

Abstract
Introduction: Plasma glial fibrillary acidic protein (GFAP) may be associated with amyloid burden, neurodegeneration, and stroke but its specificity for Alzheimer's disease (AD) in the general population is unclear. We examined associations of plasma GFAP with amyloid and tau positron emission tomography (PET), cortical thickness, white matter hyperintensities (WMH), and cerebral microbleeds (CMBs). Methods: The study included 200 individuals from the Mayo Clinic Study of Aging who underwent amyloid and tau PET and magnetic resonance imaging and had plasma GFAP concurrently assayed; multiple linear regression and hurdle model analyses were used to investigate associations controlling for age and sex. Results: GFAP was associated with amyloid and tau PET in multivariable models. After adjusting for amyloid, the association with tau PET was no longer significant. GFAP was associated with cortical thickness, WMH, and lobar CMBs only among those who were amyloid‐positive. Discussion: This cross‐sectional analysis demonstrates the utility of GFAP as a plasma biomarker for AD‐related pathologies.
Keywords: Alzheimer's disease, amyloid pathology astrogliosis, blood‐based biomarkers, plasma glial fibrillary acidic protein
1. BACKGROUND
Glial fibrillary acidic protein (GFAP) is an astrocytic cytoskeletal protein. Elevated plasma GFAP levels may be a consequence of abnormal astrocytic functional remodeling due to neuronal damage, also referred to as “reactive astrogliosis.” 1 Several animal and cell studies have shown that reactive astrocytes penetrate and surround amyloid plaques, possibly contributing to the amyloid‐pathological process. 2 , 3
Recent studies have shown that plasma GFAP may be associated with amyloid burden and cognitive impairment. 4 , 5 , 6 Elevated plasma and serum levels of GFAP are found in normal older adults at risk of Alzheimer's disease (AD) dementia, as estimated by brain amyloid load, 7 , 8 , 9 , 10 and correlate with worse cognitive function in AD dementia. 5 One study also reported an association between plasma GFAP and tau positron emission tomography (PET); however, the relationship was attenuated and no longer significant after adjustment for amyloid PET. 9
In addition to the association between GFAP and amyloid PET, GFAP has been associated with cerebrovascular disease. For example, studies have reported that plasma GFAP levels distinguished intracerebral hemorrhage from ischemic stroke, 11 and correlated with white matter hyperintensities (WMH) burden. 5 Fewer studies have examined associations between GFAP and other imaging markers of neurodegeneration such as cortical thickness. One study reported an association between increased plasma GFAP levels and lower parietal and temporal cortical thickness. 12
The first objective of our study was to comprehensively examine the associations between plasma GFAP and neuroimaging measures of AD (amyloid and tau PET), neurodegeneration (cortical thickness), cerebrovascular pathology (white matter hyperintensities [WMH] and cerebral microbleeds [CMBs]), and global cognition. Second, we stratified by amyloid PET status to determine whether any associations were specific to those with elevated brain amyloid.
2. METHODS
2.1. Study participants
The study included 200 individuals enrolled in the Mayo Clinic Study of Aging (MCSA), a population‐based study examining the epidemiology of cognitive decline and risk of mild cognitive impairment (MCI) among residents living in Olmsted County, Minnesota. 13 In 2004, the Olmsted County population was enumerated using the Rochester Epidemiology Project medical records‐linkage system. 14 A detailed description of the MCSA study has been described previously. 13
Participants underwent detailed clinical visits including neuropsychological testing, a physician examination, and blood draws every 15 months and a subset participated in neuroimaging. The current analyses included 200 participants without dementia who had concurrent measures of amyloid PET, tau PET, magnetic resonance imaging (MRI; for cortical thickness, WMH, and CMB) and plasma GFAP levels.
The study was approved by Mayo Clinic and Olmsted Medical Center institutional review boards. Written informed consent was obtained from all participants.
HIGHLIGHTS
Plasma glial fibrillary acidic protein (GFAP) is an astrogliosis biomarker.
Imaging measures and plasma GFAP levels were evaluated in a population‐based study.
GFAP levels in persons without dementia are associated with amyloid burden.
In amyloid‐positive individuals GFAP associated with neuroimaging biomarkers.
GFAP is a biomarker for Alzheimer's disease–related pathologies prior to dementia.
RESEARCH IN CONTEXT
Systematic Review: The authors reviewed the literature using traditional (e.g., PubMed) sources, meeting abstracts, and presentations. Several studies evaluated the use of plasma glial fibrillary acidic protein (GFAP) levels as a marker for cerebral amyloid burden and cognitive impairment but have not comprehensively compared the relationships of plasma GFAP to neuroimaging measures of multiple pathologies (Alzheimer's disease [AD], vascular, neurodegeneration).
Interpretation: Our findings show that GFAP levels in persons without dementia are associated with amyloid positron emission tomography burden. Further, plasma GFAP was associated with decreased cortical thickness, increased white matter hyperintensities, and cerebral microbleeds among those who were amyloid positive, indicating that this marker is specific for AD pathogenesis.
Future Directions: Future research is needed in an autopsy‐confirmed cohort to determine the pathological correlates of GFAP. Longitudinal studies of serial GFAP measures and neuroimaging are needed to understand how GFAP changes in relation to the development and progression of AD pathology.
2.2. Cognition function assessments and clinical diagnosis institution
The neuropsychological testing was administered by a trained psychometrist and included nine tests across four domains: memory, language, executive function, and visuospatial skills. A global cognition z‐score was computed as previously described. 13 , 15 Based on means (standard deviations [SDs]) from the MCSA 2004 enrollment cohort that consisted of individuals without dementia (n = 1969), individual test scores were converted to z‐scores. To assess global cognition, a summary score was estimated from the z‐transform of the mean of the four domain z‐scores.
A clinical consensus committee assessed each study participant to determine cognitive diagnoses blind to the diagnosis of the previous study visit. Cognitive performance was compared to age‐adjusted scores using Mayo's Older Americans Normative Studies. 16 The operational definition of MCI was based on clinical judgment including a history from the patient and informant and cognitive performance. Published criteria were used for the diagnosis of MCI: cognitive complaint, cognitive function not normal for age, essentially normal functional activities, no dementia. 17 A final diagnosis was made after considering education, occupation, visual or hearing deficits, and reviewing all other participant information. The diagnosis of dementia was based on Diagnostic and Statistical Manual of Mental Disorders, 4th Edition criteria. 18 Participants who performed in the normal range and did not meet criteria for MCI or dementia were deemed cognitively unimpaired (CU).
2.3. Plasma GFAP measurements
Participants’ blood was collected in clinic after an overnight fast. The blood was centrifuged, aliquoted, and kept at –80°C. GFAP was measured on the Quanterix Simoa HD‐X Analyzer using the Simoa Neurology 4‐Plex E Advantage kit (N4PE, item #103670) per manufacturer's instructions (Quanterix). After thawing and mixing, plasma samples were centrifuged 5 minutes × 10,000 g. Samples were diluted 1:4 using the instrument's onboard dilution protocol and run in duplicate from a single well each on a 96‐well plate. Eight‐point calibration curves and sample measurements were determined on Simoa HD‐X Analyzer software using a weighting factor 1/Y2 and a four‐parameter logistic curve fitting algorithm. The Simoa N4PE kit incorporates the Banyan GFAP assay (Banyan Biomarkers, Inc.) and quantifies plasma GFAP using a biotinylated anti‐GFAP mouse monoclonal immunoglobulin (Ig)G antibody clone (capture antibody) and an anti‐GFAP rabbit IgG polyclonal antibody (detection antibody). 19 , 20 The assay detects intact GFAP in addition to GFAP breakdown products (50 to 38 kDa). 20 Two levels of quality control material were included, flanking the samples at the front and end of each batch. Internal studies of inter‐assay imprecision at approximate concentrations of 235 and 5055 pg/mL were 7.2% and 8.3%, respectively.
2.4. Imaging
2.4.1. Structural MRI
Structural MRI was acquired using standardized magnetization prepared–rapid gradient echo (MPRAGE) sequences on 3T GE scanners (GE Medical Systems). FreeSurfer v 5.3 (version 5.3) was run on the MPRAGE scans. A temporal meta region of interest (ROI) using a cortical thickness composite of entorhinal, fusiform, inferior temporal, and middle temporal ROIs was included as our neurodegeneration marker because it has been widely shown to be impacted by aging and AD. 21 , 22
2.4.2. White matter hyperintensities
A trained imaging analyst segmented and edited WMH on two‐dimensional fluid‐attenuated inversion recovery (FLAIR) images by using a semi‐automated method, as explained previously. 23 , 24 In short, FLAIR images were used to identify possible WMH voxels using a clustering method: a sphere is placed around each identified voxel to make it visible on 3D rendering and overlapping spheres can merge, forming clusters. The FLAIR images were then aligned with the T1‐weighted image using SPM5 segmentations. Voxels associated with infarcts were removed and not included in the WMH measurement. Areas suspected as WMH masks were also removed if they occurred outside of white matter area, if they consisted of a single isolated voxel, or if they appeared without parallel abnormality seen on FLAIR. A normalization and smoothing process was applied. WMH volume is presented as the percentage of total intracranial volume (TIV).
2.4.3. Cerebral microbleeds
CMBs were recognized and computed as previously described 24 in agreement to consensus criteria 25 as homogeneous hypointense lesions in the gray or white matter, which are distinct from vessel flow voids on T2* gradient recalled echo (GRE) images. In addition to training image analysts for microbleed identification, a vascular neurologist experienced with interpreting T2* GRE images was consulted for confirmation.
2.4.4. PET imaging
PET imaging was acquired using previously described methods. 23 Amyloid PET imaging was performed out with 11C‐PiB (Pittsburgh compound B); tau PET was performed with AV‐1451, synthesized on site with precursor supplied by Avid Radiopharmaceuticals. Late‐uptake amyloid PET images were obtained 40 to 60 minutes and tau PET 80 to 100 minutes after injection. Computed tomography was used for attenuation correction. Amyloid PET and tau PET were analyzed using our in‐house fully automated image‐processing pipeline, in which image voxel values were extracted from automatically labeled regions of interest propagated from the MCALT template. 26 The standardized uptake value ratio (SUVR) values of amyloid PET and tau PET were determined by normalizing target regions of interest to the cerebellar crus gray matter. 21 A cut‐off of more than 1.48 SUVR was used to categorize participants as amyloid positive (A+). 21
Based on a voxel number‐weighted average of the median tau PET uptake in previously published regions of interest, a tau PET meta‐ROI was formed 21 and normalized to the cerebellar crus gray median. The tau PET meta‐ROI was composed of the entorhinal, amygdala, parahippocampal, fusiform, inferior temporal, and middle temporal regions of interest. This meta‐ROI was chosen because it has previously been used in CU individuals and increases with age as expected. 27 PET images were quantified using MRI scans of the participants. PET data were not corrected for partial volume.
2.5. Covariates
Demographic information included self‐reported age, sex, and years of education. Apolipoprotein E (APOE) ɛ4 genotyping was performed from a blood sample drawn at clinic examination.
2.6. Statistics
Spearman's rho was used to examine the correlations between GFAP and amyloid PET, tau PET, cortical thickness, WMH volume, and global z‐scores. Multivariable linear regression models were conducted to estimate the relationship between GFAP and each continuous outcome variable: temporal meta ROI, amyloid PET, tau PET, WMH, and global z‐score. GFAP was scaled by dividing by 100 in all models to ease interpretation of the coefficients. Each model adjusted for age and sex. Models examining cognition additionally adjusted for education and number of prior neuropsychological testing exposures to account for practice effects. Additional sensitivity analyses including APOE as a covariate were conducted. Regression analysis examining the association between plasma GFAP and tau PET was repeated after including amyloid PET as a covariate. We also fit multivariable models stratified by elevated brain amyloid (A+) for each outcome.
Because only 25% of the cohort had evident microbleeds, we chose to fit hurdle models to ascertain the relationship between GFAP and CMBs, using both the whole cohort and stratifying by amyloid status. The hurdle models consisted of two components: (1) a logistic regression model for predicting no CMB versus having at least one CMB, which constitutes the hurdle; and (2) a truncated negative binomial model for predicting number of CMB among those with CMB. For all logistic regression models, we computed odds ratios (ORs), and for the negative binomial portion of the hurdle models we computed incidence rate ratios (IRRs). The hurdle models adjusted for age and sex. In additional sensitivity analyses we included APOE as a covariate.
3. RESULTS
3.1. Patient characteristics
Demographics and clinical characteristics are summarized in Table 1. In total 200 participants were included in the study, of which 177 were CU and 23 were MCI. The mean age was 78 years, 50% were male, and 30% were APOE Ɛ4 carriers.
TABLE 1.
Participant demographics
| Overall | Amyloid – | Amyloid + | |
|---|---|---|---|
| Participants, n | 200 | 99 | 101 |
| Male, n (%) | 101 (50%) | 57 (58%) | 44 (44%) |
| Age, years | 78 (9) | 75 (9) | 81 (8) |
| Race | |||
| Asian | 1 (0.5%) | 1 (1%) | 0 (0%) |
| Black | 1 (0.5%) | 1 (1%) | 0 (0%) |
| White | 198 (99%) | 97 (98%) | 101 (100%) |
| GFAP (pg/mL) | 166 (81) | 134 (65) | 197 (83) |
| APOE Ɛ4 carriers, n (%) | 60 (30%) | 15 (15%) | 45 (45%) |
| Education, years | 14.36 (2.65) | 14.75 (2.65) | 13.98 (2.60) |
| MCI, n (%) | 23 (12%) | 10 (10%) | 13 (13%) |
| Global z‐score | 0.05 (1.10) | 0.34 (0.94) | −0.23 (1.18) |
| Temporal meta ROI | 2.80 (0.18) | 2.85 (0.16) | 2.75 (0.18) |
| Amyloid PET SUVR | 1.66 (0.45) | 1.36 (0.07) | 1.95 (0.47) |
| Tau PET SUVR | 1.22 (0.11) | 1.18 (0.09) | 1.25 (0.12) |
| White matter hyperintensities | 0.01 (0.01) | 0.01 (0.01) | 0.02 (0.02) |
| Cerebral microbleeds, n (%) | 50 (25%) | 17 (17%) | 33 (33%) |
Abbreviations: APOE, apolipoprotein E; GFAP, glial fibrillary acidic protein; MCI, mild cognitive impairment; PET, positron emission tomography; ROI, region of interest; SD, standard deviation; SUVR, standardized uptake value ratio.
Notes: Mean (SD) listed for the continuous variables and count (%) for the categorical variables. Amyloid + includes abnormal amyloid PET (>1.48 SUVR). White matter hyperintensities volume is expressed as a percentage of the total intercranial volume.
3.2. Univariate associations among GFAP, neuroimaging, and cognition
Plasma GFAP levels positively correlated with age (Spearman's rho = 0.653, P < .001) but did not differ by sex. In a univariate analysis shown in Table 2, significant Spearman correlations were found between higher GFAP levels and greater amyloid burden (rho = 0.438, P < .001), lower cortical thickness (rho = –0.426, P = .001), greater tau deposition (rho = 0.207, P = .003), greater WMH volume (rho = 0.493, P < .001), and lower cognition global z‐scores (rho = –0.195, P = .009).
TABLE 2.
Spearman correlations among plasma GFAP, neuroimaging, and global cognition
| Measure | Coefficient r | P value |
|---|---|---|
| Amyloid PET | 0.438 | <.001 |
| Tau PET | 0.207 | .003 |
| Temporal meta‐ROI cortical thickness | −0.426 | <.001 |
| WMH volume | 0.492 | <.001 |
| Global z‐score | −0.195 | .009 |
Notes: Amyloid PET, Tau PET, and WMH scores are log‐transformed. WMH volume is expressed as a percentage of the total intercranial volume.
Abbreviations: GFAP, glial fibrillary acidic protein; PET, positron emission tomography; ROI, region of interest; WMH, white matter hyperintensities.
3.3. Association between plasma GFAP and amyloid and tau PET, WMH, neurodegeneration, and cognition
Higher plasma GFAP concentrations were associated with greater amyloid PET (β = 0.111, P < .001) and tau PET SUVR (β = 0.027, P = .008) after adjusting for age and sex (Table 3). After adding amyloid to the model and examining the association between plasma GFAP and tau PET, the association was no longer significant (β = 0.01, P = .276). Higher plasma GFAP levels were associated with higher WMH (β = 0.223, P = .008), and an association approaching but not reaching significance was observed with lower temporal meta ROI cortical thickness (β = –0.032, P = .054), after adjusting for age and sex. There was no association between GFAP and global z‐score in multivariable models (β = –0.017, P = .877) and there were no associations found between plasma GFAP and specific cognitive domains (memory, attention, language, or visuo‐spatial scores) in additional analyses. Other additional analyses with APOE added as a covariate did not change any of the results.
TABLE 3.
Associations among plasma GFAP, neuroimaging measures, and global cognition using multivariable regression models
| Measure | Coefficient | Partial R2 | SE | P value |
|---|---|---|---|---|
| Amyloid PET SUVR | 0.111 | 0.112 | 0.023 | <.001 |
| Tau PET SUVR | 0.027 | 0.036 | 0.010 | .008 |
| Temporal meta ROI cortical thickness | −0.032 | 0.019 | 0.017 | .054 |
| WMH volume | 0.223 | 0.036 | 0.082 | .008 |
| Global z‐score | −0.017 | 0.000 | 0.110 | .877 |
Notes: P values adjusted for age and sex. For Global z‐score, P‐value was additionally adjusted for education and number of prior neuropsychological test exposures. Amyloid and tau PET scores are log‐transformed. Coefficients are reflected for 100 pg/mL change in GFAP. WMH volume is expressed as a percentage of the total intercranial volume.
Abbreviations: GFAP, glial fibrillary acidic protein; PET, positron emission tomography; ROI, region of interest; SUVR, standardized uptake value ratio; WMH, white matter hyperintensities.
3.4. Association between GFAP and CMBs
The hurdle models revealed that increasing GFAP levels were associated with an increased risk of having at least one CMB (OR = 1.72, P = .036) and an increased risk of having at least one lobar microbleed (OR = 1.82, P = .028). Among those with a CMB, there was no association between GFAP and the number of CMBs (IRR = 0.67, P = .32). The number of deep CMBs was insufficient to model. Additionally, adjusting for APOE did not change the results.
3.5. Amyloid‐stratified analysis
To determine whether the associations between GFAP and the imaging biomarkers were driven by elevated brain amyloid, we repeated the above analyses stratified by amyloid PET status (Table 4, Figure 1). The amyloid‐positive group contained 101 individuals (88 CU, 13 MCI), and the amyloid‐negative group contained 99 individuals (89 CU, 10 MCI). In the amyloid‐positive group, GFAP was associated with higher amyloid (β = 0.094, P = .002) and tau PET (β = 0.032, P = .016), lower temporal meta ROI (β = –0.047, P = .049), and higher WMH (β = 0.221, P = .04). In contrast, there were no significant associations between plasma GFAP and any of the outcomes in the amyloid‐negative group.
TABLE 4.
Amyloid stratified regression for cortical thickness, amyloid, tau, neurodegeneration, temporal cortical thickness, white matter hyperintensities, global cognition z‐score
| Amyloid + (n = 101) | Amyloid – (n = 99) | |||||
|---|---|---|---|---|---|---|
| Measures | Coefficient | P value | Partial R2 | Coefficient | P value | Partial R2 |
| Tau PET | 0.032 | .016 | 0.059 | −0.007 | .68 | 0.002 |
| Temporal meta‐ROI cortical thickness | −0.047 | .049 | 0.039 | 0.008 | .756 | 0.001 |
| WMH volume | 0.221 | .04 | 0.043 | 0.049 | .731 | 0.001 |
| Global Z‐score | 0.058 | .728 | 0.001 | 0.081 | .625 | 0.003 |
Notes: Amyloid and tau PET scores are log‐transformed. Coefficients are reflected for 100 pg/mL change in GFAP. Amyloid + includes abnormal amyloid PET (>1.48 SUVR). WMH volume is expressed as a percentage of the total intercranial volume.
Abbreviations: GFAP, glial fibrillary acidic protein; PET, positron emission tomography; ROI, region of interest; SUVR, standardized uptake value ratio; WMH, white matter hyperintensities.
FIGURE 1.
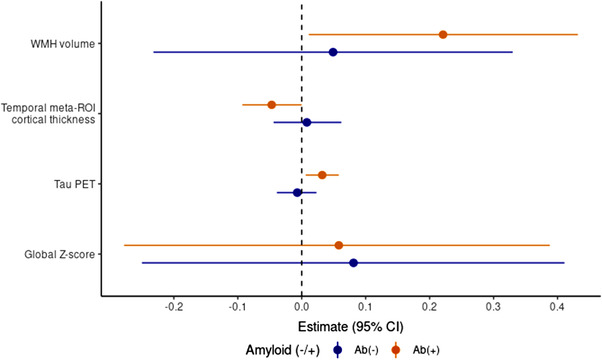
Associations between plasma glial fibrillary acidic protein (GFAP) levels and cognitive and neuroimaging outcomes. Relationships between plasma GFAP levels and cognition and neuroimaging measures stratified by amyloid beta (Ab) status (elevated Ab PET, Ab[+]; non‐elevated Ab PET, Ab[–]). Amyloid and tau PET scores are log‐transformed. Coefficients are reflected for 100 pg/mL change in GFAP. WMH volume is expressed as a percentage of the total intercranial volume. CI, confidence interval; PET, positron emission tomography; ROI, region of interest; WMH, white matter hyperintensity
There were 33 participants with a CMB among the amyloid‐positive group and 17 with a CMB among the amyloid‐negative group. In the amyloid‐positive group higher GFAP levels were associated with having at least one microbleed (OR = 2.05, P = .04) or a lobar microbleed (OR = 2.35, P = .02). Among those with a CMB, there was again no association between GFAP and the number of CMBs (IRR = 0.80, P = .63). In the amyloid‐negative group, there were too few outcomes to conduct the hurdle model.
Associations between plasma GFAP and global z‐score did not reach significance within either the amyloid‐positive or negative groups.
4. DISCUSSION
This study cross‐sectionally examined whether GFAP was associated with neuroimaging measures of AD pathology, cerebrovascular pathology, neurodegeneration, and cognition. Higher plasma GFAP levels were associated with elevated amyloid and tau PET, greater WMH, the presence of CMB, and decreased cortical thickness. Notably, the association between plasma GFAP and these neuroimaging measures was only found among those with elevated brain amyloid, suggesting the specificity of GFAP to amyloid pathology in older adults.
GFAP plays an essential role in cellular processes in astrocytes, including vesicle trafficking, neuron–astrocyte interactions, blood–brain barrier integrity, and protection of neurons from injury. 28 Elevated GFAP is thought to represent abnormal reactive astrogliosis in spatial association with amyloid plaques and neuronal damage 29 and may be viewed as an early sign of AD pathology. 7 Other biomarkers of reactive astrogliosis, such as 11C‐deprenyl PET, which measures astrocyte‐located monoamine oxidase binding, show reactive astrocytosis as an early AD phenomenon associated with cellular tissue loss in parahippocampal regions. 30 , 31 The strong association shown in our study between plasma GFAP levels and amyloid burden as measured by PET supports the link between the processes of reactive astrogliosis and amyloid deposition. Our results are consistent with previous studies showing associations between plasma GFAP and brain amyloid burden among persons with dementia, 5 , 32 MCI or subjective cognitive complaints, 4 , 33 and among CU participants. 7 , 8 , 9 A recent meta‐analysis of the associations between AD and astrocyte biomarkers, including blood GFAP, found that the association holds only when cohorts of early‐onset AD are excluded; however, all but one of the studies used enzyme‐linked immunosorbent assay detection methods instead of single‐molecule array. 34 Research by Pereira et al., 9 which included both CU and impaired participants, found that plasma GFAP levels were lowest in cognitively impaired amyloid‐negative patients, and highest in those who were cognitively impaired and amyloid‐positive, further supporting the relationship between GFAP and brain amyloid. In another study, plasma GFAP levels were higher in AD dementia patients compared to some neurodegenerative diseases (behavioral variant frontotemporal dementia, Parkinson's disease [PD]), but not compared to Lewy body dementias (LBD; dementia with Lewy bodies and PD dementia). 32 The higher levels of plasma GFAP in LBD but not PD has been attributed to increased co‐existing AD pathology including cerebral amyloid angiopathy observed in LBD but less commonly PD.
Reactive astrogliosis precedes the clinical symptoms of AD and is considered a part of the pathological cascade driving the degenerative process. Although astrocytes respond to both neurofibrillary tangles and plaques of AD, 28 the association between astrogliosis and tau tangles has been less investigated. Similar to a previous study, 9 we found an association between higher plasma GFAP and higher tau PET SUVR but the association was attenuated and no longer significant after adjustment for amyloid PET. These results further indicate that GFAP is more closely linked to amyloid pathology than tau pathology.
Higher plasma GFAP levels have also been associated with lower brain volume, specifically in parietal and temporal subregions. 12 Similarly, we observed a trend between plasma GFAP and lower cortical thickness in a temporal region. Again, after stratifying by amyloid PET status, the association only remained among those with elevated amyloid PET, further suggesting specificity to amyloid pathology. It is generally accepted that elevated amyloid deposition and neurodegeneration (quantified as cortical thinning) are associated, with amyloid beta deposition acting as an accelerator of cortical thinning. 35 , 36 Additional analysis was conducted to investigate the association between plasma GFAP levels and neurodegeneration using fluorodeoxyglucose (FDG)‐PET data, but only a subgroup of the participants (n = 131) had FDG scans, and no association was found.
Given that plasma GFAP levels may distinguish intracerebral hemorrhage from ischemic stroke 11 and correlate with WMH burden, 5 we sought to further examine the relationship between plasma GFAP and cerebrovascular pathology. Similar to the previous study, we found associations between higher plasma GFAP and greater WMH volume as well as the presence of CMBs. Again, after stratification by amyloid status, these associations only remained among those with elevated brain amyloid. It was previously shown that amyloid load on PET was associated with WMH in a topographically specific manner. 23 Because this WMH pattern is associated with lobar microbleeds, the current results may be due to the presence of cerebral amyloid angiopathy. Another study showed that GFAP concentrations were associated with increased WMH lesion burden in progranulin‐associated frontotemporal dementia. 37 In comparison, in a study which focused on CADASIL patients, a subgroup with vascular pathology rather than neurodegeneration and amyloid deposition, Chen et al. 38 found no association between GFAP levels and severity of WMH, but did demonstrate a correlation with CMBs.
There were no differences in GFAP levels between CU and MCI and there was no association found between GFAP levels and global cognition after controlling for age and sex, an association that was previously shown in AD patients. 5 Given that our participants did not have dementia, it is possible there was not enough of a range of cognitive performance scores to observe an association at this early stage and a larger sample size may be necessary. In addition, this was a cross‐sectional study, so the association between GFAP and cognition may be more discernible longitudinally.
This study offers several strengths, including well‐compiled and comprehensive information from the MCSA, which is a community‐based study. Limitations of the study include its cross‐sectional study design; longitudinal studies are needed to understand how plasma GFAP levels change over time in relation to the development and progression of AD pathology, and to determine temporality and cut points. Neuropathology studies will be necessary to determine the exact pathological processes GFAP is associated with. Finally, the cohort has predominantly European ancestry, so it is not known how broadly the cohort can be generalized to other populations.
CONFLICTS OF INTEREST
Michelle M. Mielke served as a consultant to Brain Protection Company, Biogen, and LabCorp and receives research support from the National Institutes of Health and the Department of Defense. She is a senior associate editor for Alzheimer's and Dementia: The Journal of the Alzheimer's Association. David S. Knopman serves on a Data Safety Monitoring Board for Biogen (fee paid to institution), the DIAN‐TU study (receives personal consulting fees), Agenbio (unpaid), and an endovascular carotid reconstruction study (unpaid). He is an investigator in clinical trials sponsored by Biogen, Lilly Pharmaceuticals, and the Alzheimer's Disease Cooperative Study, and receives research support from the National Institutes of Health and philanthropic funds. Prashanthi Vemuri received speaking fees from Miller Medical Communications, LLC, and receives research support from the National Institutes of Health. Jonathan Graff‐Radford receives NIH funding and serves on the editorial board for Neurology. He has received payment for speaking at the American Academy of Neurology Annual meeting. Clifford R. Jack serves on an independent data monitoring board for Roche, but he receives no personal compensation from any commercial entity. He receives research support from the National Institutes of Health, the GHR Foundation, and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Clinic. Ronald C. Petersen is a consultant for Roche, Inc., Merck, Inc., Biogen, Inc., and Eisai, Inc. He has received payment for serving on a Data Safety Monitoring Board for Genentech, receives royalties from Oxford University Press and UpToDate, and receives research support from the National Institutes of Health. Val J. Lowe consults for Bayer Schering Pharma, Piramal Life Sciences, Life Molecular Imaging, Eisai Inc., AVID Radiopharmaceuticals, and Merck Research and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, and the NIH (NIA, NCI). Nikki H. Stricker served as a consultant to Biogen. She receives research support from the National Institutes of Health and the Kevin Merszei Career Development Award in Neurodegenerative Diseases Research IHO Janet Vittone, MD. Alicia Algeciras‐Schimnich, Michelle R. Campbell, Dror Shir, Timothy G. Lesnick, Scott A. Przybelski, and Ekaterina I. Hofrenning have no conflicts to report.
AUTHOR CONTRIBUTIONS
Dror Shir participated in the design of the study and drafting, revision, and finalization of the manuscript. Jonathan Graff‐Radford, Prashanthi Vemuri, and Michelle M. Mielke devised the project; participated in the conception and design of the study; and the drafting, revision, and finalization of the manuscript. Ekaterina I. Hofrenning, Timothy G. Lesnick, and Scott A. Przybelski analyzed the data and verified the analytical methods. Val J. Lowe, David S. Knopman, Ronald C. Peterson, Clifford R. Jack, and Nikki H. Stricker contributed to the approval of the version of the manuscript to be published. Alicia Algeciras‐Schimnich and Michelle R. Campbell performed the GFAP plasma measurements.
ACKNOWLEDGMENTS
We would like to greatly thank AVID Radiopharmaceuticals, Inc., for their support in supplying AV‐1451 precursor, chemistry production advice and oversight, and FDA regulatory cross‐filing permission and documentation needed for this work. This study was supported by funding from the National Institutes of Health (RF1 AG069052‐01A1, U01 AG006786, R37 AG011378, R01 041851, R01 NS097495, and P30 AG062677) and the GHR Foundation. This study was made possible using the resources of the Rochester Epidemiology Project, which is supported by the National Institute on Aging of the National Institutes of Health under Award Number R01 AG034676. The funding source had no role in study design, collection, analysis and interpretation of data, in the writing of the report, or in the decision to submit the article for publication.
Shir D, Graff‐Radford J, Hofrenning EI, et al. Association of plasma glial fibrillary acidic protein (GFAP) with neuroimaging of Alzheimer's diseaseand vascular pathology. Alzheimer's Dement. 2022;14:e12291. 10.1002/dad2.12291
REFERENCES
- 1. Escartin C, Galea E, Lakatos A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24(3):312‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Frost GR, Li Y‐M. The role of astrocytes in amyloid production and Alzheimer's disease. Open Biol. 2017;7(12):170228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kamphuis W, Middeldorp J, Kooijman L, et al. Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer's disease. Neurobiol Aging. 2014;35(3):492‐510. [DOI] [PubMed] [Google Scholar]
- 4. Cicognola C, Janelidze S, Hertze J, et al. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimer's Res Ther. 2021;13(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elahi FM, Casaletto KB, La Joie R, et al. Plasma biomarkers of astrocytic and neuronal dysfunction in early‐ and late‐onset Alzheimer's disease. Alzheimer's Dement. 2020;16(4):681‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thijssen EH, Verberk IMW, Stoops E, Boxer AL, Teunissen CE. Amyloid, pTau, NfL, and GFAP as biomarkers for Alzheimer's disease. Alzheimer's Dement. 2020;16(S5):38179. [Google Scholar]
- 7. Chatterjee P, Pedrini S, Stoops E, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer's disease. Transl Psychiatry. 2021;11(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karikari TK, Hourregue C, Cognat E, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78(12):1471‐1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid‐β but not tau pathology in Alzheimer's disease. Brain. 2021;144(11):3505‐3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verberk IMW, Laarhuis MB & van den Bosch KA et al. Serum markers glial fibrillary acidic protein and neurofilament light for prognosis and monitoring in cognitively normal older people: a prospective memory clinic‐based cohort study. The Lancet Healthy Longevity 2021;2:E87‐95 [DOI] [PubMed]
- 11. Katsanos AH, Makris K, Stefani D, et al. Plasma glial fibrillary acidic protein in the differential diagnosis of intracerebral hemorrhage. Stroke. 2017;48(9):2586‐2588. [DOI] [PubMed] [Google Scholar]
- 12. Asken BM, VandeVrede L, Rojas JC, et al. Lower white matter volume and worse executive functioning reflected in higher levels of plasma GFAP among older adults with and without cognitive impairment. J Int Neuropsychol Soc. 2021:22:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology. 2008;30(1):58‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. St Sauver JL, Grossardt BR, Yawn BP, et al. Data resource profile: the Rochester Epidemiology Project (REP) medical records‐linkage system. Int J Epidemiol. 2012;41(6):1614‐1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vemuri P, Lesnick TG, Przybelski SA, et al. Association of lifetime intellectual enrichment with cognitive decline in the older population. JAMA Neurol. 2014;71(8):1017‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ivnik RJ, Malec JF, Smith GE, et al. Mayo's older Americans normative studies: updated AVLT norms for ages 56 to 97. Clin Neuropsychol. 1992;6(sup001):83‐104. [Google Scholar]
- 17. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183‐194. [DOI] [PubMed] [Google Scholar]
- 18. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV). 4th ed. American Psychiatric Association; 1994. [Google Scholar]
- 19. Quanterix.com . NEUROLOGY 4‐PLEX E (Aβ40, Aβ42, GFAP*, Nf‐L). Quanterix. https://www.quanterix.com/simoa‐assay‐kits/neurology‐4‐plex‐e‐ab40‐ab42‐gfap‐nf‐l‐new/ [Google Scholar]
- 20. Zoltewicz JS, Scharf D, Yang B, Chawla A, Newsom KJ, Fang L. Characterization of antibodies that detect human GFAP after traumatic brain injury. Biomark Insights. 2012;7:71‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jack CRJ, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement. 2017;13(3):205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwarz CG, Gunter JL, Wiste HJ, et al. A large‐scale comparison of cortical thickness and volume methods for measuring Alzheimer's disease severity. NeuroImage Clin. 2016;11:802‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Graff‐Radford J, Arenaza‐Urquijo EM, Knopman DS, et al. White matter hyperintensities: relationship to amyloid and tau burden. Brain. 2019;142(8):2483‐2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Graff‐Radford J, Botha H, Rabinstein AA, et al. Cerebral microbleeds: prevalence and relationship to amyloid burden. Neurology. 2019;92(3):E253‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a field guide to their detection and interpretation. Lancet Neurol. 2009;8(2):165‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Senjem ML, Gunter JL, Shiung MM, Petersen RC, Jack CR Jr. Comparison of different methodological implementations of voxel‐based morphometry in neurodegenerative disease. Neuroimage. 2005;26(2):600‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jack CRJ, Wiste HJ, Weigand SD, et al. Age‐specific and sex‐specific prevalence of cerebral β‐amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50‐95 years: a cross‐sectional study. Lancet Neurol. 2017;16(6):435‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garwood CJ, Ratcliffe LE, Simpson JE, Heath PR, Ince PG, Wharton SB. Review: astrocytes in Alzheimer's disease and other age‐associated dementias: a supporting player with a central role. Neuropathol Appl Neurobiol. 2017;43(4):281‐298. [DOI] [PubMed] [Google Scholar]
- 29. Colangelo AM, Alberghina L, Papa M. Astrogliosis as a therapeutic target for neurodegenerative diseases. Neurosci Lett. 2014;565:59‐64. [DOI] [PubMed] [Google Scholar]
- 30. Choo IH, Carter SF, Schöll ML, Nordberg A. Astrocytosis measured by 11C‐deprenyl PET correlates with decrease in gray matter density in the parahippocampus of prodromal Alzheimer's patients. Eur J Nucl Med Mol Imaging. 2014;41(11):2120‐2126. [DOI] [PubMed] [Google Scholar]
- 31. Carter SF, Schöll M, Almkvist O, et al. Evidence for astrocytosis in prodromal alzheimer disease provided by 11C‐deuterium‐L‐deprenyl: a multitracer PET paradigm combining 11C‐Pittsburgh compound B and 18F‐FDG. J Nucl Med. 2012;53(1):37‐46. [DOI] [PubMed] [Google Scholar]
- 32. Oeckl P, Halbgebauer S, Anderl‐Straub S, et al. Glial fibrillary acidic protein in serum is increased in Alzheimer's disease and correlates with cognitive impairment. J Alzheimer's Dis. 2019;67(2):481‐488. [DOI] [PubMed] [Google Scholar]
- 33. Verberk IMW, Thijssen E, Koelewijn J, et al. Combination of plasma amyloid beta(1‐42/1‐40)and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimer's Res Ther. 2020;12(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bellaver B, Ferrari‐Souza JP, Uglione da Ros L, et al. Astrocyte biomarkers in Alzheimer disease. Neurology. 2021;96(24):e2944‐55. [DOI] [PubMed] [Google Scholar]
- 35. Doré V, Villemagne VL, Bourgeat P, et al. Cross‐sectional and longitudinal analysis of the relationship between aβ deposition, cortical thickness, and memory in cognitively unimpaired individuals and in Alzheimer disease. JAMA Neurol. 2013;70(7):903‐911. [DOI] [PubMed] [Google Scholar]
- 36. Knopman DS, Lundt ES, Therneau TM, et al. Entorhinal cortex tau, amyloid‐β, cortical thickness and memory performance in non‐demented subjects. Brain. 2019;142(4):1148‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sudre CH, Bocchetta M, Heller C, et al. White matter hyperintensities in progranulin‐associated frontotemporal dementia: a longitudinal GENFI study. NeuroImage Clin. 2019;24(September):102077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen CH, Cheng YW, Chen YF, Tang SC, Jeng JS. Plasma neurofilament light chain and glial fibrillary acidic protein predict stroke in CADASIL. J Neuroinflammation. 2020;17(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]