

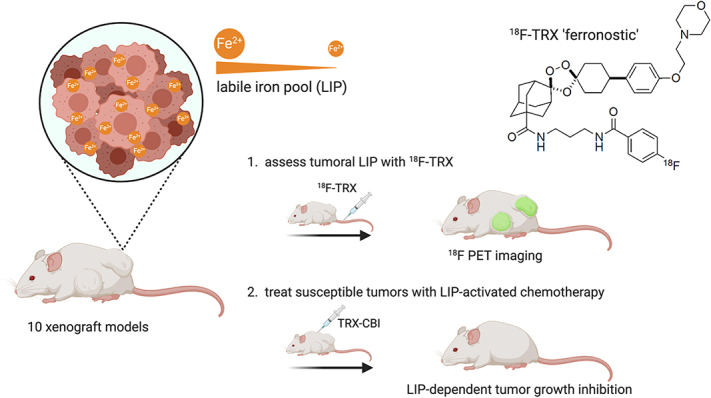
Visual Abstract
Keywords: labile iron pool, theranostics, PET
Abstract
Although cancer has been known for decades to harbor an insatiable appetite for iron, only recently has the chemistry emerged to exploit this altered state therapeutically, by targeting the expanded cytosolic labile iron pool (LIP) of the cancer cell. The state of the art includes therapies that react with the LIP to produce cytotoxic radical species (in some cases also releasing drug payloads) and molecules that exacerbate LIP-induced oxidative stress to trigger ferroptosis. Effectively implementing LIP-targeted therapies in patients will require biomarkers to identify those tumors with the most elevated LIP and thus most likely to succumb to LIP-targeted interventions. Toward this goal, we tested whether tumor uptake of the novel LIP-sensing radiotracer 18F-TRX aligns with tumor sensitivity to LIP-targeted therapies. Methods: 18F-TRX uptake was assessed in vivo among 10 subcutaneous and orthotopic human xenograft models. Glioma and renal cell carcinoma were prioritized because these tumors have the highest relative expression levels of STEAP3, the oxidoreductase that reduces ferric iron to the ferrous oxidation state, in the Broad Institute Cancer Cell Line Encyclopedia. The antitumor effects of the LIP-activated prodrug TRX-CBI, which releases the DNA alkylator CBI, were compared in mice bearing U251 or PC3 xenografts, tumors with high and intermediate levels of 18F-TRX uptake, respectively. Results: 18F-TRX showed a wide range of tumor accumulation. An antitumor assessment study showed that the growth of U251 xenografts, the model with the highest 18F-TRX uptake, was potently inhibited by TRX-CBI. Moreover, the antitumor effects against U251 were significantly greater than those observed for PC3 tumors, consistent with the relative 18F-TRX–determined LIP levels in tumors before therapy. Lastly, a dosimetry study showed that the estimated effective human doses for adult male and female mice were comparable to those of other 18F-based imaging probes. Conclusion: We report the first evidence—to our knowledge—that tumor sensitivity to an LIP-targeted therapy can be predicted with a molecular imaging tool. More generally, these data bring a new dimension to the nuclear theranostic model by showing a requirement for imaging to quantify, in situ, the concentration of a metastable bioanalyte toward predicting tumor drug sensitivity.
All cancer cells have an inescapable need for more iron than do normal tissues to support the intense energetic demands that come with indefinite proliferation (1). Cancer cells are thought to increase their intracellular supply of ferric iron (Fe3+) primarily by augmenting expression of the transferrin receptor, which binds and internalizes circulating holo-transferrin through endocytosis (Fig. 1A). Cancer cells also overexpress the oxidoreductase STEAP3 within lysosomes, which enables the reduction of ferric iron to the more soluble and bioavailable ferrous oxidation state (Fe2+). Ferrous iron is subsequently transported into the cytosol—the reservoir of cytosolic ferrous iron making up the labile iron pool (LIP)—where it can then be trafficked to ferritin for storage or to the mitochondria for conversion to the iron–sulfur cluster and heme cofactors that support essential enzyme functions in the cell (2). An expanded LIP, however, risks oxidative damage from the reactive oxygen species produced via ferrous iron–promoted Fenton reactivity with oxygen and hydrogen peroxide. Normal cells thus maintain a modest LIP, in part by storing iron in ferritin or exporting excess iron via ferroportin. To survive with an elevated LIP, cancer cells become especially reliant on cellular defense mechanisms that protect from reactive oxygen species, a state that if perturbed can lead to iron-dependent cell death, or ferroptosis (3). Thus, an expanded LIP is emerging as a defining metabolic feature that distinguishes cancer cells from normal cells.
FIGURE 1.

Expansion of LIP is a general feature of cancer and can be targeted for cancer treatment and imaging. (A) How cancer cells can adjust relative expression levels of iron regulatory proteins to expand LIP. LIP expansion comes with either increase in expression of proteins required to import ferric iron and convert it to ferrous iron (TFRC, STEAP3, DMT1) or reduction in machinery cells used to export iron through reduced expression of FPN or increased expression of FPN1 inhibitory protein HAMP. (B) Structure of artemisinin and carbon-centered radical arising from reaction of endoperoxide with iron(II)heme in blood-stage Plasmodium falciparum parasites, leading to oxidative stress from reactive oxygen species, protein alkylation, and parasite death. Structure of TRX-CBI and LIP promoted reaction leading to release of cyclopropylbenzindoline cytotoxin. (C) Structures of antimalarial artefenomel and 18F-TRX reactivity–based probe to measure LIP with PET. On exposure to LIP, 1,2,4-trioxolane reacts to produce 18F-linked carbon-centered radical, which crosslinks biomolecules and is retained within cells. CBI = cyclopropylbenzindoline; DMT1 = divalent metal transporter 1; FPN1 = ferroportin 1; HAMP = hepcidin; ROS = reactive oxygen species; TFRC = transferrin receptor; TRX = trioxolane.
Because no other metal can substitute for iron in biology, it has long been hypothesized that ferroaddiction in cancer might be exploited therapeutically (1). However, chemical strategies focusing on depriving cancer of ferric iron with chelation therapy or delivering cytotoxic payloads to cancer by targeting transferrin receptor (e.g., transferrin-CRM107) were not effective clinically (4). More recently, the emergence of ferrous iron–specific chemistry has enabled a new therapeutic approach that directly targets LIP expansion in cancer cells (5). This approach draws conceptually from the mechanism of the antimalarial artemisinin, which kills blood-stage Plasmodium falciparum parasites via reactive intermediates formed by following an initial Fenton chemistry with ferrous iron heme (6). This reaction yields a heme adduct that can no longer be detoxified by the parasite, resulting in broad oxidative damage to parasite membranes and macromolecules (Fig. 1B). The overwhelming clinical success of artemisinin has established that ferrous iron–dependent pharmacology involving the Fenton reaction can be exploited to safely treat disease in humans (including children). Newer synthetic antimalarial 1,2,4-trioxolanes (e.g., artefenomel) meanwhile inspired the design of LIP-activated trioxolane prodrugs, such as trioxolane-cyclopropylbenzindoline (TRX-CBI), that are activated by the expanded LIP within cancer cells to produce cytotoxic free radicals while releasing and activating CBI, a potent DNA alkylator (Fig. 1B) (5,7). Proof-of-concept data in preclinical models show that trioxolane caging of therapeutic payloads increases the tolerated doses of many drugs and that intratumoral LIP levels efficiently activate the prodrug, releasing the payload to realize potent antitumor effects.
In parallel with these developments, other new approaches for treating cancer by targeting iron dyshomeostasis have emerged within the past 10 years. For example, several groups have developed therapies that exacerbate LIP-associated redox stress and trigger ferroptosis. (3) These include several mechanistically unrelated drugs, such as inhibitors of the cystine/glutamate antiporter xCT (e.g., erastin) or biochemical inhibitors (e.g., RSL3) of glutathione peroxidase 4. Moreover, an emerging narrative from several groups suggests a role for ferroptosis in tumor immunology, both as a mechanism by which tumor cells productively respond to immune checkpoint inhibitors and as a vector to produce immune stimulatory signaling molecules in the tumor microenvironment (8,9). Lastly, recent data have also shown that inhibiting lysosome function with autophagy inhibitors in fact kills cancer cells by provoking acute LIP starvation (10). Collectively, these observations highlight the promise of exploiting the LIP for cancer treatment.
One crucial unmet need to address in advancing clinical trials with LIP-targeted therapies is a method to measure LIP in the tumors of cancer patients. Indeed, LIP levels in patient tumors have never been quantified, and a biomarker for LIP could be applied prospectively to define which tumors harbor the highest LIP levels and might be especially vulnerable to LIP-targeted therapies. The benefit of such a theranostic approach is now well established and in routine clinical use to select patients for endoradiotherapies such as 177Lu-DOTATATE (Lutathera; Advanced Accelerator Applications) and 131I-iobenguane (Azedra; Progenics Pharmaceuticals) (11).
Since ferrous iron rapidly oxidizes once the native cellular environment is disrupted, it cannot be reliably quantified from tumor biopsy samples. Therefore, we developed a novel radiotracer termed 18F-TRX to quantify LIP in situ with PET (12). 18F-TRX is based on the structure of artefenomel, an antimalarial with excellent pharmacokinetics that is currently in phase 2 clinical trials (Fig. 1C) (13). On reaction with the LIP in tissues, 18F-TRX fragments to generate an 18F-labeled carbon-centered radical intermediate that crosslinks cellular proteins and becomes sequestered within those cells. Thus, stable cellular accumulation of 18F is expected to be proportional to LIP concentration. We recently disclosed imaging data supportive of this mechanism of action in mice by showing that tissue retention of 18F-TRX was responsive to supplementation or withholding of iron (12). Herein, we report the first evidence—to our knowledge—suggesting that 18F-TRX can be applied to identify tumors likely to respond to an LIP-targeting drug, and these data bring a new dimension to the nuclear theranostic model by showing the value of quantifying the concentration of a metastable bioanalyte in situ and thereby predicting tumor drug sensitivity.
MATERIALS AND METHODS
General Methods
Chemicals were purchased and used without further purification. A rabbit polyclonal antibody targeting STEAP3 (catalog number 17186-1-AP) was purchased from Proteintech. A polyclonal antibody targeting β-actin (catalog number 656120) was purchased from Fisher Scientific. Isotopes were produced by the Radiopharmacy at the University of California, San Francisco. All cell lines were purchased from American Type Culture Collection and cultured according to the manufacturer’s recommendations. Both 18F-TRX and TRX-CBI were prepared according to previously reported procedures (5,12).
FerroFarRed
Cancer cells (10,000/well) were seeded into 96-well clear-bottom plates. After 24 h, the culture medium was removed, and 100 μL of Hanks balanced saline solution containing FerroFarRed (5 μM; Groyo Chemical) and Hoechst stain (1 μg/mL) were added into each well and incubated at 37°C for 50 min. The solution was removed and replaced with Hanks balanced saline solution buffer. The plate was scanned on a fluorescence microscope (IN Cell 6500 analyzer; GE Healthcare). The fluorescent intensity was quantified by the overall fluorescence strength generated by FerroFarRed dye.
Immunoblot
Cells were lysed in radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitor cocktails (ThermoFisher Scientific). The cell lysates (20 μg/sample) were separated using 4%–12% tris acetate gels (Invitrogen). The proteins were transferred to polyvinylidene difluoride membranes, blocked with 5% (w/v) bovine serum albumin solution, and immunoblotted with anti-STEAP3 antibody (1:1,000) or actin (1:4,000) for 1 h at room temperature. The blots were incubated with a secondary antibody coupled to horseradish peroxidase (1:5,000) for 30 min and developed using enhanced chemiluminescent detection reagent (ThermoFisher Scientific) and ultrasensitive film (Biorad).
Animal Studies
All animal experiments were performed under the approval of the Institutional Animal Care and Use Committee at the University of California, San Francisco. Athymic nu/nu mice (4−6 wk old) were purchased from Charles River. Cells (1 × 106) were mixed with Matrigel (Corning) and RPMI 1640 medium (1:1 v/v) and implanted into the left leg. Tumors were palpable by 2–4 wk. Orthotopic glioma tumors were established as previously described (14). Ten days after injection, tumor burden was confirmed by MRI and the mice were transferred for PET/CT studies.
For antitumor assessment studies, TRX-CBI was dissolved in a formulation comprising 5/4/1 polyethylene glycol 400/20% 2-hydroxypropylcylcodextrine in water/dimethylsulfoxide. The mice were treated with TRX-CBI (7.5 mg/kg) or saline via intraperitoneal administration. Tumor volumes were measured with digital calipers, and mouse weight was monitored twice weekly.
Small-Animal PET/CT Imaging
Data were acquired with a Siemens Inveon small-animal PET/CT scanner. For dynamic acquisitions, the mice were anesthetized beforehand with 1.5%–2% isoflurane and positioned on the scanner bed, after which approximately 30 MBq of 18F-TRX were injected via the tail vein in a volume of 100−150 μL per mouse. All PET imaging data were decay-corrected, reconstructed with CT-based attenuation correction, and analyzed with AMIDE software. Regions of interest were manually placed to calculate SUVmean data. Dosimetry was calculated as previously reported (15).
Biodistribution Studies
After radiotracer injection, the mice were euthanized with CO2 (gas) asphyxiation. Blood and tissues were collected, washed, dried, and weighed. The activity of each tissue was measured with a γ-counter. All data were decay-corrected and expressed as a percentage of the injected dose per gram of tissue.
Autoradiography
Ninety minutes after injection of 18F-TRX, the mice were anesthetized and perfused with cold phosphate-buffered saline via cardiac puncture. Tissues were immediately collected and flash-frozen in optimal-cutting-temperature compound on dry ice, sectioned to 20 μm, and mounted on glass slides. The slides were then exposed on a GE Healthcare phosphor storage screen and developed on an Amersham Typhoon 9400 phosphor imager. The autoradiography images were processed using ImageJ software.
Statistics
An unpaired, 2-tailed Student t test was used to determine statistically significant differences between 2 treatment arms. Statistically significant differences in animal survival were analyzed with a log rank (Mantel–Cox) test. Pearson correlation was performed using Prism, version 8.0 (GraphPad). In all cases, P values of less than 0.01 were reported as statistically significant.
RESULTS
We previously demonstrated the feasibility of LIP-dependent tumor detection with 18F-TRX using a small pilot group of mouse and human tumor models (12). To better understand the dynamic range of 18F-TRX uptake in tumors, we profiled 18F-TRX uptake in a larger panel of 10 subcutaneous human tumor xenografts. For this study, we chose to prioritize glioma and renal cell carcinoma, because a comparative analysis of STEAP3 expression levels across cell lines from the Broad Institute Cancer Cell Line Encyclopedia (https://portals.broadinstitute.org/ccle) showed these 2 tumor types to have the highest mean expression, and previous studies have relied on STEAP3 overexpression as a biomarker of iron dyshomeostasis in cancer (Supplemental Fig. 1; supplemental materials are available at http://jnm.snmjournals.org) (16). Biodistribution studies at 90 min after injection in mice bearing subcutaneous xenografts showed that the tracer uptake ranged from approximately 5%–1% of the injected dose per gram of tissue (Fig. 2A). We compared these data with STEAP3:actin expression levels per tumor type (Fig. 2B). A scatterplot showed a trend toward a positive correlation with 18F-TRX uptake in tumors, with a Pearson correlation r value of 0.56. However, there were some examples in which relative STEAP3 expression over the tumor panel did not correlate well with relative tumoral uptake of 18F-TRX, and the correlation did not achieve statistical significance (P = ∼0.08; Supplemental Fig. 2). We did find that the 18F-TRX data aligned with LIP levels detected using the Fe(II)-activated fluorogenic dye FerroFarRed (17), which is activated by a distinct Fe(II)-dependent reactivity, suggesting that some of the cancer cell lines use mechanisms beyond STEAP3 overexpression to expand LIP (Fig. 2C).
FIGURE 2.

Survey of 18F-TRX uptake in vivo reveals range of LIP concentrations in human glioma and renal tumors. (A) Biodistribution data collected from subcutaneous tumors in nu/nu mice at 90 min after injection of 18F-TRX (n = 4/tumor type). (B) Immunoblot for STEAP3 showing that protein expression aligns in part with 18F-TRX uptake among those tumors with highest and lowest radiotracer uptake in vivo. Comparison of STEAP3:actin ratios (n = 3/cell line) with 18F-TRX uptake per tumor cell indicated trend toward positive correlation (Pearson r = 0.56), although correlation did not achieve statistical significance (P = ∼0.08; Supplemental Fig. 2). (C) Immunofluorescence data showing relative intensity of LIP in A172 and U251 cells in vitro using FerroFarRed. Below the images is shown quantification of fluorescent intensity. *P < 0.01. %ID = percentage injected dose; a.u. = arbitrary units; RFU = relative fluorescence units; TRX = trioxolane.
We next asked whether tumors with high avidity for 18F-TRX are sensitive to LIP-targeted therapies. For these studies, we tested tumor sensitivity to TRX-CBI, an LIP-activated prodrug that releases the DNA alkylator cyclopropylbenzindoline (5). After showing that U251 cells were sensitive to the drug in vitro (Supplemental Fig. 3), we performed a 35-d antitumor assessment study treating mice bearing subcutaneous U251 xenografts with vehicle or TRX-CBI (7.5 mg/kg every 4 d). Tumor volume was significantly reduced by TRX-CBI compared with vehicle (Fig. 3A; Supplemental Fig. 3). Moreover, TRX-CBI significantly extended mouse survival, and mice treated with TRX-CBI lived past 40 d, whereas all mice receiving vehicle were euthanized by day 38 (Fig. 3B).
FIGURE 3.

Tumor model that has high uptake of 18F-TRX and is sensitive to treatment with LIP-targeted therapy. (A) Tumor volume data from antitumor assessment study show that U251, human glioma model with highest 18F-TRX uptake among tumors surveyed, is responsive to treatment with TRX-CBI. Mice (n = 6/arm) were dosed with vehicle or TRX-CBI at 7.5 mg/kg every 4 d. (B) Kaplan–Meier plot shows relative survival of mice treated with vehicle vs. TRX-CBI. Hazard ratio was 27. *P < 0.01. TRX = trioxolane.
We then asked whether tumors with significantly different levels of 18F-TRX uptake had differential sensitivity to TRX-CBI. For this study, we compared tumor responses between the U251 and PC3, as we previously showed that PC3 tumors have significantly lower 18F-TRX uptake (Fig. 4A) but subcutaneous PC3 tumors are still highly sensitive to treatment with TRX-CBI (7.5 mg/kg every 4 d) (5). Calculating the percentage change in tumor volume from day 0 showed that TRX-CBI more potently inhibited U251 tumor growth than PC3 growth (Fig. 4B). The differential sensitivity was observed despite the fact that both tumors expressed equivalent levels of STEAP3 (Fig. 4C; Supplemental Fig. 4). Moreover, we tested whether the uptake of 68Ga-citrate aligned with tumor sensitivity, as 68Ga3+ is a ferric iron mimetic that binds to transferrin in serum (18). Interestingly, PC3 tumors were significantly more avid for 68Ga3+ than were U251 tumors, further underscoring the utility and importance of measuring intracellular LIP levels specifically (with 18F-TRX) as opposed to net ferric iron import via 68Ga3+-transferrin (Fig. 4C).
FIGURE 4.

Pretreatment tumor uptake of 18F-TRX correlates with sensitivity to LIP-activated prodrug. (A) Plot of 18F-TRX uptake in nu/nu mice bearing U251 or PC3 tumors, human prostate cancer model for which we previously conducted antitumor assessment study with TRX-CBI (left); plot of uptake of FerroFarRed in U251 vs. PC3 cells, which shows that U251 has higher LIP than PC3 (middle); and representative fluorescent microscopy images (right). (B) Plot showing percentage change in PC3 and U251 tumor volume from day 0. Mice were treated with TRX-CBI at 7.5 mg/kg, every 4 d. (C) Representative immunoblot data showing that PC3 and U251 express equivalent STEAP3 (left) and biodistribution data collected 4 h after injection of 68Ga-citrate in mice bearing subcutaneous PC3 or U251 tumors (n = 4/tumor) (right). Despite higher LIP levels, U251 consumes less 68Ga3+ bound to transferrin than do PC3 tumors. *P < 0.01. %ID = percentage injected dose; a.u. = arbitrary units; RFU = relative fluorescence units; TRX = trioxolane.
Our preclinical data showing that glioma can have sufficiently high LIP levels for targeted drug delivery coincide with recent data indicating that tumors from patients with high-grade glioma overexpress STEAP3 and transferrin receptor and that genetically ablating STEAP3 potently inhibits the proliferation and survival of glioma (16). Thus, LIP-targeting therapies may be particularly well suited to glioma, and to confirm that LIP expansion is detectable with 18F-TRX PET in a more clinically relevant glioma model, we established orthotopic U87 MG tumors in mouse brains. Glioma lesions were visually obvious on PET, and region-of-interest analysis showed significantly higher 18F-TRX uptake at 90 min after injection than in involved adjacent white matter (Figs. 5A and 5B; Supplemental Fig. 5). Analysis of the radiotracer distribution within the brain with autoradiography showed 18F-TRX uptake within the tumor and low levels in normal brain (Fig. 5C).
FIGURE 5.

LIP expansion is detectable in orthotopic glioma model with 18F-TRX. (A) 18F-TRX PET/CT data showing radiotracer uptake in U87 MG tumor (arrow) implanted within right hemisphere of mouse brain. Image was acquired at 90 min after injection. (B) Quantification of 18F-TRX uptake using region-of-interest analysis of PET data from mice bearing U87 MG tumors (n = 3). Tumor uptake was compared with uptake in uninvolved normal white matter in contralateral region of brain. *P < 0.01. (C) Digital autoradiography showing distribution of radiotracer within coronal section of mouse brain. Tissue was stained with hematoxylin and eosin and merged with pseudo color image of autoradiography. %ID = percentage injected dose; DAR = digital autoradiography; H&E = hematoxylin and eosin; TRX = trioxolane.
Lastly, to assess the feasibility of human studies with 18F-TRX, we conducted a rodent dosimetry study to estimate human-equivalent organ-absorbed doses and effective doses (Table 1). Dosimetry was calculated by analyzing dynamic 90-min PET acquisitions in tumor-naïve male and female nu/nu mice. The data were fitted using the EXM module of the OLINDA/EXM package, version 1.1, to derive time-integrated activity coefficients. Organ doses were estimated using OLINDA, version 2.0, and effective doses were computed using the ICRP publication 103 tissue-weighting factors implemented in OLINDA, version 2.0. In both sexes, the effective dose was less than 0.02 mSv/MBq, or the approximate value for a routine PET acquisition with 18F-FDG (19). The organs receiving the highest absorbed dose were the heart wall, lungs, and liver.
TABLE 1.
Human-Equivalent Doses for 18F-TRX
| Absorbed dose | ||
|---|---|---|
| Organ | Female (60 kg) | Male (73 kg) |
| Adrenals | 0.0136 ± 0.0008 | 0.0141 ± 0.0005 |
| Brain | 0.0042 ± 0.0002 | 0.0030 ± 0.0006 |
| Breast | 0.0081 ± 0.0003 | |
| Esophagus | 0.0136 ± 0.0004 | 0.0112 ± 0.0013 |
| Eyes | 0.0067 ± 0.0003 | 0.0073 ± 0.0001 |
| Gallbladder wall | 0.0123 ± 0.0007 | 0.0161 ± 0.0006 |
| Left colon | 0.0116 ± 0.0004 | 0.0111 ± 0.0002 |
| Small intestine | 0.0096 ± 0.0004 | 0.0113 ± 0.0002 |
| Stomach wall | 0.0114 ± 0.0005 | 0.0117 ± 0.0008 |
| Right colon | 0.0103 ± 0.0005 | 0.0118 ± 0.0003 |
| Rectum | 0.0103 ± 0.0002 | 0.0111 ± 0.0003 |
| Heart wall | 0.0407 ± 0.0007 | 0.0206 ± 0.0089 |
| Kidneys | 0.0164 ± 0.0011 | 0.0151 ± 0.0016 |
| Liver | 0.0312 ± 0.0026 | 0.0303 ± 0.0025 |
| Lungs | 0.0261 ± 0.0008 | 0.0138 ± 0.0054 |
| Ovaries | 0.0099 ± 0.0003 | |
| Pancreas | 0.0129 ± 0.0007 | 0.0126 ± 0.0006 |
| Prostate | 0.0114 ± 0.0004 | |
| Salivary glands | 0.0074 ± 0.0003 | 0.0086 ± 0.0002 |
| Red marrow | 0.0089 ± 0.0003 | 0.0090 ± 0.0002 |
| Osteogenic cells | 0.0072 ± 0.0003 | 0.0078 ± 0.0002 |
| Spleen | 0.0102 ± 0.0004 | 0.0102 ± 0.0002 |
| Testes | 0.0088 ± 0.0002 | |
| Thymus | 0.0136 ± 0.0004 | 0.0108 ± 0.0016 |
| Thyroid | 0.0092 ± 0.0003 | 0.0095 ± 0.0004 |
| Urinary bladder wall | 0.0201 ± 0.0041 | 0.0286 ± 0.0050 |
| Uterus | 0.0104 ± 0.0002 | |
| Total body | 0.0097 ± 0.0004 | 0.0096 ± 0.0002 |
| Effective dose | 0.0126 ± 0.0003 | 0.0103 ± 0.0009 |
Data are mGy/MBq except for effective dose, which is mSv/MBq. These data were calculated using OLINDA, version 2.0, with ICRP publication 103 tissue-weighting factors based on 90-min dynamic PET acquisition in male or female C57Bl6/J mice.
DISCUSSION
Herein, we show that 18F-TRX can distinguish LIP levels among tumors, and we point to those that might be most likely to respond to LIP-targeted therapies. A survey of 10 human xenograft models revealed that glioma and renal cell carcinoma harbor a wide range of LIP concentrations. U251, the model with the highest 18F-TRX uptake, was highly sensitive to treatment with TRX-CBI, an LIP-activated prodrug. A comparison of the relative antitumor effects conferred by TRX-CBI treatment revealed that U251 tumors responded more durably than PC3 tumors, a human prostate cancer model with approximately 2-fold lower uptake of 18F-TRX. Since these data suggest a role for LIP-targeted therapies in the treatment of glioma, we further tested whether LIP expansion is evident on PET in an orthotopic model of high-grade glioma. On PET, U87 MG tumors were clearly detectable, compared with the level of uptake in uninvolved brain tissue. Lastly, a dosimetry study in mice showed the estimated effective doses to humans to be lower than those of 18F-FDG. Collectively, these observations make a strong case for human translation of 18F-TRX to collect the first measurements of LIP in tumors.
The finding that pretreatment 18F-TRX uptake in tumors is predictive of sensitivity to treatment with a prodrug that requires LIP activation provides an important confirmation that tracer uptake in tumors at extended time points after injection (i.e., 90 min) is predominantly if not entirely due to LIP-triggered probe fragmentation and cellular sequestration. On PET, we of course cannot distinguish between the different chemical species to which the radioisotope is attached, and these data provide important clarity on the unavoidable ambiguity as to whether the tumor signal is also in part derived from iron-independent tumor retention of unreacted 18F-TRX through an unknown mechanism.
Although the relative tumor LIP levels with 18F-TRX could be corroborated with the structurally and mechanistically distinct chemosensor FerroFarRed, relative STEAP3 expression did not always correlate with LIP levels. Tumors that do not overexpress STEAP3 likely use alternative mechanisms to expand the LIP, such as overexpressing HAMP to prevent ferrous iron export by ferroportin or liberating iron from cellular ferritin stores. Conversely, PC3 tumors that have high STEAP3 expression but relatively moderate LIP may express high levels of ferroportin compared with tumors such as U251. More generally, the biologic complexity of iron homeostasis suggests that LIP expansion may not be predictable for all tumor types by qualitatively assessing expression levels of a single protein biomarker using immunohistochemistry. Moreover, although it is not clear whether 68Ga3+ is a STEAP3 substrate, it is a well-established surrogate for ferric iron biodistribution. Thus, our early data showing that relative tumoral uptake of 68Ga-citrate and 18F-TRX do not align suggest that LIP levels may not be easily inferred from measurements of tumoral uptake of ferric iron. All these observations, although preliminary, converge to underscore the potential utility of a ferronostic paradigm in which direct measurement of tumoral iron levels with 18F-TRX PET is applied to prospectively identify tumors that might respond to iron-targeted therapies. We are currently expanding the scope of this study to treat additional tumor models with TRX-CBI and other LIP-activated prodrugs that release payloads mechanistically unrelated to CBI to test this hypothesis more thoroughly.
Finally, iron dysregulation occurs in many human disorders, including neurodegenerative and cardiovascular diseases and inflammation (20–22). Applying 18F-TRX in the respective patient populations to define the extent of LIP expansion in affected tissues will be an important milestone toward understanding the therapeutic potential of LIP-targeted therapies beyond oncology. We are currently working to translate 18F-TRX into the clinic to test this hypothesis.
CONCLUSION
We report evidence that tumor sensitivity to an LIP-targeted therapy can be predicted with a molecular imaging tool. These data bring a new dimension to the nuclear theranostic model by showing a requirement for imaging to quantify, in situ, the concentration of a metastable bioanalyte toward predicting tumor drug sensitivity.
DISCLOSURE
This study was supported by an American Cancer Society research scholar grant (130635-RSG-17-005-01-CCE), the CDMRP Prostate Cancer Program (W81XWH-18-1-0763, W81XWH-16-1-0435, and W81XWH1810754), and the National Institutes of Health (R01AI105106). Ryan Muir, Adam Renslo, and Michael Evans are listed as inventors on patent applications describing 18F-TRX and related radiotracers. Adam Renslo is a cofounder of and advisor to Tatara Therapeutics, Inc. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Are tumoral levels of the LIP as measured with 18F-TRX PET a predictive biomarker of tumor sensitivity to iron-targeted therapies?
PERTINENT FINDINGS: A survey of 18F-TRX uptake in 10 human xenograft models identified the glioma model U251 as having the highest level of the LIP. An antitumor assessment study showed that the ferrous iron–activated prodrug TRX-CBI potently inhibited growth. The antitumor effects were greater than those observed against PC3 tumors, human prostate cancer with approximately 2-fold lower 18F-TRX uptake.
IMPLICATIONS FOR PATIENT CARE: To our knowledge, 18F-TRX is the first translational in vivo imaging probe that can measure labile iron concentrations in tumors, a finding that could evolve into a new theranostic paradigm to identify tumors likely to succumb to iron-targeted therapies currently under development.
ACKNOWLEDGMENTS
We gratefully acknowledge Tony Huynh for technical assistance with small-animal PET/CT studies and Joseph Blecha for assistance with radiochemistry.
REFERENCES
- 1. Torti SV, Manz DH, Paul BT, Blanchette-Farra N, Torti FM. Iron and cancer. Annu Rev Nutr. 2018;38:97–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kakhlon O, Cabantchik ZI. The labile iron pool: characterization, measurement, and participation in cellular processes. Free Radic Biol Med. 2002;33:1037–1046. [DOI] [PubMed] [Google Scholar]
- 3. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weaver M, Laske DW. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J Neurooncol. 2003;65:3–13. [DOI] [PubMed] [Google Scholar]
- 5. Spangler B, Fontaine SD, Shi Y, et al. A novel tumor-activated prodrug strategy targeting ferrous iron is effective in multiple preclinical cancer models. J Med Chem. 2016;59:11161–11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meshnick SR. Artemisinin antimalarials: mechanisms of action and resistance. Med Trop (Mars). 1998;58(suppl)13–17. [PubMed] [Google Scholar]
- 7. Spangler B, Kline T, Hanson J, et al. Toward a ferrous iron-cleavable linker for antibody-drug conjugates. Mol Pharm. 2018;15:2054–2059. [DOI] [PubMed] [Google Scholar]
- 8. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19:405–414. [DOI] [PubMed] [Google Scholar]
- 9. Wang W, Green M, Choi JE, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weber RA, Yen FS, Nicholson SPV, et al. Maintaining iron homeostasis is the key role of lysosomal acidity for cell proliferation. Mol Cell. 2020;77:645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Langbein T, Weber WA, Eiber M. Future of theranostics: an outlook on precision oncology in nuclear medicine. J Nucl Med. 2019;60(suppl 2):13S–19S. [DOI] [PubMed] [Google Scholar]
- 12. Muir RK, Zhao N, Wei J, et al. Measuring dynamic changes in the labile iron pool in vivo with a reactivity-based probe for positron emission tomography. ACS Cent Sci. 2019;5:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Phyo AP, Jittamala P, Nosten FH, et al. Antimalarial activity of artefenomel (OZ439), a novel synthetic antimalarial endoperoxide, in patients with Plasmodium falciparum and Plasmodium vivax malaria: an open-label phase 2 trial. Lancet Infect Dis. 2016;16:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Izquierdo-Garcia JL, Viswanath P, Eriksson P, et al. IDH1 mutation induces reprogramming of pyruvate metabolism. Cancer Res. 2015;75:2999–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seo Y, Gustafson WC, Dannoon SF, et al. Tumor dosimetry using [124I]m-iodobenzylguanidine microPET/CT for [131I]m-iodobenzylguanidine treatment of neuroblastoma in a murine xenograft model. Mol Imaging Biol. 2012;14:735–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Han M, Xu R, Wang S, et al. Six-transmembrane epithelial antigen of prostate 3 predicts poor prognosis and promotes glioblastoma growth and invasion. Neoplasia. 2018;20:543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hirayama T, Tsuboi H, Niwa M, et al. A universal fluorogenic switch for Fe(ii) ion based on N-oxide chemistry permits the visualization of intracellular redox equilibrium shift towards labile iron in hypoxic tumor cells. Chem Sci. 2017;8:4858–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Behr SC, Villanueva-Meyer JE, Li Y, et al. Targeting iron metabolism in high-grade glioma with 68Ga-citrate PET/MR. JCI Insight. 2018;3:e93999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boellaard R, O’Doherty MJ, Weber WA, et al. FDG PET and PET/CT: EANM procedure guidelines for tumour PET imaging—version 1.0. Eur J Nucl Med Mol Imaging. 2010;37:181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. James SA, Roberts BR, Hare DJ, et al. Direct in vivo imaging of ferrous iron dyshomeostasis in ageing Caenorhabditis elegans . Chem Sci. 2015;6:2952–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. von Haehling S, Jankowska EA, van Veldhuisen DJ, Ponikowski P, Anker SD. Iron deficiency and cardiovascular disease. Nat Rev Cardiol. 2015;12:659–669. [DOI] [PubMed] [Google Scholar]
- 22. Wessling-Resnick M. Iron homeostasis and the inflammatory response. Annu Rev Nutr. 2010;30:105–122. [DOI] [PMC free article] [PubMed] [Google Scholar]