were often imprecisely named “atypical or variant RTT” or “RTT-like”. Improvements in genotyping have resulted in an increased identification of individuals with specific gene mutations (CDKL5, FOXG1, MECP2 duplication), leading to the realization that both distinct and overlapping phenotypic features exist. This study is the first to directly compare these four DEs. We focused this comparison on the shared core features to determine how similar or disparate these 4 DEs are from each other. The results indicate differences in the overall disease severity, rate of regression, and seizure occurrence as well as clear overlap between certain features. Together, these investigations will improve understanding of the natural histories and will inform refining/revisions of clinical instruments that will be necessary for each specific disorder’s clinical trial readiness.
Overall Severity and Regression:
Individuals with A-RTT, N-RTT, and MDD had lower scores on the Clinical Severity Scale then T-RTT, FOXG1, and CDD. Of note, in A-RTT there appear to be two subgroups, with mild and severe clinical severity, as previously noted and further expanded on by Cuddapah et al.5 The pattern of severity scores for the other disorders match the general impression garnered from the literature, suggesting that the two severity measures used in the Natural History Study, while originally defined for RTT and not psychometrically validated, may be useful outside of RTT.8,14,20,21 This direct comparison of four DEs using the RTT scale demonstrates the range of severities across all studied DEs. However, disease-specific severity scales are needed to be developed (and one has been developed for CDD22) and comparison of the utility of the Clinical Severity Scale to a disease-specific scale could be informative and potentially performed using the Natural History Study database.
Significant differences were also identified between the disorders with regard to the presence/absence of regression and the age of regression onset. A period of regression followed by stabilization is a defining characteristic of T-RTT and A-RTT.2 For the other disorders, less information is known about regression as most children display global developmental delay from birth. Previous estimates of the percent of individuals experiencing regression are much lower in FOXG1, CDD, and MDD.23,24 The current analysis confirms this clinical suspicion; all T-RTT and A-RTT participants reported regression, while the majority of individuals with other DEs did not regress. However, for CDD and FOXG1 individuals who did report regression, the median age of regression was lower than for T-RTT. Notably, in contrast to previous studies, age of regression was not associated with seizure onset age for any of these disorders.25,23 This difference is likely related to others reporting regression with seizure increases or severe onset, while we captured just first seizure onset which may not be severe or associated with regression. As no relationship was ascertained between seizure and regression, we next compared seizures in these four conditions.
Seizure Phenotypes:
Seizures negatively impact the quality of life and represent a significant problem for clinicians and families in all four disorders. Previous estimates of epilepsy in RTT have ranged from 48% to 94%.26,27 Much of the variation in these estimates is likely due to different sample sizes, diagnostic criteria, point-prevalence versus life-time prevalence, or participant age ranges. Additionally, studies that rely on parental questionnaires may overestimate seizure prevalence, as 12% of the spells called “seizures” by parents do not meet the clinical definition.28 In this population, the point prevalence of seizures in T-RTT was 55.7% and the estimated lifetime prevalence using survival analysis approached 75%. Tarquinio et al. found that the point prevalence with active seizures ranged from 30–44%, with an estimated lifetime prevalence of 90%.28 This somewhat differs from our results and may be due to the variability that exists in the population, especially when evaluating different age groups at different time points. The median age of seizure onset in our study was 4 years, which is consistent with previous reports of median age between 4 and 4.7 years.20,29,30 Seizures types reported were similar to the data obtained in an Italian cohort, suggesting consistency across studies.20
Compared to T-RTT, seizures in CDD are more severe and begin at a much younger age, consistent with CDD historically being associated with the early onset seizure RTT variant.31–33 The point prevalence of active seizures (87.8%), frequency of being intractable, and presence of epileptic spasms were highest in CDD, which is consistent with many previous reports.8,31,34,35
Seizures in FOXG1 began earlier than in T-RTT, but later than in CDD. Our data gives an earlier seizure onset age than reported by Mitter et al.12 This difference is likely due to the use of the mean as the measure of central tendency in Mitter et al.; whereas our data were skewed so we present a median age. If we compare means (19.6 months vs 25 months), the data are more consistent.
The age of seizure onset in MDD (median 64 months) did not differ significantly from T-RTT, however the survival analysis found a higher lifetime prevalence of seizures in MDD. Older ages of seizure onset have been reported by Caumes et al. (median 72 months in 8 subjects with MDD) and Miguet et al. (mean age of onset around 7.4 years in 59 subjects with MDD).36,37 Interestingly, a significant association existed between HC z-score and age of onset of seizures in MDD, where patients with smaller head circumferences reported earlier onset of seizures. This relationship was not observed in the other disorders and has not been reported previously in the literature. The point prevalence of seizures (40.48%) was lower than in the other three disorders, but when participants did have active seizures, they were intractable the majority of the time. Lim et al. obtained similar results; in their analysis 44% of individuals had seizures, and seizures occurred daily in nearly half of subjects with active seizures.38 In the analysis by Miguet et al., epilepsy was drug resistant 62% of the time.37
Limitations:
This first cross-comparison study of these four main disorders, which are related historically, phenotypically, and/or genetically, relied on physician input of data at the baseline Natural History Study visit. As our data was cross-sectional and the cohort of participants was relatively young, it may not fully capture the lifetime incidence of regression or seizures. This is especially true for MDD, where the age of regression was later than in the other disorders. The Natural History Study also used severity measures that were designed for RTT and may miss critical features of clinical severity for the other disorders. For example, MDD has increased infections and hospitalizations, which is not reflected in the RTT specific Clinical Severity Scale.37,39 It is also possible that the increased frequency of seizures in CDD may partially contribute to its increased severity on this scale. Overall, while the CSS is RTT specific, the scale did a good job in separating out the groups. Ideally, a scale that can be used across all DEs to allow comparisons and be easily used in a clinic and disorder specific scales to follow individuals in clinic and as potential outcome measures in studies is needed.
Additionally, and not surprisingly for a large multicenter study, with variable ways and people collecting data (coordinators, nurses, physicians), issues were noted with missing or inconsistent data. For example, three subjects did not have date of birth listed. For several A-RTT participants regression was noted on physician interview however not documented in the developmental log. As much of the data were collected retrospectively, issues with recall bias may affect the numbers reported in this study. For example, it is possible that some seizures were not recorded in the seizure log, which would lead to an underestimated lifetime prevalence of seizures. Another issue was the lack of some historical data; for 78 T-RTT participants the month of the seizure start date was unknown, thereby requiring these to be rounded to a January date. This adjustment would, on average, shift the seizure onset date by 6 months in these participants. However, with 535 T-RTT participants, the total variation would be minimal. Overall, the partially retrospective nature of any study like this requires physicians and care givers to consider age ranges, not exact ages as the accuracy of any single number is questionable.
While the main limitation of this study is related to recall issues and data entry, we simplified our analysis by not stratifying our results by sex (and a few individuals the sex was not listed highlighting data entry issues). Gender has an impact in each of these disorders. For example, in some studies males have been reported to have more severe phenotypes than females in CDD.40,23 Future analysis breaking down these populations by sex could be informative. Other issues that this study did not address are impacts of anti-seizure drug treatments on epilepsy or severity scores, role of genotype within each disorder, and other clinical variables that are shared, such as movement disorders. Future work on each of these issues within a given syndrome and across syndromes will be interesting for medical providers and caregivers of these children.
Conclusions:
This study analyzed important clinical features from one of the largest databases involving these four disorders. The results confirmed that both shared features and clear differences are present, and support classifying these DEs as distinct disorders to understand the unique clinical features and prognosis of each. The nomenclature “RTT-like”, “variant RTT”, or similar should not be used. Individuals should be ascribed to each disorder: T-RTT, A-RTT, N-RTT, CDD, FOXG1, or MDD. When diagnostic uncertainty persists, especially when genetic testing is limited or unclear, then “Developmental Encephalopathy-NOS” should be considered as the working diagnosis. This first cross comparison will be helpful to caregivers and physicians as it can focus genetic testing based on the time of presentation or severity of disease; but practically, with the shift to panel-based testing, this issue has been minimized. These data also begin to frame the discussion about approach to treatments when each disorder has different timing of particular symptoms and severity of disease. This work sets the foundation for the development of disorder specific severity scales to improve identifying the full spectra of disease. The ongoing longitudinal assessment of participants will continue to provide information for a better understanding of these developmental encephalopathies, and ultimately aid clinical trials.
Supplementary Material
Acknowledgements
The authors would like to thank the subjects and their families for participating in this study. We would also like to thank the family support groups that have encouraged families to participate in the study. We would like to thank all of the study coordinators and clinicians who have entered data and assisted families with enrolling in the study. We are very thankful for Dr. Jonathan Rollin’s for providing the LMS data from his Journal of Pediatrics 2010 paper to calculate Z-scores from our data set. Lastly, this study was funded by the National Institutes of Health, National Institutes of Child Health and Disease (3U54-HD061222-14S1) as part of the Rare Disease Research Network for all authors, the Vanderbilt IDDRC U54HD083211 for JLN.
Footnotes
Potential Conflicts of Interest
CCF, JS, DA, CG, AP, EDM, SP, JL, JN: Nothing to report
References
- 1.Swaiman KF, Ashwal S, Ferriero DM, et al. Swaiman’s pediatric neurology : principles and practice [Internet]. 2018. Available from: https://www.worldcat.org/title/swaimans-pediatric-neurology-principles-and-practice/oclc/988768908
- 2.Neul JL, Kaufmann WE, Glaze DG, et al. Rett Syndrome: Revised Diagnostic Criteria and Nomenclature [Internet]. Ann Neurol 2010;68(6):944–950.[cited 2018 Aug 8] Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3058521/pdf/nihms214759.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neul JL, Fang P, Barrish J, et al. Specific mutations in Methyl-CpG-Binding Protein 2 confer different severity in Rett syndrome. Neurology 2008;70(16):1313–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laurvick CL, de Klerk N, Bower C, et al. Rett syndrome in Australia: a review of the epidemiology. [Internet]. J. Pediatr 2006;148(3):347–52.[cited 2019 Jan 13] Available from: http://www.ncbi.nlm.nih.gov/pubmed/16615965 [DOI] [PubMed] [Google Scholar]
- 5.Cuddapah VA, Pillai RB, Shekar KV, et al. Methyl-CpG-binding protein 2 (MEPC2) mutation type is associated with disease severity in Rett Syndrome HHS Public Access [Internet]. J Med Genet 2014;51(3):152–158.[cited 2019 Jan 13] Available from: http://mecp2.chw.edu.au [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am. J. Med. Genet. Part A 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters SU, Katzenstein A, Jones D, Key AP. Distinguishing response to names in Rett and MECP2 Duplication syndrome: An ERP study of auditory social information processing. Brain Res. 2017; [DOI] [PubMed] [Google Scholar]
- 8.Guerrini R, Parrini E. Epilepsy in Rett syndrome, and CDKL5- and FOXG1-gene-related encephalopathies [Internet]. Epilepsia 2012;53(12):2067–2078.[cited 2018 Aug 8] Available from: http://doi.wiley.com/10.1111/j.1528-1167.2012.03656.x [DOI] [PubMed] [Google Scholar]
- 9.Olson HE, Demarest ST, Pestana-Knight EM, et al. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. [Internet]. Pediatr. Neurol 2019;97:18–25.[cited 2019 Oct 27] Available from: http://www.ncbi.nlm.nih.gov/pubmed/30928302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demarest ST, Olson HE, Moss A, et al. CDKL5 deficiency disorder: Relationship between genotype, epilepsy, cortical visual impairment, and development. Epilepsia 2019;60(8):1733–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vegas N, Cavallin M, Maillard C, et al. Delineating FOXG1 syndrome: From congenital microcephaly to hyperkinetic encephalopathy. Neurol. Genet 2018;4(6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitter D, Pringsheim M, Kaulisch M, et al. FOXG1 syndrome: Genotype-phenotype association in 83 patients with FOXG1 variants. Genet. Med 2018; [DOI] [PubMed] [Google Scholar]
- 13.Neul J, Glaze D, Percy A, et al. Improving Treatment Trial Outcomes for Rett Syndrome: the development of Rett-specific anchors for the Clinical Global Impression Scale HHS Public Access [Internet]. J Child Neurol 2015;30(13):1743–1748.[cited 2018 Sep 15] Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4610825/pdf/nihms-697493.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schanen C, Houwink EJF, Dorrani N, et al. Phenotypic manifestations ofMECP2 mutations in classical and atypical rett syndrome [Internet]. Am. J. Med. Genet 2004;126A(2):129–140.[cited 2019 Dec 31] Available from: http://doi.wiley.com/10.1002/ajmg.a.20571 [DOI] [PubMed] [Google Scholar]
- 15.Bebbington A, Anderson A, Ravine D, et al. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology 2008;70(11):868–875. [DOI] [PubMed] [Google Scholar]
- 16.Glaze DG, Neul JL, Kaufmann WE, et al. Double-blind, randomized, placebo-controlled study of trofinetide in pediatric Rett syndrome. Neurology 2019;92(16):e1912–e1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glaze DG, Neul JL, Percy A, et al. A Double-Blind, Randomized, Placebo-Controlled Clinical Study of Trofinetide in the Treatment of Rett Syndrome. Pediatr. Neurol 2017;76:37–46. [DOI] [PubMed] [Google Scholar]
- 18.Cole TJ, Freeman JV, Preece MA. British 1990 growth reference centiles for weight, height, body mass index and head circumference fitted by maximum penalized likelihood. Stat. Med 1998;17(4):407–429. [PubMed] [Google Scholar]
- 19.Rollins JD, Collins JS, Holden KR. United States head circumference growth reference charts: birth to 21 years. [Internet]. J. Pediatr 2010;156(6):907–913.e2.[cited 2019 Oct 28] Available from: http://www.ncbi.nlm.nih.gov/pubmed/20304425 [DOI] [PubMed] [Google Scholar]
- 20.Pintaudi M, Calevo MG, Vignoli A, et al. Epilepsy in Rett syndrome: Clinical and genetic features. Epilepsy Behav. 2010;19:296–300. [DOI] [PubMed] [Google Scholar]
- 21.Mangatt M, Wong K, Anderson B, et al. Prevalence and onset of comorbidities in the CDKL5 disorder differ from Rett syndrome [Internet]. Orphanet J. Rare Dis 2016;11(1)[cited 2018 Aug 8] Available from: http://cdkl5.childhealthresearch.org.au [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demarest S, Pestana-Knight EM, Olson HE, et al. Severity Assessment in CDKL5 Deficiency Disorder. Pediatr. Neurol 2019;97:38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fehr S, Wilson M, Downs J, et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy [Internet]. Eur. J. Hum. Genet 2013;21(3):266–273.[cited 2018 Aug 8] Available from: www.nature.com/ejhg [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kortüm F, Das S, Flindt M, et al. The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. [Internet]. J. Med. Genet 2011;48(6):396–406.[cited 2018 Dec 6] Available from: http://www.ncbi.nlm.nih.gov/pubmed/21441262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nissenkorn A, Gak E, Vecsler M, et al. Epilepsy in Rett syndrome - The experience of a National Rett Center. Epilepsia 2010; [DOI] [PubMed] [Google Scholar]
- 26.Glaze DG, Percy AK, Skinner S, et al. Epilepsy and the natural history of Rett syndrome. Neurology 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jian L, Nagarajan L, de Klerk N, et al. Seizures in Rett syndrome: An overview from a one-year calendar study. Eur. J. Paediatr. Neurol 2007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tarquinio DC, Hou W, Berg A, et al. Longitudinal course of epilepsy in Rett syndrome and related disorders. Brain 2017;140(2):306–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wold Henriksen M, Breck H, Von Tetzchner S, et al. Epilepsy in classic Rett syndrome: Course and characteristics in adult age [Internet]. Epilepsy Res. 2018;145:134–139.[cited 2018 Oct 6] Available from: 10.1016/j.eplepsyres.2018.06.012 [DOI] [PubMed] [Google Scholar]
- 30.Nissenkorn A, Levy-Drummer RS, Bondi O, et al. Epilepsy in Rett syndrome - Lessons from the Rett networked database. Epilepsia 2015;56(4):569–576. [DOI] [PubMed] [Google Scholar]
- 31.Scala E, Ariani F, Mari F, et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms [Internet]. J Med Genet 2005;42:103–107.[cited 2018 Sep 16] Available from: www.jmedgenet.com [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bahi-Buisson N, Kaminska A, Boddaert N, et al. The three stages of epilepsy in patients with CDKL5 mutations [Internet]. Epilepsia 2008;49(6):1027–1037.[cited 2018 Aug 8] Available from: http://doi.wiley.com/10.1111/j.1528-1167.2007.01520.x [DOI] [PubMed] [Google Scholar]
- 33.Bahi-Buisson N, Juliette Nectoux Ã, Haydee Ã, et al. Key clinical features to identify girls with CDKL5 mutations [Internet]. 2008;[cited 2018 Sep 14] Available from: https://academic.oup.com/brain/article-abstract/131/10/2647/1746432 [DOI] [PubMed]
- 34.Archer HL, Evans J, Edwards S, et al. CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients [Internet]. J Med Genet 2006;43:729–734.[cited 2018 Sep 16] Available from: www.jmedgenet.com [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nemos C, Lambert L, Giuliano F, et al. Mutational spectrum of CDKL5 in early-onset encephalopathies: A study of a large collection of French patients and review of the literature [Internet]. Clin. Genet 2009;76(4):357–371.[cited 2018 Sep 16] Available from: http://doi.wiley.com/10.1111/j.1399-0004.2009.01194.x [DOI] [PubMed] [Google Scholar]
- 36.Caumes R, Boespflug-Tanguy O, Villeneuve N, et al. Late onset epileptic spasms is frequent in MECP2 gene duplication: Electroclinical features and long-term follow-up of 8 epilepsy patients [Internet]. Eur. J. Paediatr. Neurol 2014;18:475–481.[cited 2018 Oct 3] Available from: 10.1016/j.ejpn.2014.03.005 [DOI] [PubMed] [Google Scholar]
- 37.Miguet M, Faivre L, Amiel J, et al. Further delineation of the MECP2 duplication syndrome phenotype in 59 French male patients, with a particular focus on morphological and neurological features. [Internet]. J. Med. Genet 2018;55(6):359–371.[cited 2019 Oct 27] Available from: http://www.ncbi.nlm.nih.gov/pubmed/29618507 [DOI] [PubMed] [Google Scholar]
- 38.Lim Z, Downs J, Wong K, et al. Expanding the clinical picture of the MECP2 Duplication syndrome [Internet]. Clin. Genet 2017;91(4):557–563.[cited 2018 Oct 3] Available from: http://doi.wiley.com/10.1111/cge.12814 [DOI] [PubMed] [Google Scholar]
- 39.Peters SU, Fu C, Suter B, et al. Characterizing the phenotypic effect of Xq28 duplication size in MECP2 duplication syndrome. Clin. Genet 2019;95(5):575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mirzaa GM, Paciorkowski AR, Marsh ED, et al. CDKL5 and ARX mutations in males with early-onset epilepsy. Pediatr. Neurol 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.