

Abstract
Cardiolipin (CL) deficiency causes mitochondrial dysfunction and aberrant metabolism that are associated in humans with the severe disease Barth syndrome (BTHS). Several metabolic abnormalities are observed in BTHS patients and model systems, including decreased oxidative phosphorylation, reduced tricarboxylic acid (TCA) cycle flux, and accumulated lactate and D-β-hydroxybutyrate, which strongly suggests that nicotinamide adenine dinucleotide (NAD) redox metabolism may be altered in CL-deficient cells. In this study, we identified abnormal NAD+ metabolism in multiple BTHS model systems and demonstrate that supplementation of NAD+ precursors such as nicotinamide mononucleotide (NMN) improves mitochondrial function. Improved mitochondrial function in the Drosophila model was associated with restored exercise endurance, which suggests a potential therapeutic benefit of NAD+ precursor supplementation in the management of BTHS patients.
Keywords: Cardiolipin deficiency, NAD+ redox, NAD+ precursors, nicotinamide mononucleotide, mitochondrial function, Barth Syndrome
Introduction
Cardiolipin (CL) is a signature phospholipid of the mitochondria. CL supports the optimal functioning of oxidative phosphorylation (OXPHOS) by physically binding with OXPHOS complexes I, III, IV, and V [5–9] and promoting their association and supramolecular organization [10–13]. The importance of CL is underscored by the rare, infantile-onset, and severe mitochondrial disease Barth syndrome (BTHS), which is caused by mutations in the CL remodeling gene TAFAZZIN [14–16]. Disruption of TAFAZZIN results in decreased incorporation of unsaturated fatty acids into CL, reduction in total CL, and accumulation of monolysocardiolipin (MLCL) [15–19].
Recently, attention has been given to metabolic abnormalities of CL-deficient cells. Loss of CL in yeast cells leads to decreased activity of the reduced tricarboxylic acid (TCA) cycle enzymes aconitase and succinate dehydrogenase and reduced synthesis of acetyl-CoA [20, 21]. In C2C12 mouse myoblast cells lacking TAFAZZIN (TAZ-KO), decreased pyruvate dehydrogenase (PDH) activity is associated with reduced flux of glucose into the TCA cycle and increased lactate production [22, 23]. In patients with BTHS, lactic acidosis is associated with metabolic decompensation, the extent of which is correlated with the severity of the clinical course of the disease [24–26].
Nicotinamide adenine dinucleotide (NAD) is a key coenzyme of energy metabolism. NAD+ is reduced to NADH by dehydrogenases or oxidoreductases in glycolysis, β-oxidation, amino acid catabolism pathways, and the TCA cycle. In mitochondria, NADH is oxidized to NAD+ by NADH dehydrogenase(s), which is coupled to the production of ATP [27–29]. Under optimal and aerobic conditions, a high NAD+/NADH ratio is maintained by the activity of NADH dehydrogenase(s) at the IMM. In CL-deficient cells, reduced OXPHOS activity is associated with the accumulation of lactate and D-β-hydroxybutyrate [23, 25, 30], which are produced via enzymes that oxidize NADH and potentially compensate for a reduced mitochondrial capacity for NADH oxidation in these cells.
A high NAD+/NADH ratio is required for optimal cellular energy production by dictating the reaction equilibria of metabolic pathways that are coupled to the reduction of NAD+ [31, 32]. Reduced NAD+ levels or NAD+/NADH ratios have been reported in several models of mitochondrial disease, aging, and heart failure [33–36]. Treatment with NAD+ precursors can increase NAD+ levels and the NAD+/NADH ratio and has been shown to be efficacious in preclinical models of aging and mitochondrial disorders [33–36]. While NAD can be synthesized de novo from tryptophan, forms of vitamin B3 such as niacin, nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR) are much more efficient NAD+ precursors. The use of niacin in patients has been limited due to its unpleasant side effect profile, which includes flushing and itching [37]. However, NMN and NR are used clinically without unpleasant side effects [38–40], and have been shown to improve mitochondrial and muscular/cardiac functions in mouse models of age-associated muscle weakness, Friedreich’s ataxia cardiomyopathy [41], and transverse aortic construction-induced heart failure [35, 41, 42].
In this study, we identified that NAD+ redox metabolism is altered in CL-deficient yeast cells lacking CL synthase (crd1Δ) and mouse TAZ-KO C2C12 cells. We further confirm that fermentation is essential to crd1Δ, likely due to the important role of this pathway in compensating for reduced mitochondrial capacity for NADH oxidation in CL-deficient cells. Importantly, we showed that supplementation with NAD+ precursors improved mitochondrial respiration in crd1Δ cells, increased mitochondrial membrane potential (ΔΨm) in TAZ-KO cells, and improved mitochondrial coupling of the Drosophila TAFAZZIN mutant (Taz889). Additionally, supplementation with NMN improved exercise endurance of Drosophila Taz889. These findings show for the first time that CL deficiency results in disrupted NAD+ metabolism, and various phenotypes associated with CL-deficiency are partially attenuated by supplementation with NAD+ precursors.
Materials and Methods
Yeast Strains and Growth Media
The yeast strains used in this study are listed in Table 1. Synthetic complete (SC) medium contained adenine (20.25 mg/L); uracil (20 mg/L); amino acids including arginine (20 mg/L), histidine (20 mg/ L), leucine (60 mg/ L), lysine (200 mg/L), methionine (20 mg/L), threonine (300 mg/L), tryptophan (20 mg/L); yeast nitrogen base that contained ammonium sulfate (5 g/L), boric acid (0.5 mg/L), copper sulfate (40 μg/L), iodide (0.1 mg/L), ferric chloride (0.2 mg/L), manganese sulfate (0.4 g/L), sodium molybdate (0.2 g/L), zinc sulfate (0.4 mg/L), monopotassium phosphate (1 g/L), magnesium sulfate (0.5 g/L), sodium chloride (0.1 g/L), and calcium chloride (0.1 g/L); and vitamin mix that contained biotin (2 μg/L), calcium pantothenate (0.4 mg/L), folic acid (2 μg/L), inositol (2 mg/L), niacin (0.4 mg/L), p-aminobenzoic acid (0.2 mg/L), pyridoxine hydrochloride (0.4 mg/L), riboflavin (0.2 mg/L), thiamine hydrochloride (0.4 mg/L). Niacin-free medium contained all the above-mentioned ingredients except niacin as indicated in the figures. Galactose (2%), sodium pyruvate (2%), glycerol (2%), and sodium lactate (2%) were added as carbon sources as indicated.
Table 1.
Strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Invitrogen |
| crd1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 crd1Δ::KanMX6 | Invitrogen |
| adh1Δ | MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 met15Δ0/MET15 LYS2/lys2Δ0 ura3Δ0/ura3Δ0 | Dharmacon |
| adh1Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 adh1Δ::KanMX6 | This study |
| adh1Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 adh1Δ::KanMX6 | This study |
| adh2Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 adh2Δ::KanMX6 | Dharmacon |
| adh2Δ | MATa his3Δ1 leu2Δ0 ura3Δ0 adh2Δ::KanMX6 | This study |
| adh3Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 adh3Δ::KanMX6 | Dharmacon |
| adh3Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 adh3Δ::KanMX6 | This study |
| adh4Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 adh4Δ::KanMX6 | Dharmacon |
| adh4Δ | MATa his3Δ1 leu2Δ0 ura3Δ0 adh4Δ::KanMX6 | This study |
| adh5Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 adh5Δ::KanMX6 | Dharmacon |
| adh5Δ | MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 adh5Δ::KanMX6 | This study |
| crd1Δadh1Δ | MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 crd1Δ::KanMX6 adh1Δ::KanMX6 | This study |
| crd1Δadh2Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 crd1Δ::KanMX6 adh2Δ::KanMX6 | This study |
| crd1Δadh3Δ | MATa his3Δ1 leu2Δ0 ura3Δ0 crd1Δ::KanMX6 adh3Δ::KanMX6 | This study |
| crd1Δadh4Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 crd1Δ::KanMX6 adh4Δ::KanMX6 | This study |
| crd1Δadh5Δ | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 crd1Δ::KanMX6 adh5Δ::KanMX6 | This study |
Mammalian Cell Lines and Growth Conditions
The TAZ-KO C2C12 myoblast strain was constructed by Lou et al. using the CRISPR-Cas9 system [23]. Growth medium consisted of DMEM (Gibco) containing 10% FBS (Atlanta Biologicals), 2 mM glutamine (Gibco), penicillin (100 units/mL) and streptomycin (100 μg/mL) (Invitrogen). Cells were grown at 37°C in a humidified incubator with 5% CO2.
Drosophila Strains and Growth Conditions
The Drosophila TAFAZZIN mutant (Taz889) was constricted from the w1118 line using the CRISPR-Cas9 system. The first 889 base pairs from the first exon of the TAFAZZIN gene were deleted. The mutated site is tagged with a red fluorescent protein (RFP) for selection. The genome editing, microinjection, validation, and stock establishment were completed by Well Genetics. Flies were raised on 10% sugar-yeast medium and housed continuously at 25°C in a 12-hour light/dark incubator.
Yeast Spotting Assay
Yeast cells were grown in SC medium to the stationary phase at 3°C, washed with sterile water, and diluted to an A550 of 0.5. 5 μL aliquots of the series of 5- or 10-fold dilutions were spotted onto solid medium and incubated at the indicated temperatures.
Yeast Growth Curve
Yeast cells in the stationary phase were inoculated into fresh medium to the concentration indicated at time zero and incubated with aeration at 230 rpm. A550 of each culture was measured at multiple time points as indicated in figures.
Measurement of Ethanol
Cells were grown to the logarithmic phase at an A550 of 0.5 to 1, collected by centrifugation, and washed twice with sterile water. Following resuspension in fresh medium, the samples were diluted with fresh medium to an A550 of 0.5. After the indicated hours of incubation at high temperature, 1 mL of culture was transferred quickly to a 1.7 mL centrifuge tube and centrifugated at maximum speed, 4°C, for 1 minute (min). 400 μL of supernatant were transferred to a new tube. To measure ethanol in the supernatant, a colorimetric assay kit (Sigma) was used according to the manufacturer’s manual.
Measurement of NAD(H)
Yeast metabolites were extracted with a modified boiling ethanol method [43]. Briefly, 200 μL of freshly harvested culture in the mid-logarithmic phase were immediately dropped into a 15 mL capped tube containing 1.3 mL of boiling ethanol buffered with 120 μL 1 M HEPES (pH 7.5) and incubated for 3 min. After cooling on ice for 3 min, the volume was reduced by evaporation using a speed vac concentrator. The residue was resuspended in 100 μL double-distilled water and centrifuged for 10 min at 5000 g at 4°C.
To extract metabolites from C2C12 cells, 7,500 cells in 100 μL medium were seeded on a treated 96-well plate (CytoOne) and incubated overnight. After treatment with vehicle (H2O) or 1 mM NMN for 5 hours, cells were washed twice with warm PBS and lysed with 0.2N NaOH containing 1% DTAB. Half of the lysate was treated with 0.4N HCl in a new well. All lysates were heated at 6°C for 15 min. After cooling down at room temperature for 10 min, samples were neutralized by HCl/Trizma or Trizma (Sigma).
Levels of NAD+, NADH, and the NAD+/NADH ratio were analyzed with the NAD/NADH-Glo™ assay kit (Promega) following the manufacturer’s instructions. Briefly, 10 μL metabolites described above were loaded onto a 384-well low-volume white plate (Corning) and mixed with 10 μL assay reagent mix. Relative light units (RLU) were read on a plate reader (Spectra Max GEMINI XPS, Molecular Devices) after a 1-hour incubation at room temperature in the dark. The linearity, sensitivity, and specificity of NAD assays were determined using NAD+ and NADH (Sigma) standards. NAD+ and NADH levels were normalized to protein levels.
Measurement of TMRM Fluorescence Intensity (FI)
TMRM fluorescence intensity was used as an indication of mitochondrial membrane potential (ΔΨm). 7,500 C2C12 cells in 100 μL medium were seeded on a 96-well plate (black wall, clear bottom, cell culture-treated; Corning) and incubated overnight. Cells were treated with vehicle (H2O) or 1 mM NMN for 5 hours, followed by incubation with 50 nM TMRM for 30 min at 30°C. After washing twice with prewarmed PBS, the FI at Ex548/Em574 and the area covered [%] by cells (confluency) of each well were read on a plate reader (Spectra Max GEMINI XPS, Molecular Devices). 10 μM FCCP was used to uncouple mitochondria and followed by twice gentle washings with prewarmed PBS. FI of each well after FCCP treatment was subtracted from the FI before FCCP, and the results were normalized to the area covered by cells (confluency × 0.32 cm2).
Measurement of Oxygen Consumption Rate (OCR)
Yeast cells in the mid-logarithmic phase were diluted with fresh medium to an A550 of 0.5. The OCR of each diluted sample was determined using the polarographic method in a closed chamber equipped with a Clark electrode (Oxytherm+ System, Hansatech) at 30°C as described [44]. 5 μM FCCP was added to uncouple OXPHOS to measure the maximum OCR. 0.2 mM KCN was added at the end to inhibit complex IV to correct for complex IV-independent oxygen utilization. OCR was analyzed with Oxygraph plus software and was defined as consumed O2 (nmol)/min/total protein (mg).
OCR and extracellular acidification rate (ECAR) of WT and TAZ-KO C2C12 cells was determined using a Seahorse Extracellular Flux Analyzer XFe96 (Agilent) following manufacturer’s instruction. In brief, 10,000 cells were seeded in DMEM containing dialyzed FBS (Hyclone) and incubated overnight. Cells were treated with the indicated concentrations of NMN for 5 hours prior a final media exchange with the Seahorse XF DMEM medium, pH 7.4, containing 10 mM glucose and 2 mM L-glutamine. After one hour of incubation at 37°C, OCR and ECAR was measured. 1 μM oligomycin A, 1 μM FCCP, and mixture of 0.5 μM rotenone and antimycin A were used in accordance with the manufacturer’s instructions. Rates were normalized to total protein.
Measurement of Drosophila Endurance
Drosophila were collected within 48 hours of enclosing (age day 1–2). Flies were transferred to fresh vials containing food mixed with vehicle (H2O) or 10 mM NMN at age day 5 and once a day for the following five days. The endurance was measured at age day 10. The Power Tower system was used to induce endurance exercise as described previously [45–47]. In brief, the Power Tower stimulates flies to climb by knocking them to the bottom of their container every 8 seconds. The endurance is determined by the time to fatigue, defined as the time when 80% of flies within a vial have stopped climbing.
Measurement of Respiratory Control Ratio (RCR) in Drosophila Mitochondria
Mitochondria were extracted with a glass-Teflon Dounce homogenizer with modifications [48, 49]. Briefly, 60 flies of each biological replicate were placed on ice for 1 min and placed in a glass tube containing 500 μL isolation buffer (0.32 M sucrose, 10 mM EDTA, 10 mM Tris HCL, 2% fatty-acid free BSA, pH 7.2). Following homogenization, the brie was filtered through a nylon filter (pore size = 10 μM, Sigma) and 1 mL of isolation buffer was used to wash the filter. The homogenate was centrifuged for 5 min at 300 g at 4°C. The supernatant was transferred to a new tube and was centrifuged for 10 min at 6000 g at 4°C. The supernatant was removed, and the pellet was resuspended in 30–50 μL respiration buffer (120 mM KCL, 5 mM KH2PO4, 3 mM HEPES, 1 mM EGTA, pH 7.2, BSA free). The protein concentration was measured using the BCA assay (Thermo Scientific). All respiration measurements of isolated mitochondrial preparations were performed within two hours after isolation.
To measure RCR, 10 μL isolated mitochondria were loaded on a chamber equipped with a Clark electrode (Oxytherm+ System, Hansatech) and containing 990 μL respiration buffer with 0.3% BSA at 25°C. Pyruvate and malate were at a final concentration of 10 μM. 125 nΜ ADP was added to stimulate state III respiration and 2.5 μM oligomycin was added to stimulate state IV respiration. RCR was calculated from state III respiration relative to state IV.
Real-Time Quantitative PCR (RT-qPCR) Analysis
Mid-logarithmic phase cells grown at 30°C were stressed for two hours at 38°C and harvested at 4°C. Total RNA was extracted using hot phenol [50] and purified using the RNeasy Mini Plus kit (Qiagen). A first-strand cDNA synthesis kit (Roche Applied Science) was used for complementary DNA (cDNA) synthesis. RT-qPCR was performed using Brilliant III Ultra-Faster SYBR Green qPCR Master Mix (Agilent Technologies). Two technical replicates each of three biological replicates were included for each reaction. RNA levels were normalized to ACT1. Relative values of mRNA transcripts are shown as fold change relative to the indicated controls. The primers for RT-qPCR are listed in Table S1. Primer concentrations were optimized to ensure efficiencies between 95% and 105% and not 5% higher or lower than the efficiency of ACT1 primers.
Statistical Analysis
Shapiro-Wilk normality testing, unpaired t-test, and/or ANOVA were performed using Prism version 9 (GraphPad) as indicated ( p>0.05; *, p≤0.05; **, p≤0.01; ***, p≤0.001; ****, p≤0.0001).
Results
Aerobic fermentation is essential for maintaining NAD+ redox balance in crd1Δ cells
In accordance with a previous report that heat stress results in uncoupling of crd1Δ mitochondria [51], we observed that crd1Δ cells display a growth defect when grown at elevated temperature with galactose as the sole carbon source (Fig. 1A). This is accompanied by a significant decrease in the NAD+/NADH ratio when compared to that of WT under the same growth conditions (Fig. 1B). Elimination of niacin, the commonly supplied NAD+ precursor, from synthetic complete (SC) medium results in decreased growth of crd1Δ (Fig. 1C), indicating a reliance of crd1Δ on this exogenously supplied NAD+ precursor.
Figure 1. The NAD(H) redox balance is disrupted in crd1Δ.
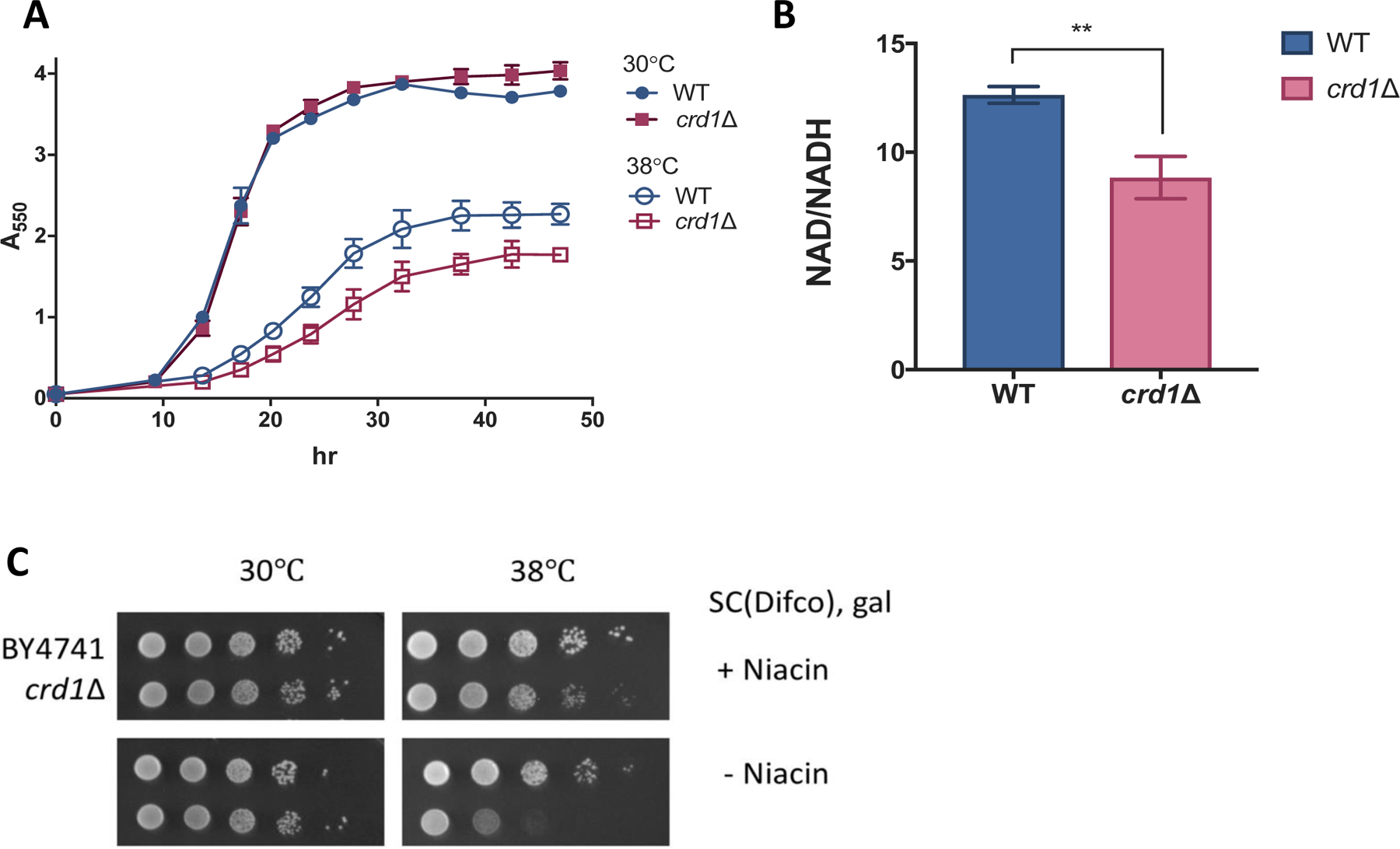
(A) Precultures in the stationary phase were inoculated to a concentration of A550=0.05 in fresh SC-galactose medium and incubated with aeration. A550 was measured at indicated times. Data shown are mean ± S.D. (n=3). (B) The NAD+/NADH ratio was measured in cells grown in galactose medium to the mid-logarithmic phase as described in Materials and Methods. Data shown are mean ± S.D. (n=3). Unpaired t test, p=0.0033 (C) 10-fold serial dilutions of WT and crd1Δ cells spotted on SC-galactose medium with or without niacin. Plates were incubated at 38.5 ± 0.5 °C for 4 days.
Increased aerobic fermentation is commonly observed in cells with mitochondrial defects. It is thought to compensate for the reduced mitochondrial capacity for NADH oxidation and ATP production [52]. To assess fermentation in WT and crd1Δ cells grown under conditions of mitochondrial stress, we measured ethanol production during exposure to heat stress for two-and five-hours. As seen in Fig. 2A, crd1Δ produced significantly more ethanol than did the WT control cells when galactose was the sole carbon source. For use in glycolysis, galactose is first converted to G-6-P by the Leloir pathway (Fig. S1). during which three ATP molecules are consumed [3, 4]. Because the conversion of G-6-P to pyruvate yields three ATP molecules (one consumed and four generated), the net glycolytic yield of ATP from galactose is zero. Therefore, increased fermentation by crd1Δ cells under these conditions cannot contribute to ATP production. More likely, aerobic fermentation in crd1Δ under these conditions is important for NAD+ redox balance. Conversely, no differences in ethanol production during exposure to heat stress were observed between crd1Δ and WT cells when glucose was the sole carbon source (Fig. 2B), a condition that normally drives fermentation and inhibits OXPHOS in S. cerevisiae [1]. Surprisingly, ethanol production by yeast cells lacking tafazzin (taz1Δ) was similar to that of WT cells during five-hour heat stress in galactose (Fig. S2), likely due to the relatively mild mitochondrial defects of taz1Δ when compared to those of crd1Δ [53].
Figure 2. crd1Δ increases ethanol production in response to heat stress in galactose.
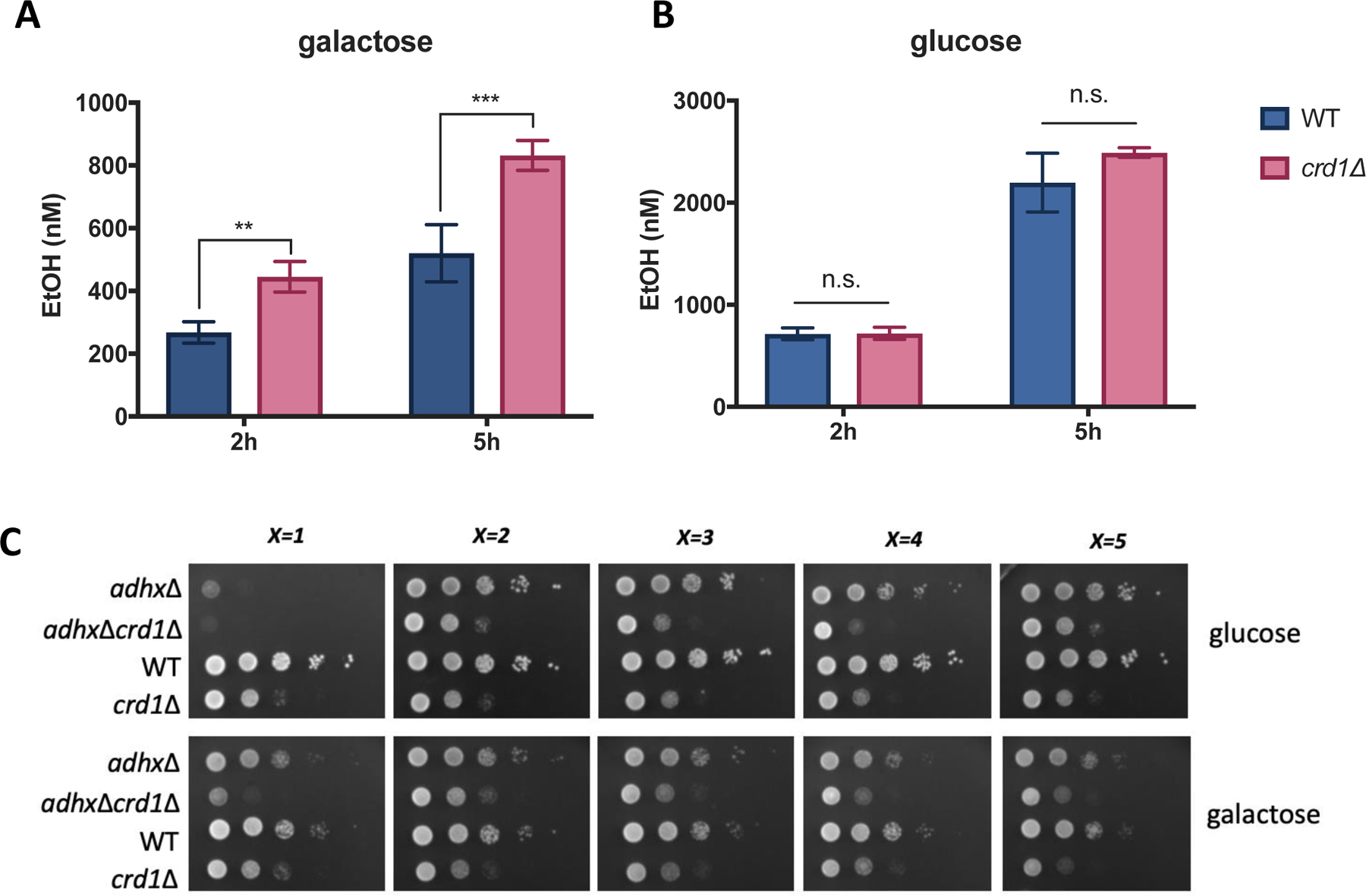
Cells were grown at 30°C in SC-glucose medium to the mid-logarithmic phase, washed twice with sterile water, resuspended in fresh (A) SC-galactose (n=4) or (B) SC-glucose (n=3) medium, and diluted to an A550 of 0.5. After shaking incubation for two or five h at 39°C, supernatant from 1 mL of culture was collected by centrifugation. Ethanol contained in the medium was measured as described in Materials and Methods. Data shown are mean ± S.D. Unpaired t test; **, p≤0.01, ***, p≤0.001. (C) WT, crd1Δ, adhxΔ (x=1, 2, 3, 4, 5), and crd1ΔadhxΔ cells were pre-cultured in SC-glucose medium to the stationary phase and diluted to an A550 of 0.5. 5 μL aliquots of a series of 10-fold dilutions were spotted on SC-glucose or SC-galactose plates and incubated at 39 °C for three days.
The increased ethanol production by crd1Δ in galactose (Fig. 2A) suggests an important role of the fermentation pathway in the mutant. Among the 13 alcohol dehydrogenase (ADH) enzymes that have been characterized in S. cerevisiae, cytosolic Adh1 is the major enzyme responsible for ethanol production during aerobic fermentation [54, 55]. Therefore, the activity of Adh1 may be essential for the survival of crd1Δ cells. To test this possibility, we individually deleted ADH1–5 genes from crd1Δ to determine if crd1Δ is synthetically lethal with loss of ADH activity. Of the five double mutants, only adh1Δcrd1Δ exhibited severely decreased growth on galactose compared to controls (Fig. 2C), indicating that Adh1 activity is required for the survival of crd1Δ under these conditions. ADH genes are highly regulated at the transcriptional level [55] and ADH1 expression is increased significantly in WT, crd1Δ, and taz1Δ cells by five- to seven- fold in response to heat stress (Fig. S3). However, ADH gene expression was not increased to a greater extent in crd1Δ compared to WT, suggesting that increased ethanol production by crd1Δ may result from regulation at the level of translation or enzymatic activity.
Exogenous supplementation with NAD+ precursors decreases ethanol production and improves mitochondrial function in crd1Δ cells
Yeast has been shown to assimilate exogenous NMN, similar to mammalian cells (Fig. 3A) [56]. We observed that supplementation of NMN rescues the sensitivity of crd1Δ cells to niacin depletion (Fig. 1C, Fig. 3B), suggesting that NMN can be used as an NAD+ precursor similar to niacin in these cells. Additionally, pretreatment with NMN decreased ethanol production during exposure to heat stress in both WT and crd1Δ cells (Fig. 3B). Because exogenous supplementation with NAD+ precursors improved crd1Δ cell growth on galactose while reducing ethanol production, we tested the ability of NMN to improve mitochondrial function in crd1Δ cells. NMN significantly improved the growth of crd1Δ cells in non-fermentable media containing either 2% glycerol and 2% lactate (Fig. 4A) or 2% pyruvate (Fig. 4B, Fig. S4A) as the carbon sources. Next, we assayed oxygen consumption of crd1Δ and WT cells grown in the glycerol lactate medium (Fig. 4C&D). Surprisingly, crd1Δ cells exhibited only a mild decrease in basal and maximum oxygen consumption rate (OCR) compared to WT, which was not significant (p=0.133 and 0.131, respectively) (Fig. 4C). Nevertheless, we observed a 20.4% increase in the maximum OCR of crd1Δ cells when treated with NMN (Fig. 4D), suggesting that exogenous supplementation of NAD+ precursors increased electron supply activities and improved respiratory capacity.
Figure 3. NMN decreases ethanol production by WT and crd1Δ during heat stress.
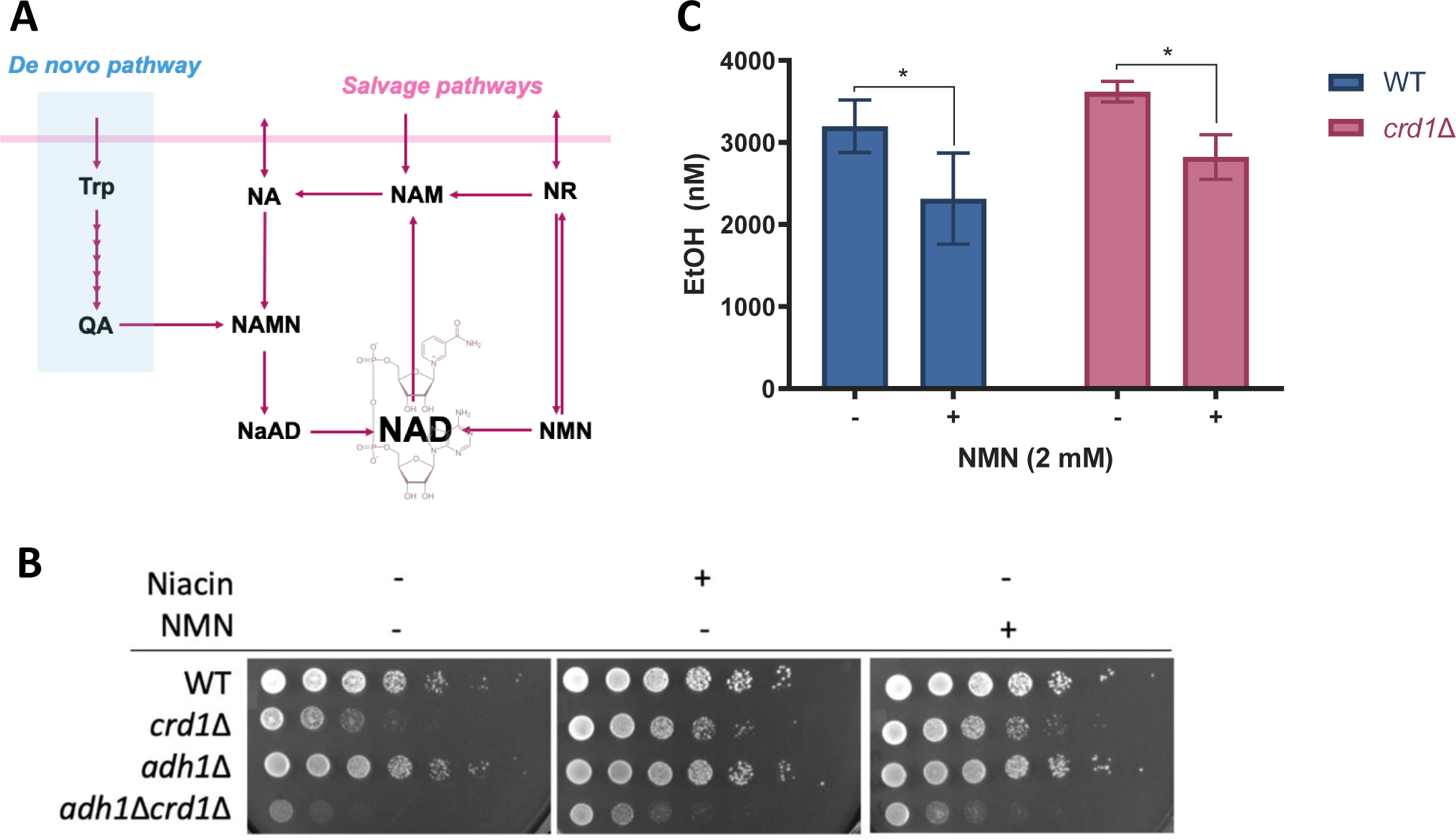
(A) NAD+ is synthesized de novo from tryptophan (Trp) by the kynurenine pathway or from forms of vitamin B3 such as nicotinic acid (NA), nicotinamide (NAM), nicotinamide riboside (NR), and nicotinamide mononucleotide (NMN) through salvage pathways. QA, quinolinic acid. NAMN, nicotinic acid mononucleotide. NaAD, deamido-NAD. (B) WT, crd1Δ, adh1Δ, and crd1Δadh1Δ were pre-cultured in SC-glucose medium to the stationary phase and diluted to an A550 of 0.5. 5 μL aliquots of a series of 5-fold dilutions were spotted on SC-galactose plates containing 0.4 mg/L niacin or 0.1 mM NMN and incubated at 38 °C for three days. (C) WT and crd1Δ cells were grown in 4 mL SC-galactose medium to the mid-logarithmic phase at 30°C. 4 mL fresh medium was added to each sample before transfer to 39°C. After 1.5 hours, cells were washed twice with sterile water, resuspended and diluted to A550=0.45 with fresh medium containing 2 mM NMN or vehicle (water), followed by 6 hours heat stress before samples were collected and measured as described in Materials and Methods. Data shown are mean ± S.D. (n=4). One-way ANOVA; *, p≤0.01.
Figure 4. NMN supplementation rescues the respiratory growth defect of crd1Δ and improves its respiratory capacity.
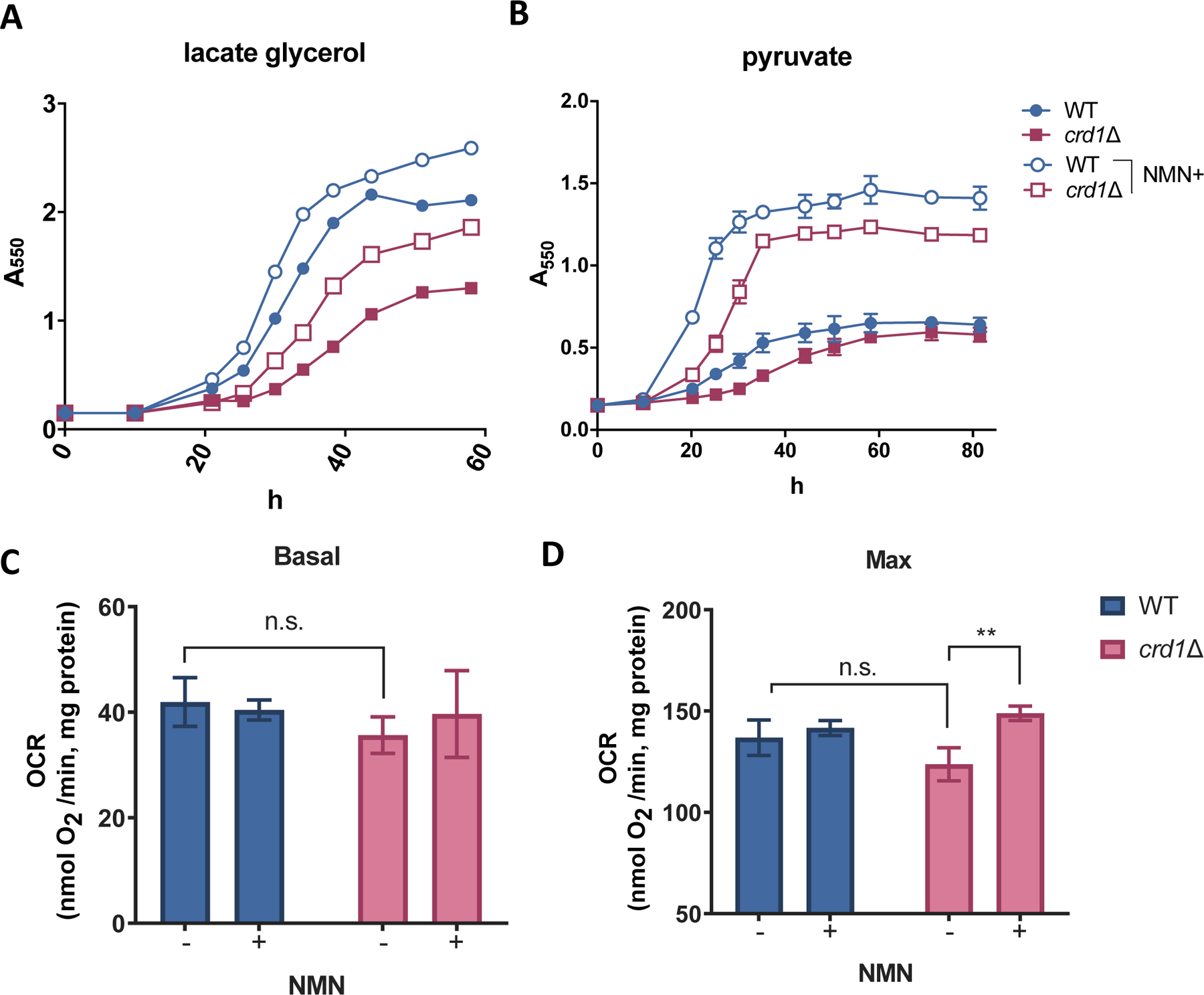
Pre-cultures in the stationary phase were inoculated to fresh SC medium containing (A) 2% glycerol and 2% sodium lactate or (B) 2% sodium pyruvate to concentrations at A550=0.15. 2 mM NMN or vehicle (water) was supplied as treatment. A550 was determined at the indicated times during the incubation at 30.0 °C. The data represent the results of two independent experiments. WT and crd1Δ cells were grown to the mid-logarithmic phase in SC-lactate glycerol medium in the presence or absence of supplemented 2 mM NMN. 1 mL of each culture that was diluted to A550=0.5 with fresh medium was used for measuring (C) basal and (D) maximum oxygen consumption rates (OCRs) as described in Materials and Methods. Data shown are mean ± S.D. (n=3). One-way ANOVA, p=0.0015.
NMN increases NAD+ content, NAD+/NADH ratio, and ΔΨm of TAZ-KO C2C12 myoblast cells
We next sought to determine if mouse TAZ-KO C2C12 cells exhibited alterations in NAD+ redox similar to those in yeast cells. Surprisingly, WT and TAZ-KO cells exhibited similar NAD+/NADH ratios (Fig. 5A). However, the pools of NAD+ and NADH were decreased in TAZ-KO cells (Fig. 5B). Both NMN and nicotinamide (NAM) are efficient NAD+ precursors for cultured C2C12 cells, which exhibit reduced growth without some form of vitamin B3 supplementation (Fig. 5C). In TAZ-KO cells, NMN supplementation significantly increased the NAD+/NADH ratio (Fig. 5A) by increasing the pool of NAD+ without significantly altering the content of NADH (Fig. 5B). NMN supplementation of WT cells did not significantly alter the NAD+ pool nor the NAD+/NADH ratio, suggesting that the effect of NMN supplementation may be specific for cells harboring a mitochondrial defect. Although NMN supplementation did not affect OCR or ECAR in WT or TAZ-KO cells (Fig. S5), NMN supplementation significantly increased ΔΨm in TAZ-KO cells, as indicated by an ~36.7% increase in the TMRM fluorescence intensity (FI) (Fig. 5D), suggesting an improvement in mitochondrial function.
Figure 5. NMN increases the NAD+/NADH ratio and mitochondrial membrane potential (ΔΨm) in TAZ-KO C2C12 cells.
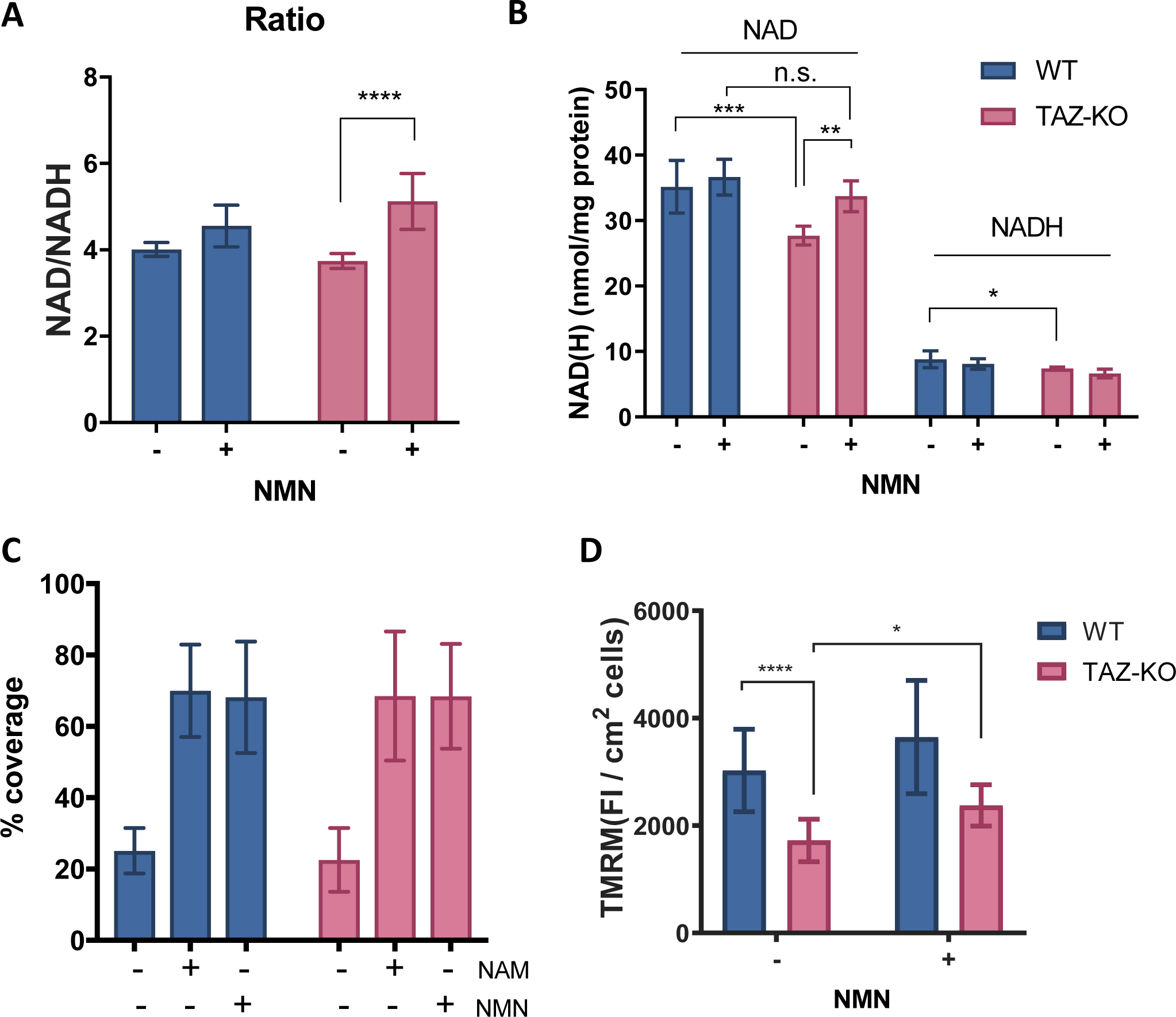
5.2×10^4 WT or TAZ-KO C2C12 cells were seeded in a 24-well plate for overnight incubation. After supplementation with 1 mM NMN for 4 hours, the cells were lysed for the measurement of (A) NAD+/NADH ratio and (B) NAD(H) levels as described in Materials and Methods. NAD(H) levels were normalized to protein levels. Data shown are mean ± S.D. (n=6). (C) WT or TAZ-KO cells were collected, washed twice, and suspended in nicotinamide and phenol red-free DMEM containing 10% dialyzed FBS. 2,000 cells were seeded into each well on a 96-well cell culture plate and treated with vehicle (water), 4 mg/L NAM, or 10.95 mg/L NMN. The % coverage by cells in each well after five days was determined with a plate reader. Data shown are mean ± S.D. (n=7–12). (D) WT and TAZ-KO cells grown in a 96-well plate were treated with 1 mM NMN for 5 hours when the confluency reached ~50%. The fluorescence intensity (FI) of TMRM associated with ΔΨm was measured as described in Materials and Methods. Data shown are mean ± S.D. (n=18). One-way ANOVA; *, p≤0.05; **, p≤0.01; ***, p≤ 0.001; ****, p≤0.0001.
NAD+ precursor supplementation improves the endurance of the Drosophila Taz889 mutant
The leg muscles of Drosophila resemble mammalian skeletal muscles with respect to myofibril morphology [57], and Drosophila has been an effective model to study muscle physiology. Disruption of TAFAZZIN in flies (Taz−/−) results in reduced CL levels, accumulation of MLCL, and decreased climbing ability [58]. The Taz889 mutant displays all the phenotypes of the Taz−/− Drosophila model (paper in review). We determined the effect of NMN on energy expenditure in the Taz889 flies with the Power Tower training protocol, which measures endurance by repetitive induction of a climbing stimulus [45–47]. Supplementation with NMN for five days in adult Taz889 flies increased exercise endurance to WT levels (Fig. 6A). NMN did not significantly affect the performance of WT flies. In agreement with the improved exercise phenotype, coupled mitochondrial respiration was improved by NMN supplementation (Fig. 6B). These findings indicate that NMN improves the energy metabolism and energy expenditure of CL-deficient flies, likely by improving mitochondrial function.
Figure 6. NMN significantly increases exercise endurance and mitochondrial respiratory control ratio (RCR) of Taz889 flies.
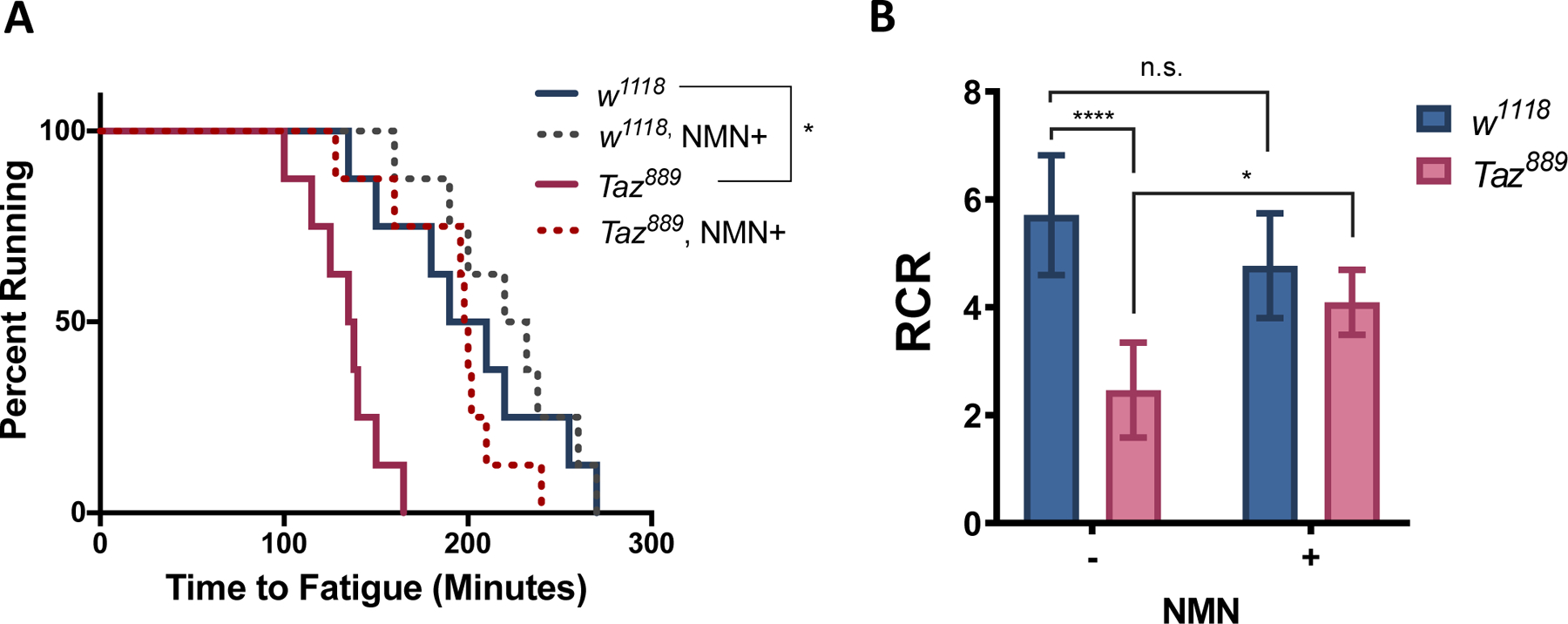
Fly food containing 0 or 10 mM NMN was supplied to WT and Taz889 flies beginning on age day 5. (A) Exercise endurance was assessed on age day 10 in eight vials (20 flies per vial) as described in Materials and Methods. At least two separate cohorts for each treatment were tested and analyzed with log-rank test. (*, p≤0.05) (B) Mitochondrial RCR was assessed on age day 10 as described in Materials and Methods. Data shown are mean ± S.D. (n=6). One-way ANOVA; *, p≤0.05; ****, p≤0.0001
Discussion
The current study demonstrates for the first time that CL deficiencies lead to alterations in NAD+ redox metabolism. Supplementation with NAD+ precursors improves mitochondrial function in various preclinical models of CL deficiency, suggesting that there may be a potential therapeutic benefit in the management of BTHS patients.
As the primary site for oxidation of NADH under aerobic conditions, the mitochondrial ETC is critical for maintaining an optimal intracellular NAD+/NADH ratio, and alterations in ETC function can greatly impact cellular metabolism [59, 60]. It has been well-established that CL is required for optimal OXPHOS activity and that CL deficiency in BTHS leads to decreased ETC function [61–63]. In agreement with these findings, we observed that depletion of CL in yeast results in aberrant NAD+ metabolism. Furthermore, the fermentation pathway is critical for the survival of crd1Δ, likely by compensating reduced NADH oxidation by the ETC.
In TAZ-KO cells, both NAD+ and NADH were reduced, however the NAD+/NADH ratio was comparable to WT. This finding differs from the observed accumulation of NADH in BTHS lymphoblasts [65]. Additionally, although NMN supplementation significantly increased mitochondrial membrane potential in the TAZ-KO cells, OCR and ECAR were not changed by NMN supplementation. One possible explanation for this discrepancy is that these cells were in their actively dividing state. When induced to differentiate, lactate production in TAZ-KO C2C12 cells is increased by three-fold when compared to controls [23], which is likely more stressful to mitochondria. Nonetheless, the significant decrease in the NAD+ content of the TAZ-KO cells in our study is suggestive of decreased biosynthesis and/or increased consumption. Although it is well documented that NAD+ content declines with aging and in mitochondrial disorders [33, 66–68], the mechanism for the decreased NAD+ content in CL-deficient cells remains unclear. Sirtuins and PARPs consume NAD+ as a substrate [69, 70] and influence mitochondrial function by regulating mitochondrial biosynthesis [71]. BTHS lymphoblasts exhibit increased mitochondrial mass, likely to compensate for the loss of mitochondrial function resulting from CL deficiency [62]. Therefore, decreased NAD+ levels in TAZ-KO cells could potentially be caused by increased consumption by sirtuins and PARPs. NAD kinase (NADK) also consumes NAD+ and NADH, generating NADP+ and NADPH. NADP+ is an important coenzyme in anabolism and antioxidative stress. In humans, NADP+ produced by NADK appears to be reduced immediately to NADPH [72], which is required for glutathione-dependent cellular antioxidant defense. It is possible that TAZ-KO cells consume NAD(H) to generate NADP(H) in response to accumulated ROS that results from CL deficiency [73].
The NAD+ precursors NMN and NR have been shown to improve mitochondrial performance by boosting NAD+ levels and/or increasing the NAD+/NADH ratio [42, 74–76]. An increased NAD+/NADH ratio has been shown to favor the oxidase activity but not the reductase activity of lactate dehydrogenase (LDH) [31], reducing the production of lactate by the fermentation pathway. In agreement, NMN treatment reduced ethanol production by WT and crd1Δ yeast cells. Importantly, increased ETC activity may also reduce the reliance of CL-deficient cells on aerobic fermentation. In accordance with this, we observed that NMN supplementation increased mitochondrial respiratory capacity of crd1Δ cells, suggesting an improvement in ETC activity and/or mitochondrial integrity. Furthermore, NMN increases the NAD+ content and NAD+/NADH ratio of TAZ-KO cells without elevating NADH levels, improving the mitochondrial integrity and polarization.
Drosophila has been an excellent model for studying mitochondrial disorders due to its relatively short life cycle, high content of mitochondria in flight muscles, and spontaneous flying and climbing behavior [46, 58, 77, 78]. The endurance of Drosophila reflects their energy expenditure and metabolism. Consistent with the previously observed decreased climbing ability in Taz−/− Drosophila [58], Taz889 Drosophila exhibited decreased endurance. NMN restored the climbing performance of Taz889 flies to almost WT levels, which is in accordance with an elevated RCR in the mutant, suggesting that NMN improves the exercise endurance of Taz889 flies by increasing their mitochondrial coupling. These findings suggest that promoting NAD+ synthesis using these precursors may prove beneficial in the management of skeletal muscle sequelae in BTHS.
In summary, the current study describes aberrant NAD+ metabolism resulting from CL deficiency and highlights the potential for NAD+ precursors to improve mitochondrial function in BTHS and other CL-associated mitochondrial disorders. The specific mechanisms whereby NAD+ precursors act to improve mitochondrial performance in CL-deficient cells are yet to be elucidated. Future studies should determine whether NAD+-responsive proteins such as sirtuins and PARPs increase mitochondrial biosynthesis and/or stress tolerance in CL-deficient cells following supplementation with NAD+ precursors [79, 80]. Additionally, it remains to be determined how CL deficiency affects subcellular pools of NAD+ and/or the NAD+ redox balance of individual subcellular compartments.
Supplementary Material
Highlights:
Aberrant NAD+ metabolism is observed in multiple preclinical model systems of Barth syndrome (BTHS)
Supplementation with nicotinamide mononucleotide (NMN), an NAD+ precursor, attenuates mitochondrial dysfunction associated with cardiolipin deficiency
Improved mitochondrial function in the Drosophila model of BTHS was associated with restored exercise endurance
Acknowledgements
Funding sources: this work was supported by the National Institutes of Health grants [R01HL117880, R01GM134715, R01AG059683, R21NS121276, and 19PRE34380493].
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Author Statement
Jiajia Ji: conceptualization, methodology, formal analysis, writing-original draft
Deena Damschroder: methodology, formal analysis
Denise Bessert: methodology
Pablo Lazcano: methodology
Robert Wessells: formal analysis, project administration
Miriam L. Greenberg: conceptualization, project administration, writing - reviewing and editing
Christian A. Reynolds: conceptualization, formal analysis, project administration, writing -reviewing and editing
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Deken RH, The Crabtree effect: a regulatory system in yeast. J Gen Microbiol, 1966. 44(2): p. 149–56. [DOI] [PubMed] [Google Scholar]
- 2.Marroquin LD, et al. , Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci, 2007. 97(2): p. 539–47. [DOI] [PubMed] [Google Scholar]
- 3.Lee JH, et al. , Optimization of the enzymatic one pot reaction for the synthesis of uridine 5’-diphosphogalactose. Bioprocess Biosyst Eng, 2010. 33(1): p. 71–8. [DOI] [PubMed] [Google Scholar]
- 4.Holden HM, Rayment I, and Thoden JB, Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem, 2003. 278(45): p. 43885–8. [DOI] [PubMed] [Google Scholar]
- 5.Fry M and Green DE, Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J Biol Chem, 1981. 256(4): p. 1874–80. [PubMed] [Google Scholar]
- 6.Robinson NC, Functional binding of cardiolipin to cytochrome c oxidase. J Bioenerg Biomembr, 1993. 25(2): p. 153–63. [DOI] [PubMed] [Google Scholar]
- 7.Eble KS, et al. , Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J Biol Chem, 1990. 265(32): p. 19434–40. [PubMed] [Google Scholar]
- 8.Lange C, et al. , Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J, 2001. 20(23): p. 6591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ozawa T, Tanaka M, and Wakabayashi T, Crystallization of mitochondrial cytochrome oxidase. Proc Natl Acad Sci U S A, 1982. 79(23): p. 7175–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren M, Phoon CK, and Schlame M, Metabolism and function of mitochondrial cardiolipin. Prog Lipid Res, 2014. 55: p. 1–16. [DOI] [PubMed] [Google Scholar]
- 11.Zhang M, Mileykovskaya E, and Dowhan W, Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem, 2002. 277(46): p. 43553–6. [DOI] [PubMed] [Google Scholar]
- 12.Acehan D, et al. , Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys J, 2011. 100(9): p. 2184–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfeiffer K, et al. , Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem, 2003. 278(52): p. 52873–80. [DOI] [PubMed] [Google Scholar]
- 14.Barth PG, et al. , An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci, 1983. 62(1–3): p. 327–55. [DOI] [PubMed] [Google Scholar]
- 15.Bione S, et al. , Transcriptional organization of a 450-kb region of the human X chromosome in Xq28. Proc Natl Acad Sci U S A, 1993. 90(23): p. 10977–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bione S, et al. , A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet, 1996. 12(4): p. 385–9. [DOI] [PubMed] [Google Scholar]
- 17.Vreken P, et al. , Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun, 2000. 279(2): p. 378–82. [DOI] [PubMed] [Google Scholar]
- 18.Schlame M, et al. , Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol, 2002. 51(5): p. 634–7. [DOI] [PubMed] [Google Scholar]
- 19.Valianpour F, et al. , Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J Lipid Res, 2005. 46(6): p. 1182–95. [DOI] [PubMed] [Google Scholar]
- 20.Patil VA, et al. , Loss of cardiolipin leads to perturbation of mitochondrial and cellular iron homeostasis. J Biol Chem, 2013. 288(3): p. 1696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raja V, et al. , Loss of Cardiolipin Leads to Perturbation of Acetyl-CoA Synthesis. J Biol Chem, 2017. 292(3): p. 1092–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, et al. , Cardiolipin-induced activation of pyruvate dehydrogenase links mitochondrial lipid biosynthesis to TCA cycle function. J Biol Chem, 2019. 294(30): p. 11568–11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lou W, et al. , Loss of tafazzin results in decreased myoblast differentiation in C2C12 cells: A myoblast model of Barth syndrome and cardiolipin deficiency. Biochim Biophys Acta Mol Cell Biol Lipids, 2018. 1863(8): p. 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donati MA, et al. , Barth syndrome presenting with acute metabolic decompensation in the neonatal period. J Inherit Metab Dis, 2006. 29(5): p. 684. [DOI] [PubMed] [Google Scholar]
- 25.Ferri L, et al. , New clinical and molecular insights on Barth syndrome. Orphanet J Rare Dis, 2013. 8: p. 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yen TY, et al. , Acute metabolic decompensation and sudden death in Barth syndrome: report of a family and a literature review. Eur J Pediatr, 2008. 167(8): p. 941–4. [DOI] [PubMed] [Google Scholar]
- 27.Small WC and McAlister-Henn L, Identification of a cytosolically directed NADH dehydrogenase in mitochondria of Saccharomyces cerevisiae. J Bacteriol, 1998. 180(16): p. 4051–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luttik MA, et al. , The Saccharomyces cerevisiae NDE1 and NDE2 genes encode separate mitochondrial NADH dehydrogenases catalyzing the oxidation of cytosolic NADH. J Biol Chem, 1998. 273(38): p. 24529–34. [DOI] [PubMed] [Google Scholar]
- 29.De Santis A and Melandri BA, The oxidation of external NADH by an intermembrane electron transfer in mitochondria from the ubiquinone-deficient mutant E3–24 of Saccharomyces cerevisiae. Arch Biochem Biophys, 1984. 232(1): p. 354–65. [DOI] [PubMed] [Google Scholar]
- 30.Powers C, et al. , Diminished Exercise Capacity and Mitochondrial bc1 Complex Deficiency in Tafazzin-Knockdown Mice. Front Physiol, 2013. 4: p. 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerdan S, et al. , The redox switch/redox coupling hypothesis. Neurochemistry International, 2006. 48(6–7): p. 523–530. [DOI] [PubMed] [Google Scholar]
- 32.Ying W, NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal, 2008. 10(2): p. 179–206. [DOI] [PubMed] [Google Scholar]
- 33.Massudi H, et al. , Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One, 2012. 7(7): p. e42357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clement J, et al. , The Plasma NAD(+) Metabolome Is Dysregulated in “Normal” Aging. Rejuvenation research, 2019. 22(2): p. 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee CF, et al. , Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation, 2016. 134(12): p. 883–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee CF, et al. , Targeting NAD(+) Metabolism as Interventions for Mitochondrial Disease. Sci Rep, 2019. 9(1): p. 3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rolfe HM, A review of nicotinamide: treatment of skin diseases and potential side effects. J Cosmet Dermatol, 2014. 13(4): p. 324–8. [DOI] [PubMed] [Google Scholar]
- 38.Conze DB, Crespo-Barreto J, and Kruger CL, Safety assessment of nicotinamide riboside, a form of vitamin B3. Hum Exp Toxicol, 2016. 35(11): p. 1149–1160. [DOI] [PubMed] [Google Scholar]
- 39.Trammell SA, et al. , Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun, 2016. 7: p. 12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Remie CME, et al. , Nicotinamide riboside supplementation alters body composition and skeletal muscle acetylcarnitine concentrations in healthy obese humans. The American Journal of Clinical Nutrition, 2020. 112(2): p. 413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin AS, et al. , Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight, 2017. 2(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gomes AP, et al. , Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell, 2013. 155(7): p. 1624–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gonzalez B, Francois J, and Renaud M, A rapid and reliable method for metabolite extraction in yeast using boiling buffered ethanol. Yeast, 1997. 13(14): p. 1347–55. [DOI] [PubMed] [Google Scholar]
- 44.Lou W, et al. , Genetic re-engineering of polyunsaturated phospholipid profile of Saccharomyces cerevisiae identifies a novel role for Cld1 in mitigating the effects of cardiolipin peroxidation. Biochim Biophys Acta Mol Cell Biol Lipids, 2018. 1863(10): p. 1354–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tinkerhess MJ, et al. , Endurance training protocol and longitudinal performance assays for Drosophila melanogaster. J Vis Exp, 2012(61). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sujkowski A and Wessells R, Using Drosophila to understand biochemical and behavioral responses to exercise. Exercise and sport sciences reviews, 2018. 46(2): p. 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Damschroder D, et al. , Drosophila Endurance Training and Assessment of Its Effects on Systemic Adaptations. Bio-protocol, 2018. 8(19): p. e3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holmbeck MA, et al. , A Drosophila model for mito-nuclear diseases generated by an incompatible interaction between tRNA and tRNA synthetase. Dis Model Mech, 2015. 8(8): p. 843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferguson M, et al. , Age-associated decline in mitochondrial respiration and electron transport in Drosophila melanogaster. Biochem J, 2005. 390(Pt 2): p. 501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kohrer K and Domdey H, Preparation of high molecular weight RNA. Methods Enzymol, 1991. 194: p. 398–405. [DOI] [PubMed] [Google Scholar]
- 51.Ma L, et al. , The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Delta mutant. Implications for Barth syndrome. J Biol Chem, 2004. 279(43): p. 44394–9. [DOI] [PubMed] [Google Scholar]
- 52.Pfeiffer T and Morley A, An evolutionary perspective on the Crabtree effect. Front Mol Biosci, 2014. 1: p. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gu Z, et al. , Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol Microbiol, 2004. 51(1): p. 149–58. [DOI] [PubMed] [Google Scholar]
- 54.Leskovac V, Trivic S, and Pericin D, The three zinc-containing alcohol dehydrogenases from baker’s yeast, Saccharomyces cerevisiae. FEMS Yeast Res, 2002. 2(4): p. 481–94. [DOI] [PubMed] [Google Scholar]
- 55.de Smidt O, du Preez JC, and Albertyn J, The alcohol dehydrogenases of Saccharomyces cerevisiae: a comprehensive review. FEMS Yeast Res, 2008. 8(7): p. 967–78. [DOI] [PubMed] [Google Scholar]
- 56.Lu SP, Kato M, and Lin SJ, Assimilation of endogenous nicotinamide riboside is essential for calorie restriction-mediated life span extension in Saccharomyces cerevisiae. J Biol Chem, 2009. 284(25): p. 17110–17119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Avellaneda J, et al. , Myofibril and mitochondria morphogenesis are coordinated by a mechanical feedback mechanism in muscle. Nat Commun, 2021. 12(1): p. 2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Y, et al. , A Drosophila model of Barth syndrome. Proc Natl Acad Sci U S A, 2006. 103(31): p. 11584–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Duchen MR, Surin A, and Jacobson J, Imaging mitochondrial function in intact cells. Methods Enzymol, 2003. 361: p. 353–89. [DOI] [PubMed] [Google Scholar]
- 60.Yang Y and Sauve AA, NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta, 2016. 1864(12): p. 1787–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barth PG, et al. , X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): respiratory-chain abnormalities in cultured fibroblasts. J Inherit Metab Dis, 1996. 19(2): p. 157–60. [DOI] [PubMed] [Google Scholar]
- 62.Xu Y, et al. , Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab Invest, 2005. 85(6): p. 823–30. [DOI] [PubMed] [Google Scholar]
- 63.Dudek J, et al. , Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res, 2013. 11(2): p. 806–19. [DOI] [PubMed] [Google Scholar]
- 64.Jiang F, et al. , Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J Biol Chem, 2000. 275(29): p. 22387–94. [DOI] [PubMed] [Google Scholar]
- 65.Gonzalvez F, et al. , Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim Biophys Acta, 2013. 1832(8): p. 1194–206. [DOI] [PubMed] [Google Scholar]
- 66.Camacho-Pereira J, et al. , CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab, 2016. 23(6): p. 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yaku K, Okabe K, and Nakagawa T, Simultaneous measurement of NAD metabolome in aged mice tissue using liquid chromatography tandem-mass spectrometry. Biomed Chromatogr, 2018. 32(6): p. e4205. [DOI] [PubMed] [Google Scholar]
- 68.Frederick DW, et al. , Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab, 2016. 24(2): p. 269–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blander G and Guarente L, The Sir2 family of protein deacetylases. Annu Rev Biochem, 2004. 73: p. 417–35. [DOI] [PubMed] [Google Scholar]
- 70.Bai P, Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol Cell, 2015. 58(6): p. 947–58. [DOI] [PubMed] [Google Scholar]
- 71.Prolla TA and Denu JM, NAD+ deficiency in age-related mitochondrial dysfunction. Cell Metab, 2014. 19(2): p. 178–80. [DOI] [PubMed] [Google Scholar]
- 72.Agledal L, Niere M, and Ziegler M, The phosphate makes a difference: cellular functions of NADP. Redox Rep, 2010. 15(1): p. 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.He Q, et al. , Mitochondria-targeted antioxidant prevents cardiac dysfunction induced by tafazzin gene knockdown in cardiac myocytes. Oxid Med Cell Longev, 2014. 2014: p. 654198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sims CA, et al. , Nicotinamide mononucleotide preserves mitochondrial function and increases survival in hemorrhagic shock. JCI Insight, 2018. 3(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Q, et al. , Increasing ovarian NAD(+) levels improve mitochondrial functions and reverse ovarian aging. Free Radic Biol Med, 2020. 156: p. 1–10. [DOI] [PubMed] [Google Scholar]
- 76.Schondorf DC, et al. , The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep, 2018. 23(10): p. 2976–2988. [DOI] [PubMed] [Google Scholar]
- 77.Sohal RS, Aging changes in insect flight muscle. Gerontology, 1976. 22(4): p. 317–33. [DOI] [PubMed] [Google Scholar]
- 78.Sanchez-Martinez A, et al. , Modeling human mitochondrial diseases in flies. Biochim Biophys Acta, 2006. 1757(9–10): p. 1190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Srivastava S, Emerging therapeutic roles for NAD(+) metabolism in mitochondrial and age-related disorders. Clin Transl Med, 2016. 5(1): p. 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Canto C, Menzies KJ, and Auwerx J, NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab, 2015. 22(1): p. 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Raja V, et al. , Cardiolipin-deficient cells depend on anaplerotic pathways to ameliorate defective TCA cycle function. Biochim Biophys Acta Mol Cell Biol Lipids, 2019. 1864(5): p. 654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ye C, et al. , Deletion of the cardiolipin-specific phospholipase Cld1 rescues growth and life span defects in the tafazzin mutant: implications for Barth syndrome. J Biol Chem, 2014. 289(6): p. 3114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.