

Abstract
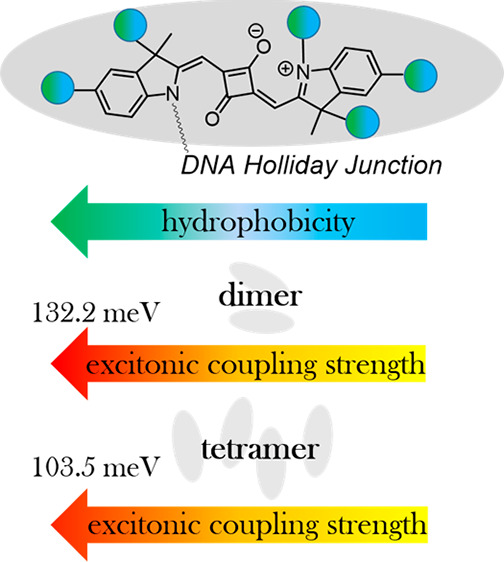
Control over the strength of excitonic coupling in molecular dye aggregates is a substantial factor for the development of technologies such as light harvesting, optoelectronics, and quantum computing. According to the molecular exciton model, the strength of excitonic coupling is inversely proportional to the distance between dyes. Covalent DNA templating was proved to be a versatile tool to control dye spacing on a subnanometer scale. To further expand our ability to control photophysical properties of excitons, here, we investigated the influence of dye hydrophobicity on the strength of excitonic coupling in squaraine aggregates covalently templated by DNA Holliday Junction (DNA HJ). Indolenine squaraines were chosen for their excellent spectral properties, stability, and diversity of chemical modifications. Six squaraines of varying hydrophobicity from highly hydrophobic to highly hydrophilic were assembled in two dimer configurations and a tetramer. In general, the examined squaraines demonstrated a propensity toward face-to-face aggregation behavior observed via steady-state absorption, fluorescence, and circular dichroism spectroscopies. Modeling based on the Kühn–Renger–May approach quantified the strength of excitonic coupling in the squaraine aggregates. The strength of excitonic coupling strongly correlated with squaraine hydrophobic region. Dimer aggregates of dichloroindolenine squaraine were found to exhibit the strongest coupling strength of 132 meV (1065 cm–1). In addition, we identified the sites for dye attachment in the DNA HJ that promote the closest spacing between the dyes in their dimers. The extracted aggregate geometries, and the role of electrostatic and steric effects in squaraine aggregation are also discussed. Taken together, these findings provide a deeper insight into how dye structures influence excitonic coupling in dye aggregates covalently templated via DNA, and guidance in design rules for exciton-based materials and devices.
Introduction
When neighboring dye molecules (chromophores) are excited, their local excited states can couple with a molecular exciton (also known as a Frenkel exciton) with excitation energy shared between the dyes in a wave-like manner. Observation of excitonic coupling and delocalization enabling efficient energy transfer in natural photosynthetic dye aggregates motivated the development of exciton-based applications and devices that encompass artificial photosynthesis,1−3 organic optoelectronics,4,5 and nanoscale computing.6−10 The functionality of these applications is governed by the ability to control and enhance the strength of excitonic coupling. According to the Kasha molecular exciton model,11,12 the excitonic hopping parameter Jm,n(13−15) characterizes the strength of the Coulombic interaction responsible for the electronic transitions’ interaction and thereof can quantitatively describe the strength of excitonic coupling. To facilitate the discussion, we treat the Coulombic interaction approximately as a dipole–dipole interaction following the approach of McRae and Kasha.12 In this case, the magnitude of Jm,n is inversely proportional to the cube of the distance between transition dipole moments constituted in the dyes of an aggregate (eq 1). As such, an organized and dense dye packing is an essential prerequisite for strong excitonic coupling that enables exciton delocalization. The magnitude of Jm,n is given by
| 1 |
where μm and μn are the transition dipoles for the dyes at sites m and n, Rm,n is the vector connecting dyes at sites m and n, ε0 is the permittivity of the vacuum, and ε is the relative dielectric constant of the medium.
With the dye concentration approaching the solubility limit in a solvent, dyes can be brought in proximity and form a non-covalent complex maintained by weak van der Waals forces, that is, a molecular aggregate. Aggregation of free dyes in solution occurs spontaneously and can result in dimers, a mixture of monomers and dimers, and higher-order aggregates.16,17 To gain more precise control over the number and position of dyes, various templates have been employed to assemble dye aggregates. Among different types of templates, DNA has been proven to be a powerful tool in organizing dyes into aggregates. The versatility of DNA as a template in the field of DNA nanotechnology stems from the relatively straightforward DNA self-assembly that relies on the complementary nature of four Watson–Crick base pairs allowing one to predict the shape of the template and location of dyes.18−21 DNA can template dyes into dye–dye aggregates whether they are covalently or non-covalently attached to the DNA. A non-covalent method when dyes non-covalently bind to DNA is indeed simpler in implementation. While non-covalent binding to DNA was employed to create extended arrays of dyes,22−27 the number of dyes in the aggregate and their exact position remained elusive.
The covalent DNA templating is based on attaching dyes via linkers to nucleobases or the backbone of single-stranded DNA, followed by self-assembly of complementary strands into double-stranded DNA. As a result, the number of dyes per DNA construct is strictly defined, with the predictable location of the dyes relative to the DNA backbone.28−47 The advantage of covalent DNA templating over dye spacing was demonstrated by Nicoli et al. who examined how base pair (bp) separation corresponding to ∼3.5 Å increments affects dimerization ability of Cy3 dyes covalently attached to DNA.28 Furthermore, Cunningham et al. studied excitonic coupling as a function of bp separation and estimated the excitonic coupling strength in a Cy3 dimer to be 33.0 meV (266 cm–1) and 18.6 meV (150 cm–1) for 0 and 1 bp separation, respectively.32 A diversity of dye aggregates covalently templated to DNA can be further extended by utilizing more complex DNA architectures such as a branched 3-way junction, a 4-way junction, DNA nanobreadboards, and DNA origami.20 The examples of high-order DNA templates include Probst’s work on the aggregates of alkynylpyrene and perylenediimide covalently templated to the branch point of a three-way DNA junction, and Mazuski’s work on Cy5 dimers attached to a DNA wire of two cross-linked DNA duplexes.48,49 An additional special feature of DNA covalent templating that offers distinct advantage over both spontaneous aggregation and aggregation via non-covalent DNA templating has been recently demonstrated by Barclay et al. with a dimer aggregate of water-soluble squaraine rotaxane created via covalent tethering to a DNA Holliday Junction (DNA HJ).50 This work uncovered the ability of DNA covalent templating to bring dyes of varying aqueous solubilities in proximity and promote their aggregation. Consequently, aggregation of a broader scope of dye chemical structures can be explored via covalent DNA templating.
In the past few years, our group has created dye aggregates via covalent templating to linear DNA and 4-arm DNA, that is, DNA HJ.7,51−53 In the DNA HJ, dyes are covalently attached to the four strand branch point allowing dye aggregation in a dimer, trimer, or a tetramer form. Initially starting with the aggregates of Cy5, we recently extended toward aggregates of commercial indolenine squaraine dyes.50,54 Squaraines structurally resemble the Cy5 dye, but feature a squarate moiety incorporated into the center of a pentamethine bridge between heterocyclic rings. Like Cy5, squaraines exhibit excellent spectral characteristics evident in their strong absorption in the visible region with a molar extinction coefficient in the range of 200,000–250,000 cm–1 M–1 and strong emission. We demonstrated that both Cy5 and squaraine dyes are capable of strong excitonic coupling and delocalization in their dimer and tetramer aggregates. However, squaraines offer advantages over Cy5 that include increased rigidity and photostability. In addition, squaraines can be synthesized with a diverse number and structure of substituents. While a dye’s photophysical properties stem mainly from the chromophore core structure, varying the structure of side substituents can be used to fine-tune dye packing and, hence, enhance excitonic coupling.
While aggregation of many dye families was investigated in aqueous solution,23,55−62 the studies mainly focused on the dye propensity to aggregate rather than their ability toward strong excitonic coupling. To our knowledge, only one report by the group of Armitage attempted to evaluate the influence of a dye peripheral structure on the excitonic coupling in cyanine aggregates templated via non-covalent binding to DNA. This study demonstrated that, in general, hydrophobic dyes have a tendency to exhibit strong excitonic coupling. However, a direct correlation between dye hydrophobicity, intermolecular distances, and the resulting strength of excitonic coupling in dye aggregates has not been provided. This study was also conducted with four cyanine dyes of similar hydrophobicity constraining the ability to fully reveal the influence of dye structures on the photophysical properties of their aggregates. This limitation presumably originates from the narrow range of dyes suitable for non-covalent DNA templating (as well as for spontaneous aggregation) excluding both insoluble and highly water-soluble dyes. The former tends to form higher-order aggregates complicating the analysis of precipitates from solution, while the latter, if they aggregate at all, have a large monomer population and require high dye concentrations that impede spectral measurements at high optical density. Also, a narrow set of explored structural variations for the same set of dyes is often due to the limitations of synthetic structural modifications.
In this paper, taking advantage of covalent DNA templating, we examine the influence of dye hydrophobicity on the aggregation behaviors and excitonic coupling strength within a wide range of indolenine-based squaraine dyes. Owing to the diversity of synthetically accessible indolenine squaraine structures, we chose six squaraines that differ by variations in the polarity and number of their substituents, including methyl, chloro, sulfo, and sulfobutyl groups. The squaraines were further categorized as overall hydrophobic or hydrophilic based on their relative water solubility. The dyes were covalently tethered to single-stranded DNA used to form a DNA HJ. The DNA HJ was used to template the dyes in the form of dimer (both transverse and adjacent) and tetramer aggregates. Both strength of excitonic coupling and aggregate geometry were quantified via Kühn–Renger–May modeling of the dye aggregate optical properties. We correlated hydrophobicity in terms of a partitioning coefficient derived via density functional theory (DFT) calculations with a center-to-center distance and strength of excitonic coupling. Structural modification of squaraine side substituents allowed us to vary the center-to-center distance between the dyes on the Angstrom level. Our findings indicate that dye hydrophobicity appears to play a predominant role in the intermolecular distance and strength of excitonic interactions in DNA-templated squaraine dye aggregates. The aggregates with the strongest excitonic coupling observed in this work originate from a dichloro derivative, which is the most hydrophobic and with minimal dye sterics that would frustrate aggregation. We believe that our findings will facilitate a deeper understanding of how dye structures influence aggregate optical properties, particularly covalently templated via DNA, and may serve as a stepping stone toward design rules for exciton-based materials and devices.
Methods
Dye Synthesis
Custom indolenine squaraine dyes were obtained from SETA BioMedicals, (Urbana-Champaign, IL). The synthesis of indolenine squaraines NHS-esters SQ-Sl3 and SQ-Sl5 was reported previously.62 The synthetic procedures for new indolenine squaraines NHS-esters SQ-H2, SQ-Cl2, SQ-Me2, and SQ-Sl2 are reported in Section S1.
Synthesis of DNA Constructs
DNA oligomers internally functionalized with a custom squaraine (SETA BioMedicals, Urbana-Champaign, IL) via a nucleosidic C6 dT sequence modifier and purified via dual high-performance liquid chromatography were purchased from Integrated DNA Technology, Inc (Coralville, IA). Non-functionalized DNA oligomers purified by standard desalting were purchased from Integrated DNA Technology, Inc. All DNA oligomers were rehydrated in ultrapure water (Barnstead Nanopure, Thermo Scientific) to prepare 100 μM stock solutions. Concentrations of the DNA samples were determined spectroscopically on a NanoDrop One Microvolume UV–Vis spectrometer (Thermo Scientific) using calculated extinction coefficients. DNA HJs were prepared by combining equimolar amounts of complimentary oligomers in 1× TBE, 15 mM MgCl2 buffer solution to a final DNA concentration 1.5 μM. All DNA samples were annealed using a Mastercycler Nexus PCR cycler (Eppendorf) according to the following protocol: 4 min at 95 °C, followed by cooling ramps: 0.1 °C per 15 s from 94 to 64 °C, and 10 °C per 1 min from 64 °C to room temperature. For the fluorescence measurements, the DNA samples were further diluted to a 0.5 μM DNA concentration.
Optical Characterization
The UV–Vis spectra were recorded in duplicates at room temperature on a dual-beam Cary 5000 UV–Vis–NIR spectrophotometer (Agilent Technologies) in a quartz cuvette with a 1 cm path length (Starna). The absorbtion spectra were monitored over a 230–800 nm wavelength range. Circular dichroism (CD) measurements were performed on a JASCO-J810 spectropolarimeter. DNA samples (120 μL) were transferred into a 1 cm path length quartz cuvette (Jasco). The spectra were recorded over a 230–800 nm wavelength range (three scans per sample were averaged) at a speed of 200 nm min–1. The steady-state fluorescence spectra were obtained using a Horiba PTI QuantaMaster 400 spectrofluorometer (Horiba Scientific) in a 1 cm path length quartz cuvette (Starna) and monitored as a function of wavelength. The fluorescence spectra were corrected for the wavelength dependence of the detection system response using the correction curve provided by the manufacturer. The fluorescence spectra were scaled by the absorptance at the excitation wavelength.
KRM Modeling
The experimental absorption and CD spectra were simultaneously fitted with theoretical spectra via a Holstein-like Hamiltonian (Section S8). Due to a larger subpopulation of the optical monomer in the SQ-Sl5 transverse dimer, which was evident from the fluorescence suppression, the absorption and CD spectra for modeling the SQ-Sl5 transverse dimer were recorded at 5 °C in order to complete aggregation. The minimum allowed distance between transition dipole moments was restricted to 3.4 Å (whose choice was guided by the van der Waals radius of carbon 1.7 Å doubled and a distance of the π–π stacking of 3.35 Å). The extended dipole approximation when two point charges of opposite sign are separated by a chromophore length was employed. To account for the potential influence of the DNA environment on covalently linked dyes, the transition dipole moment of the monomer squaraine dye is extracted from modeling experimental absorption of a single dye attached to DNA HJ, that is, the monomer. The characteristic excitonic hopping parameter constant J0 is derived from the monomer transition dipole moment. The excitonic hopping parameter Jm,n is equal to J0 times a geometrical factor that depends on the distance and orientation of dyes m and n, and that has units of inverse volume. Hence, J0 sets the values of the Jm,n and the distances Rm,n between dyes when the aggregate geometry is determined by fitting the Frenkel–Holstein model to the absorbance and CD spectra. Deriving the value of J0 from the monomer transition dipole moment enables a more accurate determination of Jm,n and Rm,n than can be achieved if J0 was treated as a fitting parameter.
The weight of the absorption and CD spectra was controlled via five types of weights. The choice of weighs was guided by the insights from a combination of experimental data. For the aggregates with a pronounced CD signal, the weight of the CD was emphasized, while for the aggregates with either weak or no CD signal, the weight of absorption and CD was kept equal. The theoretical spectra were generated for the theoretical dye configurations found via stochastic gradient search. The goodness of fit was accessed by the several goodness fit parameters (Section S8). The positions of dye transition dipole moments within an aggregate were extracted from the best fit in terms of Cartesian coordinates and zenith and azimuthal angles.
Density Functional Theory Calculations
Solvation energies were determined via DFT calculations as previously described.63−65 Chemical structures of squaraine dyes were created in Avogadro66 software, where dye geometries were initially approximated using the universal force field (UFF).67 Next, the molecular structures in the gas state were optimized using the M06-2X exchange correlation functional68 and the 6-31+** basis set in Gaussian 16.69 The implicit continuum solvent model density (SMD)70 was used for water and n-octanol solvents. The solvation energy ΔG was calculated by taking the difference in ground-state energies calculated using SMD water and vacuum given as
where Esolv is the ground-state energy calculated in a solvent and Ev is the vacuum or gas-phase ground-state energy.
Results
Molecular Design
Six indolenine squaraine dyes with varying numbers and structures of side substituents were chosen to systematically investigate the influence of hydrophobicity on the excitonic coupling and delocalization in squaraine molecular aggregates covalently templated by DNA HJ. The hydrophobicity of the unsubstituted SQ-H2 was alternated via disubstitution of the indolenine rings at the 5- and 5′-positions with chloro, methyl, and sulfo groups to afford SQ-Cl2, SQ-Me2, and SQ-Sl2, respectively (Figure 1a). Furthermore, the indolenine nitrogen in SQ-Sl2 was substituted with a N-sulfobutyl chain to afford SQ-Sl3, which in turn was equipped with two sulfobutyl chains attached to the tertiary carbons of indolenine rings to afford SQ-Sl5. All squaraines were functionalized with the N-pentyl-NHS ester for attachment to oligonucleotides. The synthesis of the NHS-ester squaraines is reported in Section S1.
Figure 1.

(a) Chemical structures of indolenine squaraine (bold) attached to C6 thymine sequence modifier T*(gray). (b) Schematic representation of immobile four-arm DNA HJs templating squaraine monomer SQ-A, adjacent dimer SQ-BC, transverse dimer SQ-AC, and a tetramer SQ-ABCD. The strands comprising DNA HJ are labeled A, B, C and D.
To promote aggregation of squaraines tethered to DNA, we utilized the same immobile DNA HJ template employed previously to create aggregates of a commercially available indolenine squaraine.54 The N-pentyl-NHS ester squaraines were covalently attached to 26-bp oligonucleotides via esterification reactions with the amino C6 thymine sequence modifier (T*) placed in the center of oligonucleotides (Section S1). As a result, the core of a squaraine dye was tethered to the DNA backbone via a single flexible linker constituting the N-pentyl fragment “L” of squaraine and the aliphatic fragment of T*(Figure 1a), with an overall length, if fully extended, of about 2.1 nm. Four non-homologous oligonucleotide strands A, B, C, and D were combined in solution to form an immobile 4-arm HJ (Figure 1b). A reference squaraine monomer SQ-A was formed by combining squaraine-labeled strand A with unlabeled strands B, C, and D. In a similar manner, SQ-labeled partially complementary strands B and C or non-complementary strands A and C were combined with the corresponding unlabeled strands to create an adjacent SQ-BC dimer and a transverse SQ-AC dimer, respectively. The tetramer was formed by combining all four SQ-labeled oligonucleotides. To ensure DNA HJ self-assembly, oligonucleotide strands were annealed at 95 °C for 4 min, followed by slow cooling to room temperature. The 1× TBE buffer solution was supplemented with 15 mM MgCl2 to promote a stacked conformation of the DNA HJ. The stacked conformation of DNA HJ in the presence of MgCl2 was observed previously for the analogous DNA HJ templating of commercially available indolenine-based squaraine aggregates.54 The formation of squaraine aggregates was assessed by analytical non-denaturing polyacrylamide gel electrophoresis. The gel imaging indicated well-formed squaraine-labeled DNA HJs (Section S2, Figure S1).
Relative hydrophobicity of custom squaraines was evaluated via their partitioning between n-octanol and water in terms of log Po/w. The log Po/w was calculated following eq 2
| 2 |
where ΔGo is the solvation energy in n-octanol and ΔGw is the solvation energy in water, R = 8.31 J*mol–1 K–1, and T = 273.15 K. The values of ΔGo and ΔGw were obtained via DFT calculations as previously described.63,64 The more positive log Po/w (ΔGo < ΔGw) is, the more hydrophobic the dye is. Conversely, the more negative log Po/w (ΔGo > ΔGw) is, the greater the propensity of the dyes to be soluble in water, and the more hydrophilic the dye is.
The values of log Po/w were determined for dye structures without a linker and for the dye structures containing a pentyl fragment of the linker denoted as “L” in Figure 1a and Table 1. Based on the obtained values of log Po/w (Table 1), custom squaraines were divided into two groups: overall hydrophobic squaraines SQ-H2, SQ-Cl2, and SQ-Me2 (log Po/w > 0) and overall hydrophilic squaraines SQ-Sl2, SQ-Sl3, and SQ-Sl5 (log Po/w < 0). When not accounting for the contribution of the linker, the hydrophobic dyes SQ-H2, SQ-Cl2, and SQ-Me2 exhibited comparable hydrophobicity in terms of log Po/w being in the range 4.30–5.31. With the linker, the difference in hydrophobicity of SQ-H2, SQ-Cl2, and SQ-Me2 became more apparent increasing in the order of SQ-H2-L < SQ-Cl2-L < SQ-Me2-L. The hydrophobicity of hydrophilic squaraines without contribution of the linker increased in order SQ-Sl5 < SQ-Sl3 < SQ-Sl2, which is in accordance with the experimental aqueous solubilities previously observed for the N-pentylcarboxy derivatives of SQ-Sl5 and SQ-Sl2 dyes in phosphate buffer.62 Inclusion of the linker strongly increased hydrophobicity of SQ-Sl3-L and SQ-Sl5-L and resulted in the hydrophobicity order SQ-Sl3-L < SQ-Sl5-L < SQ-Sl2-L.
Table 1. Squaraine Solvation Energy in Water and n-Octanol and log Po/w.
| free dye | ΔGw, kJ/mol | ΔGo, kJ/mol | log Po/w |
|---|---|---|---|
| SQ-Cl2 | –82.2 | –110 | 5.31 |
| SQ-H2 | –79.3 | –102 | 4.30 |
| SQ-Me2 | –76.5 | –104 | 5.24 |
| SQ-Sl2 | –181 | –161 | –3.66 |
| SQ-Sl3 | –244 | –211 | –6.26 |
| SQ-Sl5 | –320 | –258 | –11.82 |
| SQ-Me2-L | –73.4 | –136 | 11.94 |
| SQ-Cl2-L | –77.8 | –116 | 7.33 |
| SQ-H2-L | –75.5 | –108 | 6.26 |
| SQ-Sl2-L | –176 | –170 | –1.07 |
| SQ-Sl3-L | –236 | –215 | –3.97 |
| SQ-Sl5-L | –304 | –290 | –2.75 |
Thermal Denaturation
To characterize overall stabilities of DNA HJs templating transverse and adjacent squaraine dimers, we performed thermal denaturation experiments. The melting temperature of the unlabeled DNA HJ was used as a reference control (Tm = 60.0 °C).54 With the exception of the SQ-H2 transverse dimer, both adjacent and transverse dimers of hydrophobic dyes melted at a higher temperature than unlabeled HJ, indicative of dye–dye interactions having an overall stabilizing effect on the DNA–dye construct. In contrast, dimers of hydrophilic dyes melted at a lower temperature than unlabeled HJ. The HJs templating SQ-Cl2 dimers were characterized by the highest melting points. Notably, the melting temperatures of HJs increased as dye hydrophobicity increased (Section S3). As such, a hydrophobic effect can be considered as a major contributor of aggregation of dyes covalently templated by DNA.
Steady-State Optical Characterization
First, the squaraine monomers, as reference samples, were examined via steady-state absorption, emission, and circular dichroism (CD) spectroscopies (Figure 2 and 3, Sections S4–S7). All squaraine monomers covalently attached to DNA HJ exhibited electronic absorption spectra characteristic of the squaraine dye family. Both hydrophilic and hydrophobic squaraines templated via DNA HJs exhibited extinction coefficient values similar to those of the corresponding free dyes. However, hydrophobic squaraines were characterized with slightly lower extinction coefficient values (257,000–264,000 M–1 cm–1) compared to those of hydrophilic squaraines containing sulfo groups (282,000–304,000 M–1 cm–1). The attachment of hydrophobic SQ-Cl2, SQ-Me2, and SQ-H2 to DNA HJ resulted in a 10–13 nm red shift of their absorption maxima compared with those of free dyes (Table 2). A similar spectral shift of 9 nm observed previously for free dyes SQ-H2 and SQ-Cl2 upon transitioning from polar methanol to nonpolar dichloromethane was attributed to the solvatochromic effect.62 In our case, the red shift is likely to be caused by the squaraine being located around hydrophobic DNA. Squaraines containing sulfo groups also showed a red shift of absorption maxima upon attachment to DNA. While for SQ-Sl2, the red shift was 12 nm and comparable with the red shift of hydrophobic squaraines, SQ-Sl3 and SQ-Sl5 were only red-shifted 5 and 3 nm, respectively. Squaraines SQ-Sl2, SQ-Sl3, and SQ-Sl5 have been previously covalently attached to the IgG antibody,62 and exhibited small red shifts of 6, 5, and 4 nm, respectively, observed upon transitioning from an aqueous environment to a less-polar protein environment. Based on this comparison and chemical structures, non-covalent binding with DNA is less likely for hydrophilic and bulkier dyes SQ-Sl3 and SQ-Sl5.
Figure 2.

(Left column) Acquired steady-state absorption spectra normalized at the dye peak maximum of the hydrophilic squaraine-DNA constructs in 1× TBE, 15 mM MgCl2 at room temperature. (right column) Acquired CD of the hydrophilic squaraine-DNA constructs in 1× TBE, 15 mM MgCl2 at room temperature. The squaraine-DNA construct concentration was 1.5 μM.
Figure 3.

(Left column) Acquired steady-state absorption spectra normalized at the dye peak maximum of the hydrophobic squaraine-DNA constructs in 1× TBE, 15 mM MgCl2 at room temperature. (right column) Acquired CD of the hydrophobic squaraine-DNA constructs in 1× TBE, 15 mM MgCl2 at room temperature. The squaraine-DNA construct concentration was 1.5 μM.
Table 2. Experimental Absorption Properties of Squaraines and Squaraine Aggregate Template via DNA HJ.
| absorption
peak maximum, nm |
|||||
|---|---|---|---|---|---|
| dye | afree dye | dHJ monomer | dtrans dimer | dadj dimer | dtetramer |
| SQ-H2 | b628 | 638 | 606; 632 | 592; 629 | 583; 631 |
| SQ-Cl2 | b633 | 645 | 600; 633 | 596; 633 | 575; 633 |
| SQ-Me2 | b635 | 648 | 602; 647 | 596; 637 | 583; 640 |
| SQ-Sl2 | 628 | 640 | 607; 630 | 598; 630 | 587; 635 |
| SQ-Sl3 | c631 | 636 | 596; 632 | 594; 631 | 594; 633 |
| SQ-Sl5 | c636 | 638 | 602; 637 | 600; 636 | 600; 634 |
As carboxylic acid in phosphate buffer, pH = 7.4.
In methanol.
From [ref. (29)].
Measurements were carried out in 1× TBE, 15 mM MgCl2 containing 1.5 μM DNA construct at room temperature.
The squaraine monomers exhibited strong fluorescence emission as a near mirror image of absorption with clear (0–0) and (0–1) emission transitions, and small Stokes shifts in the range of 24–39 meV or 194–315 cm–1 (Figure S5 and Table S2). No specific correlation between substituents and Stokes shifts was observed indicating that the introduction of the side substituents does not noticeably affect the rigidity of the indolenine squaraine core.
In the CD spectra, all constructs exhibited a well-defined couplet in the 260–280 nm region indicative of a well-formed duplex DNA (Section S7). Because squaraine dyes are achiral, they are not expected to produce a CD signal in the visible region. Each monomer of SQ-H2, SQ-Me2, SQ-Sl2, SQ-Sl3, and SQ-Sl5 did not induce a signal in the visible region of the CD spectra. However, the monomers of SQ-H2 and SQ-Cl2 exhibited a very weak induced CD signal, which indicates that these dyes may interact with DNA by intercalating between base pairs or binding to the minor or major groove resulting in restriction of their conformational freedom and induced chirality.
The aggregation ability of hydrophilic and hydrophobic squaraine aggregates covalently attached to the DNA HJ was evaluated by steady-state absorption, emission, and CD spectroscopies, and compared within the adjacent dimer, transverse dimer, and tetramer series.
Hydrophilic squaraines exhibited noticeable spectral changes in dimer configurations with respect to their monomers (Figure 2 and Table 2). The spectral changes included (1) a blue shift of both low- and high-energy absorption bands that we, respectively, assign as A1 and A2; and (2) intensification of the A2 band with the suppression of the A1 band, that is, an increase of the A2/A1 ratio. The blue shift for adjacent and transverse dimers increased in the order of SQ-Sl5 < SQ-Sl2 < SQ-Sl3. Both dimers of SQ-Sl2 and adjacent dimers of SQ-Sl3 and SQ-Sl5 exhibited a more intense high-energy band (A2 > A1), while transverse dimers SQ-Sl5 and SQ-Sl3 had more intense low-energy band (A2 < A1). In accordance with the Kasha’s exciton theory,11,71 these spectral changes indicate that hydrophilic squaraines covalently attached to DNA HJ formed dimer aggregates with the spectral signatures of excitonic coupling and delocalization. This observation was not anticipated especially for SQ-Sl3 and SQ-Sl5 as these dyes in free form were shown not to aggregate at concentrations of 0.2 and 6 mM, respectively, in aqueous solution.62 The blue shift in the dimers of hydrophilic squaraines is consistent with a face-to-face orientation of transition dipole moments, that is, a H-aggregate, while similar intensities of A1 and A2 bands indicate the involvement of vibronic coupling11,71 and the overall intermediate coupling regime.11 The adjacent and transverse dimers of hydrophilic squaraines were also characterized by CD spectroscopy (Figure 2 (right column)). Achiral SQ-Sl2 and SQ-Sl3 dyes exhibited an exciton-induced bisignate CD signal in their transverse and adjacent dimers indicating on a chiral orientation of transition dipole moments in the aggregates. Though the CD signal originates from chirality of optical system, the absence of chirality does not necessarily mean a lack of excitonically coupled transition dipole moments (TDM), but might be attributed to an aggregate having a mirror plane in the orientation of transition dipole moments. This situation takes place when transition dipole moments are coplanar, which might be the case for the SQ-Sl3 transverse dimer and SQ-Sl5 dimers.
Upon dimer assembly via DNA HJ, hydrophobic squaraines demonstrated profound spectral changes evident in a blue shift and in an increase of the A2/A1 absorption band ratio with respect to the monomers (Figure 3, Table 2). The blue shift of the high A2 and low A1 energy bands increased in the order of SQ-H2 < SQ-Me2 < SQ-Cl2, and for each dye the shift was larger in the adjacent dimer than in the transverse dimer. The A2/A1 ratio increased in the order of SQ-H2 < SQ-Me2 ≤ SQ-Cl2, where A2 was greater than A1 for all hydrophobic squaraine adjacent dimers and for the SQ-Cl2 transverse dimer. For the transverse dimers of SQ-H2 and SQ-Me2, A2 was smaller than A1. Evident from these changes, all hydrophobic squaraines formed H-type dimers with increasing excitonic coupling strengths as the hydrophobicity increases. Thus, the two most hydrophobic squaraines—SQ-Cl2 and SQ-Me2—appeared to exhibit the strongest excitonic coupling based on the blue shift, as well as nearly complete suppression of the low-energy band A1 (Tables 2 and 3). While adjacent dimers of SQ-Cl2 and SQ-Me2 showed nearly identical spectral shapes, the peak extinction coefficient of the high-energy absorption band A2 of SQ-Cl2 was considerably larger than that of SQ-Me2 (Figure S4). Overall, the magnitude of excitonic coupling estimated from spectral profiles closely follows the order of relative hydrophobicity of hydrophobic squaraines. Although both adjacent and transverse dimers show signatures of excitonic coupling and delocalization, such signatures are apparently diminished for the hydrophobic dyes in the transverse dimer configuration suggesting weaker excitonic coupling and more oblique aggregates. The difference in the absorption properties between transverse and adjacent dimers suggests that the adjacent configuration of dye attachment is more favorable for promoting strong excitonic coupling in indolenine squaraine-based dimer aggregates. The exciton-induced CD spectra of hydrophobic squaraines (Figure 3, right) further supported the formation of dimer aggregates.
Table 3. Spectral Characteristics of Squaraine Aggregates.
| adj
dimer |
trans
dimer |
tetramer |
||||
|---|---|---|---|---|---|---|
| dye | aA2/A1 | bFS, % | A2/A1 | FS, % | A2/A1 | FS, % |
| SQ-H2 | 1.26 | 90.5 | 0.82 | 83.8 | 1.48 | 95.8 |
| SQ-Cl2 | 3.72 | 92.6 | 1.99 | 90.4 | 4.06 | 96.9 |
| SQ-Me2 | 3.70 | 84.8 | 0.78 | 75.1 | 2.33 | 95.5 |
| SQ-Sl2 | 1.24 | 87.3 | 1.17 | 87.3 | 1.41 | 95.4 |
| SQ-Sl3 | 1.27 | 69.5 | 0.81 | 74.7 | 1.22 | 88.1 |
| SQ-Sl5 | 1.04 | 78.7 | 0.72 | 48.4 | 1.17 | 87.1 |
A1 and A2 are relative intensities of low-energy and high-energy absorption bands in the absorption spectra of squaraine dimers recorded at a 1.5 μM dye-DNA concentration in 1× TBE, 15 mM MgCl2.
FS is fluorescence suppression.
Fluorescence emission measurements were performed to gain insights on the structure and dynamics of the squaraine dimer excited states (Tables 3 and S2). The fluorescence from the squaraine dimer solutions occurred approximately at the same wavelength as the fluorescence of the corresponding monomer solutions; however, the intensity of aggregate fluorescence was strongly suppressed (Table 3). A similar fluorescence behavior between monomer and dimer emission was previously observed for the dimers of commercial indolenine squaraines, and was assigned to the presence of optical monomers, that is, a subpopulation of dyes attached to DNA in pairs, but not forming an aggregate.50 Based on our experimental observations and the preceding study, we assign the observed emission in dimer samples to the strongly emissive optical monomers. The emission from the dimers may still occur, but it would be very weak and not measurable for the following reasons. According to the selection rules in the Kasha’s theory on the molecular aggregates,11 the radiative decay, that is, fluorescence, is prohibited from the high excited state in the H-aggregates. Moreover, strong fluorescence quenching in dye aggregates has been recently associated with the new non-radiative decay pathways available in strongly coupled aggregates.32,50,53,72 Thus, the extent of fluorescence suppression (FS), calculated in Table 3, can provide insights, at least qualitatively, into the strength of excitonic coupling (i.e., “weak” or “strong”) between dyes in squaraine dimers and the presence of subpopulation of optical isomers. The strongest FS of >90% was observed for the dimers of SQ-Cl2 suggesting a strong excitonic coupling. Other dimers (with the exception of transverse dimer SQ-Sl5) exhibited smaller FS in the range of 70–87%. The dimers of hydrophobic dyes SQ-H2, SQ-Cl2, and SQ-Me2 exhibited stronger FS in the adjacent configuration than in the transverse configuration. In contrast, the hydrophilic dyes SQ-Sl2 and SQ-Sl3 showed stronger FS in the transverse configuration. While the fluorescence in the SQ-Sl5 adjacent dimer was suppressed by 79% and similarly to other adjacent dimers, the SQ-Sl5 transverse dimer exhibited significantly smaller FS of 48% indicative of a larger subpopulation of SQ-Sl5 optical monomers. Note that electrophoretic analysis (Figure S1) showed a minor amount (and comparable to other squaraine dimers) of unhybridized single strand labeled with SQ-Sl5, suggesting that the observed fluorescence emission of the SQ-Sl5 transverse dimer sample should not be attributed to the SQ-Sl5 monomer attached to a single strand in solution.
One of the special features of the DNA HJ as a template is that it allows one to create dye tetramers. Upon tetramer formation, pronounced spectral changes were observed for all hydrophobic dyes SQ-Cl2, SQ-Me2, and SQ-H2 and a hydrophilic SQ-Sl2 indicative of the strong excitonic coupling and delocalization among the four dyes (Figures 2 and 3, Table 2 and 3). In particular, the high-energy band carrying the most oscillator strength was further blue-shifted by 613 cm–1 for SQ-Cl2, 374 cm–1 for SQ-Me2, 313 cm–1 for SQ-Sl2, and 261 cm–1 for SQ-H2 with respect to the corresponding high-energy band A2 in adjacent dimers. These changes in absorption, as compared to the monomer absorption, indicated the H-type packing of these dyes in the tetramer configuration. The CD spectra of tetramers were characterized by intense exciton-induced couplets (Figures 2 and 3). Moreover, the fluorescence was nearly quantitatively suppressed (96% on average) in the tetramers of SQ-H2, SQ-Cl2, SQ-Me2, and SQ-Sl2. In contrast, hydrophilic SQ-Sl3 and SQ-Sl5 squaraines did not demonstrate strong evidence of tetramer formation. The absorption peak maximum of SQ-Sl3 and SQ-Sl5 did not shift, while the absorbance extinction was almost doubled compared to their adjacent dimers (Figure S4). The FS in SQ-Sl3 and SQ-Sl5 tetramers appeared to be comparable with one observed in their adjacent dimers. Collectively, these observations indicate that SQ-Sl3 and SQ-Sl5 might mostly remain in their dimer form. The steric bulkiness of SQ-Sl3 and SQ-Sl5 may interfere with dye aggregation and/or the DNA HJ template, thus inhibiting the formation of SQ-Sl3 and SQ-Sl5 tetramers.
Theoretical Spectral Modeling
The modeling approach developed in our group, based on the theoretical approach of Kühn–Renger–May (KRM),73 has been demonstrated to be a powerful tool in extracting excitonic hopping parameter Jm,n as a quantitative metric of excitonic coupling strength, as well as the geometric parameters of the dye aggregates.51,52,54 The advantage of KRM modeling of dye aggregates stems from considering not only electronic contribution of dyes to the excitonic coupling but also their vibronic contribution. The vibronic contribution is considered via the dominant vibrational mode of each dye, which for squaraines has an energy of ca. 0.12–0.17 meV (970–1400 cm–1). To model Coulomb interaction between squaraines, we employed an extended dipole approximation,74 where the dipoles were modeled as two point charges of opposite sign and equal magnitude separated by nearly the dye length. This extended dipole approximation is particularly beneficial when dye intermolecular distances are much shorter than the dye length as this approach better accounts for the charge distribution over a large molecule.
From the KRM modeling results, we extracted excitonic hopping parameter J1,2 for each dimer and for each dye pair in a tetramer. Squaraine dimers exhibited medium to strong excitonic hopping parameter J1,2 ranging from 50.3 meV (406 cm–1) to 132.2 meV (1065 cm–1) with the strongest J1,2 observed in the SQ-Cl2 adjacent dimer. For comparison, the excitonic coupling in analogous dimers of the commercial squaraine and cyanine dyes Square 660 and Cy5 were estimated to be 65–68 and 70 meV, respectively. Michail et al. reported very similar coupling strengths (from 930 cm–1 or 118 meV to 390 cm–1 or 48 meV) in the J-aggregates of indolenine squaraine dimers, where squaraine molecules were linked via a covalent bridge of varying lengths.75 The extracted J1,2 values were plotted against dye partitioning between water and n-octanol as a measure of dye hydrophobicity (Figure 4a,b). A general trend was evident for the hydrophobic squaraines SQ-H2, SQ-Cl2, and SQ-Me2 in both adjacent and transverse dimer series, where J1,2 generally increased as hydrophobicity increased. Hydrophilic squaraines containing sulfo groups showed an opposite trend: a decrease in J1,2 upon an increase in partitioning. Squaraine SQ-Sl5, being the most hydrophilic dye, exhibited the excitonic coupling comparable to and exceeding the coupling of more hydrophobic dyes especially in the adjacent dimer configuration. Plotting partitioning accounting for the linker fragment versus J1,2 (Figure S13) resulted in the same general trends as those without linker contribution (Figure 4), with the exception of SQ-Sl5 and SQ-Cl2. To evaluate the role of hydrophobicity in the strength of excitonic coupling in squaraine tetramers, the highest value of Jm,n and the arithmetic mean Jm,nave for each squaraine tetramer was plotted against the partitioning between n-octanol and water with (Figure S13c) and without the linker fragment of that squaraine (Figure 5). With the linker contribution, an apparent trend was observed between excitonic coupling in the tetramer and dye hydrophobicity (Figures 5 and S13c). SQ-Sl3 and SQ-Sl5 were excluded from the KRM modeling as tetramers because steady-state spectroscopy did not indicate the formation of tetramers with these dyes. The absence of a reliable trend between partitioning and Jm,n indicates that effects other than hydrophobic effect influence the strength of excitonic coupling in squaraine aggregates. Among those effects are steric hindrance and electrostatic forces of various origin that we evaluated in Section S9.
Figure 4.

(a,b) KRM values of J1,2 in adjacent and transverse squaraine dimers plotted against squaraine partitioning between n-octanol and water (no linker included). (c,d) KRM values of center-to-center distance R in Å in adjacent and transverse squaraine dimers plotted against squaraine partitioning between n-octanol and water (no linker included).
Figure 5.
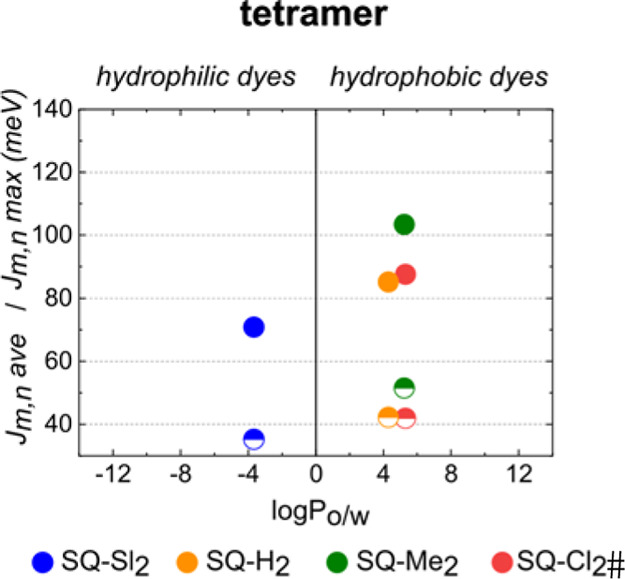
KRM values of the highest Jm,n value (solid circles) and Jm,nave (half solid circles) in squaraine tetramers plotted against partitioning between n-octanol and water as a measure of hydrophobicity (no linker included).
The geometry of squaraine dimers (Table 4) and tetramers (Tables S10–S13) was characterized by several geometric parameters derived from KRM modeling: (1) a center-to-center distance R between two TDMs, (2) the shortest distance d between TDMs, (3) oblique angle α as a quantitative measure of deviation from the parallel stacking of TDMs (α = 0°), and (4) a slip angle θs as a measure of sliding or displacement of one dye relative to another along dye’s TMD (when θs = 90°, the dyes are not displaced) (Table 4 inset). The value of R is useful, as we expect smaller R to result in an increased excitonic coupling strength (and which we expect to decrease for increasingly hydrophobic dyes).11 Among hydrophobic squaraines, R values decreased as the dye hydrophobicity increased (Figure 4c,d). Among the six squaraines, the values of R varied on the Angstrom level in the range from 3.4 to 9.5 Å. The hydrophobic squaraines generally exhibited smaller R in the adjacent dimer configuration, which is more favorable for the excitonic coupling in aggregates of these dyes. As expected, the most hydrophobic SQ-Cl2 has the smallest R in both adjacent and transverse dimers. Counterintuitively, the most hydrophilic SQ-Sl5 exhibited the smallest R among the hydrophilic squaraines. The value of α was used to characterize the amount of obliqueness with two extreme cases being α = 0°, that is, parallel TDMs and α = 90°, that is, a purely oblique dimer. In general, transverse dimers of squaraines were more oblique than adjacent dimers of analogous dyes. Adjacent dimers of SQ-Cl2 and SQ-Me2 showed nearly parallel alignment of their TDMs (Figure S10) and thereof indolenine rings were characterized by α in the range 1.35–2.4°, but differed in the slip angle or displacement. In particular, dyes in the SQ-Cl2 adjacent dimer were barely displaced (θs = 86.8°), while dyes in the SQ-Me2 dimer were noticeably displaced relative to each other (θs = 59.0°). The KRM modeling predicts the shortest distance d for the most dimers to be 3.4–3.8 Å. At this distance, molecules containing π-bond networks are known to undergo strong π–π interactions.76 The adjacent dimer of SQ-Cl2 is the only dimer, where the π–π interactions between indolenine rings can take place on both ends of the dimer based on a center-to-center distance of 3.4 Å and parallel orientation of TDMs.
Table 4. Geometric Parameters Derived by KRM Modeling of the Optical Properties of Adjacent and Transverse Dimer Aggregatesa.
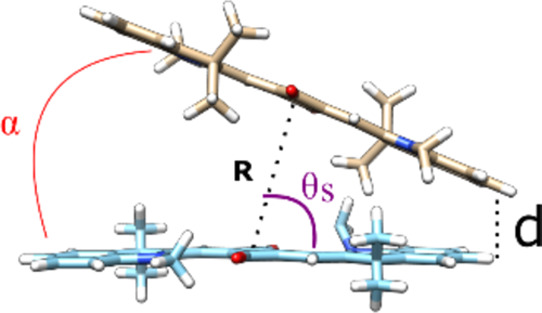
| adjacent
dimer |
transverse
dimer |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| dye | J1,2, meV | R, Å | θs, deg | α, deg | d, Å | J1,2, meV | R, Å | θs, deg | α, deg | d, Å |
| SQ-Cl2 | 132.2 | 3.40 | 86.8 | 1.35 | 3.4 | 79.6 | 5.45 | 81.8 | 15.7 | 3.5 |
| SQ-Me2 | 96.5 | 4.47 | 59.0 | 2.4 | 3.6 | 55.9 | 9.5 | 74.9 | 55.5 | 3.4 |
| SQ-H2 | 59.8 | 7.52 | 64.8 | 32.7 | 3.4 | 50.3 | 9.33 | 74.8 | 47.5 | 3.5 |
| SQ-Sl2 | 64.3 | 8.9 | 75.0 | 48.3 | 3.6 | 71.3 | 5.4 | 60.0 | 8.8 | 4.0 |
| SQ-Sl3 | 80.4 | 6.71 | 71.0 | 25.8 | 3.4 | 63.2 | 6.62 | 71.3 | 19.8 | 3.8 |
| SQ-Sl5 | 97.9 | 4.76 | 89.2 | 9.0 | 3.6 | 77.5a | 7.85 | 69.3 | 34.6 | 3.4 |
Modeling of experimental absorption and CD recorded at 5 °C.
Discussion
Three overall hydrophobic squaraine dyes with varying hydrophobicity, covalently tethered to and templated via DNA HJ, formed dimer and tetramer aggregates as was evident via drastic changes in their optical properties compared to their respective monomers. These results are in accordance with prior reports on aggregation of various dye families including cyanine dyes, where the hydrophobic effect was identified as a major driving force in both spontaneous aggregation,55−61 and aggregation via non-covalent DNA-templating in aqueous solution.23 The focus of those studies, however, was a general dye propensity toward aggregation, that is, the ability to form dimers and multimers and the associated equilibrium constants. In the work most relevant to the present study, Armitage and co-workers templated aggregates of a series four hydrophobic cyanine dyes23 via non-covalent binding to DNA. In that work, the excitonic coupling strength was estimated as twice the difference in energy between the monomer and dimer UV–Vis absorbance bands. While the study demonstrated that hydrophobic dyes tend to form aggregates with strong excitonic coupling, it was unable to provide any further insight into how a chemical structure impacts hydrophobicity, strength of excitonic coupling, and aggregate geometry parameters such as the intermolecular distance and relative orientation of dyes. In the present work, we used a combination of DFT, KRM modeling, and melting experiments to gain additional insights into correlations between the chemical structure, extent of hydrophobicity, strength of excitonic coupling, and intermolecular distance in squaraine aggregates templated by DNA. In line with our expectations, the melting experiments demonstrated that the more hydrophobic squaraines afford more stable HJs templating the squaraine aggregates suggesting a driving effect of hydrophobic forces on the aggregation of squaraines covalently templated by DNA. Moreover, our results revealed that squaraines with a more hydrophobic structure afford a denser packing, or closer intermolecular distances (Rm,n), in their dimer and tetramer aggregates. A denser packing in aggregates of more hydrophobic squaraines further manifests in larger values of excitonic hopping parameter Jm,n in accordance with the Kasha’s exciton model (eq 1), where smaller Rm,n corresponds to larger Jm,n. In the present study, the largest value of Jm,n was observed for the most hydrophobic squaraine SQ-Cl2. In addition, the correlation between hydrophobicity and packing was the most apparent when dyes were attached to partially complementary strands of DNA HJ (adjacent dimer configuration) perhaps because the sites of dye’s linker attachment to DNA are closer in space in this configuration than when dyes are attached to non-complementary strands of DNA HJ (the transverse dimer configuration).
Taking advantage of covalent DNA-templating, we were able to evaluate aggregation ability of hydrophilic dyes commonly excluded from the studies on spontaneous dye aggregation and aggregation via non-covalent DNA templating. We found that when covalently templated to DNA, “hydrophilic” dyes can form aggregates, and the excitonic coupling in aggregates of these hydrophilic dyes is comparable with that of hydrophobic dyes. These results appear counterintuitive with regard to Markova’s study, where free dyes SQ-Sl2 and SQ-Sl5 did not show signs of aggregation at the concentrations of 0.2 and 6 mM, respectively, in aqueous phosphate buffer.62 We explain this discrepancy with the ability of the DNA template to bring dyes in proximity including water-soluble dyes that otherwise stay in the free form in aqueous solution. We attempt to interpret these results in the context of a conventional understanding of a physical origin of the hydrophobic effect that is thought to be entropy-driven at room temperature.57,77 In particular, when a hydrophobic surface area is minimized upon aggregation of hydrophobic molecules that are not able to interact with water, the network of water hydrogen bonding with higher entropy is restored. The high entropy of the water structure upon aggregation of hydrophobic molecules compensates the entropy decrease of the aggregated molecules as well as the enthalpic cost of reorganization into an aggregate making the aggregation overall favorable. Conversely, hydrophilic molecules are able to form hydrogen bonds with water, so their aggregation does not afford an increase in water entropy large enough to make the aggregation favorable. However, when hydrophilic dyes are brought in proximity via covalent attachment to DNA, their reorganization into an aggregate occurs readily at a very small energetic cost. When SQ-Sl3 and SQ-Sl5 aggregate, the hydrophobic butyl chains of SQ-Sl3 and SQ-Sl5 can be involved in hydrophobic interactions in the presence of polar sulfo groups. Squaraines SQ-Sl3 and SQ-Sl5 possess the largest surface area among the squaraines in this study (Section S9), which might explain stronger coupling in their aggregates than expected from their overall hydrophilic nature. In this regard, aggregation of DNA-templated hydrophilic dyes can be compared to protein folding, where overall hydrophilic amino acids such as lysine were shown to participate in initial protein folding via hydrophobic interactions between their non-polar alkyl chains.78
Factors beyond hydrophobicity, such as dye sterics and electrostatic interactions, may additionally influence dye aggregation. The steric influence becomes apparent in the attempt to aggregate SQ-Sl3 and SQ-Sl5 into tetramers. Though sulfobutyl chains appear to be accommodated in the dimers of SQ-Sl3 and SQ-Sl5, their bulkiness strongly hinders the formation of SQ-Sl3 and SQ-Sl5 tetramers attached to DNA HJ. We evaluated influence of electrostatic interactions such as dipole–dipole interactions and induced-dipole interactions including dispersion via a static dipole moment and polarizability (Section S9). The results showed that while electrostatic forces do not appear to be major contributors in excitonic coupling, forces involving induced dipoles still play a more apparent role for hydrophobic squaraines. Hydrophobic squaraines SQ-H2, SQ-Me2, and SQ-Cl2 have comparable hydrophobicity and surface area, though differ in polarizability. Higher polarizability of SQ-Cl2 and SQ-Me2 than that of SQ-H2 suggests stronger induced-dipole electrostatic forces in the dimers of the former dyes, and may explain the increase in J1,2 in adjacent dimers of SQ-Me2 and SQ-Cl2 compared to those of SQ-H2. Furthermore, having comparable polarizabilities, SQ-Cl2 dimers exhibit significantly stronger excitonic coupling than dimers of SQ-Me2. From the KRM modeling, we found that in the adjacent dimer SQ-Cl2, TDMs, and hence the indolenine rings, are parallel and lack the displacement. According to the pioneering model of Hunter and Sanders,79 the lack of displacement suggests less electrostatic repulsion between the static charge distributions in π-systems and, hence, stronger π–π interactions stabilizing a dimer. Examination of the electrostatic potential surfaces revealed slightly decreased electron density in the electron-rich indolenine rings of SQ-Cl2 due to the electron-withdrawing effect of chlorine atoms presumably allowing SQ-Cl2 to align without displacement (Section S9). In contrast, electron-donating methyl groups slightly increase the electron density of indolenines in SQ-Me2 leading to the displacement in its dimer. Further insights into how these parameters influence aggregate geometry and excitonic coupling strength may require a similar study specifically targeting a set of dyes tailored to vary steric or electrostatic interactions.
Conclusions
Six examined indolenine squaraines with varying degrees of hydrophobicity demonstrated aggregate behavior, excitonic coupling, and exciton delocalization when covalently templated as adjacent and transverse dimers by DNA HJ in aqueous buffer. While aggregation of hydrophobic dyes was anticipated, the observed aggregation of hydrophilic dyes was not necessarily expected. The excitonic coupling was evident via spectral changes that indicated the excitonic coupling increased as the dye hydrophobicity increased. The KRM modeling supported the same trend for the hydrophobic dyes with higher values of the extracted hopping parameter J1,2 for dyes with increased hydrophobicity. For hydrophilic dyes, the surface area served as a better predictor of aggregation and excitonic coupling strength for aggregates created by proximate covalent attachment to a template. Collectively, our results demonstrate that alternating hydrophobic structure of dyes covalently attached to DNA affords control of intermolecular distances and hence excitonic coupling strength in dye aggregates on a finer level than base pair separation. We identified adjacent dimers of the strongly hydrophobic SQ-Cl2 dye that we ranked to be the best performing based on its extracted hopping parameter J1,2 up to 132.2 meV (1065 cm–1). Additional factors such as dye sterics and electrostatic factors play a role in tuning the dye alignment once dyes are brought together by the hydrophobic effect. Overall, we believe our findings will provide foundational guidance in rational design of dye aggregates that exhibit strong excitonic interactions and that are templated by DNA, including in larger aggregate networks assembled via more complex DNA scaffolds such as bricks and tiles.
Acknowledgments
This research was supported wholly by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Materials Sciences and Engineering Division and DOE’s Established Program to Stimulate Competitive Research (EPSCoR) program under Award DE-SC0020089, except as follows: the circular dichroism spectrometer was made available through the Biomolecular Research Center (BRC) at Boise State, which is supported in part by the National Institutes of Health award no. P20GM109095, MJ Murdock Charitable Trust, and Idaho State Board of Education. We thank Dr. Natalya Hallstrom for the assistance with PAGE electrophoresis of squaraine-DNA constructs.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcc.1c08981.
Chemical synthesis of custom squaraine dyes; PAGE gel electrophoresis; absorption, fluorescence, and circular dichroism measurements; KRM modeling; and electrostatic surface potentials (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Brixner T.; Hildner R.; Köhler J.; Lambert C.; Würthner F. Exciton Transport in Molecular Aggregates - from Natural Antennas to Synthetic Chromophore Systems. Adv. Energy Mater. 2017, 7, 1700236. 10.1002/aenm.201700236. [DOI] [Google Scholar]
- Wasielewski M. R. Self-Assembly Strategies for Integrating Light Harvesting and Charge Separation in Artificial Photosynthetic Systems. Acc. Chem. Res. 2009, 42, 1910–1921. 10.1021/ar9001735. [DOI] [PubMed] [Google Scholar]
- Scholes G. D.; Rumbles G. Excitons in Nanoscale Systems. Nat. Mater. 2006, 5, 683–696. 10.1038/nmat1710. [DOI] [PubMed] [Google Scholar]
- Ostroverkhova O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. 10.1021/acs.chemrev.6b00127. [DOI] [PubMed] [Google Scholar]
- Yurke B.; Kuang W. Passive Linear Nanoscale Optical and Molecular Electronics Device Synthesis from Nanoparticles. Phys. Rev. A: At., Mol., Opt. Phys. 2010, 81, 033814. 10.1103/physreva.81.033814. [DOI] [Google Scholar]
- Graugnard E.; Kellis D. L.; Bui H.; Barnes S.; Kuang W.; Lee J.; Hughes W. L.; Knowlton W. B.; Yurke B. DNA-Controlled Excitonic Switches. Nano Lett. 2012, 12, 2117–2122. 10.1021/nl3004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B. L.; Kellis D. L.; Davis P. H.; Lee J.; Kuang W.; Hughes W. L.; Graugnard E.; Yurke B.; Knowlton W. B. Excitonic and Logic Gates on DNA Brick Nanobreadboards. ACS Photonics 2015, 2, 398–404. 10.1021/ph500444d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis D. L.; Rehn S. M.; Cannon B. L.; Davis P. H.; Graugnard E.; Lee J.; Yurke B.; Knowlton W. B. DNA-Mediated Excitonic Upconversion Fret Switching. New J. Phys. 2015, 17, 115007. 10.1088/1367-2630/17/11/115007. [DOI] [Google Scholar]
- Wang S.; Lebeck A. R.; Dwyer C. Nanoscale Resonance Energy Transfer-Based Devices for Probabilistic Computing. IEEE Micro 2015, 35, 72–84. 10.1109/mm.2015.124. [DOI] [Google Scholar]
- Sawaya N. P. D.; Rappoport D.; Tabor D. P.; Aspuru-Guzik A. Excitonics: A Set of Gates for Molecular Exciton Processing and Signaling. ACS Nano 2018, 12, 6410–6420. 10.1021/acsnano.8b00584. [DOI] [PubMed] [Google Scholar]
- Kasha M. Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates. Radiat. Res. 1963, 20, 55–70. 10.2307/3571331. [DOI] [PubMed] [Google Scholar]
- McRae E. G.; Kasha M.. The Molecular Exciton Model. In Physical Processes in Radiation Biology; Augenstein L., Mason R., Rosenberg B., Eds.; Academic Press: New York, 1964; pp 23–42. [Google Scholar]
- Abramavicius D.; Mukamel S. Exciton Dynamics in Chromophore Aggregates with Correlated Environment Fluctuations. J. Chem. Phys. 2011, 134, 174504. 10.1063/1.3579455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramavicius D.; Palmieri B.; Mukamel S. Extracting Single and Two-Exciton Couplings in Photosynthetic Complexes by Coherent Two-Dimensional Electronic Spectra. Chem. Phys. 2009, 357, 79–84. 10.1016/j.chemphys.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramavicius D.; Palmieri B.; Voronine D. V.; Šanda F.; Mukamel S. Coherent Multidimensional Optical Spectroscopy of Excitons in Molecular Aggregates; Quasiparticle Versus Supermolecule Perspectives. Chem. Rev. 2009, 109, 2350–2408. 10.1021/cr800268n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelley E. E. Molecular, Nematic and Crystal States of I: I-Diethyl--Cyanine Chloride. Nature 1937, 139, 631. 10.1038/139631b0. [DOI] [Google Scholar]
- Jelley E. E. Spectral Absorption and Fluorescence of Dyes in the Molecular State. Nature 1936, 138, 1009–1010. 10.1038/1381009a0. [DOI] [Google Scholar]
- Seeman N. C. DNA in a Material World. Nature 2003, 421, 427–431. 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]
- Rothemund P. W. K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- Ke Y.; Ong L. L.; Shih W. M.; Yin P. Three-Dimensional Structures Self-Assembled from DNA Bricks. Science 2012, 338, 1177–1183. 10.1126/science.1227268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman N. C. Structural DNA Nanotechnology: An Overview. Methods Mol. Biol. 2005, 303, 143–166. 10.1385/1-59259-901-X:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Silva G. L.; Armitage B. A. DNA-Templated Formation of a Helical Cyanine Dye J-Aggregate. J. Am. Chem. Soc. 2000, 122, 9977–9986. 10.1021/ja002184n. [DOI] [Google Scholar]
- Stadler A. L.; Renikuntla B. R.; Yaron D.; Fang A. S.; Armitage B. A. Substituent Effects on the Assembly of Helical Cyanine Dye Aggregates in the Minor Groove of a DNA Template. Langmuir 2011, 27, 1472–1479. 10.1021/la104329c. [DOI] [PubMed] [Google Scholar]
- Seifert J. L.; Connor R. E.; Kushon S. A.; Wang M.; Armitage B. A. Spontaneous Assembly of Helical Cyanine Dye Aggregates on DNA Nanotemplates. J. Am. Chem. Soc. 1999, 121, 2987–2995. 10.1021/ja984279j. [DOI] [Google Scholar]
- Garoff R. A.; Litzinger E. A.; Connor R. E.; Fishman I.; Armitage B. A. Helical Aggregation of Cyanine Dyes on DNA Templates: Effect of Dye Structure on Formation of Homo- and Heteroaggregates. Langmuir 2002, 18, 6330–6337. 10.1021/la025742f. [DOI] [Google Scholar]
- Boulais É.; Sawaya N. P. D.; Veneziano R.; Andreoni A.; Banal J. L.; Kondo T.; Mandal S.; Lin S.; Schlau-Cohen G. S.; Woodbury N. W.; et al. Programmed Coherent Coupling in a Synthetic DNA-Based Excitonic Circuit. Nat. Mater. 2018, 17, 159–166. 10.1038/nmat5033. [DOI] [PubMed] [Google Scholar]
- Banal J. L.; Kondo T.; Veneziano R.; Bathe M.; Schlau-Cohen G. S. Photophysics of J-Aggregate-Mediated Energy Transfer on DNA. J. Phys. Chem. Lett. 2017, 8, 5827–5833. 10.1021/acs.jpclett.7b01898. [DOI] [PubMed] [Google Scholar]
- Nicoli F.; Roos M. K.; Hemmig E. A.; Di Antonio M.; de Vivie-Riedle R.; Liedl T. Proximity-Induced H-Aggregation of Cyanine Dyes on DNA-Duplexes. J. Phys. Chem. A 2016, 120, 9941–9947. 10.1021/acs.jpca.6b10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markova L. I.; Malinovskii V. L.; Patsenker L. D.; Häner R. Synthesis and Properties of Squaraine-Modified DNA. Org. Biomol. Chem. 2012, 10, 8944–8947. 10.1039/c2ob26787j. [DOI] [PubMed] [Google Scholar]
- Malinovskii V. L.; Wenger D.; Häner R. Nucleic Acid-Guided Assembly of Aromatic Chromophores. Chem. Soc. Rev. 2010, 39, 410–422. 10.1039/b910030j. [DOI] [PubMed] [Google Scholar]
- Li S.; Langenegger S. M.; Häner R. Control of Aggregation-Induced Emission by DNA Hybridization. Chem. Commun. 2013, 49, 5835–5837. 10.1039/c3cc42706d. [DOI] [PubMed] [Google Scholar]
- Cunningham P. D.; Kim Y. C.; Díaz S. A.; Buckhout-White S.; Mathur D.; Medintz I. L.; Melinger J. S. Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122, 5020–5029. 10.1021/acs.jpcb.8b02134. [DOI] [PubMed] [Google Scholar]
- Lee W.; von Hippel P. H.; Marcus A. H. Internally Labeled Cy3/Cy5 DNA Constructs Show Greatly Enhanced Photo-Stability in Single-Molecule Fret Experiments. Nucleic Acids Res. 2014, 42, 5967–5977. 10.1093/nar/gku199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markova L. I.; Malinovskii V. L.; Patsenker L. D.; Häner R. J- Vs. H-Type Assembly: Pentamethine Cyanine (Cy5) as a near-Ir Chiroptical Reporter. Chem. Commun. 2013, 49, 5298–5300. 10.1039/c3cc42103a. [DOI] [PubMed] [Google Scholar]
- Asanuma H.; Fujii T.; Kato T.; Kashida H. Coherent Interactions of Dyes Assembled on DNA. J. Photochem. Photobiol., C 2012, 13, 124–135. 10.1016/j.jphotochemrev.2012.04.002. [DOI] [Google Scholar]
- Heussman D.; Kittell J.; Kringle L.; Tamimi A.; von Hippel P. H.; Marcus A. H. Measuring local conformations and conformational disorder of (Cy3)2 dimer labeled DNA fork junctions using absorbance, circular dichroism and two-dimensional fluorescence spectroscopy. Faraday Discuss. 2019, 216, 211–235. 10.1039/c8fd00245b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kringle L.; Sawaya N. P. D.; Widom J.; Adams C.; Raymer M. G.; Aspuru-Guzik A.; Marcus A. H. Temperature-Dependent Conformations of Exciton-Coupled Cy3 Dimers in Double-Stranded DNA. J. Chem. Phys. 2018, 148, 085101. 10.1063/1.5020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma H.; Shirasuka K.; Takarada T.; Kashida H.; Komiyama M. DNA–Dye Conjugates for Controllable H* Aggregation1. J. Am. Chem. Soc. 2003, 125, 2217–2223. 10.1021/ja021153k. [DOI] [PubMed] [Google Scholar]
- Häner R.; Samain F.; Malinovskii V. L. DNA-Assisted Self-Assembly of Pyrene Foldamers. Chem.—Eur. J. 2009, 15, 5701–5708. 10.1002/chem.200900369. [DOI] [PubMed] [Google Scholar]
- Kashida H.; Asanuma H.; Komiyama M. Alternating Hetero H Aggregation of Different Dyes by Interstrand Stacking from Two DNA-Dye Conjugates. Angew. Chem., Int. Ed. Engl. 2004, 43, 6522–6525. 10.1002/anie.200460870. [DOI] [PubMed] [Google Scholar]
- Ikeda S.; Okamoto A. Hybridization-Sensitive on-Off DNA Probe: Application of the Exciton Coupling Effect to Effective Fluorescence Quenching. Chem.—Asian J. 2008, 3, 958–968. 10.1002/asia.200800014. [DOI] [PubMed] [Google Scholar]
- Sohail S. H.; Otto J. P.; Cunningham P. D.; Kim Y. C.; Wood R. E.; Allodi M. A.; Higgins J. S.; Melinger J. S.; Engel G. S. DNA Scaffold Supports Long-Lived Vibronic Coherence in an Indodicarbocyanine (Cy5) Dimer. Chem. Sci. 2020, 11, 8546–8557. 10.1039/d0sc01127d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart S. M.; Chen W. J.; Banal J. L.; Bricker W. P.; Dodin A.; Markova L.; Vyborna Y.; Willard A. P.; Häner R.; Bathe M.; et al. Engineering Couplings for Exciton Transport Using Synthetic DNA Scaffolds. Chem 2021, 7, 752–773. 10.1016/j.chempr.2020.12.020. [DOI] [Google Scholar]
- Cunningham P. D.; Khachatrian A.; Buckhout-White S.; Deschamps J. R.; Goldman E. R.; Medintz I. L.; Melinger J. S. Resonance Energy Transfer in DNA Duplexes Labeled with Localized Dyes. J. Phys. Chem. B 2014, 118, 14555–14565. 10.1021/jp5065006. [DOI] [PubMed] [Google Scholar]
- Garo F.; Häner R. A DNA-Based Light-Harvesting Antenna. Angew. Chem., Int. Ed. Engl. 2012, 51, 916–919. 10.1002/anie.201103295. [DOI] [PubMed] [Google Scholar]
- Fujii T.; Kashida H.; Asanuma H. Analysis of Coherent Heteroclustering of Different Dyes by Use of Threoninol Nucleotides for Comparison with the Molecular Exciton Theory. Chem.—Eur. J. 2009, 15, 10092–10102. 10.1002/chem.200900962. [DOI] [PubMed] [Google Scholar]
- Kashida H.; Tanaka M.; Baba S.; Sakamoto T.; Kawai G.; Asanuma H.; Komiyama M. Covalent Incorporation of Methyl Red Dyes into Double-Stranded DNA for Their Ordered Clustering. Chem.—Eur. J. 2006, 12, 777–784. 10.1002/chem.200500111. [DOI] [PubMed] [Google Scholar]
- Probst M.; Wenger D.; Biner S. M.; Häner R. The DNA Three-Way Junction as a Mould for Tripartite Chromophore Assembly. Org. Biomol. Chem. 2012, 10, 755–759. 10.1039/c1ob06400b. [DOI] [PubMed] [Google Scholar]
- Mazuski R. J.; Díaz S. A.; Wood R. E.; Lloyd L. T.; Klein W. P.; Mathur D.; Melinger J. S.; Engel G. S.; Medintz I. L. Ultrafast Excitation Transfer in Cy5 DNA Photonic Wires Displays Dye Conjugation and Excitation Energy Dependency. J. Phys. Chem. Lett. 2020, 11, 4163–4172. 10.1021/acs.jpclett.0c01020. [DOI] [PubMed] [Google Scholar]
- Barclay M. S.; Roy S. K.; Huff J. S.; Mass O. A.; Turner D. B.; Wilson C. K.; Kellis D. L.; Terpetschnig E. A.; Lee J.; Davis P. H.; et al. Rotaxane Rings Promote Oblique Packing and Extended Lifetimes in DNA-Templated Molecular Dye Aggregates. Chem. Commun. 2021, 4, 19. 10.1038/s42004-021-00456-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B. L.; Kellis D. L.; Patten L. K.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System. J. Phys. Chem. A 2017, 121, 6905–6916. 10.1021/acs.jpca.7b04344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B. L.; Patten L. K.; Kellis D. L.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates. J. Phys. Chem. A 2018, 122, 2086–2095. 10.1021/acs.jpca.7b12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff J. S.; Davis P. H.; Christy A.; Kellis D. L.; Kandadai N.; Toa Z. S. D.; Scholes G. D.; Yurke B.; Knowlton W. B.; Pensack R. D. DNA-Templated Aggregates of Strongly Coupled Cyanine Dyes: Nonradiative Decay Governs Exciton Lifetimes. J. Phys. Chem. Lett. 2019, 10, 2386–2392. 10.1021/acs.jpclett.9b00404. [DOI] [PubMed] [Google Scholar]
- Mass O. A.; Wilson C. K.; Roy S. K.; Barclay M. S.; Patten L. K.; Terpetschnig E. A.; Lee J.; Pensack R. D.; Yurke B.; Knowlton W. B. Exciton Delocalization in Indolenine Squaraine Aggregates Templated by DNA Holliday Junction Scaffolds. J. Phys. Chem. B 2020, 124, 9636–9647. 10.1021/acs.jpcb.0c06480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S. B.; Shillcock M.; Jones P. Equilibrium and Kinetic Studies of the Aggregation of Porphyrins in Aqueous Solution. Biochem. J. 1976, 153, 279–285. 10.1042/bj1530279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada K.; Mitshuishi M.; Ohira M.; Miyazaki K. Positional Effects of a Trifluoromethyl Group on the Aggregation of Azo Dyes in Aqueous Solutions. J. Phys. Chem. 1993, 97, 4926–4929. 10.1021/j100121a010. [DOI] [Google Scholar]
- Mukerjee P.; Ghosh A. K. Thermodynamic Aspects of the Self-Association and Hydrophobic Bonding of Methylene Blue. Model System for Stacking Interactions. J. Am. Chem. Soc. 1970, 92, 6419–6424. 10.1021/ja00725a006. [DOI] [Google Scholar]
- Murakami K. Thermodynamic and Kinetic Aspects of Self-Association of Dyes in Aqueous Solution. Dyes Pigm. 2002, 53, 31–43. 10.1016/s0143-7208(01)00104-8. [DOI] [Google Scholar]
- Patil K.; Pawar R.; Talap P. Self-Aggregation of Methylene Blue in Aqueous Medium and Aqueous Solutions of Bu4nbr and Urea. Phys. Chem. Chem. Phys. 2000, 2, 4313–4317. 10.1039/b005370h. [DOI] [Google Scholar]
- Takahashi D.; Oda H.; Izumi T.; Hirohashi R. Substituent Effects on Aggregation Phenomena in Aqueous Solution of Thiacarbocyanine Dyes. Dyes Pigm. 2005, 66, 1–6. 10.1016/j.dyepig.2004.08.008. [DOI] [Google Scholar]
- McKerrow A. J.; Buncel E.; Kazmaier P. M. Aggregation of squaraine dyes: Structure-property relationships and solvent effects. Can. J. Chem. 1995, 73, 1605–1615. 10.1139/v95-200. [DOI] [Google Scholar]
- Markova L. I.; Terpetschnig E. A.; Patsenker L. D. Comparison of a Series of Hydrophilic Squaraine and Cyanine Dyes for Use as Biological Labels. Dyes Pigm. 2013, 99, 561–570. 10.1016/j.dyepig.2013.06.022. [DOI] [Google Scholar]
- Biaggne A.; Knowlton W. B.; Yurke B.; Lee J.; Li L. Substituent Effects on the Solubility and Electronic Properties of the Cyanine Dye Cy5: Density Functional and Time-Dependent Density Functional Theory Calculations. Molecules 2021, 26, 524. 10.3390/molecules26030524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcenas G.; Biaggne A.; Mass O. A.; Wilson C. K.; Obukhova O. M.; Kolosova O. S.; Tatarets A. L.; Terpetschnig E.; Pensack R. D.; Lee J.; et al. First-Principles Studies of Substituent Effects on Squaraine Dyes. RSC Adv. 2021, 11, 19029–19040. 10.1039/d1ra01377g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fothergill J. W.; Hernandez A. C.; Knowlton W. B.; Yurke B.; Li L. Ab Initio Studies of Exciton Interactions of Cy5 Dyes. J. Phys. Chem. A 2018, 122, 8989–8997. 10.1021/acs.jpca.8b05237. [DOI] [PubMed] [Google Scholar]
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminf. 2012, 4, 17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappe A. K.; Casewit C. J.; Colwell K. S.; Goddard W. A.; Skiff W. M. Uff, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. 10.1021/ja00051a040. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H., et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, 2016.
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Spano F. C. The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates. Acc. Chem. Res. 2010, 43, 429–439. 10.1021/ar900233v. [DOI] [PubMed] [Google Scholar]
- Huff J. S.; Turner D. B.; Mass O. A.; Patten L. K.; Wilson C. K.; Roy S. K.; Barclay M. S.; Yurke B.; Knowlton W. B.; Davis P. H.; et al. Excited-State Lifetimes of DNA-Templated Cyanine Dimer, Trimer, and Tetramer Aggregates: The Role of Exciton Delocalization, Dye Separation, and DNA Heterogeneity. J. Phys. Chem. B 2021, 125, 10240–10259. 10.1021/acs.jpcb.1c04517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn O.; Renger T.; May V. Theory of Exciton-Vibrational Dynamics in Molecular Dimers. Chem. Phys. 1996, 204, 99–114. 10.1016/0301-0104(95)00448-3. [DOI] [Google Scholar]
- Czikklely V.; Forsterling H. D.; Kuhn H. Extended Dipole Model for Aggregates of Dye Molecules. Chem. Phys. Lett. 1970, 6, 207–210. 10.1016/0009-2614(70)80220-2. [DOI] [Google Scholar]
- Michail E.; Schreck M. H.; Holzapfel M.; Lambert C. Exciton Coupling Effects on the Two-Photon Absorption of Squaraine Homodimers with Varying Bridge Units. Phys. Chem. Chem. Phys. 2020, 22, 18340–18350. 10.1039/d0cp03410j. [DOI] [PubMed] [Google Scholar]
- Tsuzuki S.; Honda K.; Uchimaru T.; Mikami M.; Tanabe K. Origin of Attraction and Directionality of the π/π Interaction: Model Chemistry Calculations of Benzene Dimer Interaction. Am. Chem. Soc. 2002, 124, 104–112. 10.1021/ja0105212. [DOI] [PubMed] [Google Scholar]
- Anslyn E. V.; Dougherty D. A.. Modern Physical Organic Chemistry; University Science Books: Sausalito, California, 2005. [Google Scholar]
- Dyson H. J.; Wright P. E.; Scheraga H. A. The Role of Hydrophobic Interactions in Initiation and Propagation of Protein Folding. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13057. 10.1073/pnas.0605504103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter C. A.; Sanders J. K. M. The Nature Of .Pi.-.Pi. Interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. 10.1021/ja00170a016. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.