

Abstract
Anticancer nanomedicines are designed to improve anticancer efficacy by increasing drug accumulation in tumors through enhanced permeability retention (EPR) effect, and to reduce toxicity by decreasing drug accumulation in normal organs through long systemic circulation. However, the inconsistent efficacy/safety of nanomedicines in cancer patients versus preclinical cancer models have provoked debate for nanomedicine design criteria. In this study, we investigate nanomedicine design criteria in three types of preclinical cancer models using five clinically used nanomedicines, which identifies the factors for better clinical translations of their observed clinical efficacy/safety compared to free drug or clinical micelle formulation. When those nanomedicines were compared with drug solution or clinical micelle formulation in breast tumors, long and short-circulating nanomedicines did not enhance tumor accumulation by EPR effect in transgenic spontaneous breast cancer model regardless of their size or composition, although they improved tumor accumulations in subcutaneous and orthotopic breast cancer models. However, when tumors were compared to normal breast tissue, nanomedicines, drug solution and clinical micelle formulation showed enhanced tumor accumulation regardless of the breast cancer models. In addition, long-circulating nanomedicines did not further increase tumor accumulation in transgenic mouse spontaneous breast cancer nor universally decrease drug accumulations in normal organs; they decreased or increased accumulation in different organs, potentially changing the clinical efficacy/safety. In contrast, short-circulating nanomedicines decreased blood concentration and altered drug distribution in normal organs, which are correlated with their clinical efficacy/safety. A reappraisal of current nanomedicine design criteria is needed to ensure consistent clinical translation for improvement of their clinical efficacy/safety in cancer patients.
Keywords: Nanomedicine, Nanoparticle, Enhanced permeability retention (EPR), Long/short circulation, Delivery efficiency, Preclinical cancer models, Clinical efficacy and adverse events
1. Introduction
Although different anticancer nanomedicines have been designed with various features, they intend to improve anticancer efficacy and reduce toxicity based on two basic design criteria [1]: (1) Nanomedicines improve anticancer efficacy by increasing drug accumulation in tumors through enhanced permeability retention (EPR) effect, which is due to the leaky vasculature and lack of lymphatic drainage [2–4], (2) Nanomedicines reduce toxicity by decreasing drug accumulation in normal organs through long systemic circulation, which is usually achieved by surface modification of nanoparticles to decrease clearance by reticulum endothelial system (RES) [5,6]. The long circulation nanomedicines achieve high plasma concentration, which in turn further enhance tumor accumulation through EPR effect to improve efficacy [5,6].
In the past few decades, hundreds of anticancer nanomedicines have been developed based on these design criteria [7–11]. However, the inconsistent efficacy/safety profiles of nanomedicines in preclinical cancer models compared to clinical cancer patients has provoked heated debate on nanomedicine design criteria based on the following clinical observations [4,12–17]: (1) Most (with few exceptions) of nanomedicines failed to improve anticancer efficacy in clinical trials compared with their drug solutions despite rigorous and reproducible evidence of enhanced efficacy in preclinical xenograft models [2,4,7,8,12–14,16,18]. Additionally, most of the successful nanomedicines were approved for clinical use by comparing the nanomedicines in combination with standard care vs. standard care alone in human cancer patients, which lacks head-to-head comparison with free drugs. (2) The efficacy profiles of successful nanomedicines compared with drug solution or clinical micelle formulation in clinical cancer patients are inconsistent with these two nanomedicine design criteria. In eight Phase III clinical trials comparing nanomedicines with their respective drug solution (doxorubicin) or clinical micelle formulation (Taxol), only two trials showed that nanomedicines have superior efficacy in certain cancer types but not in other cancer types (Supplemental table 1). For instance, the long-circulating stable nanomedicine Doxil (PEGylated liposome, 83 nm) demonstrated superior clinical efficacy in AIDS-related Kaposi’s sarcoma compared with doxorubicin solution (Overall Response, OR, 45.9% in Doxil group vs 24.8% in Doxorubicin + bleomycin + vincristine group, p < 0.001) [19,20], but showed similar efficacy in breast cancer (Overall Survival, OS, 21 months in Doxil group vs 22 months in Doxorubicin group) [21,22], and did not have head-to-head comparison with doxorubicin solution in ovarian cancers or myeloma [23–26]. These clinical finding with Doxil compared to the doxorubicin solution is perplexing and counter to the pre-clinical data that led to these costly Phase 3 studies. In contrast, short-circulating fast-release nanomedicine Abraxane (albumin nanoparticle of Paclitaxel, 138 nm) showed superior efficacy in breast cancer compared to Taxol (Cremophor EL micelle of paclitaxel) (response rate was 21.5% with Abraxane vs 11.1% with Taxol, P = 0.003; median time to progression (TTP) was 23.0 weeks with Abraxane vs. 16.9 weeks with Taxol, p = 0.006) [27–31]. Abraxane was also observed to have small but superior efficacy in non-small cell lung cancer (Objective Response Rate, ORR, 33% in Abraxane group vs 25% in Taxol group, p = 0.005) [30,32,33[96]]. However, Abraxane did not show superior efficacy in gastric cancer (Median overall survival, OS, 11.1 months in Abraxane group vs. 10.9 months in Taxol group) [34]. Abraxane is approved for treating pancreatic cancer [30,35–38], but no head-to-head comparison with Taxol has been performed in phase III trial since Taxol is not approved for use in the pancreatic cancer patients. In contrast, a short circulating Genexol-PM (PEG-PLA nanoparticle of paclitaxel) showed non-inferior efficacy vs. Taxol in breast cancer (objective response rate ORR, 39.1% in Genexol-PM group vs 24.3% in Taxol group, pnon-inferiority = 0.021; overall survival OS 28.8 months in Genexol-PM group vs. 23.8 months in Taxol group, p = 0.52; progression free survival PFS 8 months in Genexol-PM group vs. 6.7 months in Taxol group, p = 0.26) [39,40]. (3) Nanomedicines do not universally decrease toxicity in clinical cancer patients, but rather have unique toxicity profiles [7,9,21,22,30,31,39]. For instance, PEGylated Doxil reduced cardiotoxicity (Doxil 3.9% vs. doxorubicin 18.8%), but increased hand-foot-syndrome (Doxil 48% vs. doxorubicin 2%), increased rash (Doxil 10% vs doxorubicin 2%), increased abnormal pigmentation (Doxil 21% vs doxorubicin 6%), and increased Erythema (18% vs 3%) in comparison with doxorubicin. These differences are not explained by RES evasion because non-PEGylated liposome Myocet also reduced cardiotoxicity (Myocet 6% vs. Doxorubicin 21%, p < 0.0002), slightly increased hand-foot syndrome (grade 2) that is significantly less than Doxil [22], While Abraxane decreased neutropenia (Abraxane 9% vs. Taxol 22%) [27,30,41,42], it increased neuropathy (Abraxane 10% vs. Taxol 2%) [43,44], increased gastrointestinal toxicity, such as nausea (Abraxane 30% vs Taxol 22%), vomiting (Abraxane 18% vs Taxol 10%), diarrhea (Abraxane 27% vs Taxol 15%) in comparison with Taxol [27,31]. In contrast, Genexol-PM increased neutropenia in comparison with Taxol (Genexol-PM 68.6% vs Taxol 40.2%, p < 0.01) [39].
The debate on the nanomedicine design criteria has been lasted for more than a decade and is largely focused EPR in tumors, which may have mixed two different questions with two distinct clinical implications as described below [7–11,45,47–49]. The debate is less focused on the long circulation of nanomedicine design, which seems to be accepted as a general requirement for anticancer nanomedicine to achieve high plasma concentration. The inconsistent efficacy/safety of anticancer nanomedicine in cancer patients compared with preclinical models demands clarification of the following questions to improve anticancer nanomedicine design: (1) Is the EPR effect present in both preclinical and human cancers? (2) Does nanomedicine enhance drug accumulation in the tumor through EPR effect in comparison with free drugs, improving anticancer efficacy in both preclinical cancer models and human cancer patients? (3) Should long circulation nanomedicines be used as a general design criterion to improve anticancer efficacy by further enhancing EPR effect in tumors while also reducing drug toxicity by decreasing accumulation in normal organs? (4) What factors may be responsible for the unique clinical efficacy/safety profiles of clinical successful anticancer nanomedicines?
To address these questions, we performed a comprehensive study to compare the delivery efficiency of five clinically approved nanomedicines in tumor and normal tissues in three preclinical cancer models. The data of these anticancer nanomedicines vs. drug solution or clinical micelle formulation in three preclinical cancer models were analyzed in relation to their observed clinical efficacy/safety profiles of both nanomedicines from phase III clinical trials. Two categories of nanomedicines were used in this study: (1) Long-circulating and stable PEGylated and non-PEGylated doxorubicin liposomes Doxil and Myocet respectively. Both formulations achieved higher plasma concentration than clinically used free doxorubicin solution. (2) Short-circulating and fast release nanomedicines, Abraxane (albumin nanoparticle of paclitaxel), Genexol-PM (PEG-PLA polymeric nanoparticle of paclitaxel), and Paclical (all-trans retinoic acid micellar nanoformulation of paclitaxel) [97], all achieved lower plasma concentration than clinical micelle formulation (Taxol). Clinical micelle formulation Taxol was used in this study as a reference formulation since there is no clinical free paclitaxel solution available. The five anticancer nanomedicines were selected in our studies since phase III clinical trials have been conducted for head-to-head comparison with their respective free drug or clinical micelle formulation. Three preclinical breast cancer models, which include transgenic mouse spontaneous breast cancer, orthotopic and subcutaneous breast cancer with the same cancer type and same genetic background. These data provide insight why nanomedicine design criteria in different preclinical cancer models may have successful or poor clinical translation for their clinical efficacy/safety in cancer patients.
2. Materials and methods
2.1. Materials
Taxol (paclitaxel injection, micelle formulation using Cremophor EL) was purchased from the Hospital Pharmacy of University of Michigan (Ann Arbor, MI). Abraxane (albumin-bound nanoparticle of paclitaxel) was provided by Celgene Corporation (Summit, NJ, U.S.A.). Paclical (The micellar formulation of paclitaxel encapsulated in the proprietary retinoid compound XR-17) was procured from the Russian Federation courtesy of Celgene Corporation, and Genexol-PM (polymer formulation of paclitaxel) was procured from the South Korea courtesy of Celgene Corporation. Doxorubicin hydrochloride solution and Doxil (PEGylated liposome from Janssen Oncology, Raritan, NJ, U.S.A.) were purchased from the Hospital Pharmacy of the University of Michigan. Myocet (Non-PEGylated liposomes of paclitaxel, GP-Pharm, Spain) was procured from the EU courtesy of Celgene Corporation.
2.2. Three types of breast cancer models
All animal procedures used in this study were approved by the University Committee on Use and Care Animals at the University of Michigan. We developed three types of breast cancer models, which have same genetic background and cancer cells to minimize variability. MMTV-PyMT transgenic spontaneous breast cancer model: The female MMTV-PyMT mice (FVB/NJ background) [50] used in this study were 8–12 weeks old with tumor sizes of 150–500 mm3. The breeding colonies were established by crossing FVB/NJ females (Stock No. 001800) with hemizygous FVB/N-Tg (MMTV-PyMT) 634Mul/J males (Stock No: 002374) from The Jackson Laboratory (Bar Harbor, ME, USA). Orthotopic breast cancer model and subcutaneous breast cancer models: PyMT breast cancer cells (1 × 106 cells/0.1 mL/mouse) isolated from MMTV-PyMT mouse tumors suspended in a Matrigel/RPMI-1640 (50/50) mixture were inoculated into the FVB/NJ female mice through subcutaneous (left inguinal fold) or orthotopic injection (fourth mammary fat pad).
The mice were injected through tail vein with four formulations of paclitaxel (Taxol-injectable paclitaxel with Cremophor EL, Abraxane, Genexol-PM, Paclical, 10 mg/kg), or three formulations of doxorubicin (Doxorubicin-injectable clinical used solution, Doxil, Myocet, 5 mg/kg) once average tumor sizes reached ~150 mm3. Mouse tumor size were monitored and calculated based on the following formula: volume = ½ × length × width × width. Serial samples of blood, plasma, tumor, brain, fat, heart, intestine, kidney, liver, lung, muscle, pancreas, spleen, stomach were collected from pre-dose, 0.083, 1, 4, 7, 24, and 72 h (doxorubicin formulations only) post dose for drug concentration analysis, imaging analysis, and immunostaining, 3 mice/time point/group.
2.3. Characterization of EPR effect via Evans Blue tumor accumulation
When tumors (in subcutaneous, orthotopic, and transgenic spontaneous breast cancer models) reached about 200 mm3, the tumor-bearing mice were intravenously administered the Evans Blue at a dose of 30 mg/kg. After 24 h, all mice were sacrificed, and the tumor masses together with the fad pads were dissected, weighed, photographed, and homogenized by PBS (pH 7.4). Subsequently, the supernatant was collected after centrifugation at 13,300 rpm for 10 min, then mixed with actone at a volume of 3:7 for extraction. After incubation for 30 h in an incubator, 200 μL of each sample were withdrawn and the content of Evans Blue in the supernatant was quantified using a microplate reader (OD = 620 nm). Results are presented as the amount of Evans Blue normalized per mg of tissues weight.
2.4. Liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis of drug concentration in plasma, blood, tumor, and other tissues
The total drug is measured in our study including released and unreleased drug, as well as protein bound drug in plasma to illustrate the questions below, unlike other studies provided the detail analysis of three different types of drugs [51,52], The LC-MS/MS analysis of paclitaxel was performed using docetaxel as internal standard (IS). LC-MS/MS analysis of doxorubicin was performed using daunarubicin as internal standard (IS). The analytical assay was validated according FDA guidance for linearity (2–5000 ng/mL), matrix effect, recovery, low detection limit, Quality control (QC) in different biological matrix, including plasma, blood, tumor, and each different organ homogenates. LC-MS/MS was performed using ABI-5500 Qtrap (Sciex, Ontario, Canada) mass spectrometer with electrospray ionization source was interfaced with Shimadzu high performance liquid chromatography (HPLC) system with Xbridge C18 column (50 × 2.1 mm ID, 3.5 μm). The mass spectrometer was operated in a positive mode with multiple reaction monitoring (MRM) for analysis. The MRM transitions were monitored at m/z 854.4 to 286.1 for paclitaxel and m/z 808.0 to 226.0 for internal standard. The MRM transitions were monitored at m/z 544.1 to 397.2 for doxorubicin and m/z 528.5 to 321 for internal standard.
2.5. Pharmacokinetic (PK) and mass balance analysis
The PK analysis from all formulations were compiled and calculated with Phoenix/WinNonlin (version 6.4; Pharsight, Mountain View, CA, U.S.A.) The plasma/blood and tissue concentration-time data were compiled. Efficiency of paclitaxel and doxorubicin delivery by different formulations was evaluated by comparing concentrations of paclitaxel or doxorubicin in plasma and tissues at each time point. The amounts of paclitaxel or doxorubicin were calculated as the products of the corresponding concentrations and the blood volumes or tissue weights. The percentage of injected dose in each tissue was calculated using the amount of paclitaxel or doxorubicin in each tissue per injected dose, which is calculated by the formula: drug percentage of dose= (drug concentration * tissue weight or blood (plasma) volume)/dose *100%. Phoenix32 software was used to calculate PK parameters for different groups. In brief, plasma data from Taxol, Abraxane, Genexol-PM and Paclical was fitted using both 1-comp and 2-comp model. To minimize AIC, 1/(pred*pred) weighting in 1-compartment model was selected to calculate parameters including V1, K10, AUC, Cmax, C1, and T1/2. In order to calculate AUC and Cmax for different tissues, non-compartment method was used.
2.6. Mass spectrometry imaging to visualize nanomedicine distribution in tumor tissues
Cryosectioned tumors of 10 μm thickness were mounted onto precooled glass plates or metal imaging plates. Then the slice was sprayed with TiO2 matrix suspension including 200 ng/mL reference compound D5-paclitaxel. MS is performed under negative mode to form in source fragment ions at M/Z 284.098 and M/Z 289.127, respectively. Cryosectioned tumors were imaged at a spatial resolution of 50 μm in negative ion mobility mode. A MALDI-ion mobility mass spectrometer Synapt G2-Si Qtof (Waters Corporations, USA) was used for imaging of the tissue samples. It is equipped with a 355 nm Nd:YAC laser with a 100–2500 Hz repetition rate. MS spectra were acquired with an automatic scan rate under sensitivity mode with positive or negative ionization method. The MALDI source settings was set with 0.5 scans per pixel, 1000 or 2000 HZ laser firing rate and 400 laser energy. HDI-maging software V1.4 from Waters was used to process and display ions distribution inside the tissue sections.
2.7. Immunohistochemistry (IHC) staining of blood vessels in tumor tissues
IHC staining of blood vessels in frozen section of primary tumor slides was performed by IVAC facility at the University of Michigan. Briefly, the cryosectioned tumor samples were incubated with anti-CD31 antibody (Dianova, Cat# DIA310, done SZ31) after post-fixation in pre-chilled acetone at −20 °C for 10 min. The detection was performed by a horseradish peroxidase biotin-free polymer based commercial detection system, disclosure with diaminobenzidine (DAB) chromogen, and nuclear counterstaining with hematoxylin. Slides were scanned on Aperio At-2.
2.8. Immunostaining and confocal imaging of blood vessels and macrophage in tumor tissues
Cryosectioned tumors of 10 μm thickness were mounted onto precooled glass plates. Immunostaining of blood vessels and macrophage in frozen section of primary tumor slides was performed with Alexa Fluor® 594 anti-mouse CD31 (BioLegend, clone MEC13.3, Cat# 102520) and Alexa Fluor® 647 anti-mouse F4/80 (BioLegend, Clone BM8, Cat# 123122) antibodies after post-fixation in IHC Zinc fixative (BD Pharmingen™) for 45 s. All immunostained slides were mount in ProLong™ Diamond Antifade Mountant with DAPI (Molecular Probes™, P36971) and imaged with Leica SP8 inverted MP confocal.
The fluorescent-labeled paclitaxel albumin formulation was prepared by dissolving Paclitaxel 10 mg, fluorescent-labeled paclitaxel (Invitrogen™ Paclitaxel, Oregon Green™ 488 Conjugate (Oregon Green™ 488 Taxol, Flutax-2), Cat: P22310) 200 μg into chloroform (2 mL), and mixed with 20 mL 0.5% Human serum albumin solution (Albumin (Human) U.S.P. Albutein® 5%, GRIFOLS, Lot No: B3ADB01432), then dispersed vigorously by a rotor-stator homogenizer (Ultra-Turrax T25, 8 K rpm – 12 K rpm, 5min) to generate milk-like emulsion. The emulsion was then processed by a high-pressure homogenizer (Nano DeBEE) with the parameters set as pressure = 15,000psi – 20,000psi, condensation temp = 5 °C, cycles = 6. Remaining organic solvent in the product was removed by a rotatory evaporator. The final nano-suspension was filtered with 0.22 μm membrane, and then directly put into 10 mL vials (2mL/vial) for lyophilization (primary drying temp = −5 °C for 30 h and secondary drying temp = 30 °C for 6 h). The vials were filled with nitrogen before sealing and stored at −20 °C. Fluorescent-labeled paclitaxel and paclitaxel in final formulation is about 1:125, which was determined using LC-MS/MS. Simple mix of paclitaxel: fluorescent-labeled paclitaxel (1:125) was used as control. The size distribution and dilution stability are measured by dynamic light scattering (DLS) analysis (Zetasizer Nano ZSP, MALVERN).
2.9. Immunohistochemistry (IHC) and immunostaining of skin tissues
The skin was dissected from the mice 20 h-post administration of Doxil and doxorubicin and then severally cryosectioned into two consecutive sections of 10 μm thickness. Thereof, one slide used to acquire the fluorescent signals of Doxil/doxorubicin was fixed immediately in IHC Zinc fixative (BD Pharmingen™) for 45 s and mount in ProLong™ Diamond Antifade Mountant with DAPI (Molecular Probes™, P36971), followed by imaging by a Nikon A1SI confocal. The other consecutively cryosectioned slides used to obtain the histological structure were stained with 0.1% Mayers Hematoxylin (Sigma; MHS-16) for 10 min, followed by dipping in 0.5% Eosin 12 times post cool running of ddH2O for 5 min. After being equilibrated by of ddH2O and ethanol gradient elution, the slides were mount and imaged with an Olympus IX83 inverted microscope.
2.10. Dilution stability of Abraxane, Doxil and Myocet
Stability of Abraxane, Doxil and Myocet against dilution was evaluated in terms of the changes of particle size and Zeta potential. The nanoformulation solution of Abraxane, Doxil, and Myocet were prepared at the concentration of 0.6 mg/mL (paclitaxel), 2 mg/mL, and 2 mg/mL, respectively and recognized as the 0 times dilution. Dilution was achieved through addition of saline to the nanoformulation solution of Abraxane at 10, 20, 50 folds, and 20, 50, 100, 500, 1000, 5000 and 10000 fold for Doxil, and Myocet. Then the changes of particles size were measured (within 1 min) and recorded immediately by dynamic light scattering (DLS) using a Zetasizer Nano (Malvern, Worcestershire, UK).
2.11. Statistical analysis
Statistical analysis was conducted using GraphPad Prism 7.0 software. One-way ANOVA was used if the comparison involved more than two groups. Two-tailed Student’s t-test was used to compare the statistical difference between two groups. A P-value < 0.05 was considered significant.
3. Results
3.1. When nanomedicines were compared with drug solution or clinical micelle formulation in tumors, nanomedicines did not enhance tumor accumulation in transgenic spontaneous breast cancers although they did in subcutaneous and orthotopic breast cancers
To clarify first debate question regarding nanomedicine design criteria based on EPR effect in tumor, we first tested if nanomedicine could enhance accumulation by EPR effect vs. drug solution or clinical micelle formulation in three different breast cancer mouse models. Although this type of comparisons have been confirmed in mouse subcutaneous xenograft cancer models previously, the enhanced tumor accumulation of nanomedicines in mouse subcutaneous xenograft cancer models rarely translated to better clinical anticancer efficacy in human solid cancer patients. In addition, there is limited data comparing tumor accumulation of nanomedicines vs. their drug solution or clinical micelle formulations in human. Further, it is not clear what types of preclinical cancer models can mimic the human cancer conditions for nanomedicine tumor accumulations.
To investigate if long circulating stable nanomedicines can enhance tumor accumulation vs free drug, the delivery efficiency of PEGylated liposome Doxil and non-PEGylated liposome Myocet vs. clinical doxorubicin solution was evaluated. These studies were carried out using orthotopic and subcutaneous preclinical breast cancer models as well as a spontaneous transgenic mouse model of breast cancer (MMTV-PyMT). This transgenic mouse model was established by overexpression of large T antigen in the mammary epithelium and chosen because it more closely mimics disease progression in humans [50,53]. These breast cancers were developed in multiple foci in the mammary glands from 6 to 14 weeks of age. We observed premalignant lesions-hyperplasia at 4–6 weeks of age, adenoma/MIN (advanced premalignant lesions) between 8 and 9 weeks of age, early carcinoma from 8 to 12 weeks of age, and late carcinoma or advanced invasive carcinoma stage at 10 weeks of age. We used mice aged 8–12 weeks in our study. Furthermore, we developed the orthotopic and subcutaneous mouse models in the mice with the same genetic background (FVB/NJ) as the PyMT mice to limit the variability. The mouse orthotopic breast cancer was generated by implanting PyMT breast cancer cells in the mammary fat pad of FVB/NJ mice. The mouse subcutaneous breast cancer model was established by implanting the PyMT cancer cells subcutaneously in the flank of FVB/NJ mice.
As shown in Fig. 1, PEGylated liposome Doxil had higher plasma concentration and longer t1/2 compared to non-PEGylated liposome Myocet, and both liposomal doxorubicin formulations had higher plasma concentration and longer t1/2 than that of doxorubicin solution in all three cancer models (Fig. 1A,D,E). Both Doxil and Myocet showed significantly higher tumor accumulation (2 to 5-fold) in subcutaneous (Fig. 1B) and orthotopic breast cancer models (Fig. 1E) compared to doxorubicin solution. These results agree with previous studies demonstrating enhanced tumor accumulation and superior efficacy in subcutaneous xenograft models [16,17]. However, Doxil and Myocet failed to increase drug tumor accumulation in transgenic PyMT mice when compared with doxorubicin solution (Fig. 1H). Additionally, in comparison with non-PEGylated liposome Myocet, PEGylated liposome Doxil showed longer circulation time in the plasma, which indeed further improved tumor accumulation only in the orthotopic and subcutaneous breast cancer mouse models; however, Doxil did not show higher tumor accumulation than Myocet in clinically relevant spontaneous breast cancer (Fig. 1H). Furthermore, the tumor/plasma ratios of Doxil and Myocet are 100- or 25-fold lower than the tumor/plasma ratios of doxorubicin solution, which suggests long-circulating stable nanomedicine reduces tissue penetration in the tumor. The enhanced tumor accumulation through EPR of the long circulating stable liposomal nanomedicines vs. drug solution is not observed in clinically relevant spontaneous breast cancer, but only seen in the subcutaneous or orthotopic cancer models.
Fig. 1.
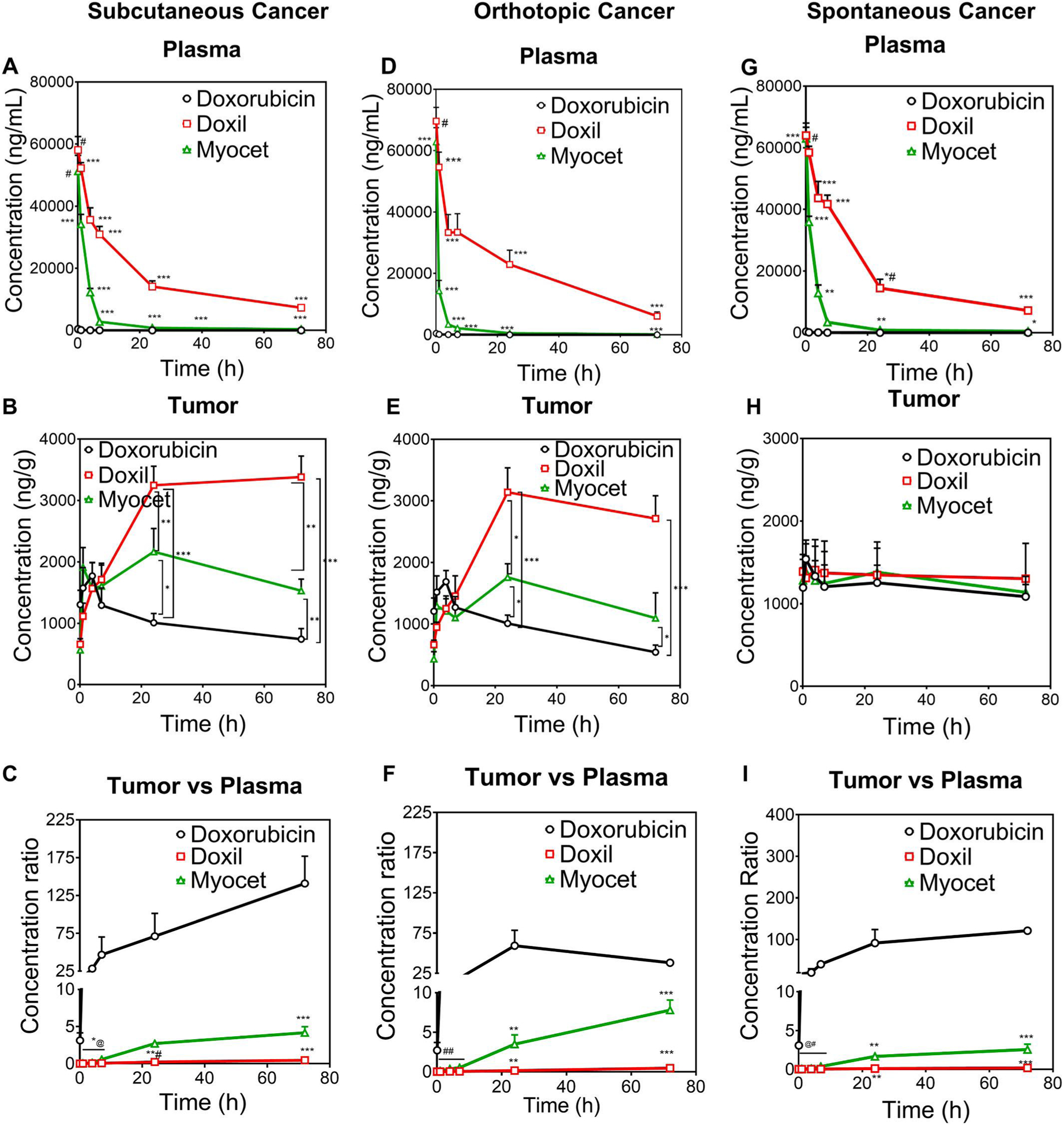
The enhanced tumor accumulation by EPR for long-circulating stable liposomal nanoformulation vs. free doxorubicin was achieved only in subcutaneous and orthotopic breast cancers, but not in transgenic spontaneous breast cancers. Drug concentration in plasma, tumor, and tumor/plasma concentration ratio in subcutaneous breast cancer model (A, B, C), orthotopic breast cancer model (D, E, F), and MMTV-PyMT spontaneous breast cancer model (G, H, I) after IV dose of doxorubicin, Doxil, and Myocet (5 mg/kg). *p < 0.1, **p < 0.01 and ***p < 0.001 vs other two groups; #p > 0.05 Doxil vs Myocet, P < 0.001 vs Dox; *#p < 0.01 vs Myocet, P < 0.001 vs DOX; *@: 1st time point: p < 0.01 Doxil vs Dox, p > 0.05 Doxil vs Myocet, p < 0.01 Myocet vs Dox; 2nd time point: p < 0.001 Doxil and Myocet vs Dox, p < 0.05 Doxil vs Myocet; 3rd time point: p < 0.001 Doxil and Myocet vs Dox; p < 0.01 Doxil vs Myocet; 4th time point: p < 0.05 Dox vs Doxil, p < 0.001 Dox vs Myocet, p < 0.05 Doxil vs Myocet; **#p < 0.05 Doxil vs Dox; p < 0.001 Doxil vs Myocet; ##: 1st time point: p < 0.01 Doxil and Myocet vs Dox and P < 0.05 Doxil vs Myocet; 2nd time point: p < 0.05 between each other; 3rd time point: p < 0.01 Doxil and Myocet vs DOX, P < 0.0001 Doxil vs Myocet; 4th time point: p < 0.0001 between each other; @#:1st time point: p < 0.001 Doxil and Myocet vs Dox; p > 0.05 Myocet vs Doxil; 2nd time point<0.01 Doxil and Myocet vs DOX and p < 0.05 Doxil vs Myocet; 3rd time point: p < 0.05 Doxil vs DOX; P < 0.01 Doxil vs Myocet; p < 0.05 Myocet vs Doxil; 4th time point: p < 0.001 Doxil vs DOX, p < 0.01 Myocet vs DOX and Doxil.
In order to further assess if short circulating fast-release nanomedicines can enhance tumor accumulation vs. clinical micelle formulation, we compared the delivery efficiency of three paclitaxel nanomedicines vs. clinical micelle formulation Taxol in the same three mouse models as described above. As shown in Fig. 2, although Abraxane, Genexol-PM, and Paclical showed 3 to 5-fold lower plasma concentration and short circulation time (Fig. 2A, D, G), they showed higher (1.5 to 2-fold) tumor accumulation in subcutaneous and orthotopic breast cancer in comparison with Taxol (Fig. 2B, E). However, these nanomedicines (Abraxane, Genexol-PM, Paclical) decreased tumor accumulation than Taxol in clinically relevant transgenic spontaneous breast cancer model (Fig. 2H). In addition, no significant differences in tumor accumulation were observed among three different nanomedicines (Abraxane, Genexol-PM, or Paclical in all three different breast cancer models (Fig. 2 B, E,H), regardless of their size 138, 20, 30 nm or composition Albumin, PEG-PLA, all-tans retinoid acid respectively. These data are in contrast with previous reports that small size nanoparticles have better EPR effect for tumor accumulation in subcutaneous cancer models [54,55]. Furthermore, the tumor/plasma ratios of Abraxane, Genexol-PM, and Paclical were slightly higher than Taxol in subcutaneous and orthotopic breast cancer (Fig. 2C,F); yet the tumor/plasma ratios of all four formulations are similar in transgenic spontaneous breast cancers (Fig. 2I), which suggests similar tumor tissue penetration. These data suggest that enhanced tumor accumulation of short-circulating fast-release paclitaxel nanomedicines vs. micelle formulation Taxol is not observed in the clinically relevant spontaneous breast cancer model, but only seen in subcutaneous and orthotopic breast cancer models.
Fig. 2.
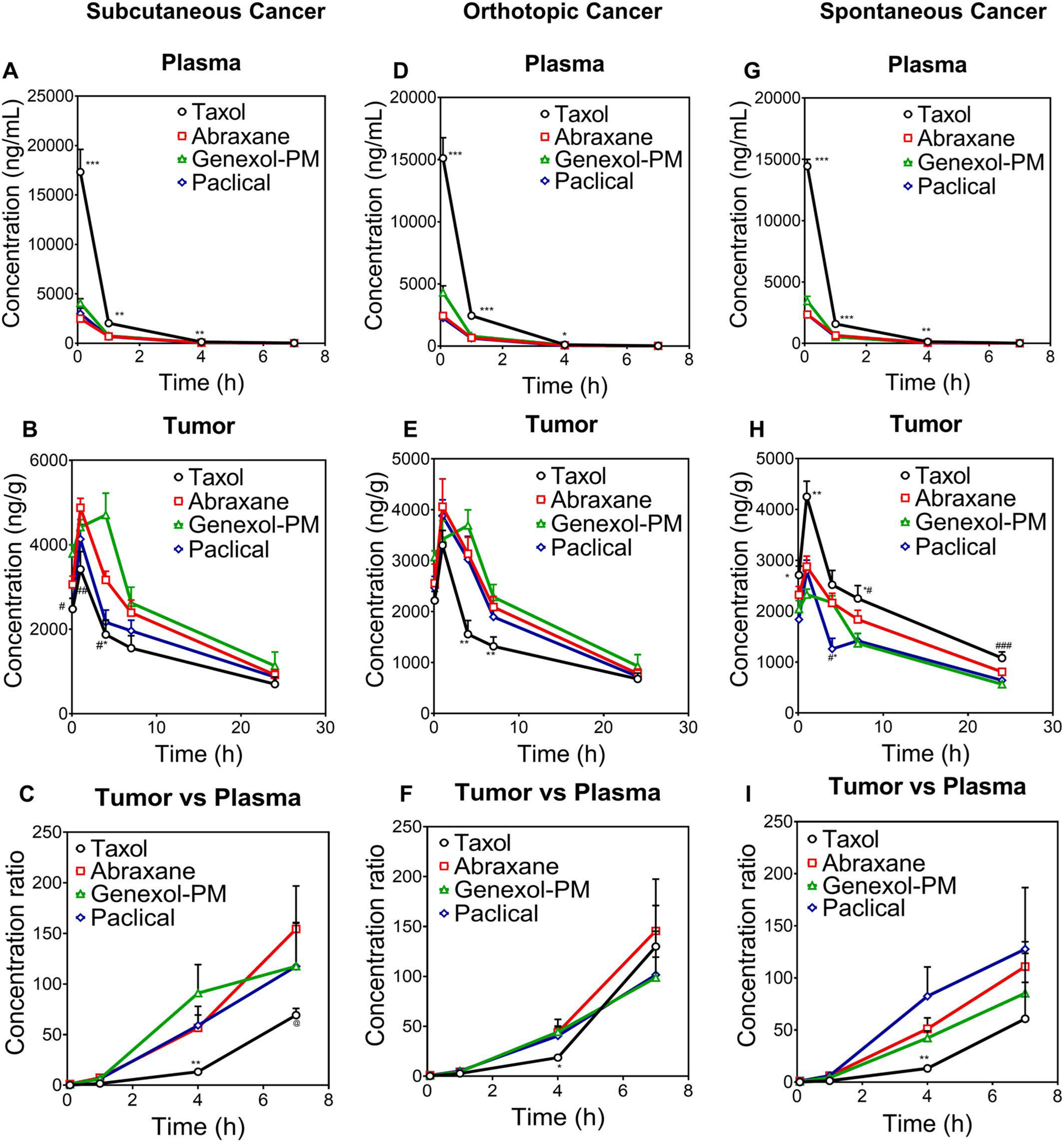
The enhanced accumulation of nanoformulations (Abraxane, Genexol-PM, and Paclical) vs. Taxol was achieved only in subcutaneous and orthotopic breast cancers but was reduced in transgenic spontaneous breast cancers. Drug concentration in plasma, tumor, and tumor/plasma concentration ratio in subcutaneous breast cancer model (A, B, C), orthotopic breast cancer model (D, E, F) and MMTV-PyMT spontaneous breast cancer model (G, H, I) after IV dose of paclitaxel (Taxol®), Abraxane®, Genexol-PM®, and Paclical® (10 mg/kg). ***: P < 0.001 Taxol vs all other groups; **p < 0.01 Taxol vs all other groups; *p < 0.05 Taxol vs all other groups: #P < 0.05 Taxol vs Abraxane, P < 0.01 Taxol vs Genexol-PM.; ##P < 0.01 Taxol vs Abraxane, P < 0.05 Taxol vs Genexol-PM; #*P < 0.01 Paclical vs Taxol; *#p < 0.01 Taxol vs Genexol-PM and Paclical; ###p < 0.05 Taxol vs Abraxane and P < 0.01 Taxol vs Genexol-PM and Paclical. @P < 0.05 Taxol vs Abraxane.
3.2. When breast tumors were compared with normal breast tissues, nanomedicines, drug solution and clinical micelle formulation enhanced tumor accumulation in three different breast cancer models
To clarify the debate questions regarding nanomedicine design criteria based on EPR effect, we tested if nanomedicine could enhance tumor accumulation by EPR effect vs. normal breast tissues in three different breast cancer mouse models. We first tested tumor accumulation of small molecules (Evans blue and doxorubicin) in tumors vs. normal breast tissues in three different mouse breast cancer models with the same cancer type and same genetic background (transgenic mouse spontaneous, orthotopic, subcutaneous breast cancers). To our surprise, the tumor accumulations of these two small molecules in breast cancer tissues were higher than that of normal breast tissues. As shown in Fig. 3A and B, Evans blue was accumulated 2 to 4-fold higher in the breast cancer tissues than normal breast fat pads in three different cancer models at 24 h. The tumor accumulation of Evans blue in transgenic mouse spontaneous breast cancers is slightly lower (2-fold), than that in orthotopic breast cancers (4-fold) (Fig. 3A and B), and that in subcutaneous breast cancer (6.5-fold) (Supplemental Fig. 1A). Similarly, doxorubicin solution showed 2–6-fold higher breast tumor accumulation vs. normal breast fat pads in both spontaneous and orthotopic breast cancers at various time points from 0.5 to 24 h (Fig. 3C and D). These data suggest that small molecules may also have preferred accumulation in the breast tumors vs. normal breast tissues.
Fig. 3.
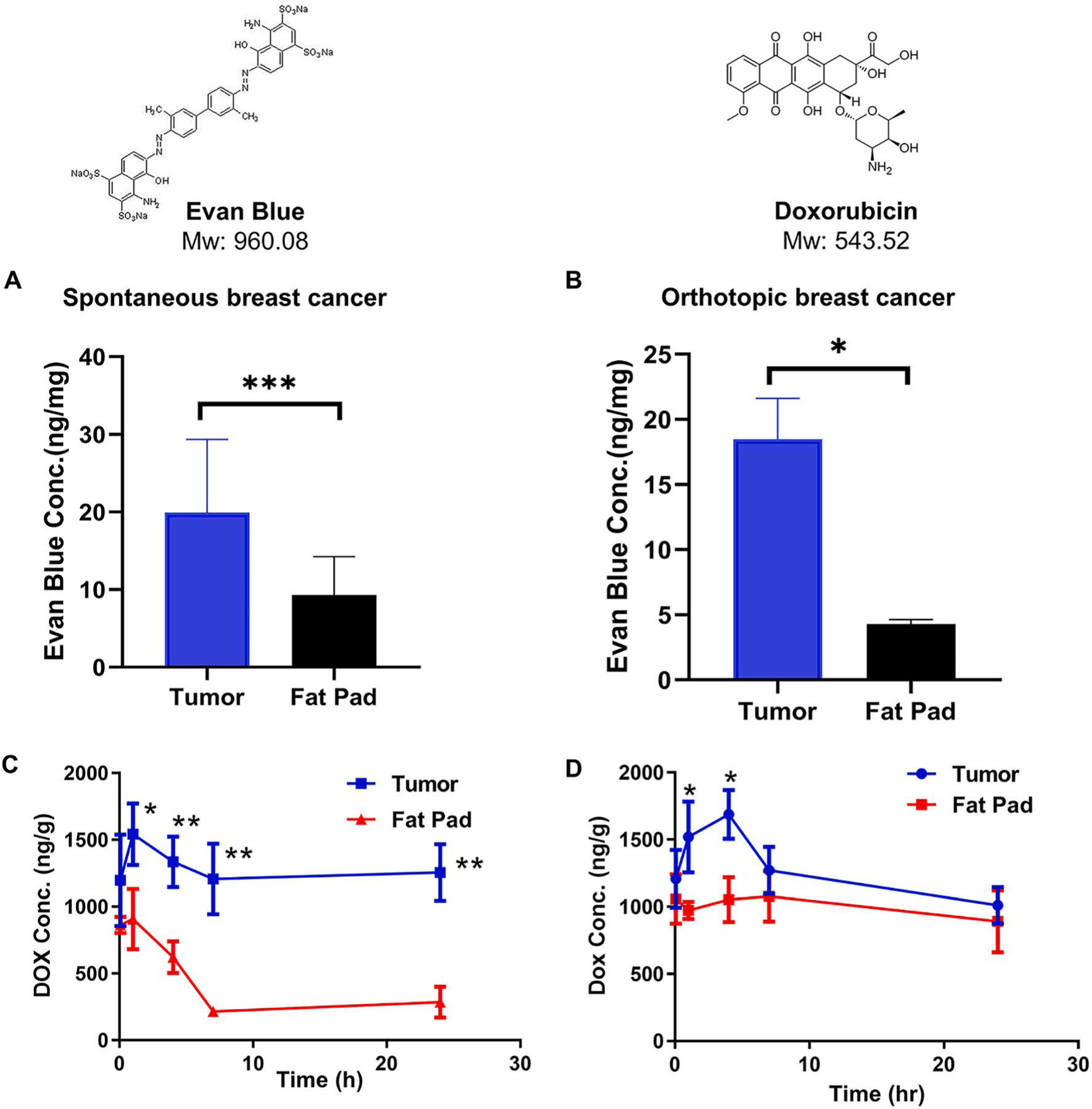
The enhanced accumulation by EPR effect in breast cancers vs. normal breast tissues were observed for small molecule Evans blue (A, B), Doxorubicin (C, D) in spontaneous and orthotopic breast cancer models. (A, B) The tumors tissues were collected 24 h after i.v. injection of Evans blue (30 mg/kg) and measured Evans Blue concentration using a microplate reader (OD = 620 nm). Drug concentration in tumor in MMTV-PyMT spontaneous or orthotopic breast cancer model at different time point after IV dose of doxorubicin solution (5 mg/kg). *p < 0.05, **p < 0.01, ***p < 0.001 tumor vs fat pad.
We then investigated the accumulation of long-circulating stable nanomedicines in preclinical breast tumor models vs. normal breast tissues using PEGylated liposomal formulation of doxorubicin (Doxil, 83 nm) and Myocet, a non-PEGylated liposomal formulation of doxorubicin (Myocet, 120 nm) (supplemental table S2). As shown in Fig. 4 (Top panel, A to D), in comparison with normal breast fat pads, both Doxil and Myocet have a 3 to 10-fold increase in tumor accumulation regardless of preclinical mouse tumor models.
Fig. 4.
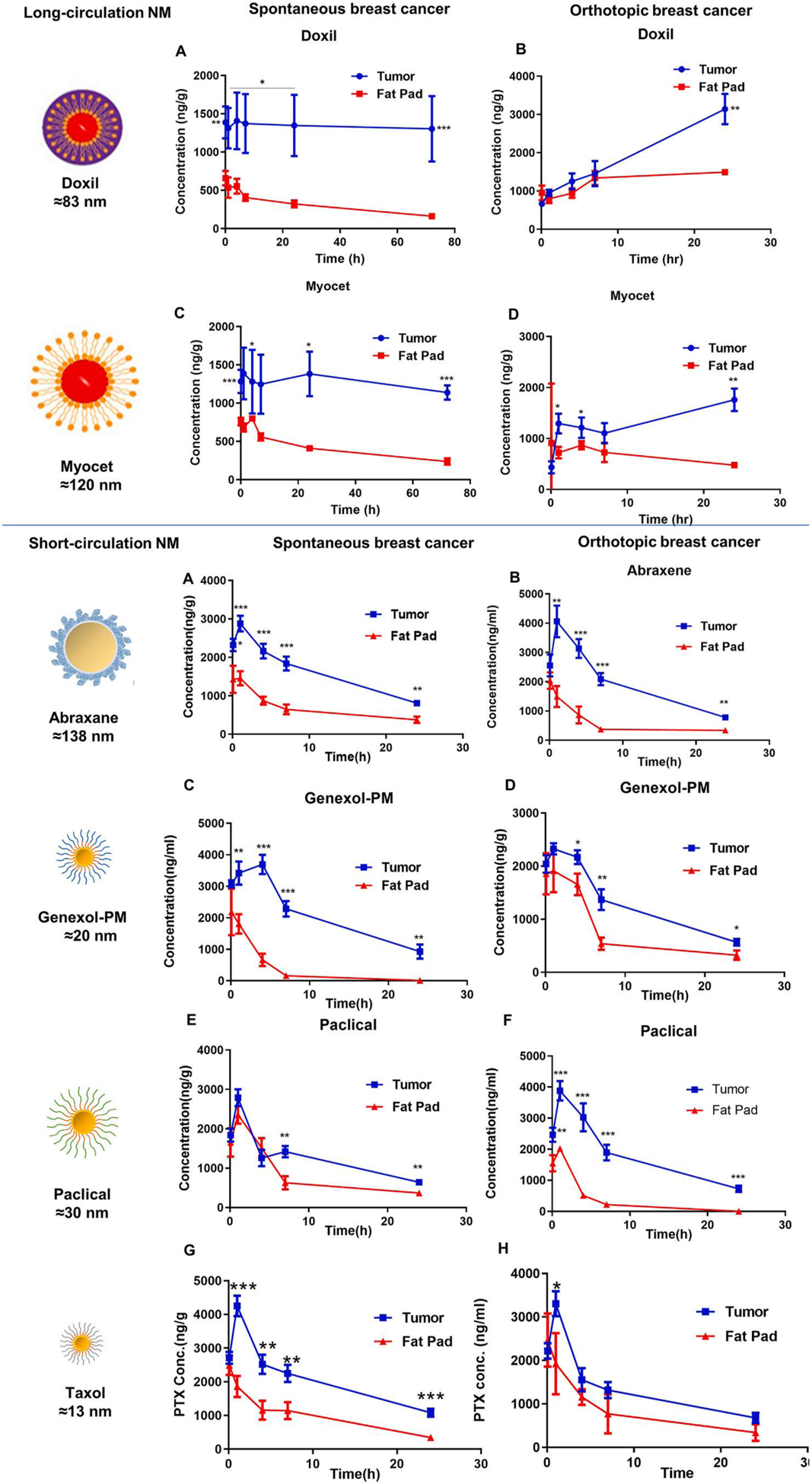
The enhanced accumulation by EPR effect in mouse transgenic spontaneous breast cancer and orthotopic breast cancer vs. normal breast tissues were observed for long-circulating stable doxorubicin liposomal nanoformulations (left panel) and short-circulating fast release nanomedicines of paclitaxel (right panel). Top panel: Drug concentration in tumor and fat pad in MMTV-PyMT spontaneous or orthotopic breast cancer model after IV dose of Doxil (A, B), and Myocet (C, D) (5 mg/kg). *p < 0.05, **p < 0.01, ***p < 0.001 tumor vs fat pad. Bottom panel: Drug concentration in tumor and fat pad in MMTV-PyMT spontaneous or orthotopic breast cancer model after IV dose of Abraxane (A,B), Genexol-PM (C,D), and Paclical (E,F), Taxol (G,H) (10 mg/kg). *p < 0.05, **p < 0.01, ***p < 0.001 tumor vs fat pad.
We also investigated the EPR effect of short-circulating fast-release nanomedicines in three breast cancer models vs. normal breast tissue. We used Abraxane, an albumin nanoparticle formulation of paclitaxel (138 nm), Genexol-PM, a PEG-PLA polymeric nanoparticle formulation of paclitaxel (20 nm), Paclical, an all-trans retinoic acid micellar nanoformulation of paclitaxel (30 nm), and Taxol, Cremophor EL (CrEL) formulation (13 nm) in our investigation. As shown in Fig. 4 (Bottom panel A to H), all four short-circulating fast release nanomedicines of paclitaxel enhanced drug accumulation in the tumor compared to normal breast fat pads in three types of breast cancer models. These data suggest that long-circulating stable nanomedicines, short-circulating fast release nanomedicines, clinical micelle formulation all enhanced tumor accumulation by EPR effect in three different types of breast tumors in comparison with normal breast tissues. In addition, long-circulating stable nanomedicines achieved longer drug retention in tumor compare with short-circulating fast release nanomedicines as shown by the results that the tumor drug concentration of Doxil/Myocet remains up to 72 h, while those of Abraxane/Genexol-PM/Paclical decreased after 2–7 h (Fig. 4). EPR described both enhanced tumor permeability and retention of drugs. Both stable and fast-release nanomedicine has enhanced permeability (accumulation) compared to normal fatpad. But stable nanomedicine tends to have more tumor retention. All these data is consistent with EPR effect vs. normal tissues as it first discovered [45].
3.3. Long or short-circulating nanomedicines have distinct tumor penetration and delivery efficiency to different cell types in tumor microenvironment, which is associated with clinical efficacy
The enhanced tumor accumulation of Doxil and Myocet vs. free doxorubicin in subcutaneous and orthotopic breast cancers did not translate to better anticancer efficacy in breast cancer patients. In contrast, both Doxil and Myocet showed similar anticancer efficacies in comparison with doxorubicin solution [21,22,56], and had similar tumor accumulation as free doxorubicin in transgenic spontaneous breast cancer models. However, the ratios of tumor/plasma of Doxil and Myocet were 100- and 25-fold lower than the tumor/plasma ratio of free doxorubicin in all three preclinical cancer models (Fig. 1C,F,I). These data differences suggest Doxil and Myocet may have lower tumor penetration. In order to test the tumor penetration, we utilized fluorescent imaging to visualize the localization of the nanomedicines in transgenic mouse spontaneous breast cancer tissues. Doxorubicin solution is distributed throughout the tumor tissues (Fig. 5A), while Doxil (Fig. 5B) and Myocet (Fig. 5C) have less tumor tissue penetration staying dose to the blood vessels. Doxil and Myocet penetrated approximately 50% compared to that of free doxorubicin (Fig. 5D).
Fig. 5.
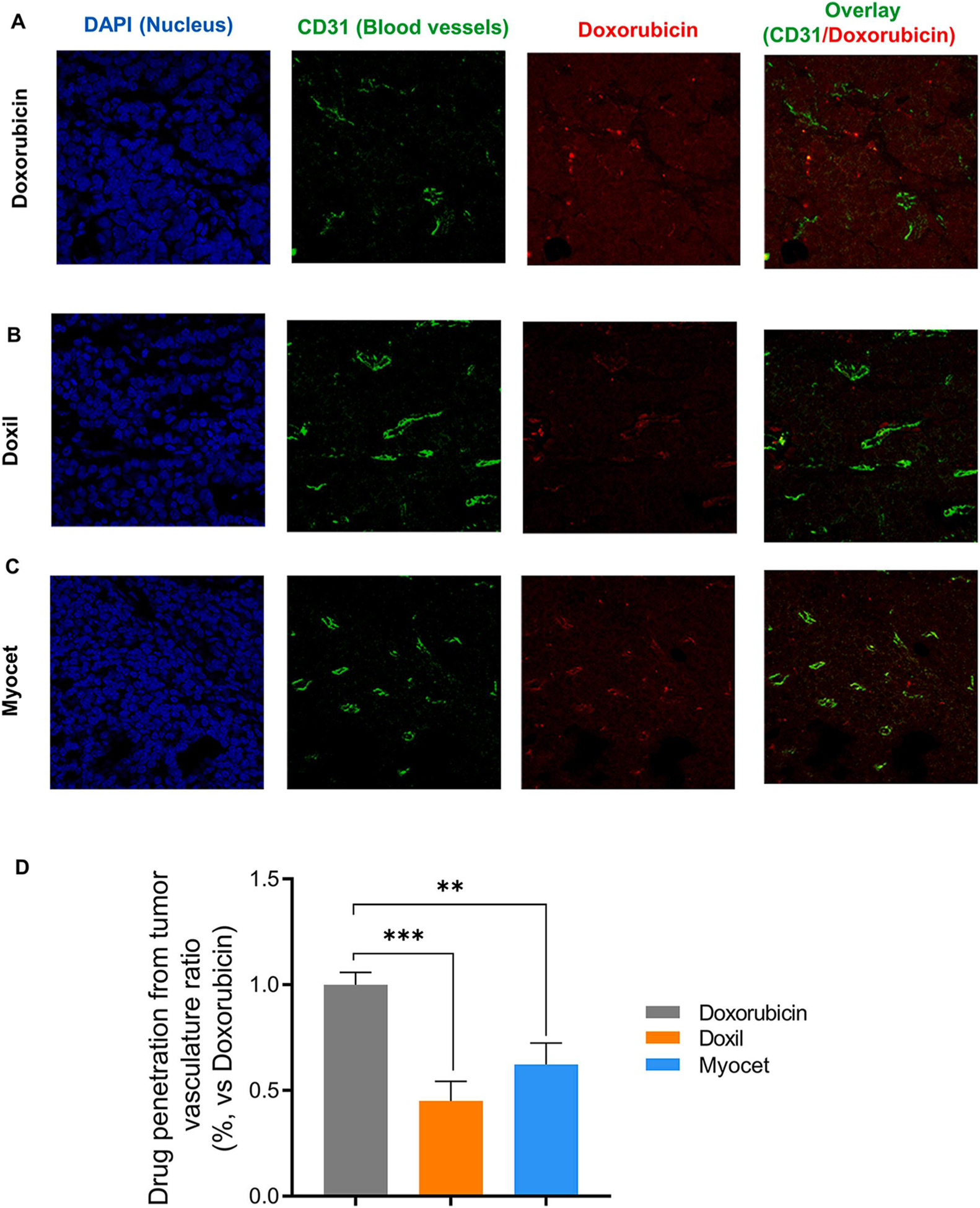
Both PEGylated and Non-PEGylated liposome decreases tumor penetration as measured by confocal fluorescent imaging. Doxorubicin (A), Doxil (B) and Myocet (C) distribution (Red) in PyMT breast tumor tissues, that is overlaid with blood vessels (green, anti-CD31 staining) and nuclear staining (Blue, DAPI staining) after IV dose (5 mg/kg). (D). Average distance of doxorubicin distributed away from blood vessels as measured by Imaging analysis from A-C. **p < 0.01; ***p < 0.001.
It is worth noting that although tumor penetration of Doxil and Myocet is decreased compared to doxorubicin solution, they showed similar clinical efficacy in breast cancer patients [21,22,56]. These findings are consistent with our previous data from Fig. 1H that showed Doxil, Myocet, and doxorubicin solution have similar total drug accumulation at the tumor in transgenic spontaneous breast cancer. These data suggest that Doxil and Myocet may achieve a locally controlled release deposit at the tumor site, which helps to maintain the similar clinical efficacy observed in breast cancer patients [21,22,56,57].
In contrast, the tumor delivery properties of three short-circulating fast-release paclitaxel nanomedicines were different from that of long-circulating and stable nanomedicines. Surprisingly, three nanoformulations (Abraxane, Genexol-PM, Paclical) had low tumor accumulation than micelle Taxol in transgenic mouse spontaneous breast cancer, although their tumor accumulation was slightly higher in subcutaneous and orthotopic breast cancer (Fig. 2B,E,H). However, the tumor/plasma ratios of all four paclitaxel formulations in spontaneous breast cancer showed no difference (Fig. 2I), which suggest that they may have similar tumor tissue penetration. Therefore, we also investigated the localization of paclitaxel in the tumor tissues from all four formulations. Since paclitaxel does not fluoresce, we utilized Mass Spectrometry Imaging (MSI) with a 50–100 μm resolution to directly visualize paclitaxel localization. MS imaging showed four short-circulating fast-release paclitaxel formulations (Abraxane, Genexol-PM, and Paclical, Taxol) have similar tissue penetration in the tumor tissue sections, where they penetrated outside of blood vessels as stained by anti-CD31 antibody staining (Fig. 6A–D).
Fig. 6.
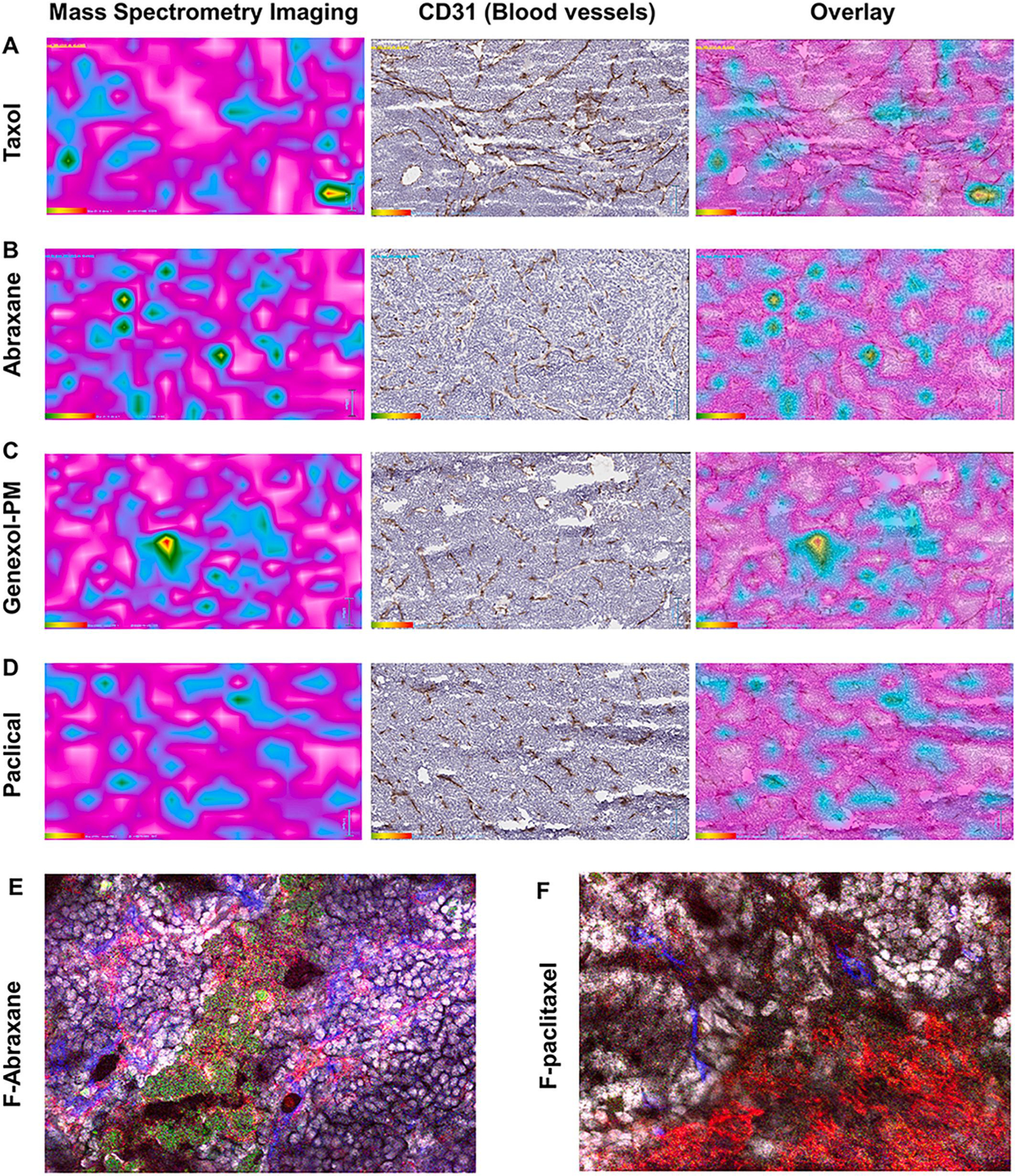
Observation of paclitaxel nanoformulation distribution in tumor tissues. Drug Tumor penetration was visualized by Mass spectrometry imaging (MSI) to show localization of paclitaxd in tumor (Cyan color) after IV dose (50 mg/kg) of (A) Taxol, (B) Abraxane, (C) Genexol-PM and (D) Paclical in transgenic spontaneous breast cancer mice (left column). Cyan: The paclitaxel in the tumor tissues from all four nanoformulations was detected by MS imaging. Brown: The blood vessel of the same slides was stained by anti-CD31 antibody (middle column). The MSI was overlaid with blood vessel staining (right column). Bar = 50 μm. The localization of albumin nanoparticle encapsulated with fluorescent-labeled paclitaxd (E) and free fluorescent-labeled paclitaxel (F) in tumor microenvironment. Green: fluorescent-labeled paclitaxel. Red: macrophage labded with anti-F4/80 antibody. Blue: blood vessel labeled with anti-CD31 antibody.
Obviously, these data raised an important question: since Abraxane has lower tumor accumulation (the absolute concentration in Fig. 2H) but similar tumor tissue penetration (the ratio of tumor/plasma in Fig. 2I) to that of Taxol, why did Abraxane had superior anticancer efficacy in breast cancer patients since Abraxane (260 mg/m2 infusion 30 min) and Taxol (175 mg/m2 infusion 3 h) has similar plasma AUC in human patients? This discrepancy may suggest other mechanisms for Abraxane’s superior efficacy to Taxol in breast cancer [58]. It has been reported that paclitaxel exerts part of its antitumor efficacy through modulation of tumor associated macrophages by inducing an M1 phenotype [59–61]. We endeavored to find out if Abraxane was co-localizing with tumor associated macrophages. We used fluorescent-labeled paclitaxel coupled with fluorescent imaging to visualize the localization of Abraxane/Paclitaxel in different cell types. We generated an albumin nanoformulation with the fluorescent-labeled paclitaxel: paclitaxel at a 1:125 ratio, which enabled sub-micron resolution using confocal fluorescent imaging. The size distribution and stability of the fluorescent-labeled albumin nanoformulation is similar to Abraxane with a size distribution of 135.67 ± 0.750 nm and PDI of 0.133 ± 0.014 (Fig. S2). The imaging showed that albumin nanoformulation of fluorescence labeled paclitaxel was co-localized with macrophages, while free fluorescence-labeled paclitaxel is evenly distributed throughout the tumor (Fig. 6E and F). It has been reported that macrophages internalize Abraxane via micropinocytosis, which may enable paclitaxel to act on endosomal TLR4 complexes, therefore result in a positive feedback signaling, promoting further uptake of drug and enhancing its M1-activating effects [59]. This data further suggests Abraxane delivers paclitaxel to the TAMs to enhance the clinical efficacy in metastatic breast cancer patients [58–61].
3.4. Long-circulating nanomedicines do not universally decrease normal tissue distribution, but change (decrease, increase, no change) the tissue distribution to alter efficacy/safety
One of the promises of designing long-circulating nanomedicines is better safety through decreasing normal tissue distribution. We carried out biodistribution studies of Doxil and Myocet, two long-circulating nanomedicine formulations of doxorubicin to test whether normal tissue distribution decreases. In all organs, Doxil and Myocet significantly decreased the tissue penetration since Doxil’s tissue/plasma ratio is 50 to 300-fold lower compared to doxorubicin (Fig. S5). Similarly, Myocet’s tissue/plasma ratio is decreased 25-fold compared to free doxorubicin (Fig. S5).
Long circulating stable nanoformulations do not universally decrease normal tissue distributions but alter the distribution differently among all normal organs.
There are four different distribution/penetration patterns for Doxil and Myocet in normal tissues (Fig. 7A, B, S4, S5). (a) In the organs of elimination, the liver, spleen, lung, and kidney, the distribution of Doxil and Myocet is higher than doxorubicin solution. This is because of the Reticuloendothelial System (RES) clears or filters particles in the size range 100–200 nm (Fig. 7A, S4). (b) Among eleven non-elimination organs, skin (dermal tissue) is the only tissue with significantly higher distribution of Doxil and Myocet than doxorubicin solution (Fig. 7B, D, S4). (c) In the three organs (stomach, uterus, and muscle), Doxil, Myocet, and doxorubicin solution showed similar distribution (Fig. 7B, S4). (d) In five organs (heart pancreas, intestine, fat pad, bone), long circulating nanomedicines decreased the distribution compared to doxorubicin, where heart has the lowest distribution of Doxil (Fig. 7B, S4). Clearly, long circulating nanomedicines did not universally decrease drug distribution in normal organs. The largest increase in tissue distribution for Doxil and Myocet is in the skin, the largest decrease is in the heart. These changes in tissue distribution are associated with increased hand-foot-syndrome, but decreased cardiomyopathy.
Fig. 7.
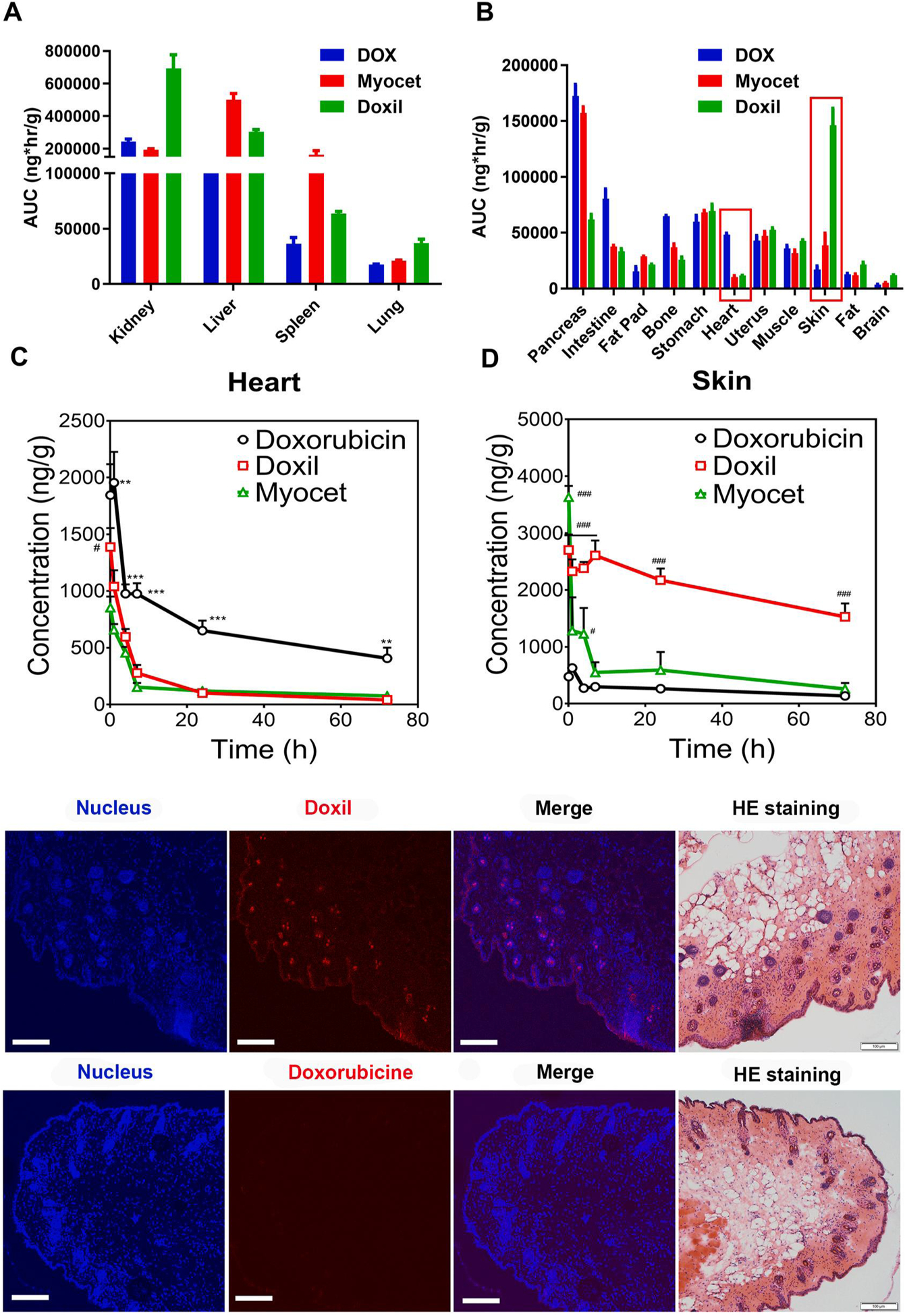
Long circulating and stable nanoformulations of doxorubicin alter tissue distribution differently in various organs. AUC0–72h of different DOX formulations in elimination related organs (A, B) and elimination non-related organs (C, D). Drug concentration in skin (A) and heart (B) in subcutaneous breast cancer model after IV dose of doxorubicin, Doxil, and Myocet (5 mg/kg). The statistical analysis results of Fig A and B are shown in supplementary materials (Table S4). #p < 0.01 DOX vs Myocet; **p < 0.01 DOX vs other two groups; ***p < 0.001 DOX vs other two groups; #p < 0.05 vs DOX; ###p < 0.001 vs DOX. (E) Confocal imaging to visualize detail localization of Doxil and doxorubicin in epidermis and dermis of the skin. Localization of Doxil throughout epidermis and dermis including hair follicles. Minimal presence of doxorubicin in epidermis and dermis tissues. Nuclear staining was performed using DAPI (Ex = 405 nm). The Doxil and doxorubicin were visualized at the Ex of 488 nm. Bar represents 100 μm H&E staining was performed in an adjacent section from series sections of the same tissue.
Long circulating stable nanomedicines (Doxil and Myocet) reduce drug distribution (absolute concentration) and penetration (tissue/plasma ratios) in the heart to reduce cardiotoxicity, while they increase drug distribution in the dermal tissues that is associated with its efficacy/safety.
Among all tissues, Heart distribution for both Doxil and Myocet is 4–5 fold lower than doxorubicin solution (Fig. 7C). This data suggested that the low heart drug distribution and retention for both Doxil and Myocet might be responsible for their low cardiotoxicity compared to free drug (Doxil 3.9% vs. doxorubicin 18.8%; Myocet 6% vs. Doxorubicin 21%) [15,21].
PEGylated doxorubicin liposome drastically increased tissue distribution in the dermal and epidermal tissues, which may be associated with increased hand-foot-syndrome but superior efficacy in treating AIDS-related Kaposi’s sarcoma (ARKS). Doxil has 6-fold higher accumulation than doxorubicin solution in the skin tissues from 15 to 72 h (Fig. 7D). Furthermore, confocal fluorescent imaging showed that Doxil was localized throughout the dermis and epidermis of the skin (Fig. 7E). In sharp contrast, doxorubicin only showed minimal presence in dermis and epidermis of the skin tissue. Our data is consistent with the higher incidences of skin related side effect in the clinical patients: increased hand-foot syndrome (Doxil 48% vs. doxorubicin 2%), increased rash (Doxil 10% vs doxorubicin 2%), and increased abnormal pigmentation (Doxil 21% vs doxorubicin 6%), and increased erythema (Doxil 18% vs. Doxorubicin 3%) [15,19,21]. It is worth noting that Doxil shows superior efficacy in treating AIDS-related Kaposi’s sarcoma (ARKS) in comparison with doxorubicin solution (OR Doxil 45.9% vs Doxorubicin + bleomycin + vincristine 24.8%, p < 0.001) but not in other types of cancers [20]. Given that ARKS arises from the spindle cells located in the dermis tissue, the unique localization of Doxil may be partially responsible for its superior clinical efficacy in ARKS compared to doxorubicin.
3.5. Short-circulating fast-release nanomedicines decrease blood concentration and alter the tissue distribution in normal organs, which may relate to the reduced adverse events in blood compartment but increased toxicity in other organs
Short-circulating nanomedicines of paclitaxel increased penetration in many organs compare to Taxol.
The physico-chemical and pharmacokinetic properties of paclitaxel determined its low tissue distribution as shown by high plasma concentration and low tissue concentration (Fig. S7–8). Abraxane, Genexol-PM and Paclical either increase or maintain tissue vs plasma ratio compared to Taxol (Fig.S8). Particularly, Abraxane increase tissue vs plasma ratio in heart, liver, lung, muscle, pancreas and kidney; Genexol-PM increase tissue vs plasma ratio in heart, fat pad, skin, intestine, brain and stomach; Paclical increase tissue vs plasma ratio in heart, kidney, fat pad, stomach, bone, brain and fat (Fig S8).
Short-circulating nanomedicines of paclitaxel have very different tissue distribution in a carrier-dependent manner (Fig. S6–7).
Four nanomedicines of paditaxel have similar absolute drug distribution in heart, liver, kidney, and pancreases. Abraxane, Genexol-PM and Padical has lower drug concentration than Taxol in spleen, uterus, muscle and lung. Particularly, Abraxane decreased the drug concentration in fat and fat pad. Genexol-PM shows high drug concentration in stomach. Padical increased drug concentration in bone, while decreased the drug concentration in intestine and skin compared to Taxol (Fig. S6–8).
The altered tissue distribution may be related to the different clinical toxicity profile of four paclitaxel nanomedicines.
Neutropenia and neuropathy are most common adverse events of Taxol (in addition to the allergic reaction by Cremophor EL). Abraxane has less incidence of neutropenia (Abraxane 9% vs. Taxol 22%), but more incidence of neuropathy than Taxol (Abraxane 10% vs. Taxol 2%), more GI toxicity such as nausea (Abraxane 30% vs Taxol 22%), vomiting (Abraxane 18% vs Taxol 10%), and diarrhea (Abraxane 27% vs Taxol 15%) [27,30,31,42,62], Short-circulating Abraxane has 3 to 5-fold lower plasma concentration vs. Taxol (Fig. 2A, D, G). These data explain why Abraxane has lower incidence of neutropenia in comparison with Taxol. In addition, our previous study showed that Abraxane enhanced secretion to gastrointestinal track in comparison with Taxol [63], which was correlated with the increased GI toxicity in comparison with Taxol [27,31]. The clinical studies also showed Abraxane has increased incidence of neuropathy (Abraxane 10% vs. Taxol 2%) [43,44,62], which needs to be further explored for its mechanism. Thus, the normal tissue distribution should be carefully evaluated for preclinical study of nanomedicine in order to understand the potential clinical toxicity profiles [64].
3.6. Percentage of dose delivered to tumors or normal organs may not be used as nanomedicine evaluation criteria
Nanomedicine is intended to achieve a high percentage of dose delivered to the tumor, which is often used as a criterion for nanomedicine design in animal cancer models, while high percentage of dose delivered into the normal organs (such as the liver) may be a concern for toxicity. However, the calculation of the percentage dose delivered to tissue is the product of the concentration of drug in tissue and tissue weight, where tissue weight significantly influences the percentage value. In tumor tissues, most nanomedicines are only able to achieve low percentages of injected dose in tumors in previous reports [2,4,12–14]. As shown in Fig 8, the percentages of injected dose in the tumor are in the range of 0.2%–l% for all five evaluated nanomedicines in all three cancer models, which is independent of nano size, structure, composition, long/short-circulation, or tumor models, although long-circulating nanomedicines indeed showed slightly higher percentage of dose delivered in tumors than short-circulating nanomedicines. In comparison, the percentage of injected dose of small molecules (Doxorubicine) in the tumors were in the range of 0.2–0.6% (Fig. 8 A, B, C). The low percentage of dose delivered to tumors (<1%) of nanomedicines in any tumor model is inconsistent with the outstanding preclinical anticancer efficacy in the subcutaneous mouse model, or in some human cancer types [2–4,12–17]. This raised the question whether percentage of dose delivered to tumor (although it is low) is a valid criterion for nanomedicine evaluation, since it is altered by the tumor size and it is inconsistent with preclinical/clinical efficacy.
Fig. 8.
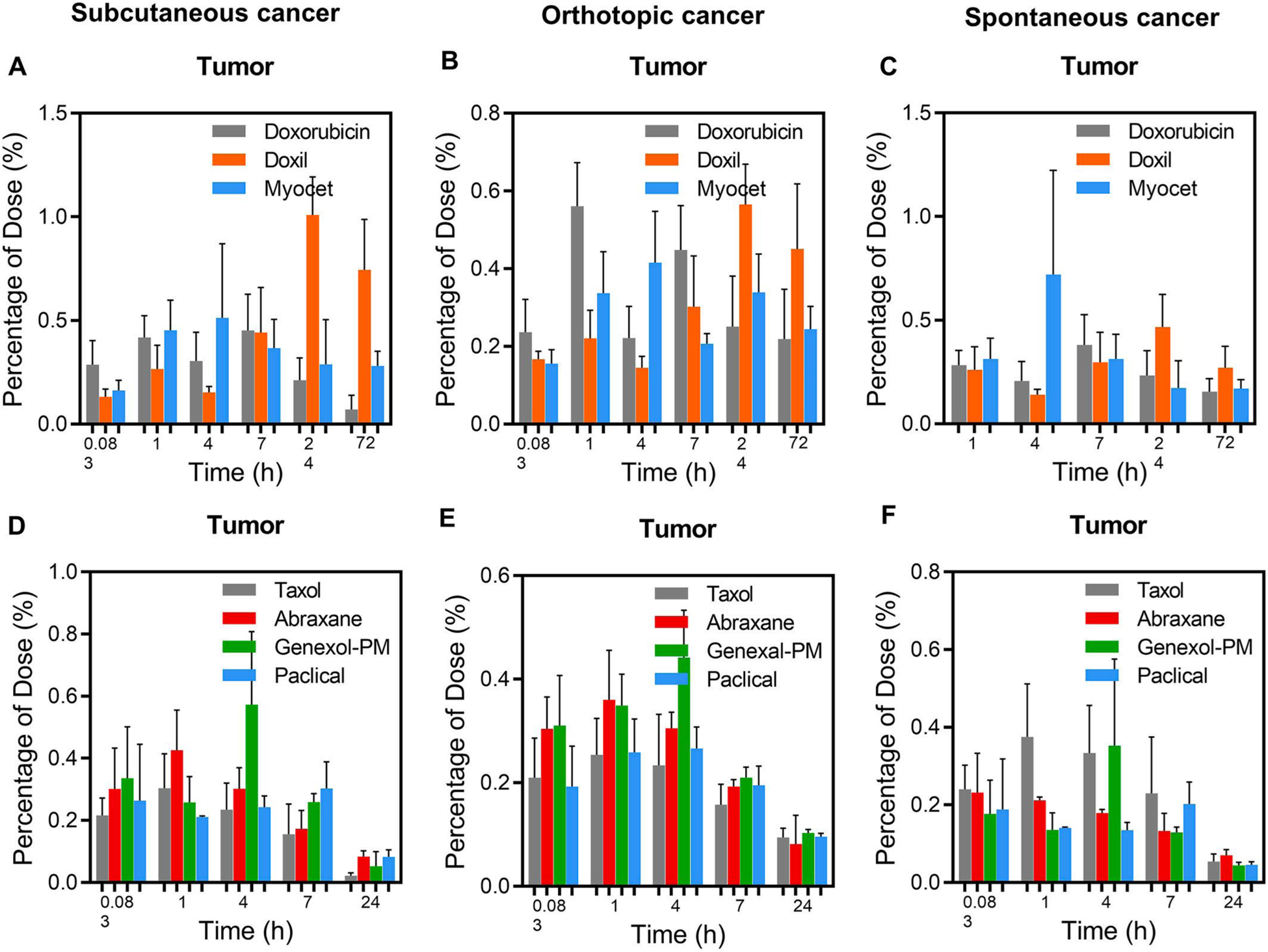
Percentage of dose injected in tumors in subcutaneous, orthotopic breast cancer, and spontaneous transgenic PyMT breast cancer models. Percentage of dose injected of long circulating nanomedicines (Doxil and Myocet) vs. free drug (doxorubicin) (10 mg/kg) in the subcutaneous breast cancer (A), orthotopic breast cancer (B), and spontaneous transgenic PyMT breast cancer model (C). Percentage of dose injected of short circulating nanomedicines (Abraxane, Genexol-PM and Paclical) vs. paclitaxel (Taxol) (10 mg/kg) in the subcutaneous breast cancer (D), orthotopic breast cancer (E), and spontaneous transgenic PyMT breast cancer model (F). The mice were IV dosed with same dose of different nanoformulations. At different time points, three mice were sacrificed to collect blood and other normal organs. The drug concentrations in tumors were measured using LC-MS/MS.
In the normal organs, due to the large mass (or volume) of blood, liver, bone, and muscle, the percentages of dose delivered to these organs are always larger than other tissues for both nanomedicines and small molecules (Fig. S9, S10). High percentage of injected dose in the liver (15–20%) is always thought to be a concern for nanomedicines due to the RES clearance by the liver [5,65,66]. However, the percentage of dose delivered to liver for small molecules is similar to nanomedicines (Fig. S9, S10). One cannot assume that high percentage of delivered dose into the liver is always detrimental for the clinical efficacy and safety profile, which is drug-dependent, formulation dependent, and pharmadynamics-dependent. This raised the question for the validity of percentage dose delivered to organs as one of the nanomedicine evaluation criterion for the concern of toxicity, since the organ mass is the major factor for the calculation.
4. Discussions
The debate for nanomedicine design based on EPR effect has lasted for more than 15 years. However, these debates may have mixed two different questions with two distinct clinical implications [1]: (1) does EPR effect exist in preclinical mouse cancer models and in human cancer patients? (2) does nanomedicine enhance accumulation by EPR effect vs. free drugs to improve clinical anticancer efficacy in comparison with free drugs? Our data showed that when breast tumors were compared with normal breast tissues, the tumor accumulation of nanomedicines, free drug solution, and clinical micelle formulation were enhanced by EPR effect in all three types of preclinical mouse cancer models (Figs. 3 and 4). As a physiological phenomenon, when tumors are compared with normal tissues, EPR effect surely exists in tumor tissues, which requires no further debate and confirmation. The EPR effect (in comparison with normal tissues) has been repeatedly confirmed not only in various preclinical cancer models using thousands of nanoparticle delivery systems [11,45,48,49], but also confirmed in human cancer patients using different nanoformulations of imaging agents (Tc99 [68–70], 64Cu [71], or In111 [72,73]). However, this EPR effect in tumors vs. normal tissues is not only observed for nanomedicines (long or short circulating), but also seen for small molecules with high protein binding as showed by previous literature [45] and our current study. EPR effects contributed to the enhanced 2 to 6-fold tumor accumulation of small molecules (Evans blue, doxorubicin) with high protein binding in comparison with normal tissues (Fig. 3). The previous study demonstrated tumor EPR effect compared to normal tissues using Evan blue with high protein binding capacity (68%) and high affinity (Ka = 4 × 105 M−1) [45]. The high protein binding capability and high affinity of Evans blue result in a longer blood circulation time, which enhances tumor accumulation via tumor EPR effect. However, it has been generally neglected that most of antitumor small molecules also have high protein binding capacity (>70–98%) despite low binding affinity. For example, doxorubicin has a high protein binding (~75%) with low binding affinity (Ka: 7.50 × 103 M−1). Once drug is administered into blood circulation, the majority of drugs are bound to plasma protein, while their distribution to whole body within seconds. In normal tissue, unbound drug diffuse to tissue where the drug can bind to tissue with different capacity and affinity in different organs, which determines the concentrations in different organs. In contrast, in tumor tissues, due to the leaky vasculature, both unbound drug and plasma protein bound drug can pass through the leaky tumor vasculature resulting a higher tumor accumulation than normal tissues. The drug can also bind to tumor tissues for high retention in tumors. Therefore, drug accumulation in the tumor is dependent on drug binding to plasma protein level, protein binding affinity, tumor tissue binding capacity and binding affinity, as well as tumor EPR effect. It is also worth noting that different small molecules may have different preferred tumor accumulation compared to normal tissues, thus it cannot be generalized that nanomedicine will have preferred tumor accumulation than free drug solution or micelle formulation, which is likely drug-dependent.
Therefore, the most relevant question for clinical efficacy is whether nanomedicines enhance drug accumulation through EPR effect in the tumor in comparison to the free drug solution or clinical micelle formulation (such as Taxol). Although enhanced accumulation of nanomedicines (both long and short circulating) vs. free drug or clinical micelle formulation in the tumors by EPR effect has been confirmed in subcutaneous and orthotopic cancer models, but the enhancement is not observed in transgenic mouse spontaneous cancers, which better mimics human cancers (Figs. 1–2). The lack of difference in the tumor accumulations in spontaneous cancers between nanomedicines and free drugs or clinical micelle formulation is because they have similar levels of enhanced tumor accumulation by EPR effect in comparison with normal tissues (Figs. 3–4). If similar situation occurs in human cancers, current nanomedicine would not expect to improve anticancer efficacy in comparison with free drugs or clinical micelle formulation. This is consistent with clinical observations for the similar efficacy between nanoformulation and free drug (Doxil vs DOX in breast cancer, Myocet vs DOX in breast cancer).
There is limited evidence to show nanomedicines enhance tumor accumulation compared to free drugs or the clinical micelle formulation in human cancer patients. Most clinical studies only compared the tumor accumulation of nanomedicines (or nanoformulations of imaging agents) in the tumor vs normal tissues but did not compare nanomedicines vs. free drugs or clinical micelle formulation in the tumor [19, 68–75]. The lack of accumulation difference between nanomedicines and free drugs or clinical micelle formulation may be one of the most important reasons for poor translation of anticancer nanomedicines from the preclinical to clinical setting [2,4,12–17]. However, a counter-point is a clinical study that showed 5.2–11.4-fold higher tumor accumulation of Doxil vs. doxorubicin solution in the lesion of AIDS-related Kaposi’s Sarcoma (ARKS) [76]. However, this study did not compare the accumulation of Doxil vs. doxorubicin solution in the normal dermal tissue, which also exhibited preferential accumulation of Doxil in the epidermis and dermis tissues as shown in our study (Fig. 7). Previous literatures suggested this comparison was also performed in normal skins, but extensive literature research only found the same set of data in sarcoma lesions [73]. Given that spindle cells of ARKS are usually spread in dermis tissues of the skin, the unique localization of Doxil, in comparison with doxorubicin, is at least partially responsible for its superior clinical efficacy in AIDS-related Kaposi’s sarcoma (ARKS). It is important to note that the spindle cells may have preferred uptake of Doxil in the ARKS lesion, so the higher tumor accumulation of nanomedicine in ARKS skin lesion may not be due to EPR effect [77].
Clearly, the enhanced tumor accumulation of nanomedicines vs. free drugs or clinical micelle formulations in subcutaneous or orthotopic mouse breast cancers may not translate to superior anticancer efficacy in human cancer patients [21,22,56,57]. The efficacy/safety profiles of five nanomedicines (vs. free drugs or clinically used standard formulation) from 14 phase III clinical trials are inconsistent with nanomedicine design criteria based on EPR effect (supplemental table 1). For instance, Doxil vs. doxorubicin showed superior efficacy in AIDS-related Kaposi’s sarcoma (ARKS (Objective response, OR Doxil 45.9% vs Doxorubicin + bleomycin + vincristine 24.8%, p < 0.001), but no difference in breast cancer patients (Overall survival, OS, Doxil 21 months vs Doxorubicin 22 months) (supplemental table 1). In contrast, Abraxane vs. Taxol showed superior efficacy in breast cancer (Response Rate, 21.5% in Abraxane vs 11.1%, in Taxol group, P = 0.003; median TTP 23.0 weeks in Abraxane vs. 16.9 in Taxol, p = 0.006), and non-small cell lung cancer (objective response rate, ORR Abraxane 33% vs Taxol 25%%, p = 0.005), but not in gastric cancer patients (Median OS, Abraxane 11.1 months vs. Taxol 10.9 months) (supplemental table l).These results also raise questions for the validity of subcutaneous or orthotopic cancer models in the evaluation of most nanomaterial since they often exhibit a high EPR effect that is different from human cancer patients [78]. Genetically engineered mouse models (GEMMs) are better suited to study the distribution of nanomedicine because of their similar vascular permeability, microenvironment, and disease progression to human tumors [78,79]. Evaluation of the delivery efficiency of the nanomedicine to the tumors in GEMMs allows for better translation from a preclinical model to a clinical setting [80,81]. It is worth noting that similar tumor distribution of free doxorubicin and liposomal Doxil may not extend to all free drugs and all nanomedicines, since different drugs and nanocarrier may have distinct tumor accumulation preference. Similarly, it also cannot be generalized that EPR can increase tumor accumulation all nanomedicine compared to free drugs. The design of nanomedicine should be drug specific and nanocarrier specific [1]. In addition, the tumor accumulation of small molecules are also dependent on tumor EPR effect, drug binding capacity and affinity to plasma protein, as well as drug binding capacity and affinity to tumor tissue.
In order to explain the poor translation for nanomedicines’ efficacy from preclinical cancer models to human patients with solid cancers, EPR heterogeneity in solid tumors was proposed and studied [4,13,14, 68–73,82,83]. For instance, Doxil showed superior clinical efficacy to treat ARKS sarcoma in comparison with doxorubicin [19,20], which is thought to be mediated by higher EPR effect of ARKS sarcoma. Doxil have no efficacy difference to treat breast, ovarian cancers in comparison to free doxorubicin [21,24,74], which may be due to the EPR heterogeneity in human solid tumor [4,8,12–14,68–73,82,83]. However, EPR heterogeneity hypothesis cannot explain why Abraxane (138 nm, albumin nanoparticle of paclitaxel) showed superior anticancer efficacy to Taxol in breast cancer, lung cancer and pancreatic cancer patients, but no efficacy difference in gastric cancer. Furthermore, small size nanomedicines were hypothesized to have better tumor accumulation and better efficacy by EPR effect [54,55]; however, small size Genexol-PM (20 nm, PEG-PLA nanoparticle of paclitaxel) only showed non-inferior anticancer efficacy in human breast cancers compared to Taxol [39,40].
Several outstanding clinical studies, using liposome-encapsulated chelated imaging agents (Tc99 [68–70], 64Cu [71] or In111 [72,73]) or ferumoxytol (FMX) iron nanoparticles [84] showed that these liposome-encapsulated imaging agents had more than 10-fold difference in tumor accumulation among different cancer patients. The higher tumor accumulation of these liposome-encapsulated imaging agents or ferumoxytol (FMX) iron nanoparticles in human cancer was correlated with the better efficacy of the nanomedicines in these patients [68,69,71,84]. However, these studies did not compare the nanoformulations vs. free cargos (or free drugs). Since tumor heterogeneity (induding EPR effect) is a common physiological phenomenon, it is reasonable to predict that small molecules would also have heterogeneous tumor accumulations. As shown in supplemental Fig. S1B, we also investigated the EPR heterogeneity using Evans blue in 26 different transgenic mouse spontaneous breast cancer lesions in comparison normal breast fat pads, which mimics human breast cancers. EPR heterogeneity was also observed in these 26 breast cancer lesions, which showed 7-fold difference of Evans blue concentration from 5.6 to 36.0 ng/mg. These EPR heterogeneity in transgenic mouse spontaneous breast cancers is similar to the EPR heterogeneity in human solid tumors with 8–35 fold differences as measured by the accumulation of liposomal imaging agents (Tc99 [68,69], 64Cu [71] or In111 [72]) or ferumoxytol (FMX) iron nanoparticles [84]. Therefore, EPR heterogeneity in human solid cancer, which surely exist, still cannot fully explain the poor translation of nanomedicines’ efficacy from preclinical cancer models to human cancer patients.
EPR heterogeneity is attributed to various factors in the tumor microenvironment such as tumor blood flow, vessel density, interstitial fluid pressure, and extracellular matrix. Strategies to enhance EPR effect including restoring obstructed tumor blood flow and improving tumor vascular permeability via vascular mediators may benefit the application of nanomedicine [85]. The clinical study to enhance EPR effect in tumors shows that angiotensin II (AT), inducing hypertension during arterial infusion of SMANCS (SX) with lipiodol, remarkably enhanced tumor delivery and therapeutic response [86]. The question remains if this enhanced EPR effect may be also observed for small molecules with high protein binding. In addition, introducing active targeting moieties on nanoparticles is another strategy widely investigated to increase nanocarrier tumor accumulation. However, despite a good efficacy in subcutaneous or orthotopic mice, prostate-specific membrane antigen (PSMA) targeted polymeric nanoparticle of docetaxel (BIND-014) failed in Phase 1 clinical trial. The question remains whether nanoformulation of a specific drug should be designed based on tumor EPR effect to improve the efficacy since some small molecule drugs may also have preferred tumor accumulation compared to normal adjacent tissues. Our data and previous study suggest that nanomedicine design may need to be drug-specific, nanocarrier specific, and cell type specific to improve success rate of nanomedicine in clinical testing [1].
The second nanomedicine design criteria is to generate long-circulating nanomedicine by decreasing RES clearance to reduce drug toxicity through decreased normal organ accumulation, and further enhance EPR effect to improve anticancer efficacy [5,6,15]. However, our data and others showed that long circulating nanomedicine design is helpful when nanomedicine can enhance tumor accumulation through strong EPR effect vs. free drug solutions in subcutaneous or orthotopic cancer models, but not in spontaneous cancer models (Fig. 1B,E,H). In addition, long-circulating stable nanomedicines do not universally decrease drug distribution in normal organs to reduce toxicity, rather alters (decreases, increases, or no changes) tissue distribution in different organs that alters safety/efficacy profiles (Fig. 7A,B, S4) [15,19–21,23,24,87]. The decreased accumulation of Doxil and Myocet in the heart tissues is associated with their decreased cardiotoxicity in contrast, the enhanced accumulation of Doxil than Myocet or free doxorubicin in the dermal tissues is correlated with increased hand-foot-syndrome, rash, and pigmentation (supplemental table 1). Conversely, the increased drug concentration in the dermal tissues leads to superior efficacy in AIDS-related Kaposi’s sarcoma [15,19,20].
In contrast to long-circulating nanomedicines, short-circulating nanomedicines are designed to rapidly disintegrate and release the free drugs [13,88]. It is unclear if these nanomedicines enhance tumor accumulation through EPR effect. For instance, although Taxol, Genexol-PM and Paclical have low CMC, drug release occurred at equilibrium between nanomedicines and free drugs when they contact with plasma protein at the concentration thousands time high above CMC [89–92]. We confirmed this property in Abraxane, which was stable above 12–30 μg/mL for several hours, but started to dissociate below 12–30 μg/ml (Fig. S3). In comparison, Doxil and Myocet remain stable during dilution (Fig. S3). Our data showed that these short-circulating fast-release nanomedicines (Abraxane, Genexol-PM, and Paclical showed some enhanced tumor accumulation vs. Taxol in subcutaneous and orthotopic mouse breast cancers, but they decreased tumor accumulation vs. Taxol in spontaneous mouse breast cancer (Fig. 2B,E,H). Pharmacokinetic analysis showed that most of drugs (95%–99% of dose) were distributed into tissue immediately after injection (Table S3). Even though these nanomedicines could release drug, it still takes minutes or hours for total drug release [91,92]. Thus, these fast-release nanomedicines would remain or at least partially remain as nanoparticle forms for tissue distribution immediately after injection, which is also supported by previous studies [63,93].
Further, the tissue distribution/penetration of short-circulating nanomedicines varies in carrier-dependent manner, which is responsible for the unique efficacy/safety profiles seen in Abraxane and Genexol-PM. Abraxane delivers the drug to tumor macrophages (Fig. 6E) contributing to its enhanced efficacy [58–61]. Therefore, the biodistribution of nanomedicines needs to be thoroughly evaluated to understand the unique efficacy and safety profiles, which can improve success rate of clinical translation from preclinical models. For instance, in comparison with Taxol, Abraxane reduced neutropenia due to lower concentration in the blood, but increased GI toxicity (nausea, vomiting, and diarrhea) due to increased secretion to GI tract [63], and increased neuropathy. In contrast, Genexol-PM has increased incidence of neutropenia in comparison with Taxol. Furthermore, bioequivalence based on similar plasma AUC cannot guarantee the same tissue biodistribution. Clinical studies have shown that Genexol-PM, Paclical and Abraxane have similar plasma pharmacokinetics [39,94,95], and we found that Genexol-PM, Abraxane and Paclitaxel has similar plasma concentration profile (Fig. 2), but the biodistribution is different among various tissues (Fig. 7, S6, S7).
5. Condusion
In summary (Fig. S11), the debate for nanomedicine design based on EPR effect may have mixed two different questions with distinct clinical implications. When nanomedicines were compared with free drug solutions or clinical micelle formulation in tumors, the long and short-circulating nanomedicines did not enhance tumor accumulation in transgenic spontaneous breast cancers, regardless their nano size and composition, although they did in subcutaneous and orthotopic breast cancers. The lack of differences in the tumor accumulation between nanomedicines vs. free drug solutions or clinical micdle formulation in spontaneous breast cancer raised the concern for the nanomedicine design based on the EPR effect for translation from preclinical cancer models to cancer patients to improve their anticancer efficacy. In contrast, when breast cancer tissues were compared with normal breast tissues, nanomedicines, free drug solutions, and clinical micelle formulations enhanced tumor accumulation by EPR effect in breast cancer tissues vs normal breast tissues in three types of preclinical mouse cancer models. Further, long-circulating nanomedicines did not further increase tumor accumulation in transgenic mouse spontaneous breast cancer, nor universally decreased tissue distribution to reduce toxicity in all normal organs. Rather, long circulation nanomedicines alter (decrease, increase, or no change) distributions in different organs, which are associated with their altered efficacy/safety in human cancer patients. In contrast, short-circulating fast-release nanomedicines decrease blood concentration that reduces hematological toxicity, and also alter the tissue distribution in normal organs that is also correlated with their altered clinical efficacy/safety profiles. These data provide insight why nanomedicine design criteria in different preclinical cancer models may have successful or poor clinical translation for their clinical efficacy/safety in cancer patients.
Supplementary Material
Funding sources
This study is partially supported by Celgene Corporation, 86 Morris Avenue, Summit, NJ 07920, USA.
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: SZ, YL MP are employees and shareholder of Bristol Myers Squibb Company (formerly Celgene Incorporation).
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.biomaterials.2021.120910.
Data availability
The raw/processed data required to reproduce these findings can be shared upon request. The complete raw datasets cannot be shared at this time as the data also forms part of an ongoing study.
References
- [1].Sun D, Zhou S, Gao W, What went wrong with anticancer nanomedicine design and how to make it right, ACS Nano 14 (10) (2020) 12281–12290. [DOI] [PubMed] [Google Scholar]
- [2].Dai Q, Wilhelm S, Ding D, Syed AM, Sindhwani S, Zhang Y, Chen YY, MacMillan P, Chan WCW, Quantifying the ligand-coated nanoparticle delivery to cancer cells in solid tumors, ACS Nano 12 (8) (2018) 8423–8435. [DOI] [PubMed] [Google Scholar]
- [3].Sykes EA, Dai Q, Sarsons CD, Chen J, Rocheleau JV, Hwang DM, Zheng G, Cramb DT, Rinker KD, Chan WC, Tailoring nanoparticle designs to target cancer based on tumor pathophysiology, Proc. Natl. Acad. Sci. U. S. A 113 (9) (2016) E1142–E1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW, Analysis of nanoparticle delivery to tumours, Nat. Rev. Mater 1 (2016) 16014. [Google Scholar]
- [5].Gabizon A, Bradbury M, Prabhakar U, Zamboni W, Libutti S, Grodzinski P, Cancer nanomedicines: closing the translational gap, Lancet 384 (9961) (2014) 2175–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Moghimi SM, Simberg D, Nanoparticle transport pathways into tumors, J. Nanoparticle Res 20 (6) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tran S, DeGiovanni PJ, Piel B, Rai P, Cancer nanomedicine: a review of recent success in drug delivery, Clin. Transl. Med 6 (1) (2017) 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST, Challenges and strategies in anti-cancer nanomedicine development: an industry perspective, Adv. Drug Deliv. Rev 108 (2017) 25–38. [DOI] [PubMed] [Google Scholar]
- [9].Wicki A, Witzigmann D, Balasubramanian V, Huwyler J, Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications, J. Contr. Release 200 (2015) 138–157. [DOI] [PubMed] [Google Scholar]
- [10].Greish K, Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting, Methods Mol. Biol 624 (2010) 25–37. [DOI] [PubMed] [Google Scholar]
- [11].Torchilin V, Tumor delivery of macromolecular drugs based on the EPR effect, Adv. Drug Deliv. Rev 63 (3) (2011) 131–135. [DOI] [PubMed] [Google Scholar]
- [12].Jain RK, Stylianopoulos T, Delivering nanomedicine to solid tumors, Nat. Rev. Clin. Oncol 7 (11) (2010) 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Danhier F, To exploit the tumor microenvironment: since the EPR effect foils in the clinic, what is the future of nanomedicine? J. Contr. Release 244 (Pt A) (2016) 108–121. [DOI] [PubMed] [Google Scholar]
- [14].Nichols JW, Bae YH, EPR: evidence and fallacy, J. Contr. Release 190 (2014) 451–464. [DOI] [PubMed] [Google Scholar]
- [15].Ngan YH, Gupta M, A comparison between liposomal and nonliposomal formulations of doxorubicin in the treatment of cancer: an updated review, Arch. Pharm. Pract 7 (1) (2016) 1–13. [Google Scholar]
- [16].Gabizon A, Shmeeda H, Barenholz Y, Pharmacokinetics of pegylated liposomal doxorubicin - review of animal and human studies, Clin. Pharmacokinet 42 (5) (2003) 419–436. [DOI] [PubMed] [Google Scholar]
- [17].Vail DM, Amantea MA, Colbern GT, Martin FJ, Hilger RA, Working PK, Pegylated liposomal doxorubicin: proof of principle using preclinical animal models and pharmacokinetic studies, Semin. Oncol 31 (6) (2004) 16–35. [DOI] [PubMed] [Google Scholar]
- [18].Weissig V, Pettinger TK, Murdock N, Nanopharmaceuticals (part 1): products on the market, Int. J. Nanomed 9 (2014) 4357–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Northfelt DW, Martin FJ, Working P, Volberding PA, Russell J, Newman M, Amantea MA, Kaplan LD, Doxorubicin encapsulated in liposomes containing surface-bound polyethylene glycol: pharmacokinetics, tumor localization, and safety in patients with AIDS-related Kaposi’s sarcoma, J. Clin. Pharmacol 36 (1) (1996) 55–63. [DOI] [PubMed] [Google Scholar]
- [20].Northfelt DW, Dezube BJ, Thommes JA, Miller BJ, Fischl MA, Friedman-Kien A, Kaplan LD, Du Mond C, Mamelok RD, Henry DH, Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi’s sarcoma: results of a randomized phase III clinical trial, J. Clin. Oncol 16 (7) (1998) 2445–2451. [DOI] [PubMed] [Google Scholar]
- [21].O’Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C, Group CBCS, Reduced cardiotoxidty and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer, Ann. Oncol 15 (3) (2004) 440–449. [DOI] [PubMed] [Google Scholar]
- [22].Harris L, Batist G, Belt R, Rovira D, Navari R, Azarnia N, Welles L, Winer E, Group TDS, Liposome-encapsulated doxorubicin compared with conventional doxorubicin in a randomized multicenter trial as first-line therapy of metastatic breast carcinoma, Cancer 94 (1) (2002) 25–36. [DOI] [PubMed] [Google Scholar]
- [23].Fink W, Zimpfer-Rechner C, Thoelke A, Figl R, Kaatz M, Ugurd S, Schadendorf D, Clinical phase II study of pegylated liposomal doxorubicin as second-line treatment in disseminated melanoma, Onkologie 27 (6) (2004) 540–544. [DOI] [PubMed] [Google Scholar]
- [24].Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ, Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan, J. Clin. Oncol 19 (14) (2001) 3312–3322. [DOI] [PubMed] [Google Scholar]
- [25].Orlowski RZ, Nagler A, Sonneveld P, Blade J, Hajek R, Spencer A, San Miguel J, Robak T, Dmoszynska A, Horvath N, Spicka I, Sutherland HJ, Suvorov AN, Zhuang SH, Parekh T, Xiu L, Yuan Z, Rackoff W, Harousseau JL, Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple mydoma: combination therapy improves time to progression, J. Clin. Oncol 25 (25) (2007) 3892–3901. [DOI] [PubMed] [Google Scholar]
- [26].Orlowski RZ, Nagler A, Sonneveld P, Bladé J, Hajek R, Spencer A, Robak T, Dmoszynska A, Horvath N, Spicka I, Sutherland HJ, Suvorov AN, Xiu L, Cakana A, Parekh T, San-Miguel JF, Final overall survival results of a randomized trial comparing bortezomib plus pegylated liposomal doxorubicin with bortezomib alone in patients with relapsed or refractory multiple myeloma, Cancer 122 (13) (2016) 2050–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J, Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer, J. Clin. Oncol 23 (31) (2005) 7794–7803. [DOI] [PubMed] [Google Scholar]
- [28].Untch M, Jackisch C, Schneeweiss A, Conrad B, Aktas B, Denkert C, Eidtmann H, Wiebringhaus H, Kummel S, Hilfrich J, Warm M, Paepke S, Just M, Hanusch C, Hackmann J, Blohmer JU, Clemens M, Darb-Esfahani S, Schmitt WD, Dan Costa S, Gerber B, Engels K, Nekljudova V, Loibl S, von Minckwitz G, German Breast G, Arbeitsgemeinschaft Gynakologische Onkologie-Breast I, Nab-paclitaxel versus solvent-based paclitaxel in neoadjuvant chemotherapy for early breast cancer (GeparSepto-GBG 69): a randomised, phase 3 trial, Lancet Oncol. 17 (3) (2016) 345–356. [DOI] [PubMed] [Google Scholar]
- [29].Hamilton E, Kimmick G, Hopkins J, Marcom PK, Rocha G, Welch R, Broadwater G, Blackwell K, Nab-paclitaxel/bevadzumab/carboplatin chemotherapy in first-line triple negative metastatic breast cancer, Clin. Breast Canc 13 (6) (2013) 416–420. [DOI] [PubMed] [Google Scholar]
- [30].Abraxane [package Insert], Celgene Corporation, Summit, NJ, 2018. [Google Scholar]
- [31].FDA, ABRAXANE® for Injectable Suspension (Paclitaxel Protein-Bound Particles for Injectable Suspension) (Albumin-bound), 2007.
- [32].Herrera DA, Ashai N, Perez-Soler R, Cheng HY, Nanoparticle albumin bound-paclitaxel for treatment of advanced non-small cell lung cancer; an evaluation of the clinical evidence, Expet Opin. Pharmacother 20 (1) (2019) 95–102. [DOI] [PubMed] [Google Scholar]
- [33].Socinski MA, Bondarenko I, Karaseva NA, Makhson AM, Vynnychenko I, Okamoto I, Hon JK, Hirsh V, Bhar P, Zhang H, Iglesias JL, Renschler MF, Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer; final results of a phase III trial, J. Clin. Oncol 30 (17) (2012) 2055–2062. [DOI] [PubMed] [Google Scholar]
- [34].Shitara K, Takashima A, Fujitani K, Koeda K, Hara H, Nakayama N, Hironaka S, Nishikawa K, Makari Y, Amagai K, Ueda S, Yoshida K, Shimodaira H, Nishina T, Tsuda M, Kurokawa Y, Tamura T, Sasaki Y, Morita S, Koizumi W, Nab-paclitaxel versus solvent-based paclitaxel in patients with previously treated advanced gastric cancer (ABSOLUTE): an open-label, randomised, non-inferiority, phase 3 trial, Lancet Gastroenterol. Hepatol 2 (4) (2017) 277–287. [DOI] [PubMed] [Google Scholar]
- [35].Giordano G, Pancione M, Olivieri N, Parcesepe P, Velocci M, Di Raimo T, Coppola L, Toffoli G, D’Andrea MR, Nano albumin bound-paclitaxel in pancreatic cancer: current evidences and future directions, World J. Gastroenterol 23 (32) (2017) 5875–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hoy SM, Albumin-bound paclitaxel: a review of its use for the first-line combination treatment of metastatic pancreatic cancer, Drugs 74 (15) (2014) 1757–1768. [DOI] [PubMed] [Google Scholar]
- [37].Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabemero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF, Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine, N. Engl. J. Med 369 (18) (2013) 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Goldstein D, El-Maraghi RH, Hammel P, Heinemann V, Kunzmann V, Sastre J, Scheithauer W, Siena S, Tabemero J, Teixeira L, Tortora G, Van Laethem JL, Young R, Penenberg DN, Lu B, Romano A, Von Hoff DD, nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial, J. Natl. Cancer Inst 107 (2) (2015). [DOI] [PubMed] [Google Scholar]
- [39].Park IH, Sohn JH, Kim SB, Lee KS, Chung JS, Lee SH, Kim TY, Jung KH, Cho EK, Kim YS, Song HS, Seo JH, Ryoo HM, Lee SA, Yoon SY, Kim CS, Kim YT, Kim SY, Jin MR, Ro J, An open-label, randomized, parallel, phase III trial evaluating the efficacy and safety of polymeric micelle-formulated paclitaxel compared to conventional cremophor EL-based paclitaxel for recurrent or metastatic HER2-negative breast cancer, Cancer Res Treat 49 (3) (2017) 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Saeki HMT, Ro J, Lin Y-C, Fujiwara Y, Nagai S, Lee KS, Watanabe J, Ohtani S, Kim SB, Kuroi K, Tsugawa K, Tokuda Y, Iwata H, Park YH, Yang Y, Nambu Y, A global phase III clinical study comparing NK105 and paclitaxel in metastatic or recurrent breast cancer patients, Ann. Oncol 28 (5) (2017) v74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ibrahim NK, Samuels B, Page R, Doval D, Patel KM, Rao SC, Nair MK, Bhar P, Desai N, Hortobagyi GN, Multicenter phase II trial of ABI-007, an albumin-bound paclitaxel, in women with metastatic breast cancer, J. Clin. Oncol 23 (25) (2005) 6019–6026. [DOI] [PubMed] [Google Scholar]
- [42].Nabholtz JM, Gelmon K, Bontenbal M, Spielmann M, Catimel G, Conte P, Klaassen U, Namer M, Bonneterre J, Fumoleau P, Winograd B, Multicenter, randomized comparative study of two doses of paclitaxel in patients with metastatic breast cancer, J. din. Oncol 14 (6) (1996) 1858–1867. [DOI] [PubMed] [Google Scholar]
- [43].Postma TJ, Vermorken JB, Liefting AJ, Pinedo HM, Heimans JJ, Paditaxel-induced neuropathy, Ann. Oncol 6 (5) (1995) 489–494. [DOI] [PubMed] [Google Scholar]
- [44].Flatters SJL, Bennett GJ, Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy, Pain 109 (1–2) (2004) 150–161. [DOI] [PubMed] [Google Scholar]
- [45].Matsumura Y, Maeda H, A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs, Canc. Res 46 (12 Pt 1) (1986) 6387–6392. [PubMed] [Google Scholar]
- [47].Stylianopoulos T, EPR-effect: utilizing size-dependent nanoparticle delivery to solid tumors, Ther. Deliv 4 (4) (2013) 421–423. [DOI] [PubMed] [Google Scholar]
- [48].Shi J, Kantoff PW, Wooster R, Farokhzad OC, Cancer nanomedicine: progress, challenges and opportunities, Nat. Rev. Cane 17 (1) (2017) 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tang L, Yang XJ, Yin Q, Cai KM, Wang H, Chaudhury I, Yao C, Zhou Q, Kwon M, Hartman JA, Dobrucki IT, Dobrudd LW, Borst LB, Lezmig S, Helferich WG, Ferguson AL, Fan TM, Cheng JJ, Investigating the optimal size of anticancer nanomedicine, Proc. Natl. Acad. Sci. U. S. A 111 (43) (2014) 15344–15349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Guy CT, Cardiff RD, Muller WJ, Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease, Mol. Cell Biol 12 (3) (1992) 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schorzman AN, Lucas AT, Kagel JR, Zamboni WC, Methods and study designs for characterizing the pharmacokinetics and pharmacodynamics of carrier-mediated agents, Methods Mol. Biol 1831 (2018) 201–228. [DOI] [PubMed] [Google Scholar]
- [52].Skoczen S, McNeil SE, Stern ST, Stable isotope method to measure drug release from nanomedicines, J. Contr. Release 220 (Pt A) (2015) 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Guerin MV, Finisguerra V, Van den Eynde BJ, Bercovid N, Trautmann A, Preclinical murine tumor models: a structural and functional perspective, Elife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K, Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size, Nat. Nanotechnol 6 (12) (2011) 815–823. [DOI] [PubMed] [Google Scholar]
- [55].Matsumoto Y, Nichols JW, Toh K, Nomoto T, Cabral H, Miura Y, Christie RJ, Yamada N, Ogura T, Kano MR, Matsumura Y, Nishiyama N, Yamasoba T, Bae YH, Kataoka K, Vascular bursts enhance permeability of tumour blood vessels and improve nanoparticle delivery, Nat. Nanotechnol 11 (6) (2016) 533–538. [DOI] [PubMed] [Google Scholar]
- [56].Batist G, Ramakrishnan G, Rao CS, Chandrasekharan A, Gutheil J, Guthrie T, Shah P, Khojasteh A, Nair MK, Hoelzer K, Tkaczuk K, Park YC, Lee LW, Reduced cardiotoxicity and preserved antitumor efficacy of liposome-encapsulated doxorubicin and cydophosphamide compared with conventional doxorubicin and cydophosphamide in a randomized, multicenter trial of metastatic breast cancer, J. Clin. Oncol 19 (5) (2001) 1444–1454. [DOI] [PubMed] [Google Scholar]
- [57].Chan S, Davidson N, Juozaityte E, Erdkamp F, Pluzanska A, Azamia N, Lee LW, Phase III trial of liposomal doxorubicin and cydophosphamide compared with epirubidn and cydophosphamide as first-line therapy for metastatic breast cancer, Ann. Oncol 15 (10) (2004) 1527–1534. [DOI] [PubMed] [Google Scholar]
- [58].Yuan H, Guo H, Luan X, He M, Li F, Burnett J, Truchan N, Sun D, Albumin nanoparticle of paclitaxel (Abraxane) decreases while Taxol increases breast cancer stem cells in treatment of triple negative breast cancer, Mol. Pharm 17 (7) (2020) 2275–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Cullis J, Siolas D, Avanzi A, Barui S, Maitra A, Bar-Sagi D, Macropinocytosis of nab-paclitaxel drives macrophage activation in pancreatic cancer, Canc. Immunol. Res 5 (3) (2017) 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wanderley CW, Colon DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA, Pereira JA, Silva CM, Silva CR, Silva RL, Speck-Hemandez CA, Mota JM, Alves-Filho JC, Lima-Junior RC, Cunha TM, Cunha FQ, Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner, Canc. Res 78 (20) (2018) 5891–5900. [DOI] [PubMed] [Google Scholar]
- [61].Yamaguchi T, Fushida S, Yamamoto Y, Tsukada T, Kinoshita J, Oyama K, Miyashita T, Tajima H, Ninomiya I, Munesue S, Harashima A, Harada S, Yamamoto H, Ohta T, Low-dose paclitaxel suppresses the induction of M2 macrophages in gastric cancer, Oncol. Rep 37 (6) (2017) 3341–3350. [DOI] [PubMed] [Google Scholar]
- [62].Scripture CD, Figg WD, Sparreboom A, Peripheral neuropathy induced by paclitaxel: recent insights and future perspectives, Curr. Neuropharmacol 4 (2) (2006) 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Li F, Yuan H, Zhang H, He M, Liao J, Chen N, Li Y, Zhou S, Palmisano M, Yu A, Pai M, Sun D, Neonatal Fc receptor (FcRn) enhances tissue distribution and prevents excretion of nab-paclitaxel, Mol. Pharm 16 (6) (2019) 2385–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Li F, Zhang H, He M, Liao J, Chen N, Li Y, Zhou S, Palmisano M, Yu A, Pai MP, Yuan H, Sun D, Different nanoformulations alter the tissue distribution of paclitaxel, which aligns with reported distinct efficacy and safety profiles, Mol. Pharm 15 (10) (2018) 4505–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tsoi KM, MacParland SA, Ma XZ, Spetzler VN, Echeverri J, Ouyang B, Fadel SM, Sykes EA, Goldaracena N, Kaths JM, Conneely JB, Alman BA, Selzner M, Ostrowski MA, Adeyi OA, Zilman A, McGilvray ID, Chan WC, Mechanism of hard-nanomaterial clearance by the liver, Nat. Mater 15 (11) (2016) 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Tavares AJ, Poon W, Zhang YN, Dai Q, Besla R, Ding D, Ouyang B, Ii A, Chen J, Zheng G, Robbins C, Chan WCW, Effect of removing Kupffer cells on nanoparticle tumor delivery, Proc. Natl. Acad. Sci. U. S. A 114 (51) (2017) E10871–E10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Arrieta O, Medina LA, Estrada-Lobato E, Ramirez-Tirado LA, Mendoza-Garda VO, de la Garza-Salazar J, High liposomal doxorubicin tumour tissue distribution, as determined by radiopharmaceutical labelling with (99m)Tc-LD, is associated with the response and survival of patients with unresectable pleural mesothelioma treated with a combination of liposomal doxorubicin and cisplatin, Canc. Chemother. Pharmacol 74 (1) (2014) 211–215. [DOI] [PubMed] [Google Scholar]
- [69].Koukourakis MI, Koukouraki S, Giatromanolaki A, Archimandritis SC, Skarlatos J, Beroukas K, Bizakis JG, Retalis G, Karkavitsas N, Helidonis ES, Liposomal doxorubicin and conventionally fractionated radiotherapy in the treatment of locally advanced non-small-cell lung cancer and head and neck cancer, J. din. Oncol 17 (11) (1999) 3512–3521. [DOI] [PubMed] [Google Scholar]
- [70].Koukourakis MI, Koukouraki S, Giatromanolaki A, Kakolyris S, Georgoulias V, Velidaki A, Archimandritis S, Karkavitsas NN, High intratumoral accumulation of stealth liposomal doxorubicin in sarcomas-rationale for combination with radiotherapy, Acta Oncol. 39 (2) (2000) 207–211. [DOI] [PubMed] [Google Scholar]
- [71].Lee H, Shields AF, Siegel BA, Miller KD, Krop I, Ma CX, LoRusso PM, Munster PN, Campbell K, Gaddy DF, Leonard SC, Geretti E, Blocker SJ, Kirpotin DB, Moyo V, Wickham TJ, Hendriks BS, 64)Cu-MM-302 positron emission tomography quantifies variability of enhanced permeability and retention of nanoparticles in relation to treatment response in patients with metastatic breast cancer, Clin. Canc. Res 23 (15) (2017) 4190–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Stewart SS HK, The biodistribution and pharmacokinetics of stealth liposomes in patients with solid tumors, Oncology 11 (1997) 5. [Google Scholar]
- [73].Harrington KJ, Mohammadtaghi S, Uster PS, Glass D, Peters AM, Vile RG, Stewart JS, Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes, Clin. Cane. Res 7 (2) (2001) 243–254. [PubMed] [Google Scholar]
- [74].Symon Z, Peyser A, Tzemach D, Lyass O, Sucher E, Shezen E, Gabizon A, Selective delivery of doxorubicin to patients with breast carcinoma metastases by stealth liposomes, Cancer 86 (1) (1999) 72–78. [PubMed] [Google Scholar]
- [75].Gabizon A, Catane R, Uziely B, Kaufman B, Safra T, Cohen R, Martin F, Huang A, Barenholz Y, Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes, Canc. Res 54 (4) (1994) 987–992. [PubMed] [Google Scholar]
- [76].Donald FJM, Northfelt W, Working Peter, Voiberding Paul A., Russell Julie, Newman Mary, Amantea Michael A., Kaplan Lawrence D., Doxorubicin encapsulated in liposome containing surface-bound polyethylene glycol: pharmacokinetics, tumor localization, and safety in patients with AIDS-related Kaposi’s sarcoma, J. CIIn. Pharmacol 36 (1996) 9. [DOI] [PubMed] [Google Scholar]
- [77].Huang SK, Martin FJ, Jay G, Vogel J, Papahadjopoulos D, Friend DS, Extravasation and transcytosis of liposomes in Kaposi’s sarcoma-like dermal lesions of transgenic mice bearing the HIV tat gene, Am. J. Pathol 143 (1) (1993) 10–14. [PMC free article] [PubMed] [Google Scholar]
- [78].Dewhirst MW, Secomb TW, Transport of drugs from blood vessels to tumour tissue, Nat. Rev. Cane 17 (12) (2017) 738–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC, Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology, Cane. Res 73 (8) (2013) 2412–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ioannidis JPA, Kim BYS, Trounson A, How to design preclinical studies in nanomedicine and cell therapy to maximize the prospects of clinical translation, Nat. Biomed.Eng 2 (11) (2018) 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].De Souza R, Spence T, Huang H, Allen C, Preclinical imaging and translational animal models of cancer for accelerated clinical implementation of nanotechnologies and macromolecular agents, J. Contr. Release 219 (2015) 313–330. [DOI] [PubMed] [Google Scholar]
- [82].Maeda H, Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity, Adv. Drug Deliv. Rev 91 (2015) 3–6. [DOI] [PubMed] [Google Scholar]
- [83].Ojha T, Pathak V, Shi Y, Hennink WE, Moonen CTW, Storm G, Kiessling F, Lammers T, Pharmacological and physical vessel modulation strategies to improve EPR-mediated drug targeting to tumors, Adv. Drug Deliv. Rev 119 (2017) 44–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ramanathan RK, Korn RL, Raghunand N, Sachdev JC, Newbold RG, Jameson G, Fetterly GJ, Prey J, Klinz SG, Kim J, Cain J, Hendriks BS, Drummond DC, Bayever E, Fitzgerald JB, Correlation between ferumoxytol uptake in tumor lesions by MRI and response to nanoliposomal irinotecan in patients with advanced solid tumors: a pilot study, Clin. Canc. Res 23 (14) (2017) 3638–3648. [DOI] [PubMed] [Google Scholar]
- [85].Fang J, Islam W, Maeda H, Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers, Adv. Drug Deliv. Rev 157 (2020) 142–160. [DOI] [PubMed] [Google Scholar]
- [86].Nagamitsu A, Greish K, Maeda H, Elevating blood pressure as a strategy to increase tumor-targeted delivery of macromolecular drug SMANCS: cases of advanced solid tumors, Jpn. J. Clin. Oncol 39 (11) (2009) 756–766. [DOI] [PubMed] [Google Scholar]
- [87].Cesarman E, Damania B, Krown SE, Martin J, Bower M, Whitby D, Kaposi sarcoma, Nat. Rev. Dis. Prim 5 (1) (2019) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Svenson S, What nanomedicine in the clinic right now really forms nanoparticles? Wiley Interdiscip Rev. Nanomed. Nanobiotechnol 6 (2) (2014) 125–135. [DOI] [PubMed] [Google Scholar]
- [89].Cho H, Gao J, Kwon GS, PEG-b-PLA micelles and PLGA-b-PEG-b-PLGA sol-gels for drug delivery, J. Contr. Release 240 (2016) 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kessel D, Properties of cremophor EL micelles probed by fluorescence, Photochem. Photobiol 56 (4) (1992) 447–451. [DOI] [PubMed] [Google Scholar]
- [91].Fan Z, Chen C, Pang X, Yu Z, Qi Y, Chen X, Liang H, Fang X, Sha X, Adding vitamin E-TPGS to the formulation of Genexol-PM: specially mixed micelles improve drug-loading ability and cytotoxicity against multidrug-resistant tumors significantly, PloS One 10 (4) (2015), e0120129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].N.C. Laboratory, Novel Method to Determine Bioequivalence of Nanomedicines, U. S. Food & Drug Administration; (Inter-Agency Award 224-16-3001S). [Google Scholar]
- [93].Chen N, Brachmann C, Liu X, Pierce DW, Dey J, Kerwin WS, Li Y, Zhou S, Hou S, Carleton M, Klinghoffer RA, Palmisano M, Chopra R, Albumin-bound nanoparticle (nab) paclitaxel exhibits enhanced paclitaxel tissue distribution and tumor penetration, Canc. Chemother. Pharmacol 76 (4) (2015) 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Borga O, Lilienberg E, Bjermo H, Hansson F, Heldring N, Dediu R, Pharmacokinetics of total and unbound paclitaxel after administration of paclitaxel micellar or nab-paclitaxel: an open, randomized, cross-over, explorative study in breast cancer patients, Adv. Ther 36 (10) (2019) 2825–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sparreboom A, Scripture CD, Trieu V, Williams PJ, De T, Yang A, Beals B, Figg WD, Hawkins M, Desai N, Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol), Clin. Cane. Res 11 (11) (2005) 4136–4143. [DOI] [PubMed] [Google Scholar]
- [96].Socinski MA, Bondarenko IN, Karaseva NA, Makhson A, Vynnichenko I, Okamoto I, Hon JK, Hirsh V, Bhar P, Iglesias J, Results of a randomized, phase III trial of nab-paclitaxel (nab-P) and carboplatin (C) compared with cremophor-based paclitaxel (P) and carboplatin as first-line therapy in advanced non-small cell lung cancer (NSCLC), J. Clin. Oncol 28 (18) (2010). [DOI] [PubMed] [Google Scholar]
- [97].A.B. Oasmia Pharmaceutical, Oasmia pharmaceutical announces positive overall survival results from phase III study of paclical/apealea for treatment of ovarian cancer. http://www.oasmia.com/news.asp?c_id=413.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw/processed data required to reproduce these findings can be shared upon request. The complete raw datasets cannot be shared at this time as the data also forms part of an ongoing study.