

Abstract
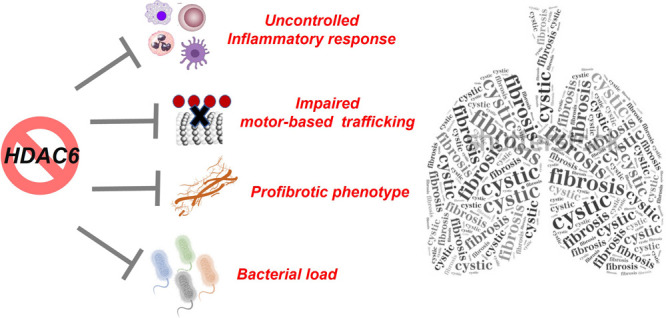
Compelling new support has been provided for histone deacetylase isoform 6 (HDAC6) as a common thread in the generation of the dysregulated proinflammatory and fibrotic phenotype in cystic fibrosis (CF). HDAC6 also plays a crucial role in bacterial clearance or killing as a direct consequence of its effects on CF immune responses. Inhibiting HDAC6 functions thus eventually represents an innovative and effective strategy to tackle multiple aspects of CF-associated lung disease. In this Perspective, we not only showcase the latest evidence linking HDAC(6) activity and expression with CF phenotype but also track the new dawn of HDAC(6) modulators in CF and explore potentialities and future perspectives in the field.
Introduction
Cystic fibrosis (CF) patients present mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which codes for a cyclic AMP-regulated chloride ion channel.1 Loss of function of this channel leads to defective transport of chloride ion across the surface of epithelial cells, which in turn interferes with the clearance of inhaled microorganisms. This lack of defense against microorganisms is responsible for severe airway infections in CF patients.2 Intensive antibiotic therapy is mandatory for maintaining lung function and quality of life and has contributed to a significant extension of the mean expected lifetime of CF patients.3 The CF-associated complex microbial flora of the respiratory tract are characterized by the coexistence of multiple bacterial species, which complicates the identification of a very efficacious antibacterial treatment. Infections triggered by Pseudomonas aeruginosa (PA) are particularly challenging and have the tendency to persist and become chronic.4 Morbimortality of CF patients is largely ascribable to chronic lung infection, inflammation, and uncontrolled fibrotic tissue rearrangement. The underlying pathways still remain underexplored; additionally, the efficacy of currently employed anti-inflammatory treatments is limited to symptomatic management of CF airway inflammation.5
The initial stage of CF-related lung disease mainly consists of prolonged airway inflammation accompanied by protracted infections, chronic inflammation, mucus hypersecretion, and oxidative stress.6−8 Evolution to chronic CF disease entails recurrent or steady infection with PA, which finally results in chronic airway inflammation, permanent pulmonary impairment, and collapse of respiratory function.9 CFTR malfunctioning is also related to a defective autophagy process that promotes chronic inflammation and oxidative stress in CF airways.10,11 Chronic inflammation is mainly associated with NFκB-mediated proinflammatory signaling (as demonstrated by increased levels of the NFκB-dependent genes KC and Mip-2) and IL-8-dependent neutrophil chemotaxis.6,8 Beyond neutrophils, other innate and adaptive immune cells are linked to increased levels of pro-inflammatory cytokines and chemokines, thus promoting CF-related lung disease;12 therefore, a deeper knowledge of the mechanisms underlying inflammatory response is of pivotal importance in CF research. Since CFTR-targeted therapies display swinging efficacy in reducing inflammation in CF airways, a complementary anti-inflammatory therapy to be employed in association with CFTR correctors and potentiators would be an added value in this context. More importantly, patients harboring CFTR mutations that are not responsive to currently available modulators would at least take advantage of innovative therapeutic options to re-establish inflammatory regulation.
It has also been demonstrated that CF fibroblasts exhibit an aberrant phenotype characterized by uncontrolled proliferation and myofibroblast differentiation, increased sensitivity to growth factors, and altered levels of proinflammatory and fibrotic mediators. In particular, CF lungs show increased release of transforming growth factor-β1 (TGF-β1), a multifunctional protein implicated in wound repair, epithelial-to-mesenchymal transition (EMT), myofibroblast differentiation, and production of various elements of connective tissue matrix.5,13
The latest studies identified the histone deacetylase (HDAC) class of enzymes as strategic components of the complex molecular machinery underlying both inflammation and fibrogenesis in CF. Accordingly, compelling new support has been provided for isoform HDAC6 as a common thread in the generation of the dysregulated proinflammatory and fibrotic phenotype in CF.14−19 HDAC6 also plays a crucial role in bacterial clearance or killing as a direct consequence of its effects on CF immune responses.18 Inhibiting HDAC6 functions thus eventually represents a novel and effective strategy to tackle multiple aspects of CF-associated lung disease. Selective HDAC6 inhibition should also avoid the common toxicities related to the currently available unselective HDAC inhibitors.
1. Current Therapeutic Strategies in Cystic Fibrosis: State of the Art and Unmet Needs
CF is characterized by a complex clinical picture. To date there is no cure for CF, and the current treatment regimen requires ad personam approaches. In general, a multidrug combination is required to control the symptomatology associated with this pathology and to ensure improved quality of life and life expectancy in CF patients.20
Currently, CF treatment includes CFTR modulators, antibiotics, bronchodilators, mucolytics, and anti-inflammatory and food supplements (because of the lack of pancreatic enzymes and vitamins in CF patients).21
CF patients also deal with chronic bacterial infections. Pulmonary infections are especially caused by PA and Staphylococcus aureus (SA). Even if primary infection is characterized by a low bacterial load, these types of infections easily become chronic because of reduced mucociliary clearance (caused by CFTR alteration), superficial airway fluid hypertonicity, and resistance toward β-lactam antibiotics, mainly due to PA strains that produce serine and metal β-lactamases.22,23 It is also important to underline how chronic infection in turn prompts pulmonary tissue destruction, thus further worsening the clinical picture for CF patients. Therefore, the role of antibiotic therapy is crucial to control PA and other CF-associated bacterial strains.24,25
PA infections in CF are generally treated with β-lactam antibiotics. Piperacillin (1, Figure 1) is among the more widely used, and it is often associated with tazobactam (2, Figure 1), aminoglycosides such as tobramycin (3, Figure 1), macrolides such as azithromycin (4, Figure 1), and carbapenems such as meropenem (5, Figure 1). Tazobactam shows efficacy in the inhibition of serine β-lactamases produced by PA.
Figure 1.

Chemical structures of compounds 1–9 used as antibiotic and anti-inflammatory drugs in CF therapy.
Fifth-generation cephalosporins such as ceftolozane (6, Figure 1), associated with tazobactam, show good activity against PA and variable activity against other Gram-negative bacteria;26 ceftaroline (7, Figure 1) is effective against methicillin-resistant SA (MRSA).27
A more difficult therapeutic approach involves bacterial strains expressing metallo β-lactamases, since no drugs are currently approved for their specific inhibition.
In addition to its effect on bacterial protein synthesis, azithromycin has important immunomodulatory and anti-inflammatory properties, which in part could explain the effectiveness of this class of antibiotics. In fact, in patients with persistent PA infection, azithromycin strongly reduces neutrophil chemiotaxis in the lung and the release of elastases by neutrophils themselves.28
Another important aspect related to the management of microbial infections in CF patients is related to nontuberculous mycobacteria (NTM) present in roughly 10% of CF patients, although only a small set of them will show NTM disease. However, patients potentially prone to develop NTM lung disease will just need careful monitoring on a precautionary basis if symptoms and radiographic outcomes are negligible.29
With regard to anti-inflammatory therapy, ibuprofen (8, Figure 1) is certainly the most widely used drug, and it is able to reduce the migration of neutrophils to the lung.30 However, its use is limited because of side effects (e.g., renal failure, gastric ulcers), which are common to all nonsteroidal anti-inflammatory drugs (NSAIDs) and are associated with the chronic intake required for CF-related inflammation treatment. Moreover, the need to monitor the blood concentration to ensure the therapeutic effect for long times is an additional deterrent to the use of ibuprofen. Another important factor to be considered when dealing with both steroidal anti-inflammatory drugs and NSAIDs is the interindividual variability, which significantly affects treatment efficacy and tolerability.
CF patients also experience the presence of thick, sticky mucus. For this reason, mucolytic therapy is considered to be beneficial to reach the lower respiratory tract, where thicker mucus is produced. Bronchitol (9, Figure 1) is the most representative and used drug of this category.
The development of CFTR modulators marked the most important breakthrough in CF therapy. Depending on the mechanism of action, they are classified as enhancers, correctors, or amplifiers. Correctors and enhancers act on mutations that involve a defect in CFTR maturation and migration on the cell membrane. Amplifiers act to increase CFTR protein production, regardless of the type of mutation; a greater amount of defective protein available would make the action of correctors and enhancers more profitable (Figure 2).31
Figure 2.

Summary of CFTR mutations and related pharmacological therapy. AONs = antisense oligonucleotides.
Symkevi is a combination of ivacaftor (10, Figure 3) and tezacaftor (11, Figure 3) and is approved for patients (>12 years old) who are homozygous for phenylalanine 508 deletion (ΔF508). This mutation is associated with a defect of CFTR transport from the nucleus to the cell membrane and with reduced stability of the channel in the cell membrane itself. CFTR correctors are basically able to increase the cell-surface expression of mutated CFTR. Ivacaftor is an effective enhancer for class III CFTR mutations (channel opening defect); it acts by improving CFTR channel opening, thus increasing chlorine transport and leading to cell osmolarity within physiological parameters.
Figure 3.

CFTR modulators (compounds 10–13) and antifibrotic drugs (compounds 14 and 15) used in CF therapy.
Tezacaftor is a CFTR corrector and improves CFTR protein folding and transport to the cell membrane. Thus, it is useful for patients bearing mutations that result defective in protein folding. Kaftrio, the last treatment approved, is a combination of ivacaftor, tezacaftor, and elexacaftor (12, Figure 3) and acts as corrector of CFTR defects, while Orkambi combines lumacaftor (13, VX809; Figure 3) and ivacaftor and is used in pediatric patients that are at least 2 years of age. CFTR modulators are essential drugs in CF therapy, but because of the large number of CFTR mutations, they unfortunately are not always effective in the entire patient population. Furthermore, their use is restricted to patients harboring the specific mutations for which their use has been approved.
Pirfenidone (14, Figure 3) and nintedanib (15, Figure 3) are used in the therapy for idiopathic pulmonary fibrosis (IPF) and represent another relevant therapeutic option to be considered in CF, with respect to CF-associated pulmonary fibrosis.32 In IPF these drugs showed efficacy in reducing fibrotic progress and pulmonary failure, delaying disease progression without reversing lung damage. However, their mechanism of action is still unclear, and therefore, more detailed studies are required for their safe and effective use in CF treatment.33
Ideally, an effective drug for CF should act independently of interindividual gene variability and should be suitable for chronic therapy and therefore characterized by a wide therapeutic window and high tolerability by the patient. Currently, none of the available drugs used in CF therapy possess all of these characteristics. Therefore, further research efforts are still necessary to identify more effective and safe therapeutic options in CF.
2. Epigenetic Regulation in Cystic Fibrosis
Epigenetic regulation is a complex mechanism involved in gene expression changes due to alterations of chromatin packaging. The main epigenetic mechanisms include histone modification and DNA methylation, among others.34,35 Specifically, acetylation, methylation, phosphorylation, and ubiquitination are the most representative histone modifications, and they are implicated in several physiopathological processes. Pairs of enzymes with opposing activity are responsible for fine-tuning these modifications, such as histone methyltransferases (HMTs) and demethylases or histone acetyltransferases (HAT) and HDACs.
These dynamic and reversible modifications regulate cell-specific gene expression, acting as promoters or repressors. In particular, histone acetylation causes gene activation, while histone methylation can silence or activate genes.36,37
To date, the role of epigenetics in CF has not yet been entirely unveiled, although several studies have pointed out the relevance of DNA methylation and histone modifications in CFTR gene regulation and protein activity.38,39 These mechanisms could be crucial for the discovery of innovative therapeutic strategies in CF treatment.
A study highlighted the relationship between CFTR mutations and microRNA (miRNA)-controlled pathways.35 miRNAs are single-stranded noncoding RNAs that regulate cell differentiation, growth, and proliferation as well as apoptosis and gene expression.40 CFTR plays a pivotal role in cellular homeostasis by regulating Cl– and HCO3– ingress and egress, and the concentrations of these ions influence miRNA expression, causing abnormal lung epithelial remodeling and altered immune response in CF patients. Altered miRNA pathways could explain major disorders associated with CFTR mutations.35 Thus, miRNAs could be a novel therapeutic target for the management of CF and CF-related pathologies such as diabetes, liver fibrosis, and pancreatic adenocarcinoma.
DNA methylation and histone deacetylation promote CFTR transcriptional inhibition. DNA methylation at promoters represses gene expression, and this mechanism is altered in CF nasal epithelial cells, blood cells, and lung macrophages.41 However, it is still unclear whether this modification is a trigger or a result of the disease.42 CFTR expression is regulated by a housekeeping promoter and distal cisregulatory elements.43 Comparison of fetal and adult tissues of lung, colon, and intestine revealed that CFTR expression was reduced during development as a result of DNA methylation. In fetal tissues, CFTR expression is not related to promoter methylation, while in adult individuals the CFTR gene is silenced, suggesting how epigenetic regulation could be correlated to the CF phenotype. In addition, mutant CFTR expression causes the activation of ROS-mediated autophagy. In this kind of cells, the CpG sequence of the CFTR gene results in hypermethylation, causing an alteration of CFTR expression.34
Similarly, histone acetylation promotes CFTR expression, but the exact mechanisms of chromatin packaging in the CFTR locus still have to be completely clarified. Histone acetylation neutralizes the positive charge on lysine residues of the amino-terminal tails of the histones, modifying the chromatin packaging and increasing the transcriptional activity.44 The basal expression of CFTR gene is connected to an inverted CCAAT element (sequence: 5′-AATTGGAAGCAAAT-3′) located between nucleotides 132 and 119 upstream of the translational start site. CFTR gene hyperacetylation of the CFTR promoter increases gene transcription as a result of major accessibility to the CCAAT sequence.45 In human lung and colon, introns 1 and 11 and the promoter result hyperacetylation, suggesting cell-type-specific gene expression of CFTR.46
Dysregulation of inflammatory mediators in the CF airway also has an epigenetic basis. A CFTR defect itself causes inflammation, but nevertheless, altered levels of histone acetylation and methylation displayed significant correlations to the increase in inflammatory markers, thus suggesting key epigenetic mechanisms underlying the CF-associated inflammatory phenotype.47−49
In conclusion, key epigenetic changes appear to be involved in CF, and they represent promising starting points for further research efforts in this field. In particular, the role of specific epigenetic enzymes, such as the HDAC class of enzymes, deserves particular attention, since multiple pathways associated with CF are closely related to HDAC expression and activity. In this review, special focus will be devoted to HDAC6 isoform because of its unique features and its multifaceted role in CF, which will be discussed in the following paragraphs.
3. Structure and Substrates of HDAC6
HDAC enzymes are responsible for acetyl group removal from histones and other protein substrates. Eighteen isoforms of HDAC enzymes have been identified to date, and they are clustered into four classes on the basis of homology to yeast HDACs. Classes I, II, and IV are zinc-dependent HDACs, while class III HDACs are NAD+-dependent. HDAC isoforms 1, 2, 3, and 8 (class I) are expressed in the nucleus of the cells of all tissues and share homology with yeast HDAC RDP3.50,51 While HDAC isoforms 1 and 2 display nuclear localization, isoforms 3 and 8 can shuttle between the cell nucleus and the cytoplasm. Class II HDACs are subdivided into Class IIA isoforms (4, 5, 7, and 9) and Class IIB isoforms (6 and 10). Class IV contains isoform 11 and shows homology with Class I and II HDACs. Class III HDACs are sirtuins (SIRT isoforms 1–7) and display homology with yeast sirtuin protein Sir2.50,52,53
HDAC6 is a cytoplasmic HDAC isoform containing 1215 amino acids that is encoded by the hdac6 gene, and the general structure and domain organization are depicted in Figure 4.54 The amino-terminal domain is characterized by a nuclear localization signal containing a high percentage of arginine and lysine residues, followed by a nuclear export signal (NES) that is rich in leucine and smooths the export of the enzyme toward its cytoplasmic localization. The center of catalytic activity is represented by two deacetylation domains, called DD1 and DD2, and HDAC6 is the only HDAC enzyme featuring two catalytic domains. The catalytic domains are followed by a serine–glutamic acid (SE14) tetradecapeptide signal that is responsible for cytoplasmic retention (Figure 4). The carboxy-terminal domain is characterized by a ubiquitin binding domain containing Zn2+ ion (ZnF-UBP) that is rich in cysteine and histidine residues and can interact with ubiquitinated proteins that undergo the degradation process. In particular, the HDAC6 carboxy-terminal domain controls the transfer of ubiquitinated proteins on the aggresomes present on the microtubules; therefore, HDAC6 binds to ubiquitin, thus preventing protein degradation via the ubiquitin proteasome.
Figure 4.
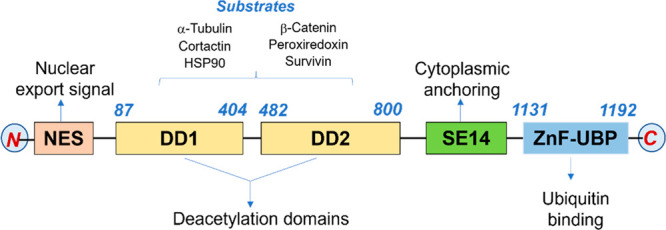
HDAC6 structure: domain organization and substrates.
The main HDAC6 substrates are non-histone proteins, namely, heat shock protein 90 (HSP90), α-tubulin, cortactin, and peroxiredoxins (Figure 4).
The first identified HDAC6 substrate was α-tubulin. Reversible acetylation of α-tubulin on residue lysine 40 plays a fundamental role in the regulation of stability and function of microtubules.55 Non-acetylated α-tubulin monomers interact with each other through electrostatic interactions, which are lost when the lysine nitrogen is acetylated, thus reducing inter-protofilament interactions and enhancing the flexibility of microtubules under mechanical stress.
Chemotactic cell movement, cell invasion, and migration are triggered by overexpression of HDAC6. Acetylated α-tubulin levels are also responsible for the fine-tuning of motor-based trafficking, which is essential for the transport of cargos along with the microtubule network (Figure 4).56 The ratio between acetylated and non-acetylated tubulin is often altered in several pathological conditions and coincides with an overexpression of HDAC6.57−59
Another crucial deacetylation substrate is HSP90, an ATP-dependent chaperone involved in the maturation of several proteins, including steroid receptors and mutant p53 proteins. HDAC6 inhibition increases acetylated HSP90 (Ac-HSP90) levels, causing its dissociation from cochaperone p23, which leads to failure of glucocorticoid receptor protein maturation, with important consequences for the transcription mechanism.60 Another consequence of the increased Ac-HSP90 levels is the dissociation of the complex between HDAC6 and heat shock transcription factor 1-(HSF1)–HSP90 which in turn triggers the activation of HSF1 and the expression of crucial cellular chaperones.61
HDAC6 also performs its deacetylating activity on other two protein substrates, namely, peroxiredoxins I (Prx I) and II (Prx II), both of which are deeply implicated in redox regulation processes.62 At low H2O2 concentrations, peroxiredoxins behave as antioxidants;63,64 on the contrary, increased levels of H2O2 cause the oxidation of their cysteine residue to the corresponding sulfonic acid, prompting the generation of high-molecular-mass protein complexes. Since acetylated Prx is more effective in reducing H2O2 levels than its non-acetylated counterpart, it is likely that inhibiting HDAC6 function may increase the antioxidant activity of these proteins.
Beyond its interaction with non-histone substrates, HDAC6 interacts with diverse proteins (e.g., ubiquitin, tau, IIp45, and EGFR) through protein–protein interactions, which modulate its deacetylase activity.14,65
Owing to its unique functional and structural characteristics, HDAC6 deserves a special role in the armamentarium of epigenetic targets. Accordingly, several research efforts have been devoted to the development of novel, potent, and selective HDAC6 inhibitors for several disease states.66−70 An increasing number of reports highlight the potential of selective HDAC6 inhibitors in noncancerous conditions and rare diseases.14 In particular, the role of this HDAC isoform, acting as a key player in diverse inflammatory states and a crucial regulator of immune response, is particularly attractive.71,72 In this context, recently reported X-ray cocrystal structures of HDAC6 in complex with selective inhibitors will further help refining the design of novel chemical entities with improved potency and selectivity features.69,73−76 This Perspective will examine the multifaceted role of HDAC6 in CF and highlight the potential of selective HDAC6 inhibitors as innovative therapeutic options for the treatment of several aspects of this disabling and lethal rare disease.
4. The Multifaceted Role of HDAC6 in Cystic Fibrosis
4.1. Impaired Microtubule Acetylation in Cystic Fibrosis
As mentioned in the previous paragraphs, defective CFTR is responsible for reduced transepithelial chloride transport, hyperabsorption of sodium, dehydration of epithelial surfaces, and altered inflammatory responses.77−79 Interestingly, mutated CFTR has also been related to perinuclear free cholesterol accumulation as a result of defective endosomal transport.80 The effectiveness of endosomal transport is a direct consequence of the microtubule network stability and the bidirectional endocytic trafficking (Figure 5).81,82 In particular, the microtubule network undergoes extensive post-translational modifications (PTMs), including acetylation, polyglutamylation, polyglycylation, and carboxy terminal cleavage. PTMs are largely responsible for fine-tuning of the microtubule network stability as well as regulation of motor proteins and recruitment of microtubule-associating proteins.83−86 Acetylation is a reversible process controlled by histone acetyltransferases, which exert their activity on residue Lys40 of α-tubulin.87−89 In contrast, the histone deacetylase enzymes are their counterparts appointed for the hydrolysis of N-acetyl groups from pertinent Lys residues. In particular, α-tubulin was the first acknowledged HDAC6 protein substrate. Accordingly, increased expression of HDAC6 promotes tubulin hypoacetylation and chemotactic cell movement (Figure 5).90−92 Acetylated α-tubulin also finely controls motor-based trafficking mediated by motor proteins, including kinesin-1 and dynein (Figure 5).93 Very recent studies also identified and characterized the disordered N-terminal region of HDAC6 as a microtubule-binding domain, with a microtubule-binding motif spanning two positively charged areas including residues Lys32 to Lys58. The deacetylase and microtubule-binding domains display crucial crosstalk that is critical for the recognition and effective in vitro and in vivo deacetylation of free tubulin dimers, thus revealing the complexity of the recognition process between tubulin and HDAC6 and demonstrating once again that domains external to the tandem catalytic core are crucial for effective substrate deacetylation.94 Therefore, perturbation of the HDAC6 function is strongly implicated in the alteration of tubulin function.
Figure 5.
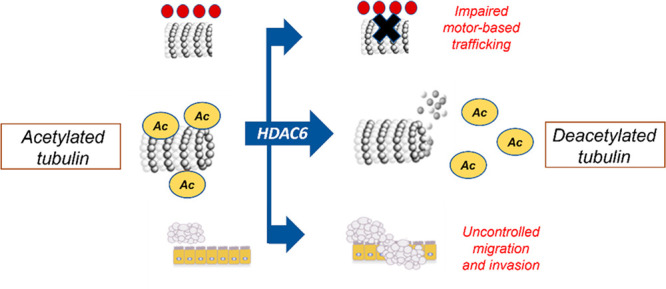
Effects of HDAC6 on α-tubulin, trafficking, and invasion.
This evidence supported the hypothesis that the microtubule acetylation status is responsible for the defective endocytic trafficking and perinuclear cholesterol accumulation detected in CF.15 The pivotal function of microtubule regulation in CF was demonstrated in cell and tissue models, and endoplasmic reticulum (ER) stress was recognized as one of the causes of the microtubule alteration in the CF epithelium.15,17
In particular, to investigate the mechanisms causing reduced trafficking in the epithelial cells in the case of defective CFTR function, the levels of Ac-tubulin were assessed in IB3 cells (human epithelial cells bearing the ΔF508 mutation) and CFTR-corrected S9 cells by densitometry analysis. As expected, reduced levels were unveiled in IB3 cells, as was perinuclear cholesterol accumulation with respect to WT S9 cells. Reduced Ac-tubulin levels were also confirmed in a primary tissue CF model, namely, the mouse nasal epithelium (MNE), where a 40% decrease in Ac-tubulin level was registered in Cftr–/– mice relative to WT mice.15
Since comparable HDAC6 expression levels were found in IB3 and S9 cells, the decreased Ac-tubulin content in CF cells and tissue was possibly ascribable to defective HDAC6 regulation in the absence of CFTR function. To assess whether HDAC6 inhibition would revert the CF phenotype, both IB3 and S9 cells were subjected to treatment with the selective HDAC6 inhibitor tubastatin A (TubA, 16; Figure 9 and Table 1) at 10 μM for 24 h. TubA-treated IB3 cells displayed increased levels of Ac-tubulin comparable to those for S9 cells as well as relocation of accumulated perinuclear cholesterol, as detected by monitoring the trafficking of fluorescent 25-[N-[(7-nitro-2,1,3-benzoxadiazol-4-yl)methyl]amino]-27-norcholesterol (NBD-cholesterol). A further confirmation that the effects of TubA were only related to selective HDAC6 inhibition was provided by knockdown of HDAC6 expression in both IB3 and S9 cells using shRNA against HDAC6. In support of the clear role of HDAC6, the silenced IB3 cells displayed increased Ac-tubulin levels, although the effects of acute HDAC6 inhibition through pharmacological treatment were more marked than the effects of gene silencing (3-fold vs 1.5-fold increase in Ac-tubulin content, respectively).15
Figure 9.

Structures of HDAC inhibitors 16–24.
Table 1. IC50 Values for HDAC Inhibitors 16–30 on HDAC6 and Other Isoforms.
| IC50 (μM) |
||
|---|---|---|
| compound | HDAC6 | other HDACs |
| tubastatin A (16)170 | 0.015 | HDAC1, 16.4 |
| HDAC2, >30 | ||
| HDAC3, >30 | ||
| HDAC8, 0.85 | ||
| sodium 4-phenylbutyrate (17)171 | HDAC1, 162 | |
| resveratrol (18)114 | 32% (100 μM)a | HDAC1, 39% (100 μM)a |
| HDAC2, 21% (100 μM) | ||
| HDAC4, 29% (100 μM) | ||
| HDAC4, 50% (100 μM) | ||
| HDAC4, 23% (100 μM) | ||
| propionic acid (19) | – | – |
| butyric acid (20)172 | – | HDAC1, 300 |
| HDAC2, 400 | ||
| HDAC7, 300 | ||
| valproic acid (21)171 | – | HDAC1, 40 |
| entinostat (22)173 | >100 | HDAC1, 0.18 |
| HDAC3, 0.74 | ||
| trichostatin A (23)174 | 0.009 | HDAC1, 0.006 |
| romidepsin (24)175 | 0.79 | HDAC1, 0.0016 |
| HDAC2, 0.0039 | ||
| SAHA (25)176 | 0.033 | HDAC1, 0.033 |
| HDAC2, 0.096 | ||
| HDAC3, 0.020 | ||
| HDAC8, 0.540 | ||
| MC2625 (26)150 | 0.01 | HDAC1, 1.42 |
| HDAC2, 1.77 | ||
| HDAC3, 0.080 | ||
| HDAC8, 0.61 | ||
| MC2780 (27)150 | 0.011 | HDAC1, 2.9 |
| HDAC2, 2.1 | ||
| HDAC3, 10.8 | ||
| HDAC8, 1.2 | ||
| CDK506 (28)68 | 0.041 | HDAC1, 11.4 |
| panobinostat (29)177 | 0.015 | HDAC1, 0.015 |
| HDAC3, 0.015 | ||
| HDAC8, 0.55 | ||
| tubacin (30)92 | 0.004 | HDAC1, 1.40 |
| HDAC2, 6.27 | ||
| HDAC3, 1.27 | ||
| HDAC8, 1.27 | ||
Overall inhibition of human HDAC enzymes in HeLa nuclear extracts.
As mentioned before, chronic ER stress has been related to decreased Ac-tubulin content in CF. ER stress in CF cells has been associated with misfolding of ΔF508 CFTR and also represents a trigger for aggresome formation.11,95−97 Accordingly, it has been shown that thapsigargin, a molecule leading to ER stress and activation of the unfolded protein response,98 is able to reduce Ac-tubulin levels and recapitulate the cholesterol accumulation phenotype in S9 cells.15 The same study also showed that IB3 cells and MNE from Cftr–/– mice display reduced Ac-tubulin levels and higher levels of GRP78, an inherent marker of ER stress.99 To further validate the role of ER stress associated with the CF phenotype, it was demonstrated that ΔF508 CFTR corrector C18100 (10 μM for 72 h) was able to increase the Ac-tubulin content to a comparable extent with respect to TubA; however, the effect on cholesterol mobilization was inferior to that exerted by TubA treatment.15 As a further effort to investigate more specific molecular mechanisms linking ER stress and tubulin acetylation status, the role of phosphatidylinositol 3-kinase p110α (PIK3CA) was examined because of its direct ability to associate with HDAC6 and respond to diverse cellular stresses.101,102 Accordingly, the use of the PIK3CA inhibitor PIK-75 (0.5 μM for 24 h) evoked a relevant increase of Ac-tubulin levels in IB3 cells and led the cholesterol distribution to a more WT phenotype.15
Interestingly, the use of 4-phenylbutyrate (4-PB, 17; Figure 9 and Table 1) as an ER stress reliever at 1 mM for 48 h103,104 resulted in a significant increase in Ac-tubulin levels and reduced perinuclear accumulation of cholesterol in IB3 cells. Since 4-PB also acts as a pan-HDAC inhibitor,105,106 the role of an HDAC6-mediated mechanism for the observed effects cannot be ruled out.
A few years later, more direct evidence was provided that HDAC6 activity influences the levels of membrane cholesterol in CF epithelium and that the electrochemical measurement of these levels is closely linked to genetic and pharmacological CFTR correction.107 In particular, the evidence demonstrating that high cholesterol levels in CF cells are correlated with the CFTR phenotype and depend on de novo cholesterol synthesis108−110 prompted the hypothesis of an adaptative response to the loss of CTFR function. On the basis of this assumption, it is reasonable to presume that electrochemical determination of membrane cholesterol levels can serve as a biomarker to observe the adjustment of intracellular events upon administration of novel therapeutics in CF.
First, the expression of HDAC6 was knocked out from a ΔF508 mouse, and then the membrane cholesterol amount was electrochemically evaluated by the use of an electrode with cholesterol oxidase. The study demonstrated that HDAC6 depletion causes an almost 2-fold increase in membrane cholesterol levels, thus restoring correct cholesterol processing. The effects of pharmacological inhibition of HDAC6 using TubA (10 μM for 24 h) on membrane cholesterol levels were also assessed in CF bronchial epithelial cells (CFBE) through a double-pulse technique.107 These experiments conclusively demonstrated that tubulin acetylation is a crucial pathway in pathophysiology of several CF phenotypes and that HDAC6 inhibition holds promising potential as innovative therapeutic option for restoring those pathways to a more WT-like pattern.
As a further support to this evidence, it was very recently demonstrated that resveratrol (RSV, 18; Figure 9 and Table 1) is able to restore intracellular transport in CF epithelial cells.111 RSV is well-characterized as an activator of SIRT1,112 although it also acts as a pan-HDAC inhibitor and antagonist of peroxisome proliferator-activated receptors PPARγ and PPARα.113,114 RSV treatment (50 μM, 24 h) in IB3 cells and control S9 cells effectively reversed the perinuclear cholesterol accumulation, suggesting that intracellular transport is restored.111
To assess whether a correctly functioning CFTR was essential for the intracellular transport correction in CF cells, RSV was tested in cells isolated from Cftr–/– mice to check the improvement of cholesterol mobilization in MNE. RSV (50 μM for 24 h) significantly lowered cholesterol accumulation in Cftr–/– MNE cells, demonstrating that CFTR function is not critical for RSV efficacy. The study also showed that RSV at the same concentration is able to trigger microtubule formation in both IB3 cells and human nasal epithelial (HNE) cells from ΔF508 patients and that these effects are not mediated by sirtuin signaling, as demonstrated by cotreatment with the SIRT1 inhibitor EX-527 (IB3 cells, 1 μM for 24 h), which did not have a significant impact on the efficacy of RSV. It was then hypothesized that the influence of RSV on microtubules could be ascribable to PPARγ activation and HDAC6 inhibition. Accordingly, cotreatment with the PPARγ inhibitor GW-9662 (IB3 cells, 20 μM, 24 h) demonstrated that the effects of RSV in promoting intracellular transport are partially mediated by PPARγ receptors. Conclusive support for the role of HDAC6 inhibition on the effects of RSV came from the evidence that treatment with RSV (50 μM, 24 h) markedly enhances tubulin acetylation in IB3 cells.111
Although additional studies are needed, the observation that RSV reverses CF cellular phenotypes more effectively than ibuprofen115 suggests that RSV could represent a valuable anti-inflammatory treatment with a multifaceted profile that is in part ascribable to its ability to inhibit HDAC6. However, other reports demonstrate that RSV is able to prevent the accumulation of acetylated tubulin resulting from mitochondrial damage, precluding inflammasome activation.116 It should also be mentioned that the main limitation of RSV is its low water solubility and the impossibility of reaching plasma levels even remotely close to the concentration of 50 μM utilized in this and other cellular studies.117
Taken together, these data validate the role of HDAC6 as a key player in microtubule acetylation status and trafficking in CF. TubA, behaving as a selective HDAC6 inhibitor, and RSV, through pan-HDAC inhibition and PPARγ activation, served as chemical probes for a deeper understanding of CF cell signaling related to tubulin acetylation status, thus providing a glimpse of a new therapeutic avenue for effective reversal of CF phenotype.
4.2. HDAC6 and Bacterial Clearance in Cystic Fibrosis
Among protein PTMs, reversible acetylation deserves a primary role among the processes involved in the responses to environmental stimuli as well as cellular homeostasis. In particular, the evidence supporting dynamic regulation of epigenetic marks by environmental cues has prompted research efforts toward the elucidation of epigenetic pathways in microbial infections.118 Accordingly, huge progress has been made lately on the bacterial regulation of the host acetylation system, with a special focus on the mechanisms engaged to evade immune response.119,120 In general, pathogenic microorganisms utilize a limited number of mechanisms involving acetylation of histone and non-histone proteins, including modulation of the activity and expression of HAT and HDAC enzymes thorough diverse bacterial effectors as well as the production of metabolites regulating the so-called “acetylome”.118 In this context, isoform HDAC1 may be considered the crucial acetylation system targeted by pathogens for evading the host immune system, as demonstrated by the efficacy of silencing of HDAC1 expression or enzymatic inhibition in restoring defense gene expression.121 More recently, it was demonstrated that the quorum-sensing signal generated by PA, namely, 2-aminoacetophenone, prompted the expression of HDAC1 in human THP-1 monocytes, thus causing hypoacetylation of histone H3K18; this in turn led to reduced induction of inflammatory cytokines and chemokines, such as TNF, IL-β, and MCP-1. The process was also demonstrated to be completely reverted by HDAC1 inhibition, thus unambiguously highlighting the role of this isoform in promoting tolerance to PA (Figure 6).122
Figure 6.

Regulation of antimicrobial responses by HDAC inhibitors.
As mentioned above, metabolites produced by pathogenic bacteria can also modulate the host acetylation system. In particular, it has been demonstrated that short-chain fatty acids (SCFAs) such as propionic acid (19, Figure 9 and Table 1) and butyric acid (20, Figure 9 and Table 1) produced by anaerobic bacteria behave as inhibitors of class I/II HDACs, thus regulating several facets of immune response (Figure 6).123 Accordingly, treatment with butyrate prompted histone H3 acetylation at the promoter region of the transcription factor FoxP3, which is crucial in the differentiation of regulatory T cells. In this context, isoforms HDAC6 and HDAC9 demonstrated a suppressive role in FoxP3 induction (Figure 6).124
Around a decade ago, pioneering studies started unveiling the effect of HDAC inhibitors on microbial infection. In particular, it was demonstrated that the use of valproic acid (VPA, 21; Figure 9 and Table 1) increased mortality in mice upon nonsevere infection by Klebsiella pneumoniae or Candida albicans(125) and that HDAC inhibition was able to reduce phagocytosis and killing of E. coli and SA by bone-marrow-derived macrophages.126
These initial observations supporting an unfavorable outcome when HDAC inhibitors are used during bacterial infection were controverted by subsequent studies. Indeed, it was clarified that the effects of HDAC inhibitors on antibacterial response strongly relied upon the isoform selectivity and timing of the treatment. Interestingly, in 2015 Ariffin et al. highlighted that only the HDAC6-selective inhibitor TubA and not MS-275 (entinostat, 22; Figure 9 and Table 1), which specifically targets class I HDACs, enhanced bacterial killing by macrophages (Figure 6).127 The same study also highlighted that acute treatment with HDAC inhibitors at the insurgence of bacterial infection boosts mitochondrial ROS production by human macrophages, which is in line with an increased antibacterial response. Almost parallel studies also showed that HDAC6-selective inhibition through the use of TubA promoted bacterial clearance, reduced pro-inflammatory cytokine production, restored innate immune cell populations in the bone marrow, and improved survival in a mouse model of sepsis (Figure 6).128,129 These data, together with the evidence linking HDAC6 to control of mitochondrial function and stimulation of mitochondrial ROS production, suggest a crucial role for HDAC6 in regulating bacterial clearance,130−132 while class I HDACs seem unlikely to exert such an effect.
While the previously described reports mainly focus on the effects of HDAC inhibitors on immune cell function, several experiments analyzing the responses of epithelial cells to bacterial challenges support the role of HDAC inhibitors as effective regulators of the production of cationic antimicrobial peptides (CAMPs) (Figure 6).133 In particular, the expression of defensins and cathelicidins, the two main classes of mammalian CAMPs, is robustly triggered by HDAC inhibitors in colonic and airway epithelial cells.134−138 Although induction of CAMPs by several HDAC inhibitors has been widely documented across several types of epithelial cells, the mechanisms underlying this effect are mostly unclear or at the stage of preliminary investigation.
Collectively, these findings underline the dual-faceted potential of HDAC6 inhibitors as anti-infective agents through either promotion of innate immune-mediated bacterial clearance or reduction of the damage induced by excessive inflammation. These data also highlight the potential to use HDAC6-selective inhibitors for treatment of chronic inflammatory diseases of the airways.
To further validate these premises, a recent study examined the outcome of Hdac6 depletion on both the CF inflammatory response and the bacterial load in a model of infection using clinical PA isolates embedded in agarose beads, which effectively recapitulates CF phenotype.18 In that study, genetic ablation was preferred over pharmacological inhibition in order to avoid contributions ascribable to off-target effects. The loss of Hdac6 was demonstrated to increase the rate of bacterial clearance in CF mice, thus restoring CF responses to bacterial challenge. These data, coupled with the limited weight loss and the regulation of neutrophil recruitment upon Hdac6 depletion demonstrated in the same study, further depict HDAC6 as a key regulator in several secondary phenotypes associated with impaired CFTR function and support the potential benefits of using HDAC6-selective inhibitors in CF patients independently of CFTR phenotype.
4.3. HDAC6 in the Regulation of Inflammatory and Fibrotic Phenotypes in Cystic Fibrosis
The complex CF phenotype is associated with several clinical manifestations, although lung disease, chronic airway inflammation, and infection are by far the main causes of morbimortality. A common thread of the end stage of CF-associated lung disease is represented by extensive pulmonary fibrosis, characterized by increased collagen deposition and tissue remodeling.
Fibrosis is generated by overgrowth of various tissues and an increased amount of myofibroblasts and it is flanked by anomalous deposition of extracellular matrix components, a process known as epithelial–mesenchymal transition. EMT is a crucial process in CF involving loss of cell–cell junctions and polarization of cell-surface molecules of epithelial cells, which thus acquire the characteristics of mesenchymal cells.5,139 Beyond their role as a key scaffold for parenchymal tissue, fibroblasts can secrete key inflammatory chemoattractants, such as chemokine C–C ligand-2 (CCL-2) and CCL-8, interleukin-16 (IL-16) and IL-8, regulated on activation normal T-cell expressed and secreted (RANTES), and monocyte chemotactic protein-1 and -2.140,141 The dysregulated fibroblast phenotype in CF was investigated in a bleomycin-induced fibrosis model in ΔF508 homozygous mice.5 An increased release of TGF-β1, a potent EMT inducer, was demonstrated in CF lungs. Members of the TGF-β family are potent inducers of EMT, and overexpression of TGF-β1 was also previously linked with a more severe CF lung phenotype.142 Moreover, CF lungs challenged with bleomycin were also shown to overexpress TIMP-1, a predictive marker of tissue remodeling reflecting a protease imbalance in damaged lungs.5,143 The study also demonstrated that upon inflammatory stimulation, mRNA and protein expression levels of proinflammatory mediators (CCL-2, TNF-α, IL-16 and IL-18) were higher in CF than in wild-type fibroblasts, thus demonstrating that dysregulated proinflammatory and fibrotic status in CF fibroblasts is an extensive and multifaceted process involving several signaling pathways and transcription factors.5
The demonstration of the close crosstalk between inflammation and fibrosis in CF opened up possibilities for novel disease-modifying treatments specifically targeting these mechanisms.
4.3.1. HDAC6 and Inflammatory Responses in Cystic Fibrosis
In the context of inflammation, increasing evidence is available highlighting the key role of HDACs and HDAC inhibitors in the regulation of chemokines and cytokines (Figure 7).144 HDAC inhibitors, in particular, have been shown to downregulate several pathways involving cytokines, chemokines, and growth factors, thus controlling key inflammatory pathways in inflammatory diseases with different origins.145,146 With respect to cytokines, the pan-HDAC inhibitors trichostatin A (TSA, 23; Figure 9 and Table 1) and romidepsin (24, Figure 9 and Table 1) were found to suppress IFN-α-induced transcriptional responses, thus regulating both innate and adaptative immunity,147,148 while the pan-HDAC inhibitors SAHA (vorinostat, 25; Figure 10 and Table 1) and TSA were demonstrated to decrease the expression of IFN-γ in various disease models.149 Interestingly, a predominant role of isoform HDAC6 in regulation of cytokines has been unveiled by several studies. Accordingly, the HDAC3/6/8 inhibitor MC2625 (26, Figure 10 and Table 1) and the HDAC6-selective inhibitor MC2780 (27, Figure 10 and Table 1) were shown to downregulate IL-1β expression in epithelial, fibroblast, and myogenic cell lines,150 and HDAC6 was identified as a key player in the regulation of NF-κB activity, which is related to IL-1β signaling.72 Selective HDAC6 inhibition by TubA was also linked to inhibition of IL-6 in an arthritis mouse model.151 However, the effects of HDAC6 inhibition on the production of the anti-inflammatory cytokine IL-10 are still controversial.150,152
Figure 7.
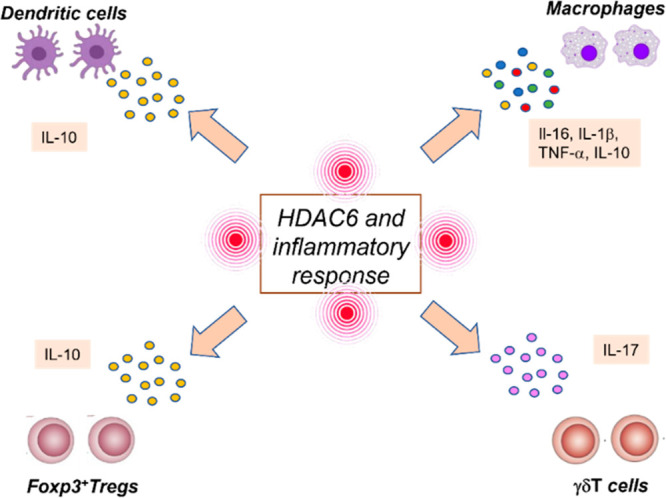
Role of HDAC6 in the regulation of inflammatory cells (macrophages, dendritic cells, γδT cells, FoxP3+ regulatory T cells) and cytokines (IL-6, IL-1β, TNF-α, IL-10, and IL-17).
Figure 10.

Structures of HDAC inhibitors 25–30.
The involvement in HDACs in chemokine signaling was also supported by several experiments. In particular, it was demonstrated that pan-HDAC inhibition induced the expression of CCL-2153 and that both HDAC6-selective inhibition with CKD506 (28, Figure 10 and Table 1) and pan-HDAC inhibition were able to decrease CCL-4 expression.154,155
HDAC6 has also been shown to play a critical role in the regulation of inflammatory cells. For example, inhibition of HDAC6 functions causes stimulation of inflammatory antigen-presenting cells with a key role in the induction of T-cell activation and T-cell tolerance.72,156,157 Moreover, depletion of HDAC6 has been demonstrated to endorse the suppressive activity of FoxP3+ regulatory T-cells in inflammation models and to increase the population of γδT cells, which are involved in the release of IL-17.158
According to these data, targeted HDAC6 inhibition has been increasingly proposed over the last year as a promising therapeutic strategy toward inflammatory disorders, including airway inflammation.71 For example, TubA has been demonstrated to efficiently relieve airway inflammatory state and hyper-responsiveness in a chronic mouse model of allergy, thus opening up the possibility for a novel therapeutic option in asthma treatment.159 Also, TubA significantly inhibited cigarette-smoke-induced airway dysfunction, thus paving the way to an innovative therapeutic strategy against chronic obstructive pulmonary disease (COPD).160
With respect to CF, the potential usefulness of HDAC inhibitors in correcting the ΔF508 CFTR variants was recently evaluated in several studies.16,161,162 In particular, the pan-HDAC inhibitors panobinostat (29, Figure 10 and Table 1) and romidepsin provided functional correction of Class II and III CFTR variants, as they were able to cause dose-dependent enhancement of the expression of ΔF508 CFTR and restore the activity of the cell-surface chloride channel in primary human bronchial epithelial cells.161 Most importantly, the effects of the combination of panobinostat and romidepsin with lumacaftor were evaluated. Panobinostat (2.5 nM) and romidepsin (0.6 nM) were combined with a higher dose of VX809 (3 μM). While the combination with romidepsin produced an additive effect on the trafficking efficiency of ΔF508 CFTR, the combination with panobinostat unveiled a synergism in improving the trafficking efficiency after immunoblotting and quantification analysis of CFTR expression in human CFBE-ΔF508 cells.161 A more inflammation-oriented study evaluated the effect of pan-HDAC inhibition in PA lipopolysaccharide (PA-LPS)-induced airway inflammation and CF lung disease.16 HDAC inhibition through SAHA was shown to control TNF-α-induced IL-8 and NF-κB reporter activity, suggesting that HDAC inhibitors regulate TNF-α-induced inflammation and CF lung disease. CFTR–/– mice treated with PA-LPS (20 μg/mouse) were used as a model for CF-induced lung infection. Intratracheal administration of SAHA (100 μg/mouse daily for 3 days) demonstrated that SAHA was able to efficiently control PA-LPS-induced IL-6 levels in mice treated for 1 day, while treatment for 2 or 3 days was shown to be detrimental. The outcome of a lower dose (50 μg/mouse) was then assessed, demonstrating that PA-LPS-induced IL-6 and myeloperoxidase levels were efficiently reduced by SAHA and highlighting its role in controlling chronic airway inflammation and neutrophil activity. Moreover, the same study demonstrated that NF-κB activation (as a marker of inflammation) and Nrf2 regulation (as a marker of antioxidant response) are effectively corrected by SAHA treatment.16 In agreement with previous studies, SAHA demonstrated the ability to restore trafficking in misfolded ΔF508 CFTR and to promote FoxP3+ T-reg activity, thus providing conclusive evidence of the role of HDAC inhibition in modulation of innate and adaptive immune response linked to pathogenesis and progression in CF-related inflammatory lung disease.16
More recent evidence pointed out the prevalent role of HDAC6 in the beneficial effect of HDAC inhibition especially with respect to the inflammatory phenotype in CF, thus supporting the evaluation of HDAC6-selective inhibitors in this context. Accordingly, it was recently demonstrated that HDAC6 depletion was able to improve CF mouse airway inflammatory responses to bacterial challenge.18 A clinical PA isolated by means of the agar bead model of infection was employed. This model nicely recapitulates the CF phenotype related to an excessive inflammatory response, with particular reference to the increased neutrophil recruitment.
The loss of Hdac6 on a CF background increased the rate of bacterial clearance, and this effect was ascribed to a crucial regulatory step in CF immune responses. Accordingly, it was shown that HDAC6 controls neutrophil recruitment in CF mice.18 Reduced cytokine production in Hdac6-depleted CF mice was also demonstrated in the same study.
4.3.2. HDAC6 and Fibrotic Remodeling in Cystic Fibrosis
Aberrant HDAC activity and/or expression is detected in fibrotic diseases, and increasing amounts of data support the role of HDACs in the initiation and evolution of fibrotic phenotype in several organs, including lungs, heart, liver, and kidneys.163
More specifically, the regulation of TGF-β1 by HDAC6 is crucial in the initiation and progression of fibrotic diseases through EMT (Figure 8).14,164,165 Recent data provided evidence that HDAC6 blockade by siRNA or tubacin (30, Figure 10 and Table 1) is able to reduce TGFβ1-induced EMT markers and SMAD3 activation in response to TGF-β1. Since SMAD3 is a key player in TGFβ1 signaling, its deactivation impairs HDAC6-dependent deacetylation of α-tubulin, highlighting the crucial importance of HDAC6 in EMT through the TGF-β1–SMAD3 signaling pathway.19 Several lines of evidence demonstrated the effectiveness of pan-HDAC inhibitors (including romidepsin, SAHA, and panobinostat) against idiopathic lung fibrosis (IPF) and fibrotic diseases, mainly based on the decrease of myofibroblast differentiation and fibroblast proliferation prompted by TGF-β1.166−168 More recent reports pointed out the efficacy of a targeted HDAC6 approach in IPF. Accordingly, TubA was shown to protect mice from lung fibrosis by repressing TGF-β1-induced collagen expression and diminished Akt phosphorylation,169 and novel indoline-based inhibitors demonstrated efficacy in a human lung model of TGF-β1-dependent fibrogenesis and the ability to inhibit fibrotic sphere formation.73
Figure 8.
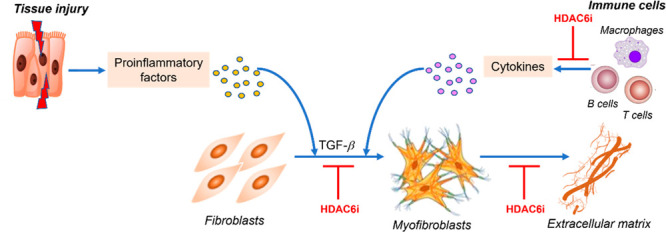
Role of HDAC6 in regulation of the fibrotic process. Injured tissues or activated immune cells induce the production of profibrotic factors, in turn prompting fibroblast differentiation into myofibroblasts, which actively synthesize extracellular matrix. HDAC6 inhibitors negatively regulate the fibrotic process by acting at different levels of the profibrotic cascade.
These data foster further investigation of HDAC6-selective inhibitors as potential therapeutic tools to tackle the fibrotic process in CF and other diseases.59,163,166,169
5. Perspectives
Increasing evidence points to unveiling a new guise for epigenetic targets as key players in many nononcological diseases, particularly rare diseases. In the context of CF, the past decade has represented a phase of hope for patients, with increased scientific awareness toward novel CF therapeutics. Accordingly, researchers in academia and pharmaceutical companies are devoting intense research efforts to leverage new technologies and scientific knowledge to successfully develop new effective therapies that are changing the patient quality and expectation of life, although there is still the need for great improvement and simplification of the therapy.
The void of knowledge on how the loss of CTFR function is linked to many of the secondary manifestations of CF remains a crucial gap to be filled. Also, there is still a lack of understanding of the molecular underpinnings and crosstalk mechanisms related to inflammatory response, immune system engagement, and microbial infections in CF.
The objective of developing a new inflammatory therapy for CF is closely linked to the necessity of identifying a novel therapeutic option that could allow the resolution of the prolonged and aggressive inflammatory phenotype associated with CF but also guarantee sustained immune response for the eradication of infections.
During the past decade, clear evidence has been provided that the protein acetylation status of the host is dynamically regulated during infection and helps establish an efficient response against pathogens. Recent evidence has also pointed out the role of microtubule acetylation as a crucial regulator of intracellular transport in CF cells and CF-related inflammatory responses.
In this frame, HDAC enzymes appear as key players because of their role in modulating the acetylation balance in several pathophysiological conditions. HDAC6, in particular, displays unique features owing to its distinctive structure, localization and nature of substrates. The concept that several CF phenotypes can be corrected by HDAC6 inhibition is of pivotal importance for those patient populations bearing CFTR mutations not approachable with currently available potentiators and/or correctors. Moreover, HDAC6 inhibitors could also flank CFTR-targeted therapies in order to improve the management of inflammation, fibrosis and infection in CF.
The effects of HDAC6 partially selective modulators and pan-HDAC inhibitors have been recently examined in several cell and animal models of rare diseases, often providing substantial proof-of-concept for their utility as innovative therapeutic option. However, none of the clinically approved HDAC displays selectivity for HDAC6. This means that they cannot work as effective chemical probes to dissect the role of HDAC6 in rare diseases. Moreover, they cannot be considered ready-to-use therapeutics for treating these patients especially due to their therapeutic index, which would not be compatible with a chronic drug administration.
In this context, the increasingly appearing X-ray cocrystal structures of HDAC6 enzyme in complex with several HDAC6 inhibitors are allowing more effective structure-based design strategies, which will certainly help medicinal chemists to drive the drug discovery trajectory toward potent, selective and safer HDAC6 inhibitors, to be employed as novel therapeutic option in several disease states.
Acknowledgments
The authors thank Fondazione Fibrosi Cistica (FFC) for financial support through Grant FFC#20/2020: Harnessing selective histone deacetylase 6 (HDAC6) inhibition to tackle inflammation and fibrotic remodeling in cystic fibrosis. We also acknowledge MIUR Grant Dipartimento di Eccellenza 2018–2022 (l. 232/2016) to the Department of Pharmacy, University of Naples Federico II.
Glossary
Abbreviation Used
- 4-PB
4-phenylbutyrate
- Ac-HSP90
acetylated HSP90
- ALI
acute lung injury
- BALF
broncho-alveolar lavage fluid
- BDNF
brain-derived neurotrophic factor
- BDNG-YFP
BDNF tagged with a yellow fluorescent protein
- CAMPs
cationic antimicrobial peptides
- CCL
chemokine C–C ligand
- CF
cystic fibrosis
- CFBE
CF bronchial epithelial cells
- CFTR
cystic fibrosis transmembrane conductance regulator
- AMP
adenosine monophosphate
- CMT
Charcot–Marie–Tooth disease
- CNS
central nervous system
- COPD
chronic obstructive pulmonary disease
- COX
cyclooxygenase
- DD
deacetylation domain
- dHMN2B
distal hereditary motor neuropathy 2B
- DNMT
DNA methyltransferase
- DRG
dorsal root ganglion
- E. coli
Escherichia coli
- EGFR
epidermal growth factor receptor
- EMT
epithelial–mesenchymal transition
- ER
endoplasmic reticulum
- FDA
U.S. Food and Drug Administration
- GlyRS
Glycyl tRNA synthetase
- GR
glucocorticoid receptor
- GRP78
78 kDa glucose-regulated protein
- HAT
histone acetyltransferase
- HD
Huntington’s disease
- HDAC
histone deacetylase
- HMN
hereditary motor neuropathies
- HMT
histone methyltransferase
- HNE
human nasal epithelial
- HSF1
heat shock transcription factor 1
- HSP90
heat shock protein 90
- HSPB1
heat-shock protein B1
- IB3
human epithelial cells bearing the ΔF508 mutation
- IFN
interferon
- IL
interleukin
- ILD
interstitial lung disease
- IPF
idiopathic pulmonary fibrosis
- iPSCs
pluripotent stem cells
- IRD
inherited retinal disorder
- JIP1
JNK-interacting protein 1
- KAT
lysine acetyltransferase
- KC
keratinocyte-derived chemokine
- LOX
lysyl oxidase
- LPS
lipopolysaccharide
- LTB4
leukotriene B4
- Lys
lysine
- MCP-1
monocyte chemoattractant protein-1
- MeCP2
X-linked methyl-CpG-binding protein 2
- Mip-2
microphage inflammatory protein-2
- miRNA
microRNA
- MNE
mouse nasal epithelium
- MRSA
methicillin-resistant Staphylococcus aureus
- NBD-cholesterol
25-[N-[(7-nitro-2,1,3-benzoxadiazol-4-yl)methyl]amino]-27-norcholesterol
- ncRNA
noncoding RNA
- NES
nuclear export signal
- NFκB
nuclear factor kappa light-chain enhancer of activated B cells
- Nrf2
nuclear factor erythroid 2-related factor 2
- NSAID
nonsteroidal anti-inflammatory drug
- NTG
nontransgenic mice
- NTM
nontuberculous mycobacteria
- p97/VCP
p97/valosin-containing protein
- PA
Pseudomonas aeruginosa
- PIK-75
PIK3CA inhibitor
- PIK3CA
phosphatidylinositol 3-kinase p110α
- PLAP
phospholipase A2 inactivating protein
- PPAR
peroxisome proliferator-activated receptor
- Prx
peroxiredoxin I
- PTM
post-translational modification
- RANTES
regulated on activation normal T-cell expressed and secreted
- ROS
reactive oxygen species
- RP
retinitis pigmentosa
- RTT
Rett syndrome
- RSV
resveratrol
- SA
Staphylococcus aureus
- SAHA
suberoylanilide hydroxamic acid
- SCFA
short chain fatty acid
- SE
serine–glutamic acid
- shRNA
shot hairpin RNA
- SIRT
sirtuin
- SMAD3
small mothers against decapentaplegic family member 3
- TGF-β
transforming growth factor-β
- TIMP-1
tissue inhibitor of metalloproteinase-1
- TSA
trichostatin A
- Tub A
tubastatin A
- UBP
ubiquitin-specific protease
- VEGF
vascular endothelial growth factor
- VIPN
vincristine-induced peripheral neuropathies
- VPA
valproic acid
- WT
wild type
- ZnF-UBP
ubiquitin C-terminus hydrolase-like zinc finger
Biographies
Simona Barone graduated in Pharmaceutical Chemistry and Technology from the University of Naples “Federico II” in 2021. Currently she is spending a five-month fellowship granted by the Italian Cystic Fibrosis Foundation at the Department of Pharmacy, University of Naples “Federico II”, where she is working on the synthesis of HDAC6-selective inhibitors as a novel therapeutic option for the treatment of cystic fibrosis.
Emilia Cassese graduated in Pharmaceutical Chemistry and Technology from the University of Naples “Federico II” in 2021. During her thesis internship, she was involved in the design and synthesis of antiviral drugs. She is currently working in the same department as a research fellow under the support of the Italian Cystic Fibrosis Foundation on the development of HDAC6 inhibitors for the treatment of cystic fibrosis.
Antonella Ilenia Alfano is currently pursuing her Ph.D. in the SPOTS Lab at the Department of Pharmacy at the University of Naples “Federico II”. She graduated in 2018 in Pharmaceutical Chemistry and Technology. Soon after she spent a six-month fellowship working on the multicomponent synthesis of biologically active compounds via visible-light photocatalysis. Her Ph.D. project is focused on the development of greener and more sustainable synthesis and derivatization of privileged scaffolds in pharmaceutical chemistry through continuous flow approaches. She is currently working on the development of compounds as novel antibacterial and anti-biofilm agents.
Margherita Brindisi received her Ph.D. in Pharmaceutical Sciences from the University of Siena. As a junior researcher in Siena, she worked on developing compounds for the treatment of cancer, viral diseases, and brain disorders. She was a postdoctoral fellow in Professor Arun Ghosh’s research group at Purdue University in 2010–2011 and a Visiting Scientist in the same group in 2016–2017. In April 2019 she was appointed as Assistant Professor at the Department of Pharmacy at University of Naples “Federico II”, where she is working on the development of novel therapeutics as modulators of epigenetic targets in rare disorders, including cystic fibrosis. In the last 5 years, she has been involved in the development of several structural templates for selective HDAC6 inhibition.
Vincenzo Summa obtained his Ph.D. in Organic Chemistry from the University of Wuppertal. In 1996 he joined Merck Research Laboratory-Italy (IRBM). He served as Director in the Medicinal Chemistry Department, leading the team that discovered Isentress, the first-in-class HIV integrase inhibitor (Galien Prize USA and Italy for the best pharmaceutical agent-2008), and Grazoprevir, a pan-genotype HCV protease inhibitor. For these discoveries, he was recognized as a Hero of Chemistry by the American Chemical Society in 2013 and 2017, respectively. In 2009, he became cofounder of IRBM Science Park and Vice President of Drug Discovery. Since 2019 he has been Professor of Medicinal Chemistry at the Department of Pharmacy, University of Naples “Federico II”. His research focuses on the identification of anti-infective agents and novel therapeutics for cancer and rare diseases.
Author Contributions
† S.B. and E.C. contributed equally to this work.
Author Contributions
‡ M.B. and V.S. are co-senior authors.
The authors declare no competing financial interest.
Special Issue
Published as part of the Journal of Medicinal Chemistry special issue “Epigenetics 2022”.
References
- Bye M. R.; Ewig J. M.; Quittell L. M. Cystic fibrosis. Lung 1994, 172 (5), 251–270. 10.1007/BF00164308. [DOI] [PubMed] [Google Scholar]
- Boucher R. C. Relationship of airway epithelial ion transport to chronic bronchitis. Proc. Am. Thorac. Soc. 2004, 1 (1), 66–70. 10.1513/pats.2306018. [DOI] [PubMed] [Google Scholar]
- Doring G.; Flume P.; Heijerman H.; Elborn J. S. Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J. Cystic Fibrosis 2012, 11 (6), 461–479. 10.1016/j.jcf.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Zobell J. T.; Young D. C.; Waters C. D.; Stockmann C.; Ampofo K.; Sherwin C. M.; Spigarelli M. G. Optimization of anti-pseudomonal antibiotics for cystic fibrosis pulmonary exacerbations: I. aztreonam and carbapenems. Pediatr. Pulmonol. 2012, 47 (12), 1147–1158. 10.1002/ppul.22655. [DOI] [PubMed] [Google Scholar]
- Huaux F.; Noel S.; Dhooghe B.; Panin N.; Lo Re S.; Lison D.; Wallemacq P.; Marbaix E.; Scholte B. J.; Lebecque P.; Leal T. Dysregulated proinflammatory and fibrogenic phenotype of fibroblasts in cystic fibrosis. PLoS One 2013, 8 (5), e64341 10.1371/journal.pone.0064341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodas M.; Vij N. The NF-kappaB signaling in cystic fibrosis lung disease: pathophysiology and therapeutic potential. Discovery Med. 2010, 9 (47), 346–356. [PMC free article] [PubMed] [Google Scholar]
- Vij N.; Mazur S.; Zeitlin P. L. CFTR is a negative regulator of NFkappaB mediated innate immune response. PLoS One 2009, 4 (2), e4664 10.1371/journal.pone.0004664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner C. J.; Schultz C.; Mall M. A. Neutrophil elastase and matrix metalloproteinase 12 in cystic fibrosis lung disease. Mol. Cell Pediatr. 2016, 3 (1), 25. 10.1186/s40348-016-0053-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani C.; Assael B. M. Cystic fibrosis: a clinical view. Cell. Mol. Life Sci. 2017, 74 (1), 129–140. 10.1007/s00018-016-2393-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodas M.; Silverberg D.; Walworth K.; Brucia K.; Vij N. Augmentation of S-Nitrosoglutathione controls cigarette smoke-induced inflammatory-oxidative stress and chronic obstructive pulmonary disease-dmphysema pathogenesis by restoring cystic fibrosis transmembrane conductance regulator function. Antioxid. Redox Signaling 2017, 27 (7), 433–451. 10.1089/ars.2016.6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciani A.; Villella V. R.; Esposito S.; Brunetti-Pierri N.; Medina D.; Settembre C.; Gavina M.; Pulze L.; Giardino I.; Pettoello-Mantovani M.; D’Apolito M.; Guido S.; Masliah E.; Spencer B.; Quaratino S.; Raia V.; Ballabio A.; Maiuri L. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12 (9), 863–875. 10.1038/ncb2090. [DOI] [PubMed] [Google Scholar]
- Nichols D. P.; Chmiel J. F. Inflammation and its genesis in cystic fibrosis. Pediatr. Pulmonol. 2015, 50 (S40), S39–S56. 10.1002/ppul.23242. [DOI] [PubMed] [Google Scholar]
- Hinz B.; Celetta G.; Tomasek J. J.; Gabbiani G.; Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 2001, 12 (9), 2730–2741. 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindisi M.; Saraswati A. P.; Brogi S.; Gemma S.; Butini S.; Campiani G. Old but Gold: Tracking the New Guise of Histone Deacetylase 6 (HDAC6) Enzyme as a Biomarker and Therapeutic Target in Rare Diseases. J. Med. Chem. 2020, 63 (1), 23–39. 10.1021/acs.jmedchem.9b00924. [DOI] [PubMed] [Google Scholar]
- Rymut S. M.; Harker A.; Corey D. A.; Burgess J. D.; Sun H.; Clancy J. P.; Kelley T. J. Reduced microtubule acetylation in cystic fibrosis epithelial cells. Am. J. Physiol.: Lung Cell. Mol. Physiol. 2013, 305 (6), L419–L431. 10.1152/ajplung.00411.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodas M.; Mazur S.; Min T.; Vij N. Inhibition of histone-deacetylase activity rescues inflammatory cystic fibrosis lung disease by modulating innate and adaptive immune responses. Respir. Res. 2018, 19 (1), 2. 10.1186/s12931-017-0705-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymut S. M.; Corey D. A.; Valerio D. M.; Erokwu B. O.; Flask C. A.; Kelley T. J.; Hodges C. A. Improved growth patterns in cystic fibrosis mice after loss of histone deacetylase 6. Sci. Rep. 2017, 7 (1), 3676. 10.1038/s41598-017-03931-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenjack J.; Hodges C. A.; Darrah R. J.; Kelley T. J. HDAC6 depletion improves cystic fibrosis mouse airway responses to bacterial challenge. Sci. Rep. 2019, 9 (1), 10282. 10.1038/s41598-019-46555-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B.; Yao T. P.; Nguyen H. T.; Zhuo Y.; Levy D. R.; Klingsberg R. C.; Tao H.; Palmer M. L.; Holder K. N.; Lasky J. A. Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J. Biol. Chem. 2008, 283 (30), 21065–21073. 10.1074/jbc.M802786200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almughem F. A.; Aldossary A. M.; Tawfik E. A.; Alomary M. N.; Alharbi W. S.; Alshahrani M. Y.; Alshehri A. A. Cystic fibrosis: overview of the current development trends and innovative therapeutic strategies. Pharmaceutics 2020, 12 (7), 616. 10.3390/pharmaceutics12070616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch R. M.; Kmit A. H.; Marson F. A.; Levy C. E.; Barros-Filho A. A.; Ribeiro J. D. Association of growth and nutritional parameters with pulmonary function in cystic fibrosis: a literature review. Rev. Paul. Pediatr. 2016, 34 (4), 503–509. 10.1016/j.rppede.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkerman-Nijland A. M.; Akkerman O. W.; Grasmeijer F.; Hagedoorn P.; Frijlink H. W.; Rottier B. L.; Koppelman G. H.; Touw D. J. The pharmacokinetics of antibiotics in cystic fibrosis. Expert Opin. Drug Metab. Toxicol. 2021, 17 (1), 53–68. 10.1080/17425255.2021.1836157. [DOI] [PubMed] [Google Scholar]
- Lomovskaya O.; Tsivkovski R.; Nelson K.; Rubio-Aparicio D.; Sun D.; Totrov M.; Dudley M. N. Spectrum of beta-lactamase inhibition by the cyclic boronate QPX7728, an ultrabroad-spectrum beta-lactamase inhibitor of serine and metallo-beta-lactamases: enhancement of activity of multiple antibiotics against isogenic strains expressing single beta-lactamases. Antimicrob. Agents Chemother. 2020, 64 (6), e00212-20. 10.1128/AAC.00212-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doring G.; Conway S. P.; Heijerman H. G.; Hodson M. E.; Hoiby N.; Smyth A.; Touw D. J. Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: a European consensus. Eur. Respir. J. 2000, 16 (4), 749–767. 10.1034/j.1399-3003.2000.16d30.x. [DOI] [PubMed] [Google Scholar]
- McCaffery K.; Olver R. E.; Franklin M.; Mukhopadhyay S. Systematic review of antistaphylococcal antibiotic therapy in cystic fibrosis. Thorax 1999, 54 (5), 380–383. 10.1136/thx.54.5.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluck D.; Lewis P.; Stayer B.; Spivey J.; Moorman J. Ceftolozane-tazobactam: A new-generation cephalosporin. Am. J. Health Syst. Pharm. 2015, 72 (24), 2135–2146. 10.2146/ajhp150049. [DOI] [PubMed] [Google Scholar]
- Duplessis C.; Crum-Cianflone N. F. Ceftaroline: A New Cephalosporin with Activity against Methicillin-Resistant Staphylococcus aureus (MRSA). Clin. Med. Rev. Ther. 2011, 3, a2466. 10.4137/CMRT.S1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giron Moreno R. M.; Garcia-Clemente M.; Diab-Caceres L.; Martinez-Vergara A.; Martinez-Garcia M. A.; Gomez-Punter R. M. Treatment of pulmonary disease of cystic fibrosis: A comprehensive review. Antibiotics (Basel) 2021, 10 (5), 486. 10.3390/antibiotics10050486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skolnik K.; Kirkpatrick G.; Quon B. S. Nontuberculous mycobacteria in cystic fibrosis. Curr. Treat. Options Infect. Dis. 2016, 8 (4), 259–274. 10.1007/s40506-016-0092-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstan M. W.; Krenicky J. E.; Finney M. R.; Kirchner H. L.; Hilliard K. A.; Hilliard J. B.; Davis P. B.; Hoppel C. L. Effect of ibuprofen on neutrophil migration in vivo in cystic fibrosis and healthy subjects. J. Pharmacol. Exp. Ther. 2003, 306 (3), 1086–1091. 10.1124/jpet.103.052449. [DOI] [PubMed] [Google Scholar]
- Marson F. A. L.; Bertuzzo C. S.; Ribeiro J. D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4 (8), e37–e38. 10.1016/S2213-2600(16)30188-6. [DOI] [PubMed] [Google Scholar]
- Knuppel L.; Ishikawa Y.; Aichler M.; Heinzelmann K.; Hatz R.; Behr J.; Walch A.; Bachinger H. P.; Eickelberg O.; Staab-Weijnitz C. A. A Novel antifibrotic mechanism of nintedanib and pirfenidone. Inhibition of collagen fibril assembly. Am. J. Respir. Cell Mol. Biol. 2017, 57 (1), 77–90. 10.1165/rcmb.2016-0217OC. [DOI] [PubMed] [Google Scholar]
- Richeldi L.; du Bois R. M.; Raghu G.; Azuma A.; Brown K. K.; Costabel U.; Cottin V.; Flaherty K. R.; Hansell D. M.; Inoue Y.; Kim D. S.; Kolb M.; Nicholson A. G.; Noble P. W.; Selman M.; Taniguchi H.; Brun M.; Le Maulf F.; Girard M.; Stowasser S.; Schlenker-Herceg R.; Disse B.; Collard H. R. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370 (22), 2071–2082. 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- Sirinupong N.; Yang Z. Epigenetics in cystic fibrosis: epigenetic targeting of a genetic disease. Curr. Drug Targets 2015, 16 (9), 976–987. 10.2174/1389450116666150416114514. [DOI] [PubMed] [Google Scholar]
- Xu W.; Hui C.; Yu S. S.; Jing C.; Chan H. C. MicroRNAs and cystic fibrosis—an epigenetic perspective. Cell Biol. Int. 2011, 35 (5), 463–466. 10.1042/CBI20100664. [DOI] [PubMed] [Google Scholar]
- Maruyama R.; Choudhury S.; Kowalczyk A.; Bessarabova M.; Beresford-Smith B.; Conway T.; Kaspi A.; Wu Z.; Nikolskaya T.; Merino V. F.; Lo P. K.; Liu X. S.; Nikolsky Y.; Sukumar S.; Haviv I.; Polyak K. Epigenetic regulation of cell type-specific expression patterns in the human mammary epithelium. PLoS Genet. 2011, 7 (4), e1001369 10.1371/journal.pgen.1001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black J. C.; Van Rechem C.; Whetstine J. R. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell 2012, 48 (4), 491–507. 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott M.; De Sario A. DNA methylation changes in cystic fibrosis: Cause or consequence?. Clin. Genet. 2020, 98 (1), 3–9. 10.1111/cge.13731. [DOI] [PubMed] [Google Scholar]
- Hutt D. M.; Herman D.; Rodrigues A. P.; Noel S.; Pilewski J. M.; Matteson J.; Hoch B.; Kellner W.; Kelly J. W.; Schmidt A.; Thomas P. J.; Matsumura Y.; Skach W. R.; Gentzsch M.; Riordan J. R.; Sorscher E. J.; Okiyoneda T.; Yates J. R. 3rd; Lukacs G. L.; Frizzell R. A.; Manning G.; Gottesfeld J. M.; Balch W. E. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 2010, 6 (1), 25–33. 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J.; Hayder H.; Zayed Y.; Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402. 10.3389/fendo.2018.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott C. J.; Suszko M.; Blackledge N. P.; Wright J. E.; Crawford G. E.; Harris A. A complex intronic enhancer regulates expression of the CFTR gene by direct interaction with the promoter. J. Cell Mol. Med. 2009, 13 (4), 680–692. 10.1111/j.1582-4934.2008.00621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoszewski R.; Rab A.; Twitty G.; Stevenson L.; Fortenberry J.; Piotrowski A.; Dumanski J. P.; Bebok Z. The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J. Biol. Chem. 2008, 283 (18), 12154–12165. 10.1074/jbc.M707610200. [DOI] [PubMed] [Google Scholar]
- Bergougnoux A.; Rivals I.; Liquori A.; Raynal C.; Varilh J.; Magalhães M.; Perez M.-J.; Bigi N.; Des Georges M.; Chiron R.; Squalli-Houssaini A. S.; Claustres M.; De Sario A. A balance between activating and repressive histone modifications regulates cystic fibrosis transmembrane conductance regulator (CFTR) expression in vivo. Epigenetics 2014, 9 (7), 1007–1017. 10.4161/epi.28967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12 (5), 599–606. 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- Pittman N.; Shue G.; Neal S. L.; Walsh M. J. Transcription of cystic fibrosis transmembrane conductance regulator requires a CCAAT-like element for both basal and cAMP-mediated regulation. J. Biol. Chem. 1995, 270 (48), 28848–28857. 10.1074/jbc.270.48.28848. [DOI] [PubMed] [Google Scholar]
- Yoshimura K.; Nakamura H.; Trapnell B. C.; Chu C.-S.; Dakemans W.; Pavirani A.; Lecocq J.-P.; Crystal R. G. Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res. 1991, 19 (19), 5417–5423. 10.1093/nar/19.19.5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrini G.; Rimessi A.; Borgatti M.; Lampronti I.; Finotti A.; Pinton P.; Gambari R. Role of cystic fibrosis bronchial epithelium in neutrophil chemotaxis. Front. Immunol. 2020, 11, 1438. 10.3389/fimmu.2020.01438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul T.; Li S.; Khurana S.; Leleiko N. S.; Walsh M. J. The epigenetic signature of CFTR expression is co-ordinated via chromatin acetylation through a complex intronic element. Biochem. J. 2007, 408 (3), 317–326. 10.1042/BJ20070282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poghosyan A.; Patel J. K.; Clifford R. L.; Knox A. J. Epigenetic dysregulation of interleukin 8 (CXCL8) hypersecretion in cystic fibrosis airway epithelial cells. Biochem. Biophys. Res. Commun. 2016, 476 (4), 431–437. 10.1016/j.bbrc.2016.05.140. [DOI] [PubMed] [Google Scholar]
- Khan N.; Jeffers M.; Kumar S.; Hackett C.; Boldog F.; Khramtsov N.; Qian X.; Mills E.; Berghs S. C.; Carey N.; Finn P. W.; Collins L. S.; Tumber A.; Ritchie J. W.; Jensen P. B.; Lichenstein H. S.; Sehested M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409 (2), 581–589. 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Delcuve G. P.; Khan D. H.; Davie J. R. Targeting class I histone deacetylases in cancer therapy. Expert Opin. Ther. Targets 2013, 17 (1), 29–41. 10.1517/14728222.2013.729042. [DOI] [PubMed] [Google Scholar]
- Bosch-Presegue L.; Vaquero A. The dual role of sirtuins in cancer. Genes Cancer 2011, 2 (6), 648–662. 10.1177/1947601911417862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcuve G. P.; Khan D. H.; Davie J. R. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin. Epigenet. 2012, 4 (1), 5. 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulya S.; Amin S. A.; Adhikari N.; Biswas S.; Jha T.; Ghosh B. HDAC6 as privileged target in drug discovery: A perspective. Pharmacol. Res. 2021, 163, 105274. 10.1016/j.phrs.2020.105274. [DOI] [PubMed] [Google Scholar]
- Nekooki-Machida Y.; Hagiwara H. Role of tubulin acetylation in cellular functions and diseases. Med. Mol. Morphol. 2020, 53 (4), 191–197. 10.1007/s00795-020-00260-8. [DOI] [PubMed] [Google Scholar]
- Cabrero J. R.; Serrador J. M.; Barreiro O.; Mittelbrunn M.; Naranjo-Suarez S.; Martin-Cofreces N.; Vicente-Manzanares M.; Mazitschek R.; Bradner J. E.; Avila J.; Valenzuela-Fernandez A.; Sanchez-Madrid F. Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Mol. Biol. Cell 2006, 17 (8), 3435–3445. 10.1091/mbc.e06-01-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H.; Yang J. J.; Hu W.; Shi K. H.; Li J. HDAC6 promotes cardiac fibrosis progression through suppressing RASSF1A expression. Cardiology 2015, 133 (1), 18–26. 10.1159/000438781. [DOI] [PubMed] [Google Scholar]
- Ke B.; Chen Y.; Tu W.; Ye T.; Fang X.; Yang L. Inhibition of HDAC6 activity in kidney diseases: a new perspective. Mol. Med. 2018, 24 (1), 33. 10.1186/s10020-018-0027-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu X.; Hu M.; Peng J.; Zhang X.; Sanders Y. Y. HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther. Adv. Chronic Dis. 2019, 10, 2040622319862697. 10.1177/2040622319862697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs J. J.; Murphy P. J.; Gaillard S.; Zhao X.; Wu J. T.; Nicchitta C. V.; Yoshida M.; Toft D. O.; Pratt W. B.; Yao T. P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18 (5), 601–607. 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Boyault C.; Zhang Y.; Fritah S.; Caron C.; Gilquin B.; Kwon S. H.; Garrido C.; Yao T. P.; Vourc’h C.; Matthias P.; Khochbin S. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007, 21 (17), 2172–2181. 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmigiani R. B.; Xu W. S.; Venta-Perez G.; Erdjument-Bromage H.; Yaneva M.; Tempst P.; Marks P. A. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. U.S.A. 2008, 105 (28), 9633–9638. 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon J. C.; Hah Y. S.; Kim W. Y.; Jung B. G.; Jang H. H.; Lee J. R.; Kim S. Y.; Lee Y. M.; Jeon M. G.; Kim C. W.; Cho M. J.; Lee S. Y. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin Isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J. Biol. Chem. 2005, 280 (31), 28775–28784. 10.1074/jbc.M505362200. [DOI] [PubMed] [Google Scholar]
- Wood Z. A.; Schroder E.; Harris J. R.; Poole L. B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28 (1), 32–40. 10.1016/S0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- Yao Y. L.; Yang W. M. Beyond histone and deacetylase: an overview of cytoplasmic histone deacetylases and their nonhistone substrates. J. Biomed. Biotechnol. 2011, 2011, 146493. 10.1155/2011/146493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. X.; Wan R. Z.; Liu Z. P. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 2018, 143, 1406–1418. 10.1016/j.ejmech.2017.10.040. [DOI] [PubMed] [Google Scholar]
- Cosenza M.; Pozzi S. The therapeutic strategy of HDAC6 inhibitors in lymphoproliferative disease. Int. J. Mol. Sci. 2018, 19 (8), 2337. 10.3390/ijms19082337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S. D.; Kozikowski A. P. A patent review of histone deacetylase 6 inhibitors in neurodegenerative diseases (2014–2019). Expert Opin. Ther. Pat. 2020, 30 (2), 121–136. 10.1080/13543776.2019.1708901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. H.; Qin-Ma; Wu H. P.; Khamis M. Y.; Li Y. H.; Ma L. Y.; Liu H. M. A Review of progress in histone deacetylase 6 inhibitors research: structural specificity and functional diversity. J. Med. Chem. 2021, 64 (3), 1362–1391. 10.1021/acs.jmedchem.0c01782. [DOI] [PubMed] [Google Scholar]
- Landucci E.; Brindisi M.; Bianciardi L.; Catania L. M.; Daga S.; Croci S.; Frullanti E.; Fallerini C.; Butini S.; Brogi S.; Furini S.; Melani R.; Molinaro A.; Lorenzetti F. C.; Imperatore V.; Amabile S.; Mariani J.; Mari F.; Ariani F.; Pizzorusso T.; Pinto A. M.; Vaccarino F. M.; Renieri A.; Campiani G.; Meloni I. iPSC-derived neurons profiling reveals GABAergic circuit disruption and acetylated alpha-tubulin defect which improves after iHDAC6 treatment in Rett syndrome. Exp. Cell Res. 2018, 368 (2), 225–235. 10.1016/j.yexcr.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran J.; Zhou J. Targeted inhibition of histone deacetylase 6 in inflammatory diseases. Thorac. Cancer 2019, 10 (3), 405–412. 10.1111/1759-7714.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F.; Lienlaf M.; Wang H. W.; Perez-Villarroel P.; Lee C.; Woan K.; Rock-Klotz J.; Sahakian E.; Woods D.; Pinilla-Ibarz J.; Kalin J.; Tao J.; Hancock W.; Kozikowski A.; Seto E.; Villagra A.; Sotomayor E. M. A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J. Immunol. 2014, 193 (6), 2850–2862. 10.4049/jimmunol.1302778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campiani G.; Cavella C.; Osko J. D.; Brindisi M.; Relitti N.; Brogi S.; Saraswati A. P.; Federico S.; Chemi G.; Maramai S.; Carullo G.; Jaeger B.; Carleo A.; Benedetti R.; Sarno F.; Lamponi S.; Rottoli P.; Bargagli E.; Bertucci C.; Tedesco D.; Herp D.; Senger J.; Ruberti G.; Saccoccia F.; Saponara S.; Gorelli B.; Valoti M.; Kennedy B.; Sundaramurthi H.; Butini S.; Jung M.; Roach K. M.; Altucci L.; Bradding P.; Christianson D. W.; Gemma S.; Prasse A. Harnessing the role of HDAC6 in idiopathic pulmonary fibrosis: design, synthesis, structural analysis, and biological evaluation of potent inhibitors. J. Med. Chem. 2021, 64 (14), 9960–9988. 10.1021/acs.jmedchem.1c00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter N. J.; Mahendran A.; Breslow R.; Christianson D. W. Unusual zinc-binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2017, 114 (51), 13459–13464. 10.1073/pnas.1718823114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraswati A. P.; Relitti N.; Brindisi M.; Osko J. D.; Chemi G.; Federico S.; Grillo A.; Brogi S.; McCabe N. H.; Turkington R. C.; Ibrahim O.; O’Sullivan J.; Lamponi S.; Ghanim M.; Kelly V. P.; Zisterer D.; Amet R.; Hannon Barroeta P.; Vanni F.; Ulivieri C.; Herp D.; Sarno F.; Di Costanzo A.; Saccoccia F.; Ruberti G.; Jung M.; Altucci L.; Gemma S.; Butini S.; Christianson D. W.; Campiani G. Spiroindoline-capped selective HDAC6 inhibitors: design, synthesis, structural analysis, and biological evaluation. ACS Med. Chem. Lett. 2020, 11 (11), 2268–2276. 10.1021/acsmedchemlett.0c00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osko J. D.; Porter N. J.; Narayana Reddy P. A.; Xiao Y. C.; Rokka J.; Jung M.; Hooker J. M.; Salvino J. M.; Christianson D. W. Exploring structural determinants of inhibitor affinity and selectivity in complexes with histone deacetylase 6. J. Med. Chem. 2020, 63 (1), 295–308. 10.1021/acs.jmedchem.9b01540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Sant’Agnese P.; Darling R. C.; Perera G. A.; Shea E. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas. AMA Am. J. Dis. Child. 1953, 86 (5), 618–619. [PubMed] [Google Scholar]
- Knowles M.; Gatzy J.; Boucher R. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N. Engl. J. Med. 1981, 305 (25), 1489–1495. 10.1056/NEJM198112173052502. [DOI] [PubMed] [Google Scholar]
- Nichols D.; Chmiel J.; Berger M. Chronic inflammation in the cystic fibrosis lung: alterations in inter- and intracellular signaling. Clin. Rev. Allergy Immunol. 2008, 34 (2), 146–162. 10.1007/s12016-007-8039-9. [DOI] [PubMed] [Google Scholar]
- Manson M. E.; Corey D. A.; Bederman I.; Burgess J. D.; Kelley T. J. Regulatory role of beta-arrestin-2 in cholesterol processing in cystic fibrosis epithelial cells. J. Lipid Res. 2012, 53 (7), 1268–1276. 10.1194/jlr.M021972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniento F.; Emans N.; Griffiths G.; Gruenberg J. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J. Cell Biol. 1993, 123 (6), 1373–1387. 10.1083/jcb.123.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bananis E.; Nath S.; Gordon K.; Satir P.; Stockert R. J.; Murray J. W.; Wolkoff A. W. Microtubule-dependent movement of late endocytic vesicles in vitro: requirements for Dynein and Kinesin. Mol. Biol. Cell 2004, 15 (8), 3688–3697. 10.1091/mbc.e04-04-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Brady S. T. Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends Cell Biol. 2015, 25 (3), 125–136. 10.1016/j.tcb.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann S.; Weber K. Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 2003, 4 (12), 938–947. 10.1038/nrm1260. [DOI] [PubMed] [Google Scholar]
- Wloga D.; Joachimiak E.; Fabczak H. Tubulin post-translational modifications and microtubule dynamics. Int. J. Mol. Sci. 2017, 18 (10), 2207. 10.3390/ijms18102207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutin M. J.; Bosc C.; Peris L.; Andrieux A. Tubulin post-translational modifications control neuronal development and functions. Dev. Neurobiol. 2021, 81 (3), 253–272. 10.1002/dneu.22774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDizet M.; Piperno G. Identification of an acetylation site of Chlamydomonas alpha-tubulin. Proc. Natl. Acad. Sci. U.S.A. 1987, 84 (16), 5720–5724. 10.1073/pnas.84.16.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. J.; Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26 (37), 5310–5318. 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- Howes S. C.; Alushin G. M.; Shida T.; Nachury M. V.; Nogales E. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol. Biol. Cell 2014, 25 (2), 257–266. 10.1091/mbc.e13-07-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C.; Guardiola A.; Shao R.; Kawaguchi Y.; Ito A.; Nixon A.; Yoshida M.; Wang X. F.; Yao T. P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417 (6887), 455–458. 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Cabrero J. R.; Serrador J. M.; Barreiro O.; Mittelbrunn M.; Naranjo-Suarez S.; Martin-Cofreces N.; Vicente-Manzanares M.; Mazitschek R.; Bradner J. E.; Avila J.; Valenzuela-Fernandez A.; Sanchez-Madrid F. Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Mol. Biol. Cell 2006, 17 (8), 3435–3445. 10.1091/mbc.e06-01-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty S. J.; Koeller K. M.; Wong J. C.; Grozinger C. M.; Schreiber S. L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. U.S.A. 2003, 100 (8), 4389–4394. 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y.; Kovacs J. J.; McLaurin A.; Vance J. M.; Ito A.; Yao T. P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115 (6), 727–738. 10.1016/S0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Ustinova K.; Novakova Z.; Saito M.; Meleshin M.; Mikesova J.; Kutil Z.; Baranova P.; Havlinova B.; Schutkowski M.; Matthias P.; Barinka C. The disordered N-terminus of HDAC6 is a microtubule-binding domain critical for efficient tubulin deacetylation. J. Biol. Chem. 2020, 295 (9), 2614–2628. 10.1074/jbc.RA119.011243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A.; Bukau B.; Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol. Cell 2010, 40 (2), 238–252. 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Johnston J. A.; Ward C. L.; Kopito R. R. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 1998, 143 (7), 1883–1898. 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs G. L.; Mohamed A.; Kartner N.; Chang X. B.; Riordan J. R.; Grinstein S. Conformational maturation of CFTR but not its mutant counterpart (ΔF508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994, 13 (24), 6076–6086. 10.1002/j.1460-2075.1994.tb06954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra J. D.; Kaufman R. J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18 (6), 716–731. 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35 (4), 373–381. 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Okiyoneda T.; Veit G.; Dekkers J. F.; Bagdany M.; Soya N.; Xu H.; Roldan A.; Verkman A. S.; Kurth M.; Simon A.; Hegedus T.; Beekman J. M.; Lukacs G. L. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9 (7), 444–454. 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Song J.; Cassidy M. G.; Rychahou P.; Starr M. E.; Liu J.; Li X.; Epperly G.; Weiss H. L.; Townsend C. M. Jr.; Gao T.; Evers B. M. PI3K p110alpha/Akt signaling negatively regulates secretion of the intestinal peptide neurotensin through interference of granule transport. Mol. Endocrinol. 2012, 26 (8), 1380–1393. 10.1210/me.2012-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone L.; Bardelli A.; Avvedimento V. E. Activation of beta-catenin by oncogenic PIK3CA and EGFR promotes resistance to glucose deprivation by inducing a strong antioxidant response. PLoS One 2012, 7 (5), e37526 10.1371/journal.pone.0037526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein R. C.; Egan M. E.; Zeitlin P. L. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J. Clin. Invest. 1997, 100 (10), 2457–2465. 10.1172/JCI119788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein R. C.; Zeitlin P. L. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am. J. Respir. Crit. Care Med. 1998, 157 (2), 484–490. 10.1164/ajrccm.157.2.9706088. [DOI] [PubMed] [Google Scholar]
- Ammerpohl O.; Trauzold A.; Schniewind B.; Griep U.; Pilarsky C.; Grutzmann R.; Saeger H. D.; Janssen O.; Sipos B.; Kloppel G.; Kalthoff H. Complementary effects of HDAC inhibitor 4-PB on gap junction communication and cellular export mechanisms support restoration of chemosensitivity of PDAC cells. Br. J. Cancer 2007, 96 (1), 73–81. 10.1038/sj.bjc.6603511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan Z.; Akhtar M.; Ekström T. J. HDAC inhibitor 4-phenylbutyrate preserves immature phenotype of human embryonic midbrain stem cells: implications for the involvement of DNA methyltransferase. Int. J. Mol. Med. 2011, 28 (6), 977–983. 10.3892/ijmm.2011.791. [DOI] [PubMed] [Google Scholar]
- Lu B.; Li L.; Schneider M.; Hodges C. A.; Cotton C. U.; Burgess J. D.; Kelley T. J. Electrochemical measurement of membrane cholesterol correlates with CFTR function and is HDAC6-dependent. J. Cystic Fibrosis 2019, 18 (2), 175–181. 10.1016/j.jcf.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D.; West R. H.; Manson M. E.; Ruddy J.; Jiang D.; Previs S. F.; Sonawane N. D.; Burgess J. D.; Kelley T. J. Increased plasma membrane cholesterol in cystic fibrosis cells correlates with CFTR genotype and depends on de novo cholesterol synthesis. Respir. Res. 2010, 11, 61. 10.1186/1465-9921-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D.; Devadoss A.; Palencsar M. S.; Fang D.; White N. M.; Kelley T. J.; Smith J. D.; Burgess J. D. Direct electrochemical evaluation of plasma membrane cholesterol in live mammalian cells. J. Am. Chem. Soc. 2007, 129 (37), 11352–11353. 10.1021/ja074373c. [DOI] [PubMed] [Google Scholar]
- West R. H.; Lu H.; Shaw K.; Chiel H. J.; Kelley T. J.; Burgess J. D. Double potential pulse chronocoulometry for detection of plasma membrane cholesterol efflux at disk platinum microelectrodes. J. Electrochem. Soc. 2014, 161 (6), B111–B116. 10.1149/2.005406jes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B.; Corey D. A.; Kelley T. J. Resveratrol restores intracellular transport in cystic fibrosis epithelial cells. Am. J. Physiol.: Lung Cell. Mol. Physiol. 2020, 318 (6), L1145–L1157. 10.1152/ajplung.00006.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M.; McDonagh T.; Heltweg B.; Hixon J.; Westman E. A.; Caldwell S. D.; Napper A.; Curtis R.; DiStefano P. S.; Fields S.; Bedalov A.; Kennedy B. K. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005, 280 (17), 17038–17045. 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- Calleri E.; Pochetti G.; Dossou K. S. S.; Laghezza A.; Montanari R.; Capelli D.; Prada E.; Loiodice F.; Massolini G.; Bernier M.; Moaddel R. Resveratrol and its metabolites bind to PPARs. ChemBioChem 2014, 15 (8), 1154–1160. 10.1002/cbic.201300754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturelli S.; Berger A.; Bocker A.; Busch C.; Weiland T.; Noor S.; Leischner C.; Schleicher S.; Mayer M.; Weiss T. S.; Bischoff S. C.; Lauer U. M.; Bitzer M. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PLoS One 2013, 8 (8), e73097 10.1371/journal.pone.0073097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymut S. M.; Kampman C. M.; Corey D. A.; Endres T.; Cotton C. U.; Kelley T. J. Ibuprofen regulation of microtubule dynamics in cystic fibrosis epithelial cells. Am. J. Physiol.: Lung Cell. Mol. Physiol. 2016, 311 (2), L317–L327. 10.1152/ajplung.00126.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misawa T.; Saitoh T.; Kozaki T.; Park S.; Takahama M.; Akira S. Resveratrol inhibits the acetylated alpha-tubulin-mediated assembly of the NLRP3-inflammasome. Int. Immunol. 2015, 27 (9), 425–434. 10.1093/intimm/dxv018. [DOI] [PubMed] [Google Scholar]
- Amri A.; Chaumeil J. C.; Sfar S.; Charrueau C. Administration of resveratrol: What formulation solutions to bioavailability limitations?. J. Controlled Release 2012, 158 (2), 182–193. 10.1016/j.jconrel.2011.09.083. [DOI] [PubMed] [Google Scholar]
- Grabiec A. M.; Potempa J. Epigenetic regulation in bacterial infections: targeting histone deacetylases. Crit. Rev. Microbiol. 2018, 44 (3), 336–350. 10.1080/1040841X.2017.1373063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne H.; Hamon M.; Cossart P. Epigenetics and bacterial infections. Cold Spring Harb Perspect Med. 2012, 2 (12), a010272. 10.1101/cshperspect.a010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon M. A.; Cossart P. Histone modifications and chromatin remodeling during bacterial infections. Cell Host Microbe 2008, 4 (2), 100–109. 10.1016/j.chom.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Garcia-Garcia J. C.; Barat N. C.; Trembley S. J.; Dumler J. S. Epigenetic silencing of host cell defense genes enhances intracellular survival of the rickettsial pathogen Anaplasma phagocytophilum. PLoS Pathog. 2009, 5 (6), e1000488 10.1371/journal.ppat.1000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhaya A.; Tsurumi A.; Maura D.; Jeffrey K. L.; Rahme L. G. A quorum-sensing signal promotes host tolerance training through HDAC1-mediated epigenetic reprogramming. Nat. Microbiol. 2016, 1, 16174. 10.1038/nmicrobiol.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa-Oliveira R.; Fachi J. L.; Vieira A.; Sato F. T.; Vinolo M. A. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5 (4), e73 10.1038/cti.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier U. H.; Wang L.; Han R.; Akimova T.; Liu Y.; Hancock W. W. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci. Signaling 2012, 5 (229), ra45. 10.1126/scisignal.2002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger T.; Lugrin J.; Le Roy D.; Goy G.; Mombelli M.; Koessler T.; Ding X. C.; Chanson A. L.; Reymond M. K.; Miconnet I.; Schrenzel J.; Francois P.; Calandra T. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood 2011, 117 (4), 1205–1217. 10.1182/blood-2010-05-284711. [DOI] [PubMed] [Google Scholar]
- Mombelli M.; Lugrin J.; Rubino I.; Chanson A. L.; Giddey M.; Calandra T.; Roger T. Histone deacetylase inhibitors impair antibacterial defenses of macrophages. J. Infect. Dis. 2011, 204 (9), 1367–1374. 10.1093/infdis/jir553. [DOI] [PubMed] [Google Scholar]
- Ariffin J. K.; das Gupta K.; Kapetanovic R.; Iyer A.; Reid R. C.; Fairlie D. P.; Sweet M. J. Histone deacetylase inhibitors promote mitochondrial reactive oxygen species production and bacterial clearance by human macrophages. Antimicrob. Agents Chemother. 2016, 60 (3), 1521–1529. 10.1128/AAC.01876-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Zhao T.; Liu B.; Halaweish I.; Mazitschek R.; Duan X.; Alam H. B. Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic model. J. Trauma Acute Care Surg. 2015, 78 (2), 378–385. 10.1097/TA.0000000000000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T.; Li Y.; Liu B.; Pan B.; Cheng X.; Georgoff P.; Alam H. B. Inhibition of histone deacetylase 6 restores innate immune cells in the bone marrow in a lethal septic model. J. Trauma Acute Care Surg. 2016, 80 (1), 34–41. 10.1097/TA.0000000000000897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Shi X.; Padmanabhan R.; Wang Q.; Wu Z.; Stevenson S. C.; Hild M.; Garza D.; Li H. Identification of novel modulators of mitochondrial function by a genome-wide RNAi screen in Drosophila melanogaster. Genome Res. 2008, 18 (1), 123–136. 10.1101/gr.6940108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamemura K.; Ogawa M.; Ohkubo S.; Ohtsuka Y.; Shitara Y.; Komiya T.; Maeda S.; Ito A.; Yoshida M. Depression of mitochondrial metabolism by downregulation of cytoplasmic deacetylase, HDAC6. FEBS Lett. 2012, 586 (9), 1379–1383. 10.1016/j.febslet.2012.03.060. [DOI] [PubMed] [Google Scholar]
- Bai J.; Lei Y.; An G. L.; He L. Down-regulation of deacetylase HDAC6 inhibits the melanoma cell line A375.S2 growth through ROS-dependent mitochondrial pathway. PLoS One 2015, 10 (3), e0121247 10.1371/journal.pone.0121247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedery R. D.; Jerse A. E. Augmentation of cationic antimicrobial peptide production with histone deacetylase inhibitors as a novel epigenetic therapy for bacterial infections. Antibiotics (Basel) 2015, 4 (1), 44–61. 10.3390/antibiotics4010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauber J.; Svanholm C.; Termen S.; Iffland K.; Menzel T.; Scheppach W.; Melcher R.; Agerberth B.; Luhrs H.; Gudmundsson G. H. Expression of the cathelicidin LL-37 is modulated by short chain fatty acids in colonocytes: relevance of signalling pathways. Gut 2003, 52 (5), 735–741. 10.1136/gut.52.5.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann J.; Halldorsson S.; Agerberth B.; Gudmundsson G. H. Phenylbutyrate induces antimicrobial peptide expression. Antimicrob. Agents Chemother. 2009, 53 (12), 5127–5133. 10.1128/AAC.00818-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Liu J.; Roschmann K. I. L.; van Egmond D.; Golebski K.; Fokkens W. J.; Wang D.; van Drunen C. M. Histone deacetylase inhibitors up-regulate LL-37 expression independent of toll-like receptor mediated signalling in airway epithelial cells. J. Inflammation (London) 2013, 10 (1), 15. 10.1186/1476-9255-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer N.; Sechet E.; Friedman R.; Amiot A.; Sobhani I.; Nigro G.; Sansonetti P. J.; Sperandio B. Histone deacetylase inhibition enhances antimicrobial peptide but not inflammatory cytokine expression upon bacterial challenge. Proc. Natl. Acad. Sci. U.S.A. 2016, 113 (21), E2993–E3001. 10.1073/pnas.1605997113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miraglia E.; Nylen F.; Johansson K.; Arner E.; Cebula M.; Farmand S.; Ottosson H.; Stromberg R.; Gudmundsson G. H.; Agerberth B.; Bergman P. Entinostat up-regulates the CAMP gene encoding LL-37 via activation of STAT3 and HIF-1alpha transcription factors. Sci. Rep. 2016, 6, 33274. 10.1038/srep33274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard T. N.; Regamey N.; Shute J. K.; Nicholson A. G.; Alton E. W.; Bush A.; Davies J. C. Airway remodelling in children with cystic fibrosis. Thorax 2007, 62 (12), 1074–1080. 10.1136/thx.2006.074641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciaky D.; Brazer W.; Center D. M.; Cruikshank W. W.; Smith T. J. Cultured human fibroblasts express constitutive IL-16 mRNA: cytokine induction of active IL-16 protein synthesis through a caspase-3-dependent mechanism. J. Immunol. 2000, 164 (7), 3806–3814. 10.4049/jimmunol.164.7.3806. [DOI] [PubMed] [Google Scholar]
- Hogaboam C. M.; Steinhauser M. L.; Chensue S. W.; Kunkel S. L. Novel roles for chemokines and fibroblasts in interstitial fibrosis. Kidney Int. 1998, 54 (6), 2152–2159. 10.1046/j.1523-1755.1998.00176.x. [DOI] [PubMed] [Google Scholar]
- Harris W. T.; Muhlebach M. S.; Oster R. A.; Knowles M. R.; Clancy J. P.; Noah T. L. Plasma TGF-beta(1) in pediatric cystic fibrosis: potential biomarker of lung disease and response to therapy. Pediatr. Pulmonol. 2011, 46 (7), 688–695. 10.1002/ppul.21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagues N.; Pawlowski V.; Sobry C.; Hanton G.; Borde F.; Soler S.; Freslon J. L.; Chevalier S. Investigation of the molecular mechanisms preceding PDE4 inhibitor-induced vasculopathy in rats: tissue inhibitor of metalloproteinase 1, a potential predictive biomarker. Toxicol. Sci. 2007, 100 (1), 238–247. 10.1093/toxsci/kfm161. [DOI] [PubMed] [Google Scholar]
- Gatla H. R.; Muniraj N.; Thevkar P.; Yavvari S.; Sukhavasi S.; Makena M. R. Regulation of chemokines and cytokines by histone deacetylases and an update on histone decetylase inhibitors in human diseases. Int. J. Mol. Sci. 2019, 20 (5), 1110. 10.3390/ijms20051110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden J. E.; Shi W.; Jankowski K.; Kan C. Y.; Cluse L.; Martin B. P.; MacKenzie K. L.; Smyth G. K.; Johnstone R. W. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013, 4, e519 10.1038/cddis.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull E. E.; Montgomery M. R.; Leyva K. J. HDAC inhibitors as epigenetic regulators of the immune system: impacts on cancer therapy and inflammatory diseases. Biomed. Res. Int. 2016, 2016, 8797206. 10.1155/2016/8797206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinzon I.; Horvath C. M. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc. Natl. Acad. Sci. U.S.A. 2003, 100 (25), 14742–14747. 10.1073/pnas.2433987100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasakova J.; Novakova Z.; Rossmeislova L.; Kahle M.; Hozak P.; Hodny Z. Histone deacetylase inhibitors suppress IFNalpha-induced up-regulation of promyelocytic leukemia protein. Blood 2007, 109 (4), 1373–1380. 10.1182/blood-2006-02-003418. [DOI] [PubMed] [Google Scholar]
- Deng R.; Zhang P.; Liu W.; Zeng X.; Ma X.; Shi L.; Wang T.; Yin Y.; Chang W.; Zhang P.; Wang G.; Tao K. HDAC is indispensable for IFN-gamma-induced B7-H1 expression in gastric cancer. Clin. Epigenet. 2018, 10 (1), 153. 10.1186/s13148-018-0589-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Liddo R.; Valente S.; Taurone S.; Zwergel C.; Marrocco B.; Turchetta R.; Conconi M. T.; Scarpa C.; Bertalot T.; Schrenk S.; Mai A.; Artico M. Histone deacetylase inhibitors restore IL-10 expression in lipopolysaccharide-induced cell inflammation and reduce IL-1beta and IL-6 production in breast silicone implant in C57BL/6J wild-type murine model. Autoimmunity 2016, 1–11. 10.3109/08916934.2015.1134510. [DOI] [PubMed] [Google Scholar]
- Vishwakarma S.; Iyer L. R.; Muley M.; Singh P. K.; Shastry A.; Saxena A.; Kulathingal J.; Vijaykanth G.; Raghul J.; Rajesh N.; Rathinasamy S.; Kachhadia V.; Kilambi N.; Rajgopal S.; Balasubramanian G.; Narayanan S. Tubastatin, a selective histone deacetylase 6 inhibitor shows anti-inflammatory and anti-rheumatic effects. Int. Immunopharmacol. 2013, 16 (1), 72–78. 10.1016/j.intimp.2013.03.016. [DOI] [PubMed] [Google Scholar]
- Licciardi P. V.; Karagiannis T. C. Regulation of immune responses by histone deacetylase inhibitors. ISRN Hematol. 2012, 2012, 690901. 10.5402/2012/690901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth L.; Roberts J. L.; Poklepovic A.; Kirkwood J.; Dent P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget 2017, 8 (47), 83155–83170. 10.18632/oncotarget.17950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi E. W.; Song J. W.; Ha N.; Choi Y. I.; Kim S. CKD-506, a novel HDAC6-selective inhibitor, improves renal outcomes and survival in a mouse model of systemic lupus erythematosus. Sci. Rep. 2018, 8 (1), 17297. 10.1038/s41598-018-35602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao T.; Zhou X.; Zheng X.; Cui Y.; Tsien J. Z.; Li C.; Wang H. Histone deacetylase inhibitor alleviates the neurodegenerative phenotypes and histone dysregulation in presenilins-deficient Mice. Front. Aging Neurosci. 2018, 10, 137. 10.3389/fnagi.2018.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B.; Liu Y.; Bai H.; Chen M.; Xie S.; Li D.; Liu M.; Zhou J. HDAC6 regulates IL-17 expression in T lymphocytes: implications for HDAC6-targeted therapies. Theranostics 2017, 7 (4), 1002–1009. 10.7150/thno.17615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F.; Lienlaf M.; Perez-Villarroel P.; Wang H. W.; Lee C.; Woan K.; Woods D.; Knox T.; Bergman J.; Pinilla-Ibarz J.; Kozikowski A.; Seto E.; Sotomayor E. M.; Villagra A. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol. Immunol. 2014, 60 (1), 44–53. 10.1016/j.molimm.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Zoeten E. F.; Wang L.; Butler K.; Beier U. H.; Akimova T.; Sai H.; Bradner J. E.; Mazitschek R.; Kozikowski A. P.; Matthias P.; Hancock W. W. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol. Cell. Biol. 2011, 31 (10), 2066–2078. 10.1128/MCB.05155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y.; Su X.; Kong L.; Li M.; Zhao X.; Yu N.; Kang J. Therapeutic effects of histone deacetylase inhibitors in a murine asthma model. Inflammation Res. 2016, 65 (12), 995–1008. 10.1007/s00011-016-0984-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam H. C.; Cloonan S. M.; Bhashyam A. R.; Haspel J. A.; Singh A.; Sathirapongsasuti J. F.; Cervo M.; Yao H.; Chung A. L.; Mizumura K.; An C. H.; Shan B.; Franks J. M.; Haley K. J.; Owen C. A.; Tesfaigzi Y.; Washko G. R.; Quackenbush J.; Silverman E. K.; Rahman I.; Kim H. P.; Mahmood A.; Biswal S. S.; Ryter S. W.; Choi A. M. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Invest. 2013, 123 (12), 5212–5230. 10.1172/JCI69636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angles F.; Hutt D. M.; Balch W. E. HDAC inhibitors rescue multiple disease-causing CFTR variants. Hum. Mol. Genet. 2019, 28 (12), 1982–2000. 10.1093/hmg/ddz026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankow S.; Bamberger C.; Calzolari D.; Martinez-Bartolome S.; Lavallee-Adam M.; Balch W. E.; Yates J. R. 3rd F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 2015, 528 (7583), 510–516. 10.1038/nature15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S.; Kang G.; Eom G. H. HDAC inhibitors: therapeutic potential in fibrosis-associated human diseases. Int. J. Mol. Sci. 2019, 20 (6), 1329. 10.3390/ijms20061329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery J. P.; Sleeman J. P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7 (2), 131–142. 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Zavadil J.; Bottinger E. P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24 (37), 5764–5774. 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- Conforti F.; Davies E. R.; Calderwood C. J.; Thatcher T. H.; Jones M. G.; Smart D. E.; Mahajan S.; Alzetani A.; Havelock T.; Maher T. M.; Molyneaux P. L.; Thorley A. J.; Tetley T. D.; Warner J. A.; Packham G.; Ganesan A.; Skipp P. J.; Marshall B. J.; Richeldi L.; Sime P. J.; O’Reilly K. M. A.; Davies D. E. The histone deacetylase inhibitor, romidepsin, as a potential treatment for pulmonary fibrosis. Oncotarget 2017, 8 (30), 48737–48754. 10.18632/oncotarget.17114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korfei M.; Stelmaszek D.; MacKenzie B.; Skwarna S.; Chillappagari S.; Bach A. C.; Ruppert C.; Saito S.; Mahavadi P.; Klepetko W.; Fink L.; Seeger W.; Lasky J. A.; Pullamsetti S. S.; Kramer O. H.; Guenther A. Comparison of the antifibrotic effects of the pan-histone deacetylase-inhibitor panobinostat versus the IPF-drug pirfenidone in fibroblasts from patients with idiopathic pulmonary fibrosis. PLoS One 2018, 13 (11), e0207915 10.1371/journal.pone.0207915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Chen C.; Finger S. N.; Kwajah S.; Jung M.; Schwarz H.; Swanson N.; Lareu F. F.; Raghunath M. Suberoylanilide hydroxamic acid: a potential epigenetic therapeutic agent for lung fibrosis?. Eur. Respir. J. 2009, 34 (1), 145–155. 10.1183/09031936.00084808. [DOI] [PubMed] [Google Scholar]
- Saito S.; Zhuang Y.; Shan B.; Danchuk S.; Luo F.; Korfei M.; Guenther A.; Lasky J. A. Tubastatin ameliorates pulmonary fibrosis by targeting the TGFbeta-PI3K-Akt pathway. PLoS One 2017, 12 (10), e0186615 10.1371/journal.pone.0186615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler K. V.; Kalin J.; Brochier C.; Vistoli G.; Langley B.; Kozikowski A. P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132 (31), 10842–10846. 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber K.; Doyon G.; Plaks J.; Fyne E.; Mellors J. W.; Sluis-Cremer N. Inhibitors of histone deacetylases: correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J. Biol. Chem. 2011, 286 (25), 22211–22218. 10.1074/jbc.M110.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Zhang J.; Jiang Q.; Zhang L.; Song W. Zinc binding groups for histone deacetylase inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33 (1), 714–721. 10.1080/14756366.2017.1417274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A.; Yamashita T.; Mariko Y.; Nosaka Y.; Tsuchiya K.; Ando T.; Suzuki T.; Tsuruo T.; Nakanishi O. A synthetic inhibitor of histone deacetylase, MS-27–275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. U.S.A. 1999, 96 (8), 4592–4597. 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigushin D. M.; Ali S.; Pace P. E.; Mirsaidi N.; Ito K.; Adcock I.; Coombes R. C. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 2001, 7 (4), 971–976. [PubMed] [Google Scholar]
- Furumai R.; Matsuyama A.; Kobashi N.; Lee K.-H.; Nishiyama M.; Nakajima H.; Tanaka A.; Komatsu Y.; Nishino N.; Yoshida M.; Horinouchi S. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62 (17), 4916–4921. [PubMed] [Google Scholar]
- Richon V. M.; Emiliani S.; Verdin E.; Webb Y.; Breslow R.; Rifkind R. A.; Marks P. A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 1998, 95 (6), 3003–3007. 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuto A.; Kirschbaum M.; Kowolik C.; Kretzner L.; Juhasz A.; Atadja P.; Pullarkat V.; Bhatia R.; Forman S.; Yen Y.; Jove R. The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph- acute lymphoblastic leukemia cells. Blood 2008, 111 (10), 5093–5100. 10.1182/blood-2007-10-117762. [DOI] [PMC free article] [PubMed] [Google Scholar]