

Abstract
While impaired ribosomal biogenesis is observed in neurodegenerative diseases its pathogenic contributions are not clear. For instance, it is well established that in rodent neurons, genetic inhibition of RNA-Polymerase 1 that transcribes rRNA results in structural disruption of the nucleolus, neuronal apoptosis, and, neurodegeneration. However, in most neurodegenerative diseases, nucleolar morphology is unaffected. It is reported here that in primary cortical neurons from newborn rats, inhibition of ribosomal biogenesis by shRNA-mediated knockdowns of several ribosomal proteins including S6, S14 or L4 resulted in p53-mediated apoptosis despite absence of structural disruption of the nucleolus. Conversely, knockdown of the RP L11, which in non-neuronal systems mediates p53 activation downstream of ribosomal stress, protected neurons against inhibition of ribosomal biogenesis but not staurosporine. Moreover, overexpression of L11 enhanced p53-driven transcription and increased neuronal apoptosis. In addition, inhibition of p53, or, L11 knockdown blocked apoptosis in response to the RNA analog 5-fluorouridine which perturbed nucleolar structure, inhibited ribosomal synthesis and activated p53. Although the DNA double strand break (DSB) inducer etoposide activated p53, nucleolar structure appeared intact. However, by activating the DNA damage response kinase ATM, etoposide increased 47S pre-rRNA levels and enhanced nucleolar accumulation of nascent RNA suggesting slower rRNA processing and/or increased Pol1 activity. In addition, shL11 reduced etoposide-induced apoptosis. Therefore, seemingly normal morphology of the neuronal nucleolus does not exclude presence of ribosomal stress. Conversely, targeting the ribosomal stress-specific signaling mediators including L11 offers a novel approach to uncover neurodegenerative contributions of de-regulated ribosomal synthesis as exemplified in DSB-challenged neurons.
Keywords: apoptosis, p53, neurons, nucleolus, ribosomal proteins, DNA damage
Introduction
Increased neuronal death is a component of many neurodegenerative diseases, including Alzheimer’s (AD), Huntington’s (HD), Parkinsons’s (PD), amyotrophic lateral sclerosis (ALS), and frontal temporal dementia (FTD) [1,2]. However, progress in understanding the mechanisms of neuronal death in cellular- and animal models of these conditions has not yet translated into neuroprotective treatments that work clinically. Hence, the existing models of neurodegeneration-associated neuronal death may be missing some important factors that determine death or survival of neurons under pro-neurodegenerative challenges.
The ribosome is a large ribonucleoprotein complex that is the principal component of the protein synthesis machinery. Biogenesis of ribosomes takes place in the nucleolus [3–5]. Dysregulation of that process triggers the ribosomal stress pathway, one of the major contributors to tumor suppression [6,7]. Though ribosomal deficits and/or impaired ribosomal biogenesis are documented in AD, HD, PD, and ALS/FTD [8–18], the role of ribosomal stress in neurodegeneration is not clear.
In proliferating cells, both decreased- and increased ribosomal biogenesis may initiate the ribosomal stress response, which includes activation of the p53 pathway [19,20,6]. The latter process relies on interactions between the p53 ubiquitin ligase MDM2 and ribosomal proteins (RPs) [6]. Specifically, when not involved in ribosome assembly, RPL11 binds and inhibits MDM2 and thus stabilizes p53 [21,19,22]. Besides MDM2, L11 may also interact with other proteins and RNAs to suppress cell growth via p53-dependent and/or independent mechanisms [6]. Thus, L11 is required for p53 activation downstream of ribosomal stress. However, the role of L11 in neuronal death has not yet been reported.
While the nucleolus is a center of ribosomal biogenesis, it also evolved to fulfill other functions including assembly of non ribosomal ribonucleoprotein complexes, non-rRNA processing, chromatin organization, and, sequestration, regulated release and degradation of various proteins with critical roles in cell cycle control, stress responses or transcriptional regulation [23–26]. When RNA-Polymerase-1 (Pol1), which initiates ribosomal biogenesis, is inhibited, nucleolar structure is disrupted leading to a state of nucleolar stress [4,27]. For instance, in cultured cortical neurons from newborn rats, the Pol1-inhibiting DNA single strand break inducer, camptothecin, or knockdown of the Pol1 co-activator Tif1a, induced apoptosis which was preceded by nucleolar stress and inhibited by the dominant-negative variant of p53 [28]. In addition, p53 activation and neuronal death were reported after knocking out the Pol1 co-activator, Tif1a, in various neuronal populations of adult murine brain including forebrain neurons [10,29,30].
However, observations of neurodegeneration-associated disruption to the nucleolar structure are relatively rare and usually documented at relatively more advanced stages of such diseases as PD or AD [10,17,18]. In contrast, greater numbers of sensitive neurons including those at early stages of PD-, AD- or ALS/FTD- pathology displayed either no nucleolar alterations or moderate reductions and/or increases of nucleolar size [15,31–36]. Therefore, one can wonder whether in the absence of nucleolar disruption, dysregulation of ribosomal biogenesis is sufficient to induce neuronal death.
In non-neuronal cell lines reduced levels of RPs of the small ribosomal subunit lead to ribosomal stress without major changes to nucleolar structure or Pol1 inhibition [19]. However, under such conditions, L11-mediated activation of p53 and cell cycle arrest are still observed, suggesting that the ribosomal stress response does not require nucleolar disruption. Therefore, the current work was initiated to determine whether in cultured cortical and hippocampal neurons from newborn rats, shRNA-mediated knockdowns of two RPSs, S6 and S14, induce p53- and L11- mediated apoptosis in the absence of nucleolar stress.
Materials and Methods
Animals.
Sprague–Dawley pregnant female rats were purchased from Harlan (Indianapolis, IN, USA). All animal experiments strictly followed the protocols that were approved by the Institutional Animal Care and Use Committee of the University of Louisville and the NIH guidelines.
Materials.
Oligonucleotides were purchased from IDT. All other reagents were obtained from Sigma (St. Louis, MO), VWR (Radnor, PA), Life Technologies-Invitrogen (Grand Island, NY) or Qiagen, MD) unless stated otherwise.
Plasmids.
The following plasmids were previously described: EF1α-LacZ (EF1α promoter-driven β-galactosidase), shTif1a, shGFP, and DN-p53 [28]; TS-p53 [37]; shLuc [38]; shS6, shS14, shL4, shL11, chicken β-actin promoter-driven-EGFP-RPL4 [39]; PG13 (p53 response element-driven luciferase reporter plasmid) [40]. To prepare the EGFP-RPL11 expression vector a cDNA encoding rat RPL11 was cloned into the chicken β-actin promoter-driven-pEGFP-C2 plasmid.
Cell culture and transfections.
Cultures of cortical and hippocampal neurons were prepared from newborn Sprague-Dawley rats at postnatal day 0 (P0) as described previously [41]. Cells were plated onto Poly-D-Lysine- and laminin-coated 12 mm diameter plastic coverslips that were produced in the lab from the electron microscopy-grade mylar masks (Electron Microscopy Sciences, Hatfield, PA, #50425). Culture medium was Basal Medium Eagle (BME, Lonza) supplemented with 10% heat-inactivated bovine calf serum (HyClone, GE Healthcare Life Sciences, Logan, UT, USA), 35 mM glucose, 1 mM L-glutamine, 100 units/ml penicillin, and 0.1 mg/ml streptomycin; on day in vitro 2 (DIV2), media was supplemented with 2.5 μM cytosine arabinoside to inhibit the proliferation of non-neuronal cells. Neurons were transfected on DIV3 using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Drug treatments.
5-Fluorouridine (5FU) and 5-Fluoro-5′-deoxyuridine (5FdU) were dissolved in water. Etoposide (Etop), staurosporine, CPT and KU55933 (ATMi) were dissolved in DMSO. Treatments were performed at DIV5 unless stated otherwise.
Immunostaining.
All stainings of cultured neurons were performed using standard protocol. Briefly, cells were fixed in 4% formaldehyde at room temperature for 20 min, permeabilized using 0.5 % NP-40 for 20 min and blocked with 5% goat serum/0.1 % Triton X-100 for 1 h. The following primary antibodies were used: chicken anti-β-galactosidase (Abcam, 1:1000), mouse anti-nucleophosmin-1(Npm)/B23 (Sigma, 1:750), mouse anti-nucleolin (Ncl)/C23 (St. Cruz, 1:200), and mouse anti-fibrillarin (Abcam, 1:750).The following secondary antibodies were used: Alexa Fluor 488 anti-mouse IgG (Invitrogen, 1:300) and Alexa Fluor 594 anti-chicken IgG (Jackson ImmunoResearch, 1:300). DNA was counterstained with 2.5 μg/mL Hoechst-33258.
Ribosome labeling.
Ribosomes were visualized with the NeuroTrace 500/525 Green Fluorescent Nissl Stain (Invitrogen, N21480, 1:500) as described previously [39]. DNA was counterstained with 2.5 μg/mL Hoechst-33258.
Quantification of the fluorescence intensity (FI) of ribosome- and nucleolar marker stainings.
Stained cells were visualized with Zeiss Observer.Z1 fluorescent microscope using a 40x lens to image nucleophosmin and nucleolin immunostaining or ribosome staining, respectively. Digitalized pictures were captured using Zeiss AxioVision (Zeiss) followed by conversion to gray scale TIFF files. Quantification of the FI at the single cell level was performed using the „Integrated Density (IntDen)” parameter in the ImageJ software. Ribosome content was evaluated by calculating FI of the perikaryal NeuroTrace Green signal that was normalized against fluorescence intensity of the Hoechst signal. A change of FI was expressed as a fold control that was defined by a ratio of an individual FI value to the average FI value of the control group as indicated for each set of experiments. Such calculations were performed for each independent experiment before pooling data for statistical analysis. Similar normalization approach was used for quantification of nucleolar Npm- or Ncl stainings except IntDen of the nucleolar signal was divided by IntDen of the total nuclear signal instead of Hoechst. Whenever territory of the nucleolus was measured an „Area” parameter was used.
Western blotting.
Western blotting was performed using standard procedures. The primary antibodies used were as follows: anti-p53 (Santa Cruz Biotechnology, rabbit antibody FL-393, catalog# sc-6243, 1:500), anti-phospho-Ser15-p53 (Cell Signaling, 1:1000), anti-phospho-Ser-1981-ATM (Cell Signaling, 1:1000), anti-γH2Ax (Cell Signaling, 1:1000), anti-β-actin (Sigma, 1:1000), anti-β-tubulin (Sigma, 1:1000), secondary antibodies were horseradish peroxidase conjugated.
Northern blotting.
Total RNA of cultured cortical neurons was isolated using RNAeasy Mini Kit (Qiagen, #74107). A total of 3μg RNA from each sample was fractionated on 1.5 % agarose/formaldehyde gel and transferred to Zetaprobe membrane (Biorad Inc, Hercules, CA). Ethidium bromide was used to stain total RNA in the gel. Preparation of oligonucleotide probes, hybridization and Phospho Imager analyses were performed as described previously [42]. Oligonucleotide probes were used as follows: rat 5’ETS 5’TAGCACCAAACGGGAAAACC3’; rat ITS1 5’GGTTATCTGAGGTGTGAGCG’3; human 5.8S was previously published [42].
Quantification of apoptosis.
To detect apoptosis in transfected neurons cells were immunostained for the transfection marker β-gal and counter-stained with 2.5 μg/mL Hoechst-33258. Cells with condensed or fragmented nuclei were scored as apoptotic. At least 150 transfected cells (i.e. showing positive immunostaining for β-gal) were analyzed for each condition in each experiment.
Transcription assay.
Luciferase and β-gal activities were measured using commercial kits (Promega) and the Berthold Orion II luminometer as previously described [43]. Transcriptional activity was expressed as a β-gal-normalized luciferase activity.
RNA extraction and qRT-PCR.
Total RNA was isolated using TRI Reagent (Sigma); cDNA was prepared using the AMV First-Strand cDNA Synthesis Kit (Invitrogen) and random hexamers. Quantitative Reverse Transcriptase-PCR (qRT-PCR) was performed on the 7900HT cycler (Applied Biosystems) using previously described primers complementary to rat 5’ETS region of 47S pre-rRNA or rat 18S rRNA [28] and SYBR green. The quantitative analysis was performed using the ΔΔCt method. As neurons are postmitotic cells with relatively low activity of ribosomal biogenesis, 2– 4 h treatments with anti-ribosomal drugs such as 5FU are not expected to significantly affect expression levels of mature rRNA including 18S [39]. Indeed, pilot experiments comparing 18S to two housekeeping mRNA amplicons including β-actin and hexokinase-1 revealed similar effects of 5FU on 47S rRNA levels (Fig. S1). Moreover, in our samples, 18S CT values were closer to CT values of 47S pre-rRNA than for the mRNA normalizers that we have tested (in all cases those were higher than 18S CTs by 7–15). Hence, the 18S amplicon appeared to be the most accurate indicator of total RNA content and, therefore, the reference of choice for our analyses.
In situ run-on assay.
Neuronal nascent RNA was labeled with the RNA precursor 5-ethynyl uridine (5-EU; Berry & Associates, Inc., Dexter, MI) which was detected using the Click-It reagent Oregon Green® azide (Invitrogen) as previously described [38].
Statistical analysis.
Data were analyzed by one- or two-way ANOVA followed by Tukey posthoc tests or by the Mann-Whitney u-test as indicated.
Results
Knockdowns of RPs induce p53-dependent apoptosis of cortical neurons.
To determine the effects of reduced ribosomal biogenesis on the survival of cultured rat cortical neurons from newborn rats, validated shRNAs expression plasmids targeting ribosomal proteins S6, S14 and L4 were used [39]. At two days after transfection, these constructs increased neuronal apoptosis from the 27% that was observed with a control shRNA targeting Renilla luciferase (shLuc) to 67, 67 and 64%, respectively (Fig. 1A–B). Co-expression of the dominant-negative mutant form of p53 (DN-p53) blocked pro-apoptotic effects of shRPs with apoptosis at 5, 10, 12 and 8% in neurons receiving shLuc, shS6, shS14 and shL4, respectively (Fig. 1A–B). Consistently with previously published data [28], shRNA against the Pol1 co-activator Tif1a induced similar levels of apoptosis that was inhibited by the DN-p53 (Fig. 1C). Finally, either shRPs or shTif1a induced apoptosis in hippocampal neurons (Fig. 1D).
Figure 1. Ribosomal protein depletion triggers neuronal apoptosis that is p53-dependent.
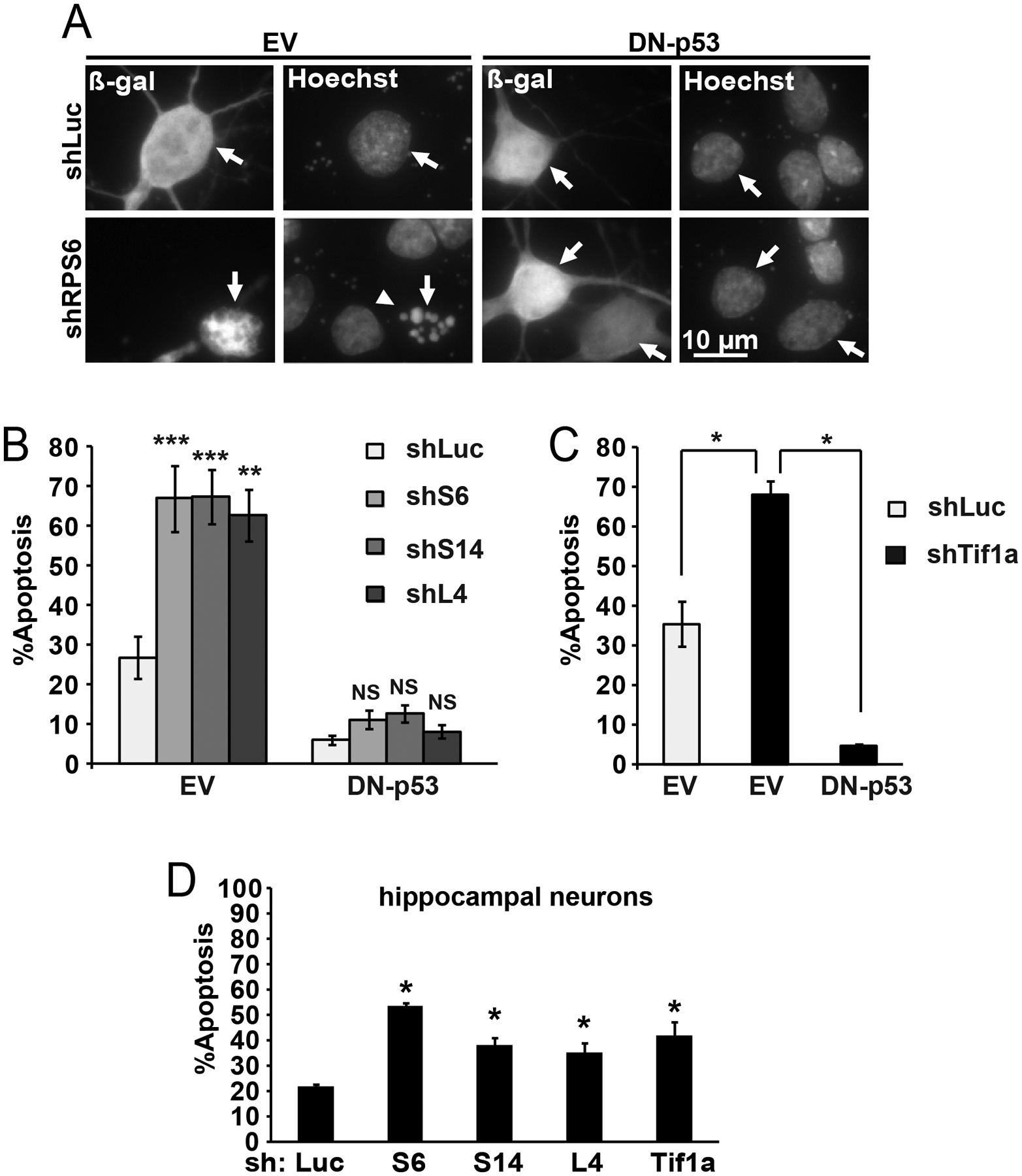
DIV3 neurons were transfected with expression vectors for β-galactosidase (β-gal) and shRNAs (0.1 and 0.3 μg plasmid DNA/5×105 cells, respectively; 0.4 μg shRNA plasmids were used in D); an shRNA targeting Renilla luciferase (shLuc) was used as a control. The dominant-negative mutant of p53 (DN-p53) or its empty vector control (EV) was added as indicated (0.1 μg plasmid DNA). All shRNAs were previously validated in rat forebrain neurons [39]. Cells were fixed after 48 h. A, Representative photomicrographs depicting transfected neurons as identified by immunofluorescence for β-gal (arrows). Apoptotic changes, including condensation and fragmentation of the nucleus, were visualized by counterstaining with Hoechst-33258 (arrowhead). B, Increased apoptosis after transfection of shRPs was prevented by the DN-p53. C, Consistently with previously published data, knockdown of the Pol1 co-activator Tif1a elicited similar apoptotic response [28]. D, In hippocampal neurons, shRPs or shTif1a also induced apoptosis. Averages ±SEM of 2 sister cultures from 3 (B) or 2 (C, D) independent experiments are shown; p>0.05, NS; p<0.05, *, p<0.001, *** as compared to shLuc-transfected controls unless indicated otherwise (u-test).
In RP-knocked down cortical neurons that were protected from apoptosis by the DN-p53, nucleolar morphology was determined by immunofluorescence staining for the nucleolar marker proteins nucleophosmin-1/B23 (Npm), nucleolin (Ncl) and fibrillarin (Fig. 2). There were no major effects of shRPs on nucleolar morphology except moderate but significant changes with shS6 (Fig. 2A, B, D). Those included nearly 20% decreases of nucleolar Npm- and Ncl signals as well as 28% increase in an average number of Npm-positive nucleoli/cell (Fig. 2D). In contrast, knockdown of Tif1a, reduced the nucleolar signals of Npm and Ncl by nearly 80% (Fig. 2B). In addition, in shTif1a-transfected neurons, Npm-positive territory of an average nucleolus declined by 55% (Fig. 2C). A twenty four % decrease of average number of Npm-positive nucleoli was also observed (Fig. 2D). Nucleolar staining of fibrillarin was maintained in shRPs- or shTif1a-transfected neurons (Fig. 2A). Such findings are consistent with a critical role of Pol1 activity for maintenance of the nucleolar granular component (GC) that is the predominant enrichment site for Npm and Ncl [3,44].
Figure 2. In neurons, ribosomal protein depletion has relatively minor effects on nucleolar structure.
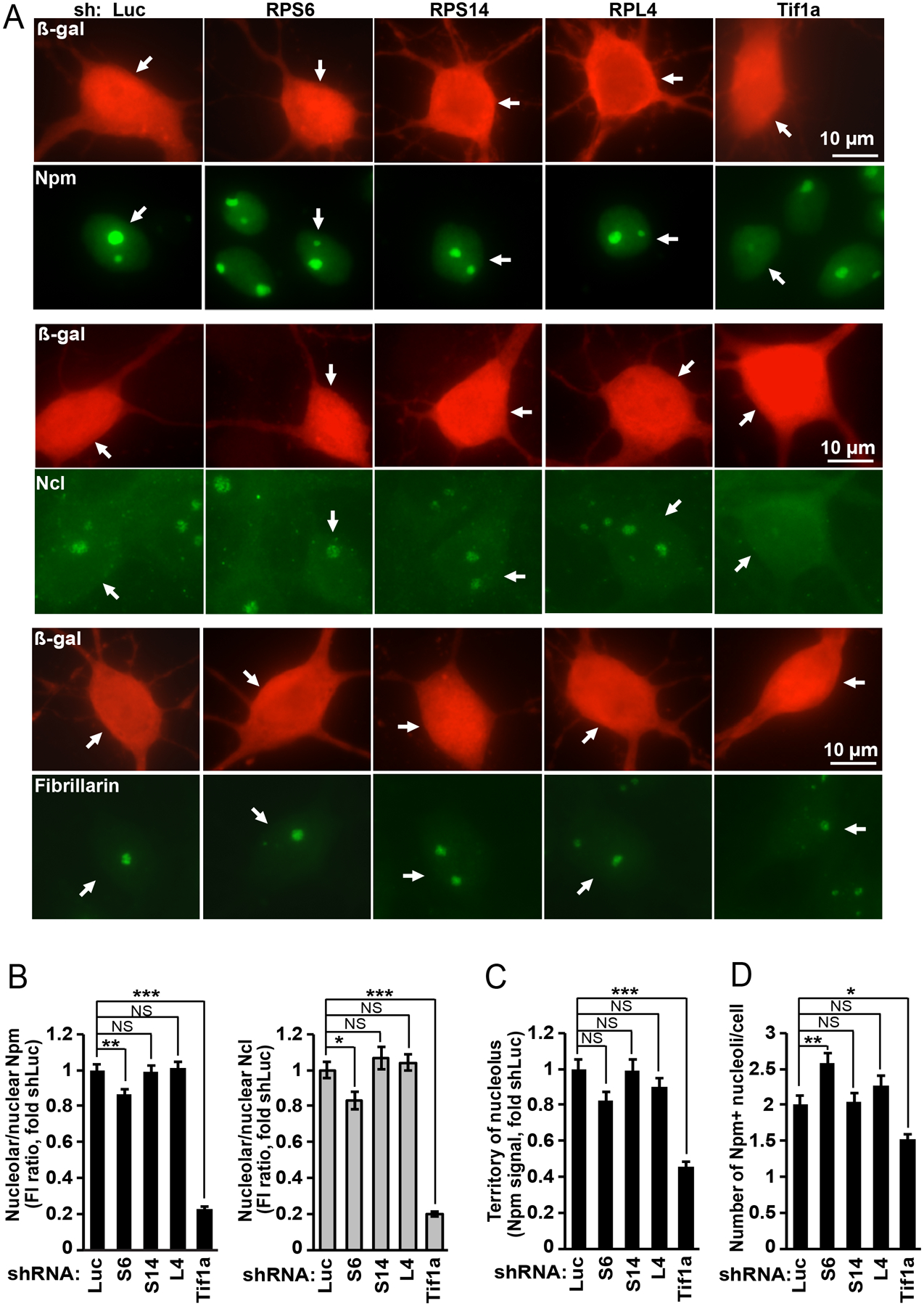
Cortical neurons were transfected as in Fig. 1; DN-p53 was included in all transfections to prevent apoptosis. After 48 h cell were fixed and nucleolar marker proteins including nucleophosmin-1/B23 (Npm), nucleolin (Ncl) and fibrillarin were detected by immunofluorescence. A, Representative images of nucleoli in transfected cells; arrows indicate the transfected (i.e. β-gal-positive) neurons. Knockdown of Tif1a but not RPs disrupted nucleolar staining of Npm and Ncl but not fibrillarin. B, Quantitative analysis of fluorescence intensity (FI) for Npm- and Ncl signals in the nucleolus and the nucleus. Decline of the nucleolar signal in shTif1a-transfected neurons suggests structural reorganization of the nucleolus including reduced size of the granular component. Of shRPs only shS6 produced relatively small decrease in nucleolar signals of Npm and Ncl. C, Territory of an average nucleolus as defined by the Npm signal was reduced by shTif1a but not shRPs. D, Average number of Npm-positive nucleoli/cell was increased or reduced by shS6 or shTif1a, respectively; shS14 or shL4 did not affect that parameter. In B-D, averages ±SEM of at least 40 cells from 2 independent experiments are shown; p>0.05, NS; p<0.05, *; p<0.001, *** (one-way ANOVA/Tukey posthoc tests).
However, shRPs or shTif1a had similar negative effects on perikaryal ribosome content with declines ranging from 19 to 30% after 3 day co-expression with the DN-p53 (Fig. 3A–B). Therefore, in forebrain neurons, p53-dependent apoptosis and reduced ribosome supply but not nucleolar disruption are common consequences of various shRNA-based strategies to inhibit ribosomal biogenesis.
Figure 3. Similar anti-ribosomal effects after neuronal depletion of RPs and the Pol1 co-factor Tif1a.
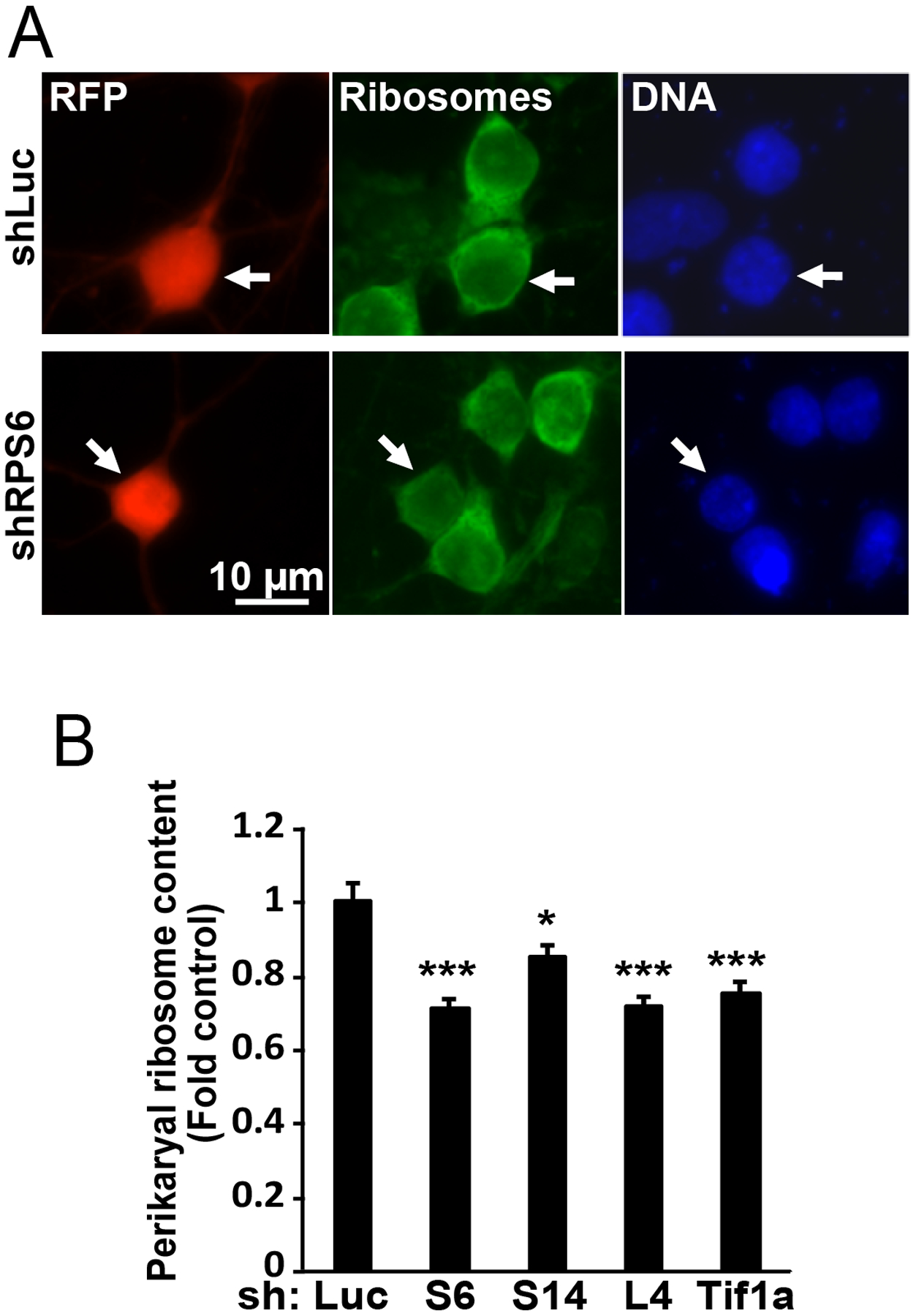
Cortical neurons were transfected as in Fig. 2 except the β-gal vector was replaced by a RFP expression vector; 48 h later cells were fixed and ribosomes and DNA were stained using NeuroTrace Green and Hoechst-33258, respectively. Representative images of transfected (i.e. RFP-positive) neurons are shown in A (arrows); quantification of perikaryal ribosome content is presented in B. Reduced perikaryal ribosome content was observed in cells that were transfected with shRPs or shTif1a. Averages±SEM of at least 45 cells from 3 independent experiments are shown; p<0.05, *; p<0.001, *** as compared to shLuc-transfected controls (ANOVA/Tukey posthoc tests in B).
Knockdown of L11 reduces apoptosis in response to nucleolar- and/or ribosomal stress.
To determine the role of L11 in ribosomal stress-induced neuronal apoptosis, two shRNAs targeting L11 were designed and validated. These constructs blocked expression of the L11-EGFP fusion protein when co-expressed in cortical neurons (Fig. 4A–B). In addition, they reduced perikaryal ribosome content in a manner similar to the shRNAs targeting S6, S14, L4 or Tif1a (Fig. 4C and Fig. 3B). However, unlike the latter constructs, shL11 did not induce apoptosis (Fig. 4D–E). Further, it reduced apoptosis in response to shS6, shS14, shL4 or shTif1a (Fig. 4D–F). Complete protection was observed only for shL4 (Fig. 4E). In other cases including depletion of S6, S14 or Tif1a, knockdown of L11 offered only partial inhibition of apoptosis (Fig. 4E–F). The protection is unlikely due to off target effects as two different shRNA constructs targeting L11 produced similar anti-apoptotic effects in Tif1A-depleted neurons (Fig. 4F).
Figure 4. Knockdown of the ribosomal protein L11 reduces apoptotic responses to ribosomal stress regardless of nucleolar disruption.
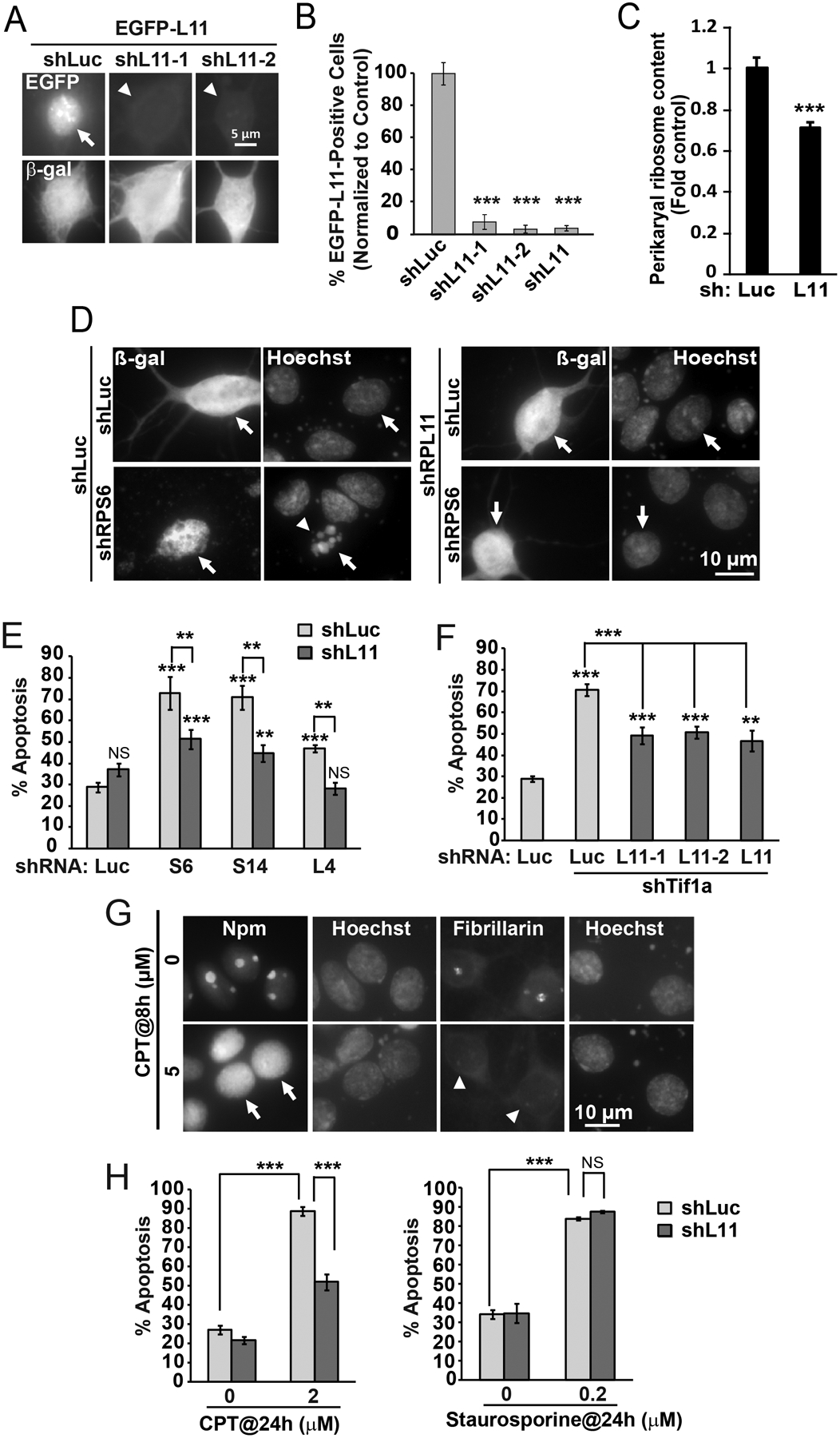
A-B, DIV3 cortical neurons were transfected with expression vectors for β-gal, EGFP-L11, and, shRNAs against L11 including shL11–1, shL11–2 or their equimolar mix that was designated as shL11 (0.1+0.1+0.4 μg plasmid DNA/5×105 cells, respectively); control was the shRNA against Renilla luciferase (shLuc). After 48 h, cells were fixed and fraction of transfected (i.e. β-gal-positive) cells that also expressed EGFP was determined by co-immunofluorescence. EGFP-L11, which in most shLuc-transfected controls was present in the nucleus and the nucleolus (arrow), was virtually undetectable in the presence of either shL11 plasmid (arrowheads). C, For ribosome content determination, cortical neurons were transfected with shL11 as described for Fig. 3. As with other RPs, knockdown of L11 resulted in declining ribosome content. D-F, Cortical neurons were transfected with expression vectors for β-gal and shRPs or shTif1a and shL11 (0.1+0.2+0.2 μg/5×105 cells); shLuc was used as a control. D, Representative images of transfected neurons that were identified by β-gal immunofluorescence (arrows) at 48 h post transfection. Note apoptotic changes in nuclear morphology of the shLuc+shS6-transfected neuron (arrowhead). E-F, Quantification of ribosomal stress-induced apoptosis revealed protection by shL11. Note similar protection against shTif1a by either shL11–1 or shL11–2 or their mix. G, DIV5 cortical neurons were treated with camptothecin (CPT) as indicated; the nucleolar markers Npm1 and fibrillarin were detected by immunofluorescence. CPT induced nucleoplasmic translocation of Npm (arrows) and loss of nucleolar fibrillarin (arrowheads) indicating disruption of the nucleolus. H, Transfections with shL11 were performed as for Fig. 1D. After 24 h, cells were treated with CPT or staurosporine for the next 24 h. Knockdown of L11 reduced CPT- but not staurosporine-induced apoptosis. In B, E, F, and H, averages ±SEM of at least 2 sister cultures from at least 3 independent experiments are shown; in C, data represent 30 cells from two independent experiments; p>0.05, NS; p<0.01, **; p<0.001, *** as compared to shLuc-transfected controls unless indicated otherwise (u-test in B, E, F, H; one-way ANOVA/Tukey posthoc tests in C).
Next, a pro-apoptotic role of L11 was examined in neurons that were treated with two chemical inducers of neuronal apoptosis, camptothecin (CPT) and staurosporine. The effects of CPT are due to DNA damage-including single strand breaks. Such lesions inhibit Pol1, thereby disrupting nucleolar integrity and activating p53-dependent neuronal apoptosis [45,28]. Conversely, staurosporine is a non-specific kinase inhibitor that induces p53-independent apoptosis in such cells [45].
Consistent with the previously demonstrated anti-nucleolar effects of CPT [28], 4 h exposure to this drug resulted in nucleoplasmic translocation of Npm1 (Fig. 4G). After 24 h, CPT increased apoptosis to 87 % as compared with 28% in vehicle-treated neurons (Fig. 4H). However, such an apoptotic response was reduced to 54% after knockdown of L11. In contrast, shL11 did not affect staurosporine-induced apoptosis (Fig. 4H). These data suggest that L11 is a specific pro-apoptotic mediator for a subset of death stimuli that kill by perturbing ribosomal biogenesis and, in consequence, activating p53.
Overexpression of L11 is sufficient to enhance p53-driven transcription and increase neuronal apoptosis.
To determine the sufficiency of L11 to regulate the neuronal p53 pathway, transcriptional activity of the temperature-sensitive mutant of p53 was determined after co-expression of L11-EGFP (Fig. 5A). Under a non-permissive temperature, TS-p53 behaves like a dominant-negative mutant [37]. When switched to a permissive temperature, it assumes wild type conformation and induces p53-mediated responses, including apoptosis of cortical neurons [37,28]. Indeed, after 8 h at permissive temperature, in TS-p53-transfected neurons, the p53RE-luciferase reporter activity was 4.96 fold higher than in neurons receiving DN-p53. Such activation was enhanced 2.5 fold when neurons co-expressed TS-p53 and L11-EGFP (Fig. 5B). This response appeared to be L11-specific, as L4-EGFP did not affect activation of TS-p53 (Fig. 5B). Therefore, in rat cortical neurons, L11 is a positive regulator of p53.
Figure 5. Overexpression of L11 is moderately pro-apoptotic.
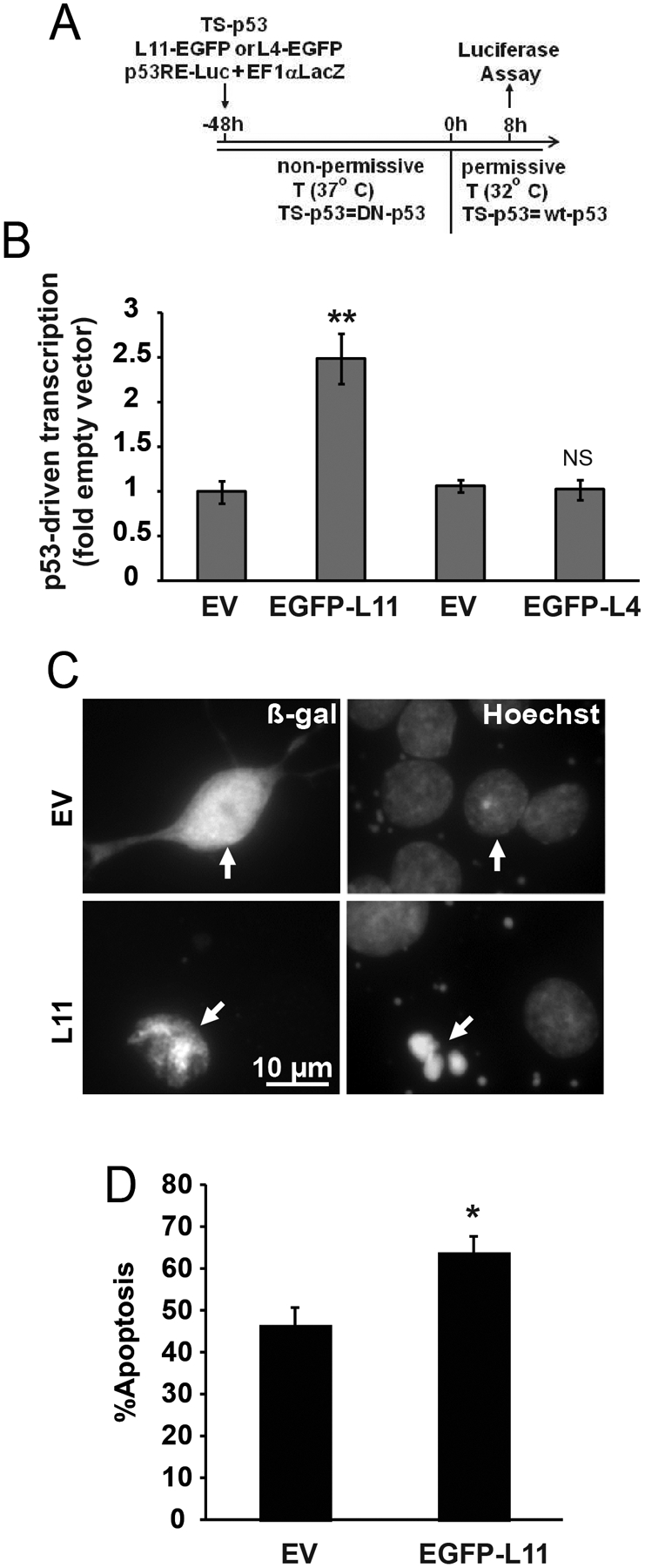
A-B, Co-expression of p53 and EGFP-L11 but not EGFP-L4 increases p53-driven transcription. Neurons were transfected with expression vectors for β-gal (EF1αLacZ), the temperature sensitive (TS) mutant form of p53, EGFP-L11 or EGFP-L4, and p53 response element-luciferase reporter construct (p53RE-Luc, 0.1+0.1+0.05+0.1 μg plasmid DNA/5×105 cells, respectively). Empty cloning vector (EV) was used as an additional control for RPL plasmids. After 8 h at the permissive temperature, β-gal-normalized luciferase activity was determined to evaluate p53-driven transcription. C-D, Moderate increase of apoptosis after overexpression of EGFP-L11. Expression vectors for β-gal and EGFP-L11 were co-transfected at 0.1 and 0.04 μg plasmid DNA/5×105 cells. After 24 h, apoptosis was evaluated in β-gal-positive neurons (arrows) using Hoechst-33258. Data represent averages ±SEM of 4- (B) or 2 (C) sister cultures from three independent experiments; p>0.05, NS; p<0.05, * ; p<0.01, *** as compared to EV-transfected controls (u-test).
The expression vector for L11-EGFP was also used to determine the sufficiency of L11 oversupply to induce cortical neuron apoptosis. At 48 h post L11-EGFP transfection apoptosis increased to 64% as compared to 47% in empty vector-transfected neurons (Fig. 5C–D). These data complement the findings of the L11 loss of function experiments from Fig. 4 by demonstrating that L11 gain of function is sufficient to enhance p53 activation and apoptosis.
Knockdown of L11 protects neurons against the RNA analog 5FU.
In several non-neuronal systems the RNA analog 5FU is a potent inhibitor of ribosomal biogenesis [46]. Therefore we examined its anti-ribosomal activity in neurons. Treatment with 5FU affected nucleolar staining of the GC marker, Npm (Fig 6A). In vehicle-treated neurons, the Npm-stained GC appeared to have a clear border suggesting a spherical shape of this nucleolar sub-compartment. In contrast, as early as 4 h after onset of 5FU exposure small protrusions that disrupted the GC sphere-like appearance were evident (Fig. 6A). In addition, at 24 h but not 4 h the nucleolus/nucleoplasm ratio of the Npm signal intensity decreased by 24% suggesting its partial release from the nucleolus (Fig. 6B). However, in 5FU-treated- and Npm-stained neurons, no significant changes of the nucleolar territory or the nucleolar number were found (Fig. 6C–D). No obvious effects of 5FU were observed on the fibrillarin-positive dense fibrillar component, either (DFC, Fig. 6A).
Figure 6. The RPL11-p53 pathway contributes to ribosomal stress-induced apoptosis in 5FU-treated neurons.
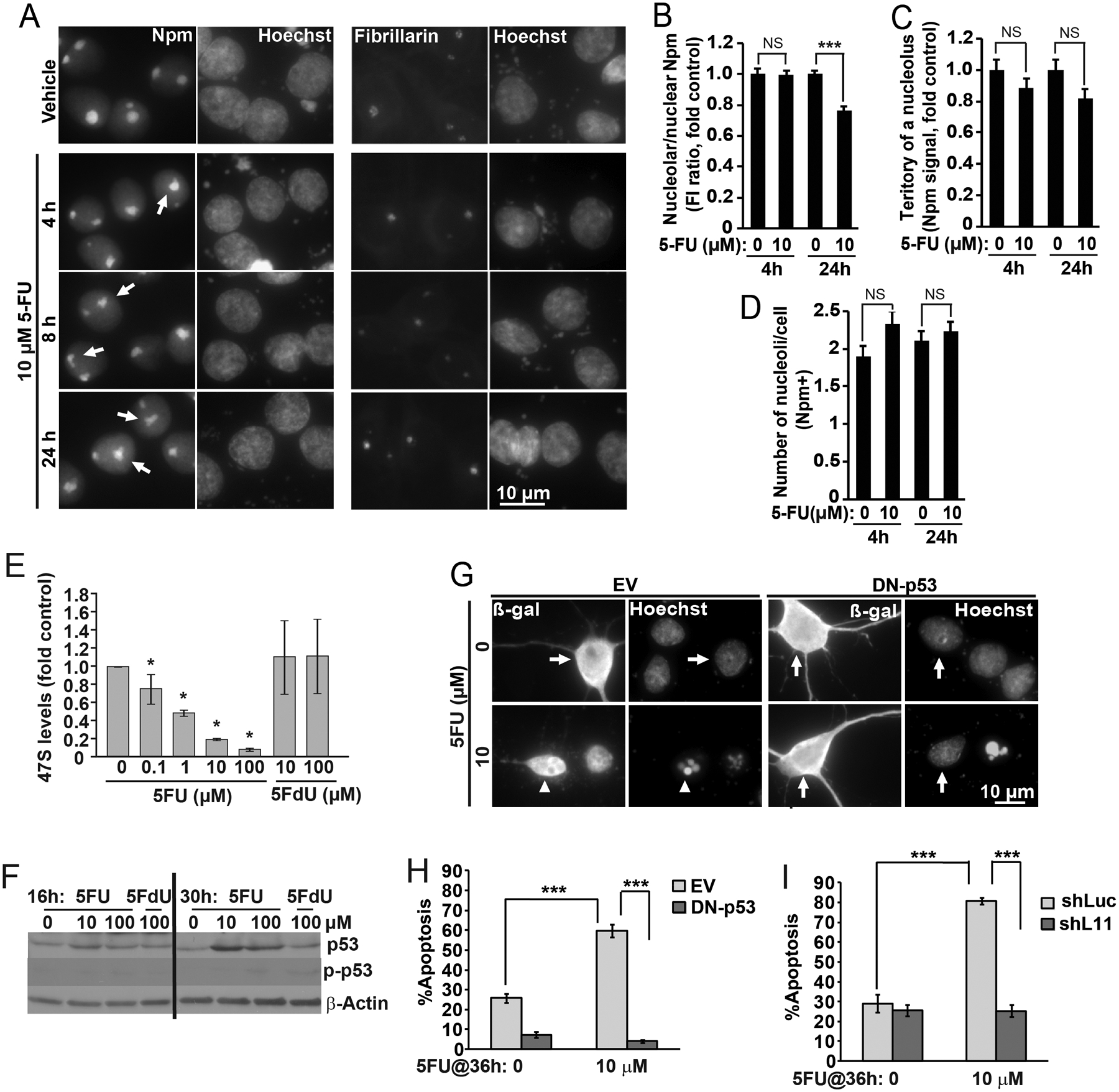
Treatments with 5-fluorouridine (5FU) and 5-fluorodeoxyuridine (5FdU) were performed at DIV5 as indicated. A, In 5FU-treated cells, Npm immunofluorescence revealed irregular shape of the nucleolar-nucleoplasm border (arrows). There were no obvious changes to fibrillarin labeling. B, Fluorescence intensity analysis revealed moderate but significant reduction of nucleolar Npm signal after 24 h 5FU treatment. C-D, Neither territory of an average nucleolus nor average number of nucleoli/cell (as defined by Npm staining) were affected by 5FU. E, After 2 h treatment, levels of 47S pre-rRNA were lower after 5FU- but not 5FdU. 47S levels were determined by qRT-PCR; 18S rRNA was used as a normalizer; similar results were obtained with other normalizers (Fig. S1). F, Increased levels of the total- but not Ser-15-phosphorylated p53 (p-p53) in 5FU-treated neurons as revealed by western blotting. Equal loading was verified by probing the membranes with an anti-β-actin antibody. G-I, DN-p53 or shL11 blocked apoptotic response to 5FU. Neurons were transfected with DN-p53 or shL11 as described for Fig. 1A–C or 1D, respectively. After 24 h, 5FU was added for the next 36 h. G, Representative images of transfected neurons. Arrows point non-apoptotic cells; arrowheads indicate a cell with apoptotic changes of nuclear morphology as revealed by Hoechst 33258 staining. H-I, Quantification of apoptosis in transfected neurons. Data represent averages ±SEM of at least 40 cells (B-D), single cultures (E) or 2 sister cultures (H-I) from 2, 3 or 3 independent experiments, respectively; p>0.05, NS; p<0.05, * ; p<0.01, *** (two-way ANOVA/Tukey posthoc tests in B-D; u-test in E, H, I); in F, similar results were obtained in 2 independent experiments.
In 5FU-treated neurons, declining pre-rRNA levels were observed (Fig. 6E). After 4 h treatment with 10 μM 5FU, 47S pre-rRNA was down by 80%. In contrast, the deoxy analog of 5FU, which can be incorporated into DNA but not RNA, did not reduce pre-rRNA levels, even if applied at 100 μM. Likewise, pre-rRNA was unaffected by treatment with ethynyl-uridine, which is used as a tool to label nascent rRNA by Click-It technology [38] (data not shown). While declining pre-rRNA levels suggest inhibition of rRNA transcription, 5FU did not cause a major nucleoplasmic release of Npm, which usually accompanies Pol1 blockade (Figs. 2, 4G and S3). Such findings indicate that in neurons 5FU may alter stability of rRNA precursors including their processing toward mature rRNA species. Indeed, Northern blot analysis revealed that despite declining levels of the primary rRNA transcript (47S) and its 5’processing products, other intermediates were still present (Fig. S2). However, there were notable exceptions including the 20S precursor of 18S rRNA and the 12S precursor of 5.8S rRNA. Thus, 5FU likely affects both Pol1 transcription and the specific downstream steps of pre-rRNA processing. Such a scenario would explain why in 5FU-treated neurons declining 47S levels coincide with the continued presence of other precursor rRNAs and the persistence of nucleolar Npm, which requires RNA binding for nucleolar localization [47].
Such a possibility is supported by an apparent correlation between precursor rRNA levels and maintenance of nucleolar integrity. Thus, in cortical neurons that were treated with the general transcriptional inhibitor actinomycin-D (ActD) sharp declines of pre-rRNA were attenuated by chemical ischemia that is expected to block not only rRNA transcription but also rRNA processing (Fig. S3). Moreover, ActD-induced nucleoplasmic release of Npm was reduced by chemical ischemia with some nucleolar Npm signal still present despite inhibition of Pol1 (Fig. S3). Therefore, simultaneous inhibition of rRNA transcription and rRNA processing may stabilize various precursor rRNAs promoting maintenance of nucleolar morphology.
Treatment with 5FU increased protein levels of p53. The increase was present at 16- or 30 h (Fig. 6F), but not at earlier time points (4 and 8 h, data not shown). Such an increase was not accompanied by increased phosphorylation of p53 at the Ser-15 residue (Fig. 6F). Therefore, 5FU activates p53 differently than DNA damaging agents, including the DSB inducer, etoposide, which induced robust phosphorylation at this site (Fig. 7A). Indeed, markers of the DNA damage response, such as autophosphorylation of ATM, or the appearance of γH2Ax, were unaffected by 5FU (data not shown).
Figure 7. ATM-dependent dysregulation of ribosomal biogenesis contributes to etoposide-induced neuronal apoptosis.
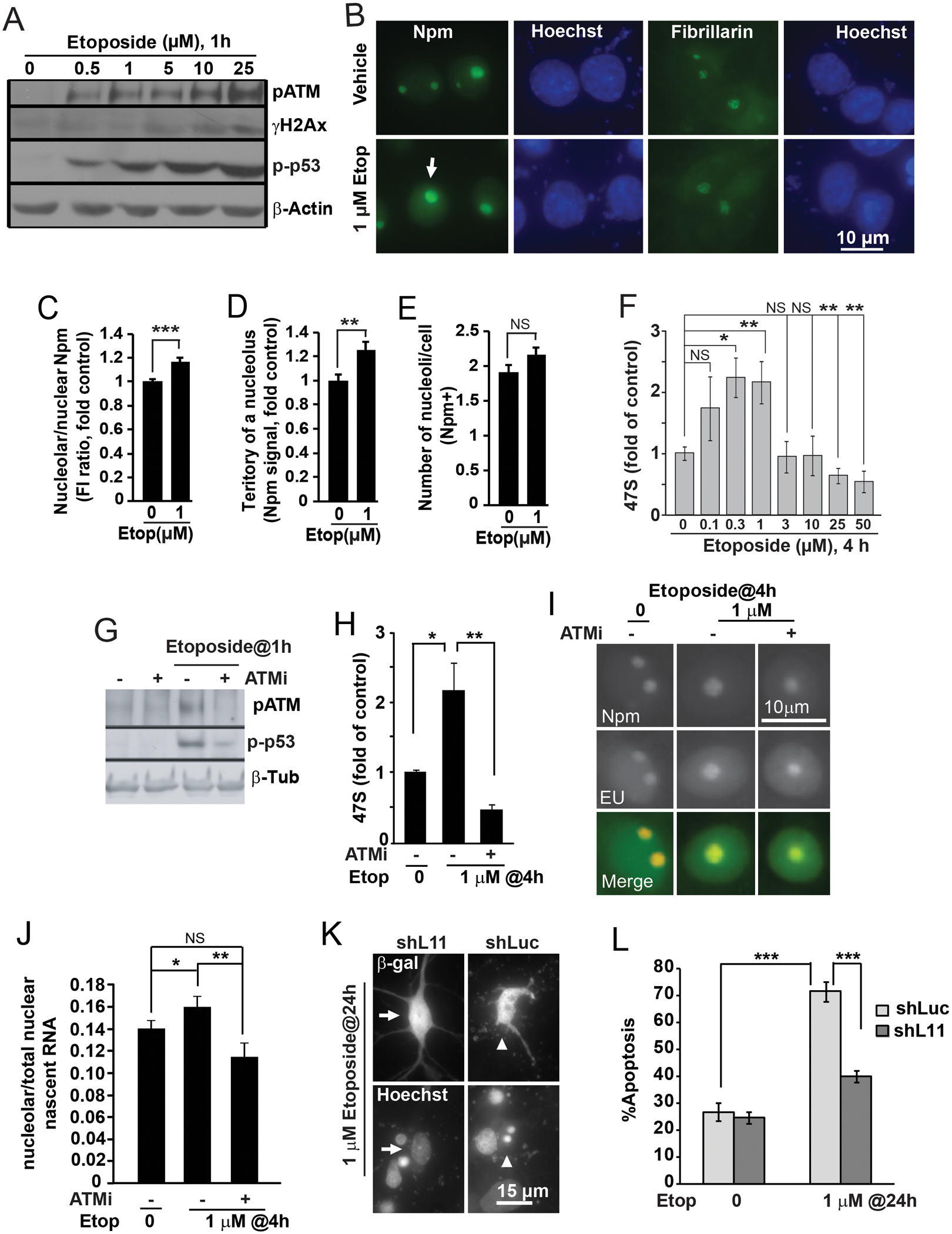
Cortical neurons were treated with etoposide on DIV5 (A-J) or DIV4 (K-L) as indicated. A, Activation of the DNA damage response by etoposide as revealed by western blotting for DNA-damage-associated phosphorylations of ATM (pS1981, p-ATM), H2Ax (pS139, γH2Ax) and p53 (pS15, p-p53). Equal protein loading was verified by reprobing the membranes with anti-β-actin antibody. Note that the etoposide-induced DNA damage response appeared even at the lowest etoposide concentration tested (0.5 μM). B, Representative images of immunofluorescence for nucleolar markers Npm and fibrillarin after 4 h treatment with 1 μM etoposide (Etop). Note apparent nucleolar enlargement as revealed by Npm staining (arrow). C, Fluorescence intensity analysis revealed moderate but significant increase of nucleolar Npm signal. D, Similar increase of territory of an average nucleolus was also present (as defined by Npm staining). E, In contrast, average number of Npm-positive nucleoli per cell was unaffected. F, Etoposide affected 47S pre-rRNA levels in a concentration-dependent manner. 47S levels were determined by qRT-PCR with normalization against 18S rRNA. Note increased pre-rRNA levels at concentrations ≤1 μM that did not disrupt nucleoli (B-E). G-J, Ribosomal biogenesis dysregulation by etoposide is ATM-dependent. G, Western blots to verify inhibition of the ATM pathway with the ATM drug inhibitor KU55933 (ATMi, 10 μM). ATMi blocked 10 μM etoposide effects on ATM autophosphorylation (pS1981) and phospho-(S15) p53; β-tubulin was used as a normalizer (β-Tub). H, After 4 h treatment with 1 μM etoposide, increases of pre-rRNA levels were attenuated by ATMi. I-J, ATMi-sensitive accumulation of nascent RNA in nucleoli of etoposide-treated neurons. In situ run on assay with the RNA precursor EU was performed using Click-It chemistry; immunostaining for Npm was also done to mark nucleoli. For quantification, EU signal intensity in the nucleolus was normalized against EU signal intensity from the whole nucleus. K-L, Knockdown of shL11 reduced etoposide-induced apoptosis. Neurons were transfected with shL11 as for Fig. 1D; 24 h later etoposide treatment was performed as indicated. K, Representative images of transfected neurons; arrows point a non-apoptotic cell; arrowheads indicate a cell with apoptotic changes of nuclear morphology. L, Quantification of apoptosis in transfected neurons. Data represent averages ±SEM of at least 40 individual cells (C-E, G), single cultures (F, H) or 2 sister cultures (L) from three independent experiments; p>0.05, NS; p<0.05, * ; p<0.01, **; p<0.001, *** (one-way ANOVA/Tukey posthoc tests in C-E, and, J; u-test in F, H, and, L); in A, and, G, similar results were obtained in at least 2 independent experiments.
However, in 5FU-treated neurons, p53 activation appeared to be pro-apoptotic. Thus, the 36 h treatment with 10 μM 5FU increased apoptosis to 61% as compared to 28% in vehicle-treated neurons (Fig. 6G, H). Such an apoptotic response was blocked by overexpression of the DN-p53 (Fig. 6G, H). Moreover, knockdown of L11 prevented 5FU-induced apoptosis (Fig. 6I). Therefore ribosomal stress is a major contributor to apoptotic toxicity of 5FU in immature cortical neurons.
In etoposide treated neurons, dysregulation of ribosomal biogenesis contributes to apoptosis.
In a previous report, it was shown that rat cortical neurons treated with the DSB inducer, etoposide, show a clear, concentration-dependent nucleolar stress at or above 25 μM [48]. However, etoposide induced both DSBs and p53-dependent apoptosis at concentrations as low as 1 μM, suggesting that DSBs are sufficient to activate the pro-apoptotic p53 pathway in the absence of nucleolar stress. As the current data suggest that ribosomal stress may occur when nucleolar morphology is apparently unaffected, the ribosomal stress contribution to etoposide-induced apoptosis was re-evaluated.
Consistent with etoposide’s ability to induce DSBs, there was a concentration-dependent increase in autophosphorylation of the DSB-response kinase, ATM, which reflects its activation (Fig. 7A). The increase was clearly present even at the lowest concentration tested (0.5 μM). In addition the DSB marker, phosphorylated histone H2Ax (γH2Ax), was elevated by etoposide treatment in a concentration-dependent manner (Fig. 7A). These changes were accompanied by increased phosphorylation of p53 at the Ser-15 residue, which is known to contribute to the ATM pathway-dependent activation of p53 by DSBs [49](Fig. 7A). However, consistent with our previous report, nucleolar staining of Npm did not reveal nucleolar disruption in neurons that were treated with 1 μM etoposide (Fig. 7B). In contrast, such cells displayed moderate increases of nucleolar Npm signal and Npm-positive territory of an average nucleolus but not number of nucleoli/cell (Fig. 7C–E).
Surprisingly, a careful dose response experiment revealed bi-directional effects of etoposide on 47S pre-rRNA. For instance, in neurons that were treated with 1 μM etoposide for 4 h, 47S levels were 2.4 fold higher than controls (Fig. 7F). Similar increases were also present with lower etoposide concentrations including 0.1 and 0.3 μM. Conversely, at 25- or 50 μM, etoposide reduced 47S levels, which is consistent with published evidence of nucleolar stress under such conditions (Fig. 7F). As many etoposide effects are mediated by the DSB-activated ATM, one could wonder whether in etoposide treated neurons, the increase of 47S is dependent on ATM. Therefore, neurons were treated with etoposide and the ATM kinase inhibitor KU55933. Inhibition of ATM attenuated the etoposide-induced phosphorylation of p53 at Ser-15, a downstream target of the DSB-ATM pathway (Fig. 7G). It also attenuated the 1 μM etoposide-mediated increase of 47S pre-rRNA, suggesting its dependence on ATM kinase activity (Fig. 7H).
In situ run-on assays revealed small but significant increases of nucleolar nascent RNA content in 1 μM etoposide-treated neurons (Fig. 7I–J). This 15% increase was prevented by ATM inhibition. Therefore, in neurons, low concentrations of etoposide may increase Pol1-mediated transcription of rRNA in an ATM-dependent manner. Alternatively, increases in pre-rRNA levels as well as greater nucleolar accumulation of nascent RNA may suggest an ATM-dependent reduction of the rRNA processing rate.
Recent studies in non-neuronal cells have revealed that the ribosomal stress pathway can be engaged by either reduced or increased ribosomal biogenesis [20]. Therefore, regardless of the mechanism by which low concentrations of etoposide elevate pre-rRNA levels, ribosomal stress may contribute to etoposide responses including apoptosis. To examine such a possibility, anti-apoptotic effects of shL11 were tested in etoposide-treated neurons (Fig. 7K–L). In cells that received a control shRNA, a 24 h treatment with 0 or 1 μM etoposide resulted in 27 or 73% apoptosis, respectively (Fig. 7L). In neurons that were transfected with shL11, such an apoptotic response was attenuated to 40% apoptosis after 1 μM etoposide treatment (Fig. 7L). Hence, in etoposide-treated neurons, the ATM-dependent dysregulation of ribosomal biogenesis triggers pro-apoptotic ribosomal stress.
Discussion
Nucleolar disruption and ribosomal stress as pro-apoptotic stimuli.
The presented data systematically document that impairment of ribosomal biogenesis is sufficient to activate p53-dependent neuronal apoptosis. Moreover, disruption of nucleolar integrity is not required for such an apoptotic response. Indeed, we observed a similar pro-apoptotic role of the p53 regulator, RPL11, regardless of the status of nucleolar structure as revealed by immunofluorescence for the GC marker Npm. These findings imply that perturbations of ribosomal biogenesis may trigger neuronal death even when the nucleolus is relatively well preserved and nucleolar transcription remains active. Hence, ribosomal stress defined as broadly as any dysregulation of ribosomal biogenesis appears to be a sufficient stimulus to activate neuronal stress response including apoptosis (Table 1).
Table 1.
Characteristics of ribosomal stress (also known as ribosomal biogenesis stress or nucleolar stress (based on [6,7,72] and additional references as indicated)
| Definition | A cellular stress response to dysregulation of ribosomal biogenesis and the following quantitative imbalance of ribosomal components | |
| Subtypes | With nucleolar disruption (proposed here to be named nucleolar stress) | Without nucleolar disruption |
| Causes | Inhibition of RNA-Pol1 due to action of drugs, toxins, DNA damaging agents or mutations that negatively affect activity of Pol1 [46] | 1) Deficits (or imbalance) of ribosomal proteins or ribosomal biogenesis factors that participate in posttranscriptional steps of ribosomal biogenesis [19,52,53] 2) drugs blocking rRNA processing (5FU) [46] 3) stimulation of ribosomal biogenesis by activated oncogenes [20] |
| Levels of rRNA precursors | All severely reduced | Often precursor–specific effects including depletion or increased accumulation; overall precursor levels may be moderately reduced, unchanged or increased |
| Localization of proteins whose nucleolar localization requires rRNA precursors (e.g. NPM, PES, BOP, MYBBP1)) | Translocation to the nucleoplasm/cell body [51,53] | Remain nucleolar [19,20,51] |
| Morphology of nucleoli | The three components of nucleolar ultrustructure (GC, FC, DFC) separate; FC/DFC proteins translocate to nucleolar caps (e.g. FBL, UBF) [73] | Size of GC may increase if rRNA processing is perturbed; number of nucleoli per cell may change (decrease or increase when ribosomal biogenesis is down or up, respectively), nucleolar boundaries may become irregular [52,53] |
| Status of rRNA transcription | Blocked | May be reduced, unchanged, or increased |
| Status of rRNA processing | N/A | Often perturbed [52] |
| Ribosomal output | Blocked | Often reduced, but may be unchanged, or increased |
| Stress response in proliferating cells with wt p53 | 5S RNP-mediated stabilization- and MYBBP1-mediated acetylation of p53, p53-dependent apoptosis [51] | 5S RNP-mediated stabilization of p53, p53-dependent cell cycle arrest and/or senescence [19,51–53] |
| Stress response in neurons | L11 (5SRNP?)-mediated stabilization of p53 and activation of p53-dependent apoptosis ([28,29] and current article) | |
In their seminal paper, Rubbi and Milner first proposed that activation of p53 is caused by functional disruption of the nucleolus [50]. Importantly, not all stressors used in that work were equally potent in altering nucleolar morphology. For instance, there was no correlation between p53 accumulation and Npm release from the nucleolus in proliferating cells treated with the established anti-ribosomal agent 5FU. Subsequent studies have provided multiple examples of p53 accumulation in response to stressors that perturb ribosomal biogenesis without structural disruption of the nucleolus. Thus, in various cell lines, depletions of either RPs of the small ribosomal subunit or several ribosomal biogenesis factors that regulate post-transcriptional stages of ribosome synthesis stabilized p53 despite normal or enlarged nucleoli [19,20,51–53]. This response resulted in cycle arrest and/or senescence but not apoptosis.
In contrast, at least in the human breast cancer cell line MCF7, nucleolar disruption appears to be necessary to steer the p53 signaling toward inducing apoptosis [51]. Thus, reduced nucleolar transcription lowers nucleolar rRNA content and consequently triggers release of nucleolar proteins, whose nucleolar residence requires RNA interactions. Among such proteins are Npm1 and the ribosomal biogenesis factor, MYBBP1. After release from the nucleolus, MYBBP1 facilitates acetylation of the TP53 K382 residue enhancing the TP53-driven transcription of pro-apoptotic genes including BAX, thus promoting apoptosis.
Currently, it is unknown why cultured neurons from neonate brain respond to ribosomal biogenesis inhibition with p53-dependent apoptosis regardless of nucleolar disruption. One possibility is that in these cells pro-apoptotic modifications of p53 may be constitutive while its stability is the primary regulator of p53 activation. Alternatively, in developing neurons, chromatin arrangement of the p53-regulated killer genes may favor their expression lowering the threshold for apoptotic activation of p53 [54]. One should also note that such a pro-apoptotic potential may be different in various types of neurons.
Nucleolar disruption and ribosomal stress in neurodegeneration.
The finding that in neurons ribosomal biogenesis inhibition may occur in the absence of nucleolar disruption, and that the latter event is not necessary to activate p53-mediated apoptosis suggests that nucleolar morphology may not be a reliable indicator of ribosomal stress in neurodegeneration. Such a notion is also consistent with our recent report of the p53-independent impairment of dendritic growth and maintenance as well as reduced protein synthesis in hippocampal neurons whose ribosomal biogenesis was perturbed without disrupting the nucleolus [39]. Therefore, normal nucleolar morphology, including nucleolar appearance of Npm, does not exclude the existence of neurotoxic ribosomal stress. Indeed, the definition of ribosomal stress as dysregulation of ribosomal biogenesis that produces quantitative imbalances of ribosomal components includes cases in which nucleolar morphology is unaffected (Table 1).
Additional parameters that determine ribosomal biogenesis may be needed to test for the presence of ribosomal stress in the degenerating nervous system. Such parameters may include the epigenetic status of rDNA, levels of rRNAs and their precursors, the expression of ribosomal biogenesis factors, and finally, cellular ribosome content. Examples of such a comprehensive approach were recently presented in the context of postmortem brain samples from patients suffering of PD or AD [17,18]. In both cases morphological changes to neuronal nucleoli were relatively modest with up to 30% affected neurons at the most advanced stages of either pathology. In contrast, decreasing levels of rRNA and/or mRNAs for some RPs were present at early/intermediate stages. For instance, at least 50% declines were reported for 28S or 18S rRNA in the CA1 region of the hippocampus at AD stage I-II or the substantia nigra at PD stage III-IV, respectively. Such observations suggest that in degenerating neurons impaired ribosomal biogenesis precedes disruption of the nucleolus.
What may cause ribosomal stress in neurodegenerative diseases? For several genetic forms of neurodegeneration, including HD, some types of familial PD, Machado–Joseph disease, or ALS/FTLD, neurodegeneration-associated mutations have been shown to impair rDNA transcription or rRNA processing [11–16]. In sporadic cases, causes of ribosomal biogenesis perturbations are not clear, but may include oxidative damage of nucleic acids and/or increased formation of RNA stress granules with consequent impairments of rRNA transcription/processing and/or translation of RPs, respectively [8,9,55–57]. In addition, recently identified neurodegeneration-associated disturbances in nuclear pore function may also contribute to ribosomal biogenesis disturbances as shown in proliferating cells that were depleted of specific components of the pore complex [58–61]. Finally, one should note that, at least in non-neuronal systems, ribosomal stress may arise as a consequence of enhanced ribosome production as exemplified by ribosomal stress in cells that overexpress oncogenes [20]. Therefore, in neurons that respond to tissue damage by hypertrophy [33], enhanced production of ribosomes may also produce ribosomal stress. It is tempting to speculate that regardless of its origins, dysregulated ribosomal biogenesis contributes to neurodegeneration by affecting translation and/or inducing the ribosomal stress response including the pro-apoptotic activation of p53.
L11 as a mediator of ribosomal stress.
To address the role of ribosomal stress in neurological diseases one should identify its specific mediators that could be targeted to block that pathway. Our data indicate that in neurons, L11 is one such mediator. This conclusion is consistent with findings from proliferating cells, in which L11 is necessary to activate p53 upon ribosomal stress [19,61–63,51,20]. However, in neurons, knocking down L11 afforded a different magnitude of protection dependent on the ribosomal stress inducer. Thus, the greatest protection was observed in 5FU-treated or shL4-transfected neurons. Partial protection was observed with DNA topoisomerase inhibitors or shRNAs that targeted Tif1a or S6. In contrast, in all these cases, inhibition of p53 offered complete protection. Therefore, besides L11 there may be additional mediators of the ribosomal stress-p53 pathway. Moreover, ribosomal stress-independent activation of the p53 pathway may have contributed to neuronal apoptosis in those cases where L11 knockdown offered only partial protection. At least in neurons exposed to DNA damaging agents, including camptothecin or etoposide, the DNA damage response could have provided an additional arm of p53 activation.
Recent work has established solid evidence that in proliferating cells, L11 activates p53 by binding to MDM2 as a component of the ribonucleoprotein complex 5S RNP [20,62–64]. Each component of that complex including L11, L5 and 5S rRNA is required for the ribosomal stress-mediated activation of p53. It has been proposed that under normal conditions 5S RNP is rapidly used for the synthesis of the large ribosomal subunit., Factors that prevent its incorporation into the large subunit (LSU) or that increase its generation without a corresponding increase in LSU assembly lead to ribosomal stress [62,63]. For instance, increased translation of L11 follows disruption of the small ribosomal subunit assembly [19]. Mutually stabilizing effects of L11 and L5 were also reported after inhibition of ribosomal biogenesis [65]. In addition, in ribosomal stress exposed cells, 5S RNP localization may change from mostly nucleolar to nucleoplasmic [63]. Such an increase in the nucleoplasm has been proposed to enable inhibitory interactions with MDM2, which resides in that compartment of the nucleus. Future research will establish whether in neurons components of 5S RNP other than L11 are required for ribosomal stress signaling and whether their nucleoplasmic levels increase upon ribosomal stress.
Role of ribosomal stress in etoposide-induced apoptosis.
Our data indicate that in neurons, ATM-dependent dysregulation of ribosomal biogenesis, but not nucleolar disruption, occurs in response to low concentrations of the DNA double strand break inducer, etoposide. Moreover, we speculate that the etoposide-induced increases in pre-rRNA levels and nascent RNA labeling in the nucleolus reveal slower rRNA processing and/or increased rRNA transcription. There were several reports from highly proliferative cell lines as well as primary cells suggesting DSB-induced-, and, ATM-mediated inhibition of nucleolar transcription with DSBs present either in cis- or in trans- to the rDNA loci [66–70]. Yet, the functional significance of such a response remains unclear. Our finding that shL11 attenuated etoposide-induced neuronal apoptosis indicates that at least in neurons, DNA damage-induced ribosomal stress contributes to the pro-apoptotic activation of p53. We speculate that in developing neurons challenged with moderate levels of DSBs such a mechanism may amplify the p53 response ensuring the elimination of damaged cells.
How ATM perturbs ribosomal biogenesis in neurons is not clear. However, rRNA processing factors, as well as components of the Pol1 transcriptional machinery, are among the putative ATM substrates that are phosphorylated in response to DNA damage in non-neuronal cells [71]. In addition, after DNA damage, an ATM recruitment factor, NBS1, translocates to the nucleoli suggesting that ATM may be able to interact with nucleolar proteins that produce ribosomes [67].
In summary, we provide evidence that ribosomal stress is sufficient to activate p53-mediated apoptosis of cortical neurons without nucleolar disruption. Therefore, the pathological dysregulation of ribosomal biogenesis may be overlooked if nucleolar integrity is the only criterion evaluated. In addition, we identify L11 as a mediator of ribosomal stress-induced neuronal apoptosis. Hence, targeting ribosomal stress-specific signal transducers, such as L11, provides an approach to determine the pathological significance of ribosomal stress in neurological diseases.
Supplementary Material
Acknowledgments.
This work was supported by NIH (NS073584 and 8P30GM103507 to MH), NSF (IOS1021860 to MH), and the Commonwealth of Kentucky Challenge for Excellence Fund. The authors wish to thank Ms. Jing-Juan Zheng for excellent technical assistance.
References:
- 1.Yuan J, Yankner BA (2000) Apoptosis in the nervous system. Nature 407 (6805):802–809. [DOI] [PubMed] [Google Scholar]
- 2.Martin LJ (2001) Neuronal cell death in nervous system development, disease, and injury (Review). International journal of molecular medicine 7:455–478 [PubMed] [Google Scholar]
- 3.Sirri V, Urcuqui-Inchima S, Roussel P, Hernandez-Verdun D (2008) Nucleolus: the fascinating nuclear body. Histochem Cell Biol 129:13–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drygin D, Rice WG, Grummt I (2010) The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu Rev Pharmacol Toxicol 50:131–156 [DOI] [PubMed] [Google Scholar]
- 5.Hetman M, Pietrzak M (2012) Emerging roles of the neuronal nucleolus. Trends Neurosci 35:305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim TH, Leslie P, Zhang Y (2014) Ribosomal proteins as unrevealed caretakers for cellular stress and genomic instability. Oncotarget 5:860–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quin JE, Devlin JR, Cameron D, Hannan KM, Pearson RB, Hannan RD (2014) Targeting the nucleolus for cancer intervention. Biochim Biophys Acta 1842:802–816 [DOI] [PubMed] [Google Scholar]
- 8.Honda K, Smith MA, Zhu X, Baus D, Merrick WC, Tartakoff AM, Hattier T, Harris PL, Siedlak SL, Fujioka H, Liu Q, Moreira PI, Miller FP, Nunomura A, Shimohama S, Perry G (2005) Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. J Biol Chem 280:20978–20986 [DOI] [PubMed] [Google Scholar]
- 9.Ding Q, Markesbery WR, Chen Q, Li F, Keller JN (2005) Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci 25:9171–9175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rieker C, Engblom D, Kreiner G, Domanskyi A, Schober A, Stotz S, Neumann M, Yuan X, Grummt I, Schutz G, Parlato R (2011) Nucleolar Disruption in Dopaminergic Neurons Leads to Oxidative Damage and Parkinsonism through Repression of Mammalian Target of Rapamycin Signaling. J Neurosci 31:453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vilotti S, Codrich M, Dal Ferro M, Pinto M, Ferrer I, Collavin L, Gustincich S, Zucchelli S (2012) Parkinson’s disease DJ-1 L166P alters rRNA biogenesis by exclusion of TTRAP from the nucleolus and sequestration into cytoplasmic aggregates via TRAF6. PLoS One 7:e35051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang H, Shin JH (2014) Repression of rRNA transcription by PARIS contributes to Parkinson’s disease. Neurobiol Dis 73C:220–228 [DOI] [PubMed] [Google Scholar]
- 13.Lee J, Hwang YJ, Boo JH, Han D, Kwon OK, Todorova K, Kowall NW, Kim Y, Ryu H (2011) Dysregulation of upstream binding factor-1 acetylation at K352 is linked to impaired ribosomal DNA transcription in Huntington’s disease. Cell Death Differ 18: 1726–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsoi H, Chan HY (2013) Expression of expanded CAG transcripts triggers nucleolar stress in Huntington’s disease. Cerebellum 12:310–312 [DOI] [PubMed] [Google Scholar]
- 15.Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507:195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL (2014) Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345:1139–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernandez-Ortega K, Garcia-Esparcia P, Gil L, Lucas JJ, Ferrer I (2015) Altered machinery of protein synthesis in Alzheimer’s: from the nucleolus to the ribosome. Brain Pathol 26: 593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-Esparcia P, Hernandez-Ortega K, Koneti A, Gil L, Delgado-Morales R, Castano E, Carmona M, Ferrer I (2015) Altered machinery of protein synthesis is region- and stage-dependent and is associated with alpha-synuclein oligomers in Parkinson’s disease. Acta Neuropathol Commun 3:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fumagalli S, Di Cara A, Neb-Gulati A, Natt F, Schwemberger S, Hall J, Babcock GF, Bernardi R, Pandolfi PP, Thomas G (2009) Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol 11:501–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishimura K, Kumazawa T, Kuroda T, Katagiri N, Tsuchiya M, Goto N, Furumai R, Murayama A, Yanagisawa J, Kimura K (2015) Perturbation of ribosome biogenesis drives cells into senescence through 5S RNP-mediated p53 activation. Cell Rep 10:1310–1323 [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y (2003) Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol 23:8902–8912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macias E, Jin A, Deisenroth C, Bhat K, Mao H, Lindstrom MS, Zhang Y (2010) An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell 18:231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pederson T, Tsai RY (2009) In search of nonribosomal nucleolar protein function and regulation. J Cell Biol 184:771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyd MT, Vlatkovic N, Rubbi CP (2011) The nucleolus directly regulates p53 export and degradation. J Cell Biol 194:689–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Latonen L, Moore HM, Bai B, Jaamaa S, Laiho M (2011) Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene 30:790–805 [DOI] [PubMed] [Google Scholar]
- 26.Guetg C, Santoro R (2012) Formation of nuclear heterochromatin: the nucleolar point of view. Epigenetics 7:811–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI (2010) The nucleolus under stress. Mol Cell 40:216–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalita K, Makonchuk D, Gomes C, Zheng JJ, Hetman M (2008) Inhibition of nucleolar transcription as a trigger for neuronal apoptosis. J Neurochem 105:2286–2299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parlato R, Kreiner G, Erdmann G, Rieker C, Stotz S, Savenkova E, Berger S, Grummt I, Schutz G (2008) Activation of an endogenous suicide response after perturbation of rRNA synthesis leads to neurodegeneration in mice. J Neurosci 28:12759–12764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kreiner G, Bierhoff H, Armentano M, Rodriguez-Parkitna J, Sowodniok K, Naranjo JR, Bonfanti L, Liss B, Schutz G, Grummt I, Parlato R (2013) A neuroprotective phase precedes striatal degeneration upon nucleolar stress. Cell Death Differ 20:1455–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wakayama I (1992) Morphometry of spinal motor neurons in amyotrophic lateral sclerosis with special reference to chromatolysis and intracytoplasmic inclusion bodies. Brain Res 586:12–18 [DOI] [PubMed] [Google Scholar]
- 32.Gertz HJ, Siegers A, Kuchinke J (1994) Stability of cell size and nucleolar size in Lewy body containing neurons of substantia nigra in Parkinson’s disease. Brain Res 637:339–341 [DOI] [PubMed] [Google Scholar]
- 33.Iacono D, Markesbery WR, Gross M, Pletnikova O, Rudow G, Zandi P, Troncoso JC (2009) The Nun study: clinically silent AD, neuronal hypertrophy, and linguistic skills in early life. Neurology 73:665–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iacono D, O’Brien R, Resnick SM, Zonderman AB, Pletnikova O, Rudow G, An Y, West MJ, Crain B, Troncoso JC (2008) Neuronal hypertrophy in asymptomatic Alzheimer disease. J Neuropathol Exp Neurol 67:578–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amlie-Wolf A, Ryvkin P, Tong R, Dragomir I, Suh E, Xu Y, Van Deerlin VM, Gregory BD, Kwong LK, Trojanowski JQ, Lee VM, Wang LS, Lee EB (2015) Transcriptomic Changes Due to Cytoplasmic TDP-43 Expression Reveal Dysregulation of Histone Transcripts and Nuclear Chromatin. PLoS One 10:e0141836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riancho J, Ruiz-Soto M, Villagra NT, Berciano J, Berciano MT, Lafarga M (2014) Compensatory Motor Neuron Response to Chromatolysis in the Murine hSOD1(G93A) Model of Amyotrophic Lateral Sclerosis. Front Cell Neurosci 8:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michalovitz D, Halevy O, Oren M (1990) Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell 62:671–680 [DOI] [PubMed] [Google Scholar]
- 38.Gomes C, Smith SC, Youssef MN, Zheng JJ, Hagg T, Hetman M (2011) RNA polymerase 1-driven transcription as a mediator of BDNF-induced neurite outgrowth. J Biol Chem 286:4357–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Slomnicki LP, Pietrzak M, Vashishta A, Jones J, Lynch N, Elliot S, Poulos E, Malicote D, Morris BE, Hallgren J, Hetman M (2016) Requirement of Neuronal Ribosome Synthesis for Growth and Maintenance of the Dendritic Tree. J Biol Chem 291:5721–5739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75:817–825 [DOI] [PubMed] [Google Scholar]
- 41.Habas A, Kharebava G, Szatmari E, Hetman M (2006) NMDA neuroprotection against a phosphatidylinositol-3 kinase inhibitor, LY294002 by NR2B-mediated suppression of glycogen synthase kinase-3beta-induced apoptosis. J Neurochem 96:335–348 [DOI] [PubMed] [Google Scholar]
- 42.Flygare J, Aspesi A, Bailey JC, Miyake K, Caffrey JM, Karlsson S, Ellis SR (2007) Human RPS19, the gene mutated in Diamond-Blackfan anemia, encodes a ribosomal protein required for the maturation of 40S ribosomal subunits. Blood 109:980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalita K, Kharebava G, Zheng JJ, Hetman M (2006) Role of megakaryoblastic acute leukemia-1 in ERK1/2-dependent stimulation of serum response factor-driven transcription by BDNF or increased synaptic activity. J Neurosci 26:10020–10032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tajrishi MM, Tuteja R, Tuteja N (2011) Nucleolin: The most abundant multifunctional phosphoprotein of nucleolus. Commun Integr Biol 4:267–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson MD, Kinoshita Y, Xiang H, Ghatan S, Morrison RS (1999) Contribution of p53-dependent caspase activation to neuronal cell death declines with neuronal maturation. J Neurosci 19:2996–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burger K, Muhl B, Harasim T, Rohrmoser M, Malamoussi A, Orban M, Kellner M, Gruber-Eber A, Kremmer E, Holzel M, Eick D (2010) Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem 285:12416–12425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitrea DM, Cika JA, Guy CS, Ban D, Banerjee PR, Stanley CB, Nourse A, Deniz AA, Kriwacki RW (2016) Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. eLife 5: e13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pietrzak M, Smith SC, Geralds JT, Hagg T, Gomes C, Hetman M (2011) Nucleolar disruption and apoptosis are distinct neuronal responses to etoposide-induced DNA damage. J Neurochem 117 :1033–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281:1674–1677 [DOI] [PubMed] [Google Scholar]
- 50.Rubbi CP, Milner J (2003) Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J 22:6068–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumazawa T, Nishimura K, Katagiri N, Hashimoto S, Hayashi Y, Kimura K (2015) Gradual reduction in rRNA transcription triggers p53 acetylation and apoptosis via MYBBP1A. Scientific reports 5:10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicolas E, Parisot P, Pinto-Monteiro C, de Walque R, De Vleeschouwer C, Lafontaine DL (2016) Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nature Commun 7:11390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langhendries JL, Nicolas E, Doumont G, Goldman S, Lafontaine DL (2016) The human box C/D snoRNAs U3 and U8 are required for pre-rRNA processing and tumorigenesis. Oncotarget doi: 10.18632/oncotarget.11148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wright KM, Smith MI, Farrag L, Deshmukh M (2007) Chromatin modification of Apaf-1 restricts the apoptotic pathway in mature neurons. J Cell Biol 179:825–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hetman M, Vashishta A, Rempala G (2010) Neurotoxic mechanisms of DNA damage: focus on transcriptional inhibition. J Neurochem 114:1537–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bentmann E, Haass C, Dormann D (2013) Stress granules in neurodegeneration--lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J 280:4348–4370 [DOI] [PubMed] [Google Scholar]
- 57.Wolozin B (2014) Physiological protein aggregation run amuck: stress granules and the genesis of neurodegenerative disease. Disc Med 17:47–52 [PMC free article] [PubMed] [Google Scholar]
- 58.Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18:1226–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525:129–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Golomb L, Bublik DR, Wilder S, Nevo R, Kiss V, Grabusic K, Volarevic S, Oren M (2012) Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Mol Cell 45:222–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Donati G, Peddigari S, Mercer CA, Thomas G (2013) 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep 4:87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sloan KE, Bohnsack MT, Watkins NJ (2013) The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep 5:237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pelava A, Schneider C, Watkins NJ (2016) The importance of ribosome production, and the 5S RNP-MDM2 pathway, in health and disease. Biochemical Society transactions 44:1086–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bursac S, Brdovcak MC, Pfannkuchen M, Orsolic I, Golomb L, Zhu Y, Katz C, Daftuar L, Grabusic K, Vukelic I, Filic V, Oren M, Prives C, Volarevic S (2012) Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. PNAS 109:20467–20472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kruhlak M, Crouch EE, Orlov M, Montano C, Gorski SA, Nussenzweig A, Misteli T, Phair RD, Casellas R (2007) The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 447:730–734 [DOI] [PubMed] [Google Scholar]
- 67.Larsen DH, Hari F, Clapperton JA, Gwerder M, Gutsche K, Altmeyer M, Jungmichel S, Toledo LI, Fink D, Rask MB, Grofte M, Lukas C, Nielsen ML, Smerdon SJ, Lukas J, Stucki M (2014) The NBS1-Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nature Cell Biol 16:792–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Sluis M, McStay B (2015) A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes & Dev 29:1151–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harding SM, Boiarsky JA, Greenberg RA (2015) ATM Dependent Silencing Links Nucleolar Chromatin Reorganization to DNA Damage Recognition. Cell Rep 13:251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim J, Sturgill D, Tran AD, Sinclair DA, Oberdoerffer P (2016) Controlled DNA double-strand break induction in mice reveals post-damage transcriptome stability. Nucleic Acids Res 44:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316:1160–1166 [DOI] [PubMed] [Google Scholar]
- 72.Golomb L, Volarevic S, Oren M (2014) p53 and ribosome biogenesis stress: the essentials. FEBS Lett 588:2571–2579 [DOI] [PubMed] [Google Scholar]
- 73.Shav-Tal Y, Blechman J, Darzacq X, Montagna C, Dye BT, Patton JG, Singer RH, Zipori D (2005) Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol Biol Cell 16:2395–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.