

Abstract
Silicosis is a devastating disease that, without effective treatment, endangers the health of miners. Therefore, studies exploring the pathogenesis of SiO2-induced pulmonary fibrosis are necessary to develop treatments for silicosis. Although macrophages are known to play a pivotal role in SiO2-induced pulmonary fibrosis, the underlying mechanism remains unknown. Here, we explored whether ferroptosis was involved in SiO2-induced pulmonary fibrosis. To this end, C57BL/6 mice and mouse macrophage (RAW264.7) cells and mouse lung fibroblast (MLF) cells were subjected to iron content, cell viability, enzyme-linked immunosorbent assay, immunofluorescence staining, histological, western blotting, quantitative reverse transcription-PCR, reactive oxygen species, and lipid peroxidation analysis. In vivo, SiO2 was found to damage the lung alveolar structure, cause infiltration of inflammatory cells, and facilitate fibrosis. Additionally, it increased the iron concentration and lipid peroxidation as well as altered the expression of ferroptosis-related genes and the mitochondrial morphology in macrophages. In vitro, ferroptosis occurred in SiO2-treated RAW264.7 cells, which showed iron overload, lipid peroxidation, and gene alterations. Furthermore, ferrostatin-1 (Fer-1) attenuated ferroptosis in SiO2-treated RAW264.7 cells by inhibiting lipid peroxidation and cell death and regulating ferroptosis-related genes expression, in addition to attenuating the secretion of pro-fibrotic cytokines and fibrosis. Collectively, SiO2 induces ferroptosis in macrophages, which leads to the secretion of pro-fibrotic cytokines and fibrosis.
Keywords: silicosis, macrophage, ferroptosis, ferrostatin-1, lipid peroxidation
Introduction
Silicosis is a serious occupational disease that endangers the health of workers worldwide [1] and is characterized by persistent pulmonary fibrosis. A study conducted in 2015 showed that the incidence of silicosis is relatively high in South Africa [2] as well as in India where 11.5 million workers were reported to be exposed to silica dust [3]. Patients with silicosis have respiratory and circulatory dysfunction accompanied by cough, sputum, chest tightness, dyspnea, chest pain, and fatigue. Treatment approaches include pulmonary lavage and antifibrosis drugs; although these treatments inhibit fibrosis development during silicosis, they fail to remove residual SiO2 in the alveolar cavity. Therefore, exploring the pathogenesis of SiO2-induced pulmonary fibrosis is necessary for developing treatments.
Macrophages have a pivotal role in the pathogenesis of SiO2-induced pulmonary fibrosis by secreting various pro-fibrotic cytokines in response to SiO2 [4]. Therefore, macrophages damage or death may initiate SiO2-induced pulmonary fibrosis. In the pathogenesis of SiO2-induced pulmonary fibrosis, macrophages damage or death occur through programmed cell death (PCD) processes such as apoptosis, autophagy, and pyroptosis. Apoptosis plays a significant role in the progression of silicosis. SiO2 binding to the scavenger receptor on the alveolar macrophage surface can trigger the upregulation of FasL, which binds Fas and leads to apoptosis. Additionally, autophagy regulates the synthesis and degradation of cellular components and plays a pivotal part in the pathogenesis of silicosis. SiO2 is found in the macrophage autophagolysosomes of patients with silicosis, and unusual autophagy activity has been observed in rats with silicosis [5, 6]. In addition, pyroptosis accompanied by rupture of the nuclear DNA and cell membrane leads to the release of a large number of inflammatory factors (such as interleukin (IL)-α, IL-1β, and IL-33) [7]. However, the pathogenesis of pulmonary fibrosis remains controversial. It was previously reported that after the inhibition of special damage or death types, pulmonary fibrosis was not significantly attenuated [8]. It indicates that other mechanisms may be involved in SiO2-induced pulmonary fibrosis.
Ferroptosis is a type of PCD discovered in 2012. The iron-mediated Fenton reaction induces reactive oxygen species (ROS) and lipid peroxidation, which causes cell membrane disruption, including mitochondria, and results in ferroptosis [9]. Studies have shown that ferroptosis occurs in various pulmonary diseases. Cigarette smoke promotes iron overload and phospholipid peroxidation, leading to ferroptosis in chronic obstructive pulmonary disease [10]. ROS accumulation and glutathione (GSH) depletion, which are closely associated with ferroptosis, strongly affect the pathogenesis of pulmonary diseases [11]. Additionally, Li observes that irradiation induces ROS and changes the concentration of inflammatory cytokines. Ferroptotic characteristic changes of mitochondria in acute radiation-induced lung injury (RILI) was observed by transmission electron microscopy (TEM). In summary, ferroptosis participates in RILI [12, 13]. Nevertheless, the role of ferroptosis in SiO2-induced pulmonary fibrosis remains unclear. Here, we explored the role of ferroptosis in SiO2-induced pulmonary fibrosis and the underlying mechanisms.
Materials and Methods
Mouse models
C57BL/6 mice (22 ± 2 g) were purchased from the Ning Xia Medical University Laboratory Animal Centre and fed in a sterile environment. The animal procedures were approved by the Ethics Committee of Ningxia Medical University (approval no. 2020-493). Mice were randomly assigned to the SiO2 group, which was treated with 100 μl SiO2 suspensions (100 mg/ml) via intratracheal instillation, or saline group as controls. The animals were anesthetized with isoflurane and sacrificed after 56 days.
Cell culture and SiO2 treatment
The RAW264.7 cells (mouse macrophage, MINGJIN BIOLOGY, Shanghai, China) and the MLF cells (mouse lung fibroblast, Procell Life Science&Technology Co., Ltd, Wuhan, China) cultured in Dulbecco’s modified Eagle medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco) in an incubator with 5% CO2 at 37°C. The SiO2 group was treated with 50 μg/cm2 SiO2 suspension for 24 h. The saline group was treated with saline. Ferrostatin-1 (Fer-1, 10 μM, a ferroptosis inhibitor, Selleck, Houston, USA) or Z-VAD-FMK (20 μM, a caspase inhibitor that inhibits apoptosis, Selleck, Houston, USA) was added to the RAW264.7 cells 1 h before SiO2 administration.
Coculture experiment
We used a 24 mm Transwell® (Corning, NY, USA). RAW264.7 cells were seeded into the upper wells and treated with SiO2 (50 μg/cm2) for 24 h. Fer-1 (10 μM) was added to the RAW264.7 1 h before SiO2 administration. MLF cells attached to the lower wells were cocultured with RAW264.7 cells in a Transwell 6-wells plate system for another 24 h. The supernatant was collected for cytokine detection, and MLF cells were collected to detect the protein and mRNA levels of α-smooth muscle actin (α-sma), matrix metalloprotein-9 (MMP-9), and collagen-1.
GSH/GSSG assay and MDA assay
The glutathione/glutathione oxidized (GSH/GSSG) and malondialdehyde (MDA) levels were measured using a GSH/GSSG assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and MDA assay kit (BestBio, Shanghai, China), respectively, as per the manufacturer’s instructions.
Iron assay
Iron concentrations in the lung tissues and cells were measured using an iron assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and iron colorimetric assay kit (Applygen Technologies, Beijing, China), respectively, as per the manufacturer’s instructions.
Cell viability assay
Cell viability was assessed using Cell Counting Kit-8 (Beyotime Biotechnology, Shanghai, China) as per the manufacturer’s instructions. Cell viability% = Absorbance of (experimental group-blank control group)/absorbance of (control group-blank control group) × 100%.
ROS measurement
Briefly, ROS levels were assessed using the dichloro-dihydro-fluorescein diacetate (DCFH-DA) probe (BestBio, Shanghai, China). RAW264.7 cells were cultured in 35 mm confocal dishes and exposed to an SiO2 suspension (50 μg/cm2) for 24 h. The DCFH-DA probe (1:1000) was added to the cells and incubated for 20 min, followed by three washes with phosphate-buffered saline (PBS). Fluorescence intensity was observed via laser scanning confocal microscopy.
Lipid peroxidation measurement
Lipid peroxidation was measured using the C11-BODIPY581/591 probe (Thermo Fisher Scientific, Waltham, MA, USA). Upon ROS-induced intracellular lipid peroxidation, the fluorescent signal of this probe shifts from red to green. The fluorescent signal was detected using laser scanning confocal microscopy (LSM800, Carl Zeiss, Jena, Germany). RAW264.7 cells were incubated for 20–30 min with C11-BODIPY581/591 (1 μM) in a confocal dish. The fluorescence of the probe was measured by simultaneous acquisition of the red (Texas red) and green (fluorescein isothiocyanate) signals.
Immunofluorescence staining
Frozen lung tissue sections or cells were fixed in 4% paraformaldehyde for 25 min at room temperature, washed with PBS for 15 min, permeabilized for 20 min with 0.3% Triton X-100, and blocked for 1 h with 1% bovine serum albumin. Anti-XCT antibody (1:100, #ab37185, Abcam, Cambridge, UK) and anti-GPX4 antibody (1:100, #ab125066, Abcam, Cambridge, UK) were incubated with the sections for 1–2 h at room temperature. The nucleus was labeled using 4′,6-diamidino-2-phenylindole (DAPI). Fluorescent images were acquired via laser scanning confocal microscopy (LSM800, Carl Zeiss, Jena, Germany).
Histology
After fixation in 4% paraformaldehyde, lung tissues were embedded in paraffin and cut into 6-μm-thick paraffin sections. The sections were stained with hematoxylin and eosin (H&E) to visualize silicosis nodules and Sirius red and Masson to visualize fibrosis. The procedures were conducted according to the manufacturer’s instructions.
Transmission electron microscopy
The lung tissues were fixed with 2.5% glutaraldehyde and 1% osmium tetroxide and cut into 60–70 nm-thick sections. After staining with uranyl acetate and lead citrate, mitochondrial morphological features were observed via TEM (JEOL, Tokyo, Japan).
Enzyme-linked immunosorbent assay
The levels of interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and transforming growth factor-β (TGF-β) in the supernatant were analyzed using enzyme-linked immunosorbent assay kits according to the manufacturer’s instructions (JONLN Reagent Co., Ltd, Shanghai, China). The absorbance was detected at 450 nm.
Western blotting
The lung tissue homogenates and cells were ground using a grinder. Total protein was extracted using the Whole Cell Lysis Assay Kit (KeyGEN BioTECH, Nanjing, China). Proteins were separated via 10%–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes. The membranes were blocked in 5% skim milk at room temperature for 2 h and washed six times (5 min each) with PBS containing Tween 20. Next, the membranes were incubated with anti-XCT (1:1000, #ab37185, Abcam, Cambridge, UK), anti-GPX4 (1:1000, #ab125066, Abcam, Cambridge, UK), anti-α-SMA antibody (1:1000, #AF1032, Affinity Biosciences, Jiangsu, China), anti-MMP-9 antibody (1:1000, #ab38898, Abcam, Cambridge, UK), anti-collagen-1 antibody (1:1000, #ab260043, Abcam, Cambridge, UK), and anti-β-actin antibody (1:5000, #SC-47778, Santa Cruz Biotechnology, Heidelberg, Germany) antibodies for 1–2 h at 37°C or overnight at 4°C, followed by washing with PBS containing Tween 20 and incubation with a goat antirabbit secondary antibody (1:5000, ZSGB-BIO, Beijing, China) at room temperature for 2–4 h. The membranes were incubated with enhanced chemiluminescence solution for 5 s to visualize the protein bands.
Quantitative reverse transcription-polymerase chain reaction
Total RNA was extracted from the lung homogenates and cell lysates using the RNA simple Total RNA Kit (TIANGEN, Beijing, China). RNA quality was detected using SimpliNano (GE Healthcare, Little Chalfont, UK). PrimeScript RT Master Mix (TaKaRa, Shiga, Japan) was used for the reverse transcription of RNA into cDNA, and then quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was performed using TB Green Premix Ex Taq II (TaKaRa, Shiga, Japan) according to the manufacturer’s instructions. The cycling conditions were as follows: 30 s at 95°C followed by 40 cycles of 95°C for 5 s and 60°C for 30 s. The following qRT-PCR primers were used: GPX4: forward, 5′-TGTGCATCCCGCGATGATT-3′, reverse, 5′-CCCTGTACTTATCCAGGCAGA-3′; XCT: forward: 5′-GGCACCGTCATCGGATCAG-3′, reverse, 5′-CTCCACAGGCAGACCAGAAAA-3′;GAPDH: forward, 5′-AGGTCGGTGTGAACGGATTTG-3′, reverse, 5′-GGGGTCGTTGATGGCAACA-3′. The cycle threshold (Ct) values were acquired, and the relative expression of XCT and GPX4 was calculated using the 2−ΔΔCt method. GAPDH was used as an internal control. All experiments were performed in triplicates.
Statistical analysis
All statistical analyses and visualization of graphs were performed using the GraphPad 8 software (GraphPad, Inc., La Jolla, CA, USA). Data are shown as mean ± standard deviation. Comparisons between two groups were performed using Student’s t-test. Multiple groups were analyzed using one-way analysis of variance followed by Tukey’s multiple comparisons test. P < 0.05 was considered to indicate statistically significant results.
Results
SiO2 promoted lung injury and fibrosis in mice
H&E staining revealed that SiO2 injured the normal structure, thickened the alveolar wall, and caused infiltration of inflammatory cells in the lungs. Silicosis nodules were detected in SiO2-induced lung tissues, whereas no significant histological changes were observed in the saline group (Fig. 1A). Masson and Sirius red staining were performed to observe lung fibrosis. Compared to that in the saline group, collagen deposition obviously increased in the SiO2 group (Fig. 1B and C). α-sma and collagen-1 expression are related to lung fibrosis, and western blot analysis revealed that SiO2 upregulated the expression of these proteins (Fig. 1D and E). qRT-PCR analysis showed that the mRNA levels of α-sma and collagen-1 were markedly increased (Fig. 1F and G). Collectively, SiO2 facilitated lung injury and fibrosis in mice.
Figure 1.
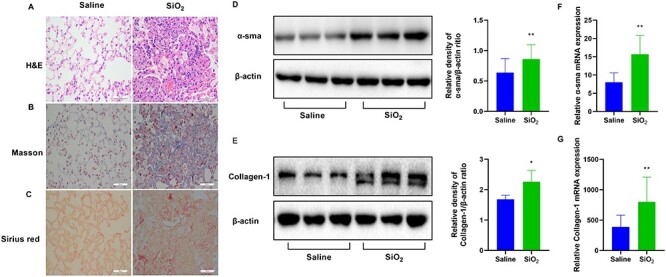
SiO2 promotes lung injury and fibrosis in mice. To induce lung injury and fibrosis, mice were intratracheally injected with SiO2 (100 mg/ml). Representative images of (A) hematoxylin and eosin (H&E) staining, (B) Masson staining, and (C) Sirius red staining of the lung tissue sections. Scale bars, 50 μm. Western blotting and quantification of the (D) α-smooth muscle actin (α-sma) and (E) collagen-1 proteins in the indicated groups. mRNA expression of (F) α-sma and (G) collagen-1 in the indicated groups. *P < 0.05 vs. saline group; **P < 0.01 vs. saline group.
Ferroptosis occurred in mice after treatment with SiO2
Next, we examined whether ferroptosis occurred in SiO2-treated mice. Iron is necessary for ferroptosis for inducing the Fenton reaction. Thus, ferroptosis is largely characterized by excessive iron levels. SiO2 increased iron concentration in the SiO2-treated group (Fig. 2A). Lipid peroxidation is a well-known biomarker of ferroptosis. MDA is an important endogenous metabolite of lipid peroxidation. We estimated MDA levels in the lung tissues using the MDA assay kit and immunofluorescence. As shown in Figure 2B and C, MDA levels were increased in the SiO2 group. GSH and GSSG are characteristic markers of ferroptosis. We observed that the GSH/GSSG ratio was decreased in SiO2-treated mice (Fig. 2D). Based on the importance of GPX4 and XCT in the regulation of ferroptosis, we measured the protein and mRNA expression of these molecules in mice. GPX4 expression was decreased and XCT expression was increased in SiO2-treated mice (Fig. 2E–H). Ferroptosis is known to cause typical changes in the mitochondrial morphology by reducing cristae, and rupturing the outer membrane. To identify which cell types involved in ferroptosis, we scanned all cell types with TEM in the lungs of mice. Compared to that in the saline group, we observed reduced numbers of cristae, and a ruptured outer membrane in macrophages from the SiO2-treated lung tissues (Fig. 2I). These results indicated that ferroptosis occurred in macrophages. Therefore, we focused ferroptosis in macrophages in further experiments.
Figure 2.
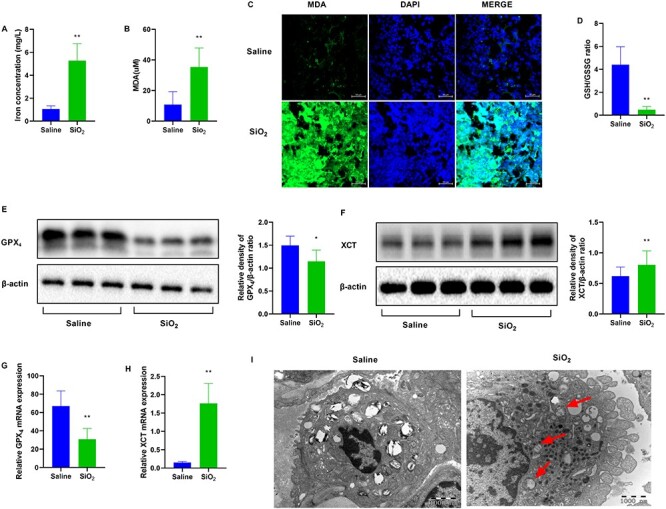
SiO2 induces ferroptosis in mice. Mice were intratracheally injected with SiO2 (100 mg/ml) for 56 days. n = 6 per group. (A) Iron concentration in the lung tissue sections from the indicated groups. (B) Malondialdehyde (MDA) levels for peroxide formation and (C) MDA staining. Scale bars, 50 μm. (D) The glutathione/glutathione oxidized (GSH/GSSG) assay. Western blotting and quantification of the (E) GPX4 and (F) XCT proteins in lung tissues from saline and SiO2 groups. mRNA expression of (G) GPX4 and (H) XCT in lung tissues from mice exposed to saline or SiO2. (I) Mitochondrial morphological changes were detected via transmission electron microscopy (TEM). Typical mitochondrial morphology in ferroptosis is indicated with red arrows. Scale bars, 1000 nm. *P < 0.05 vs. saline group; **P < 0.01 vs. saline group.
Ferroptosis was observed in SiO2-treated RAW264.7 cells
To examine whether SiO2-induced ferroptosis occurred in vitro, we used mouse macrophages RAW264.7. Cell viability showed that the number of dead RAW264.7 cells was higher in the SiO2-treated group than in the saline group (Fig. 3A). Iron concentration increased in the SiO2 group (Fig. 3B). ROS and lipid peroxidation, which are well-known biomarkers of ferroptosis, were measured in RAW264.7 cells after SiO2 treatment using a DCFH-DA probe, MDA, GSH/GSSG, and the C11-BODIPY581/591 assay kit. MDA concentration was increased and the GSH/GSSG ratio was decreased in SiO2-treated cells (Fig. 3C and D). The number of positive cells stained by DCFH-DA probe was increased in the SiO2 group (Fig. 3E). The number of unoxidized positive cells stained with C11-BODIPY581/591 was decreased, whereas that of oxidized positive cells was increased after treatment with SiO2 (Fig. 3F), indicated increased lipid peroxidation. Additionally, GPX4 expression was decreased and XCT expression was increased in SiO2-treated cells (Fig. 3G–L). These findings indicated that SiO2 stimulated ferroptosis in RAW264.7 cells.
Figure 3.
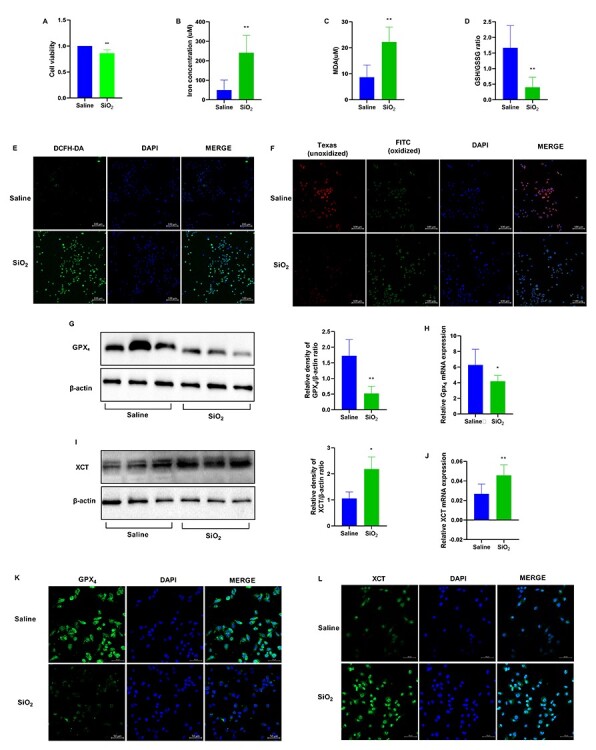
SiO2 induces ferroptosis in RAW264.7 cells. RAW264.7 cells were treated with saline or SiO2 suspension at 50 μg/cm2 for 24 h. n = 3 per group. (A) Cell viability of RAW264.7 cells detected with CCK-8 assay. (B) Iron concentration in RAW264.7 cells in the indicated groups. (C) Malondialdehyde (MDA) levels and (D) The glutathione/glutathione oxidized (GSH/GSSG) assay. (E) Reactive oxygen species (ROS) was observed using DCFH-DA. Scale bars, 100 μm. (F) Lipid peroxidation was measured using the C11-BODIPY581/591 probe. Scale bars, 100 μm. Western blotting and quantification of the (G) GPX4 and (I) XCT proteins in RAW264.7 cells in the saline and SiO2 groups. mRNA expression of (H) GPX4 and (J) XCT in RAW264.7 cells in the saline and SiO2 groups. Representative images of RAW264.7 cells showing the expression of (K) GPX4 and (L) XCT. Scale bars, 50 μm. *P < 0.05 vs. saline group; **P < 0.01 vs. saline group.
Fer-1 protected against oxidative stress and attenuated ferroptosis in RAW264.7 cells, whereas Z-VAD-FMK did not
Fer-1 is a classic ferroptosis inhibitor and Z-VAD-FMK is a classic apoptosis inhibitor. The number of dead cells was remarkably decreased in the SiO2 + Fer-1 group, which was not affected by Z-VAD-FMK treatment (Fig. 4A). MDA concentration was increased in SiO2-treated RAW264.7 cells and was attenuated in Fer-1-treated cells; However, Z-VAD-FMK caused no significant change (Fig. 4B). The GSH/GSSG ratio, recovered after Fer-1 treatment but not after Z-VAD-FMK treatment (Fig. 4C). ROS levels were reduced in the presence of Fer-1, but not in the presence of Z-VAD-FMK (Fig. 4D). Lipid peroxidation was reduced in RAW264.7 cells pretreated with Fer-1, whereas Z-VAD-FMK had no effect (Fig. 4E). Fer-1 treatment, but not Z-VAD-FMK treatment, significantly increased the expression of GPX4 and downregulated that of XCT (Fig. 4F–I). However, Fer-1 was not found to regulate iron concentration. The function of Fer-1 may not depend on iron metabolism while regulating lipid peroxidation. These datas suggested that ferroptosis occurred in SiO2-treated RAW264.7 cells, and that Fer-1 protected against oxidative stress and attenuated ferroptosis in RAW264.7 cells, whereas Z-VAD-FMK did not.
Figure 4.
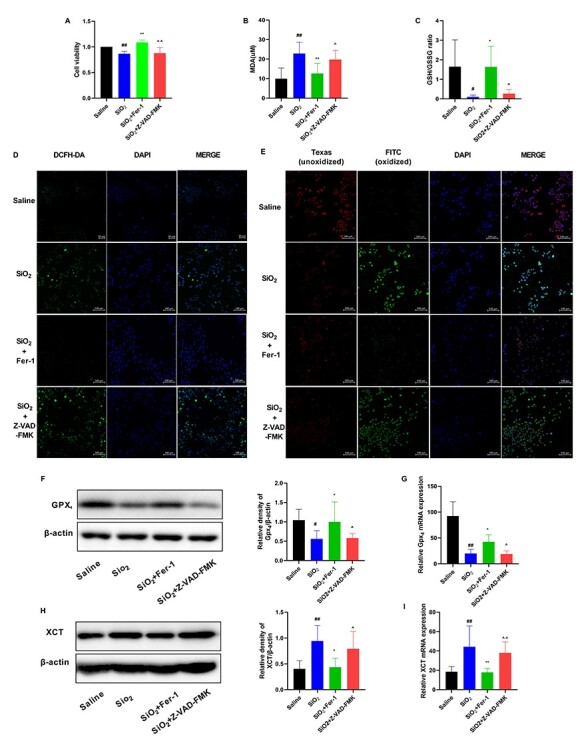
Fer-1 protects against oxidative stress and attenuates ferroptosis in RAW264.7 cells. RAW264.7 cells were treated with saline or an SiO2 suspension at 50 μg/cm2 for 24 h. Fer-1 (10 μM) or Z-VAD-FMK (20 μM) was added to RAW264.7 cells 1 h before SiO2 administration. (A) Cell viability of RAW264.7 cells detected with CCK-8 assay. (B) Malondialdehyde (MDA) levels and (C) The glutathione/glutathione oxidized (GSH/GSSG) assay. (D) ROS was observed using DCFH-DA probe. Scale bars, 100 μm. (E) Lipid peroxidation was measured using the C11-BODIPY581/591 probe. Scale bars, 100 μm. Western blotting and quantification of the (F) GPX4 and (H) XCT proteins in the indicated groups. mRNA expression of (G) GPX4 and (I) XCT in the indicated groups. #P < 0.05 vs. saline group; ##P < 0.01 vs. saline group. *P < 0.05 vs. SiO2 group; **P < 0.01 vs. SiO2 group. ^P < 0.05 vs. SiO2 + Fer-1 group; ^^P < 0.01 vs. SiO2 + Fer-1 group.
Fer-1 alleviated SiO2-induced fibrosis in vitro
Fibroblasts act as effector cells of pulmonary fibrosis by secreting extracellular matrix proteins. Thus, we examined fibroblasts in vitro. We established a coculture system with RAW264.7 cells (mouse macrophage) and MLF cells (mouse lung fibroblast) (Fig. 5A), and the supernatant and MLF cells were collected. IL-1β, TNF-α, and TGF-β are critical pro-fibrotic cytokines. The results of enzyme-linked immunosorbent assay (ELISA) showed that Fer-1 effectively inhibited the secretion of pro-fibrotic cytokines in the supernatant (Fig. 5B–D). Furthermore, we measured the expression of α-sma, collagen-1, and MMP-9 in MLF cells. Western blot analysis showed that the expression of α-sma, collagen-1, and MMP-9 was decreased by Fer-1 (Fig. 5E), which is consistent with the qRT-PCR data (Fig. 5F–H). These findings indicated that Fer-1 might alleviate fibrosis in MLF cells by inhibiting ferroptosis in SiO2-treated RAW264.7 cells, which secreted pro-fibrotic cytokines.
Figure 5.
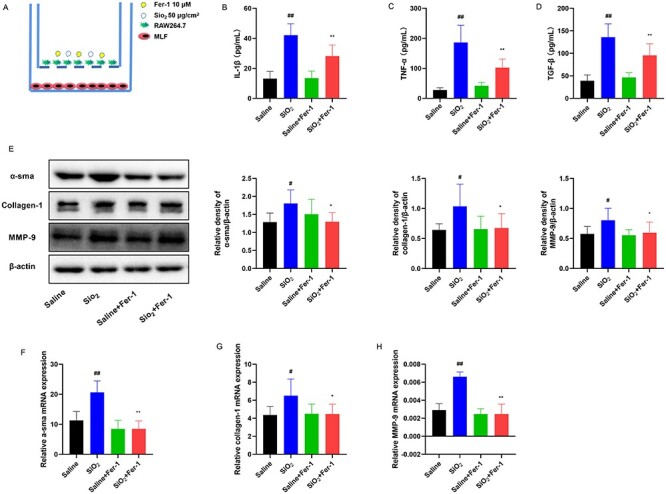
Fer-1 alleviates SiO2-induced fibrosis in vitro. RAW264.7 cells seeded into the upper wells were treated with SiO2 (50 μg/cm2) for 24 h. Fer-1 (10 μM) was added to RAW264.7 cells 1 h before SiO2 administration. MLF cells attached to the lower wells were cocultured with RAW264.7 cells in a Transwell 6-wells plate system for another 24 h. The supernatant was collected to detect cytokines, and MLF cells were collected to detect the protein and mRNA levels of α-sma, collagen-1, and MMP-9. n = 3 per group. (A) The coculture system of RAW264.7 cells and MLF cells. Levels of (B) IL-β, (C) TNF-α, and (D) TGF-β were measured in the supernatant by ELISA after 24 h of coculture. (E) Western blotting and quantification of the α-sma, collagen-1, and MMP-9 proteins in the indicated group. mRNA expression of (F) α-sma, (G) collagen-1, and (H) MMP-9 in the indicated group. #P < 0.05 vs. saline group; ##P < 0.01 vs. saline group. *P < 0.05 vs. SiO2 group; **P < 0.01 vs. SiO2 group.
Discussion
In vivo, we found that SiO2 damaged the alveolar structure, caused infiltration of inflammatory cells, and facilitated fibrosis. SiO2 increased the iron concentration and lipid peroxidation and altered the expression of ferroptosis-related genes, such as GPX4 and XCT, and the mitochondrial morphology in macrophages. These results indicated that ferroptosis occurred in macrophages. In vitro, we observed that ferroptosis occurred in SiO2-treated RAW264.7 cells, which showed iron overload, lipid peroxidation, and gene expression alterations. In addition, Fer-1 attenuated ferroptosis in SiO2-treated RAW264.7 cells by inhibiting lipid peroxidation and cell death and suppressing the expression of ferroptosis-related genes. Furthermore, Fer-1 alleviated fibrosis in MLF cells by inhibiting the ferroptosis in SiO2-treated RAW264.7 cells that secreted pro-fibrotic cytokines. Taken together, SiO2 induced ferroptosis in macrophages that secreted pro-fibrotic cytokines and resulted in fibrosis.
Iron overload can accelerate the accumulation of lipid peroxides, which depends on the Fenton reaction, and influences ferroptosis sensitivity [14]. Ferroptosis requires transferrin and the transferrin receptor to import iron from the extracellular environment [15]. Knocking out the key gene of iron metabolism, iron responsive element binding protein 2 (IREB2), has been reported to decrease ferroptosis [9]. In our study, SiO2 upregulated iron concentration to trigger ferroptosis in SiO2-induced pulmonary fibrosis. Ferroptosis is also defined as a form of regulated cell death, which is induced by lipid peroxidation. We predicted that SiO2 induces iron overload and triggers ROS production via the Fenton reaction, leading to lipid peroxidation and ferroptosis development. Taken together, both iron overload and lipid peroxidation greatly affect ferroptosis in SiO2-induced pulmonary fibrosis.
GPX4 and XCT are the main proteins involved in ferroptosis. GPX4 is a selenoprotein glutathione peroxidase [16]. Silencing of GPX4 increases embryonic mortality in postcoitum, with concomitantly increased cell death. GPX4 has been identified as a key regulator of ferroptosis [17–19]. In this study, GPX4 expression was reduced in SiO2-induced models. In agreement with previous findings, the reduced expression of GPX4 was found to induce ferroptosis. XCT imports cystine and exports glutamate simultaneously to supply glutathione that attenuates oxidative stress. Inhibition of XCT can result in GSH depletion and ferroptosis [20]. Interestingly, we observed increased expression of XCT after treatment with SiO2in vivo and in vitro, contrasting the results of previous studies. SiO2 may stimulate a negative feedback mechanism to suppress ferroptosis via the rapid consumption of GSH. Moreover, the function of XCT is unclear, although it is known to be related to ferroptosis, but silencing of the XCT gene cannot trigger ferroptosis in vivo [21]. Possibly, SiO2-induced models may initiate a protective mode against ferroptosis by increasing the expression of XCT. Altogether, SiO2 regulates GPX4 and XCT, suggesting that ferroptosis is involved in SiO2-induced pulmonary fibrosis.
Fer-1 is a synthetic compound that inhibits ferroptosis and generally accepted hydroperoxyl radicals as a scavenger. In vitro, by scavenging lipid peroxides, Fer-1 can inhibit ferroptosis. In this study, Fer-1 significantly attenuated the SiO2-induced MDA content, GSH/GSSG, ROS, and lipid peroxidation and inhibited SiO2-induced cell death. Furthermore, Fer-1 alleviated fibrosis in MLF cells by inhibiting the ferroptosis of SiO2-treated RAW264.7 cells that secreted pro-fibrotic cytokines. These results suggest that SiO2 induces ferroptosis in SiO2-induced pulmonary fibrosis.
Our study has many limitations; for example, the causes of increased iron concentration and alterations of ferroptosis-related genes are unclear. Additionally, the precise mechanism of how SiO2 induces ferroptosis in silicosis must be determined. Our further studies will focus on pathways regulating ferroptosis-related genes.
Conclusions
In this study, after macrophages engulfed SiO2, SiO2 increased the intracellular iron levels, which induced ROS and lipid peroxidation through the Fenton reaction. After the exhaustion of GSH, oxidative stress was increased, which might regulate ferroptosis-related genes, eventually resulting in ferroptosis. Ferroptotic macrophages secreting pro-fibrotic cytokines initiated the fibrosis process through an interaction with fibroblasts (Fig. 6). In conclusion, our findings indicate that SiO2-induced ferroptosis in macrophages promotes the development of pulmonary fibrosis in silicosis models.
Figure 6.
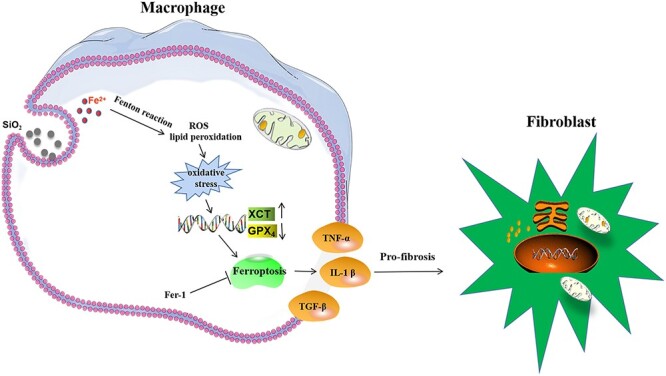
Summary illustration showing a relationship between SiO2-induced ferroptosis in macrophages and pulmonary fibrosis.
Authors’ contributions
Zhihong Liu designed the current study. Rui Bao participated in data collection and organization. Taiyang Liu and Yue Sun wrote the manuscript together. Qiushi Wang, Wei Hao, Yaoyang Liu, Sirong Chang, Meng Wang, and Yuanyuan Li performed the statistical analysis and made critical comments. The author(s) read and approved the final manuscript.
Conflicts of Interest Statement
The authors declare that they have no competing interests.
Acknowledgments
This research was funded by the National Natural Science Foundation of China grant numbers 81960018 and 82060264, Science and Technology Department of Ningxia grant number 2020BEG03008, and Ningxia Medical University Scientific Research Project grant number XT2018012. We are thankful to Ningxia Medical University for funding this work. We would like to thank Editage (www.editage.cn) for English language editing.
Contributor Information
Taiyang Liu, School of Public Health and Management, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China; NHC KEY Laboratory of Metabolic Cardiovascular Diseases Research, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China.
Rui Bao, School of Public Health and Management, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China; NHC KEY Laboratory of Metabolic Cardiovascular Diseases Research, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China.
Qiushi Wang, School of Public Health and Management, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China; NHC KEY Laboratory of Metabolic Cardiovascular Diseases Research, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China.
Wei Hao, School of Public Health and Management, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China; NHC KEY Laboratory of Metabolic Cardiovascular Diseases Research, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China.
Yaoyang Liu, School of Public Health and Management, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China; NHC KEY Laboratory of Metabolic Cardiovascular Diseases Research, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China.
Sirong Chang, School of Public Health and Management, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China; NHC KEY Laboratory of Metabolic Cardiovascular Diseases Research, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China.
Meng Wang, School of Public Health and Management, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China; NHC KEY Laboratory of Metabolic Cardiovascular Diseases Research, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China.
Yuanyuan Li, School of Public Health and Management, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China; NHC KEY Laboratory of Metabolic Cardiovascular Diseases Research, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China.
Zhihong Liu, School of Public Health and Management, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China; NHC KEY Laboratory of Metabolic Cardiovascular Diseases Research, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China.
Yue Sun, School of Public Health and Management, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China; NHC KEY Laboratory of Metabolic Cardiovascular Diseases Research, Ningxia Medical University, No. 1160, Shengli Street, Xingqing District, Yinchuan 75000, Ningxia, China.
References
- 1. Sitong D, Li C, Yiping Let al. . Dioscin alleviates crystalline silica-induced pulmonary inflammation and fibrosis through promoting alveolar macrophage autophagy. Theranostics 2019;9:1878–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Knight D, Ehrlich R, Fielding Ket al. . Trends in silicosis prevalence and the healthy worker effect among gold miners in South Africa: a prevalence study with follow up of employment status. BMC Public Health 2015;15:1258–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jindal SK. Silicosis in India: past and present. Curr Opin Pulm Med 2013;19:163–8. [DOI] [PubMed] [Google Scholar]
- 4. Song L, Weng D, Dai Wet al. . Th17 can regulate silica-induced lung inflammation through an IL-1beta-dependent mechanism. J Cell Mol Med 2014;18:1773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bois RM. Strategies for treating idiopathic pulmonary fibrosis. Nat Rev Drug Discov 2010;9:129–40. [DOI] [PubMed] [Google Scholar]
- 6. Wang M, Jin Y, Chen Set al. . The study of autophagy in alveolar macrophages of patients with coal workers’ pneumoconiosis. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2015;33:41–4. [PubMed] [Google Scholar]
- 7. Chen S, Jin Y-L, Yao S-Qet al. . Autophagy in lung tissue of rats exposed to silica dust. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2013;31:607–10. [PubMed] [Google Scholar]
- 8. Stegmann KA, De Souza JB, Riley EM. IL-18-induced expression of high-affinity IL-2R on murine NK cells is essential for NK-cell IFN-gamma production during murine Plasmodium yoelii infection. Eur J Immunol 2015;45:3431–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dixon SJ, Lemberg KM, Lemprecht MRet al. . Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yoshida M, Minagawa S, Araya Jet al. . Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun 2019;10:3145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Y, Tang M. PM2.5 induces ferroptosis in human endothelial cells through iron overload and redox imbalance. Environ Pollut 2019;254:112937–47. [DOI] [PubMed] [Google Scholar]
- 12. Li X, Zhuang X, Qiao T. Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem Biophys Res Commun 2019;519:240–5. [DOI] [PubMed] [Google Scholar]
- 13. Li X, Duan L, Yuan Set al. . Ferroptosis inhibitor alleviates radiation-induced lung fibrosis (RILF) via down-regulation of TGF-β1. J Inflamm (Lond) 2019;16:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stockwell BR, Angeli JPF, Bayir Het al. . Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017;171:273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gao M, Monian P, Quadri Net al. . Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 2015;59:298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Imai H, Matsuoka M, Kumagai Tet al. . Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol 2017;403:143–70. [DOI] [PubMed] [Google Scholar]
- 17. Sakai O, Uchida T, Imai H, Ueta T. Glutathione peroxidase 4 plays an important role in oxidative homeostasis and wound repair in corneal epithelial cells. FEBS Open Bio 2016;6:1238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sakai O, Uchida T, Roggia MFet al. . Role of glutathione peroxidase 4 in glutamate-induced oxytosis in the retina. PLoS One 2015;10:e0130467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Imai H, Hirao F, Sakamoto Tet al. . Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun 2003;305:278–86. [DOI] [PubMed] [Google Scholar]
- 20. Jiang L, Kon N, Li Tet al. . Ferroptosis as a p53-mediated activity during tumor suppression. Nature 2015;520:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang H, An P, Xie Eet al. . Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017;66:449–65. [DOI] [PMC free article] [PubMed] [Google Scholar]