

Abstract
Chemoimmunotherapy with fludarabine, cyclophosphamide and rituximab (FCR) can induce long-term remissions in patients with chronic lymphocytic leukemia. Treatment efficacy with Bruton's tyrosine kinase inhibitors was found similar to FCR in untreated chronic lymphocytic leukemia patients with a mutated immunoglobulin heavy chain variable (IGHV) gene. In order to identify patients who specifically benefit from FCR, we developed integrative models including established prognostic parameters and gene expression profiling (GEP). GEP was conducted on n=337 CLL8 trial samples, “core” probe sets were summarized on gene levels and RMA normalized. Prognostic models were built using penalized Cox proportional hazards models with the smoothly clipped absolute deviation penalty. We identified a prognostic signature of less than a dozen genes, which substituted for established prognostic factors, including TP53 and IGHV gene mutation status. Independent prognostic impact was confirmed for treatment, β2-microglobulin and del(17p) regarding overall survival and for treatment, del(11q), del(17p) and SF3B1 mutation for progression-free survival. The combination of independent prognostic and GEP variables performed equal to models including only established non-GEP variables. GEP variables showed higher prognostic accuracy for patients with long progression-free survival compared to categorical variables like the IGHV gene mutation status and reliably predicted overall survival in CLL8 and an independent cohort. GEP-based prognostic models can help to identify patients who specifically benefit from FCR treatment. The CLL8 trial is registered under EUDRACT-2004-004938-14 and clinicaltrials gov. Identifier: NCT00281918.
Introduction
Chemoimmunotherapy with fludarabine, cyclophosphamide and rituximab (FCR) was defined as the standard first-line therapy for patients with chronic lymphocytic leukemia (CLL) who are eligible for intensive treatment.1,2
There is prognostic impact of recurrent genetic alterations and NOTCH1 mutations were identified as a predictive marker for reduced benefit of FCR over FC.3,4,5,6,7 While substantial treatment benefit has been established for FCR in distinct patient populations, 1 high efficacy of novel targeted compounds such as the Bruton's tyrosine kinase (BTK) inhibitor ibrutinib was recently reported in previously untreated patients,8,9 and for cohorts with genetic high-risk subgroups or refractory populations.10,11,12,13,14 However, progression-free survival (PFS) in previously untreated patients ≤70 years old with a mutated immunoglobulin heavy chain variable (IGHV) gene was similar for the treatment with BTK inhibition or FCR.14 Therefore, identification of young and fit patients who specifically benefit from the treatment with FCR is needed to optimize long-term outcomes, in particular in the light of toxicity and cost associated with lifelong ibrutinib treatment. Additional biological characterization, such as gene expression profiling (GEP), may be helpful for further refinement of prognostic models leading to an increased prognostic accuracy and precise segregation of patients with a high treatment efficacy of FCR. Established markers mostly constitute categorical variables or consensus cut-offs, in the case of IGHV mutation status, and therefore may not fully reflect the underlying biology. In addition, established prognostic markers may loose some of their impact with novel treatments.
Since such large-scale studies on randomized trials are scarce, we performed GEP on 337 baseline patient samples from the CLL8 trial and modeled different scenarios for the combined use with established prognostic factors. We identified less than a dozen genes substituting for the prognostic impact of distinct recurrent alterations for PFS and overall survival (OS). Our results provide the basis for refined prognostic models and rational treatment selection.
Methods
Patients and samples
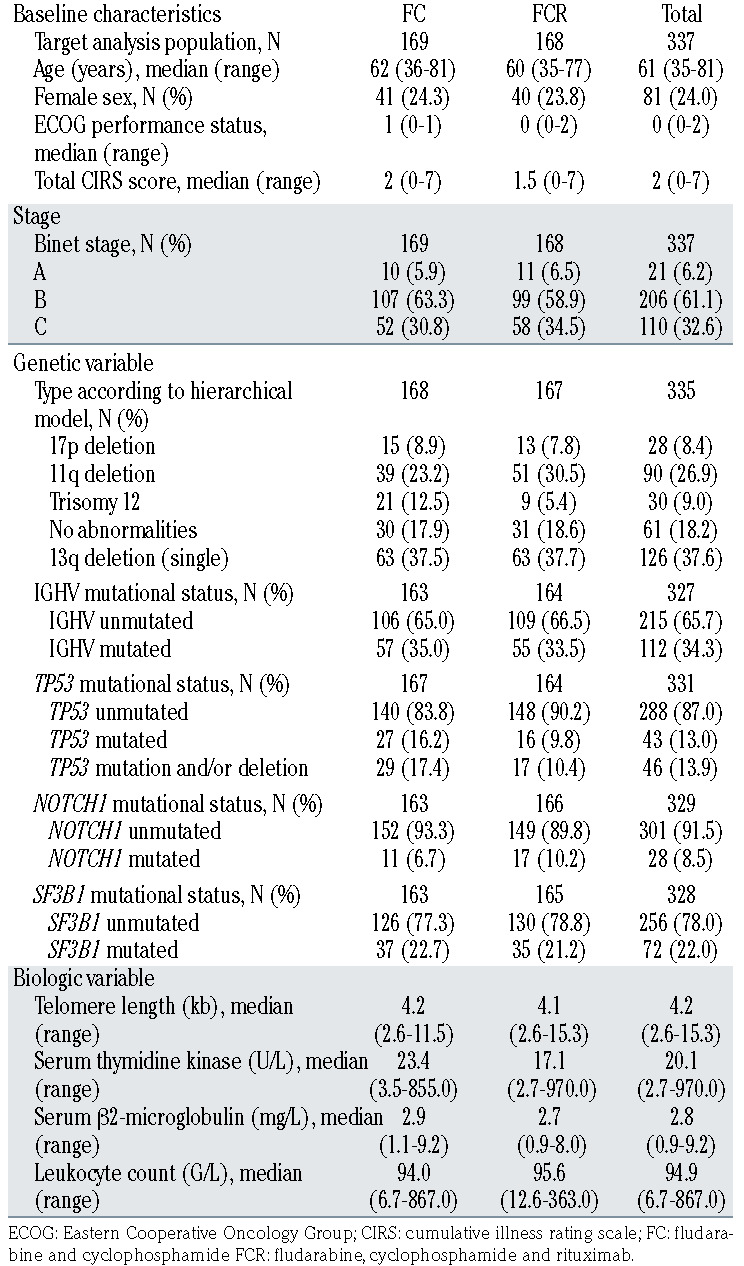
The study was conducted on peripheral blood samples from 337 previously untreated CLL patients (Table1) collected at enrolment on the CLL8 trial, a prospective, international, multi-center trial comparing first-line treatment with FC or FCR in a 1:1 randomized fashion. Further details for the study are provided online at the ClinicalTrials.gov (CTG) homepage (www.clinicaltrials.gov#NCT00281918).1 Ficoll density gradient centrifugation for isolation of mononuclear cells followed by an immunomagnetic tumor cell enrichment via CD19 (Midi MACS, Miltenyi Biotec®, Bergisch Gladbach, Germany) was performed on all samples. Data on genomic aberrations del(13q), trisomy 12, del(11q), del(17p) and mutation status for IGHV, TP53, SF3B1 and NOTCH1 was assessed as previously described.5 Informed consent and ethics committee approval was obtained in accordance with the Declaration of Helsinki for all patients.
RNA isolation, quality assessment and gene expression profiling on Exon ST 1.0 arrays
Total RNA was extracted from whole cell lysate according to the Allprep DNA/RNA mini kit (Qiagen). Quality control was performed using the Agilent 2100 Bioanalyzer with the RNA 6000 Nano LabChip (Agilent Technologies). In order to ensure accuracy and reproducibility, samples with an RNA integrity number (RIN) less than 7.0 were excluded from further analysis.
Samples were analyzed for mRNA expression using the Affymetrix GeneChip® Human Exon 1.0 ST Array (Affymetrix, Santa Clara, CA, USA). Further details are provided in the Online Supplementary Appendix.
Normalization of expression data
Raw Affymetrix data files were preprocessed by the robust multichip average (RMA) algorithm using the aroma.affymetrix R package (2008).15 Within RMA normalization, background correction and quantile normalization was conducted. Aroma.affymetrix was applied to generate GEP values summarized on the exon/probe set level and on the transcript level using the ‘core’ probe set definition according to Affymetrix. ‘Core’ refers to probe sets that are supported by the most reliable evidence from RefSeq and full-length mRNA GenBank records containing complete coding sequences information. We further assessed and excluded the presence of potential batch effects induced by external factors such as time point and location of sampling as well as time point of labeling and hybridization. Quality control was further conducted with "Relative Log Expression” (RLE) and "Normalized Unscaled Standard Errors” (NUSE), where we also did not find any abnormalities indicating potential batch effects.
Statistical analyses
Data was analyzed to evaluate improvement of prognostication for PFS and OS by using GEP in addition to prognostic factors del(17p), del(11q), trisomy 12, del(13q), IGHV mutation status, SF3B1, NOTCH1, TP53 mutations, β2-microglobulin (β2-m), thymidine kinase (TK), white blood cell count (WBC), Eastern Cooperative Oncology Group (ECOG) performance status, study medication (FC or FCR), sex and age. For the following analyses, missing values in the clinical data were imputed using chained equations.16 The algorithm imputes the missing values using a model with all other clinical variables as predictors, thus generating ’plausible’ synthetic values. As the percentage of missingness for each variable was low (maximum of 16 missing values in 337 patients), a single imputation method was adequate. Furthermore, a non-specific filtering was performed selecting the 500 genes with highest variability over all samples. The final model was built by sparsed Cox proportional hazards model using the smoothly clipped absolute deviation (SCAD) penalty.17 The “reference model” for our analysis is a Cox proportional hazards model including variables with confirmed prognostic impact: age (continuous), sex (male or female), study medication (FC or FCR), ECOG performance status (1 or 2 vs. 0), WBC, TK and β2-m (all continuous), IGHV/ NOTCH1/ SF3B1 mutation status (all unmutated vs. mutated), del(11q), del(13q), del(17p), trisomy 12 and TP53 mutation (all present or absent). The analysis is based on updated results from the CLL8 trial.1
Table 1.
Patient characteristics of the CLL8 gene expression profiling cohort.
Models investigated for possible improvement of prognostication using GEP included, first: the combination of all above-mentioned confirmed prognostic variables without penalization and a subset of the GEP data selected by SCAD penalization (referred to as “fixed model”), and secondly: the combination of confirmed prognostic variables and GEP data in which all variables were equally penalized (“equally penalized model”) allowing for substitution of the confirmed prognostic variables with equally strong prognostic GEP variables. For internal validation bootstrap subsampling with 1.000 subsamples equal to 63.2% of the original sample size was used.18 The prognostic value of the final model was evaluated on the basis of the time-dependent Brier score (as implemented in the R-package pec).19 The Brier score was used to estimate the prediction error at a given time point. Resulting prediction error curves show the time-dependent Brier score over 60 months of follow-up and the integrated Brier score (IBS) was used to summarize prediction accuracy. For external validation the apparent error was calculated. For visualization purposes, survival curves were calculated by means of the Stone-Beran estimator20 using symmetrical nearest neighborhoods around the lowest, the median, and the highest observed values of the prognostic variable combinations using the R-package prodlim,21 both for OS and PFS. Statistical analysis was performed with the R environment for statistical computing, version 3.3.1, using the R packages survival, version 2.39-5, prodlim, version 1.5.7, mice, version 2.25, ncvreg, version 3.6-0, pec, version 2.4.9 and bootstrap, version 2015.2. For validation, the prognostic gene signature established on the CLL8 cohort was tested in an array-based GEP training set of an independent cohort (n=149 unsorted CLL samples from treatmentnaive [83%] and pretreated [17%] patients).22 Unmutated IGHV was reported in 49.3% and del(17p) in 8.6% of tested samples. Further details on cohort characteristics are provided in a previous publication.22
Results
Gene expression profiling variables substitute established prognostic markers in multivariate models
We first established multivariate models for variables for which the prognostic impact was confirmed in previous studies and is herein referred to as the “reference model”. Results are shown in the Online Supplementary Table S1A for OS and in Online Supplementary Table S1B for PFS, respectively.
In order to evaluate the impact for OS including a signature consisting of GEP variables selected in the penalized Cox model (Online Supplementary Table S2A), we tested various combinations of confirmed prognostic variables and GEP. Only model combinations including genetic markers with prognostic impact achieved prediction error estimates similar to the confirmed prognostic variables used in the “reference model” (Figure 1A). Using the “fixed model”, penalization of GEP resulted in selection of only one GEP variable (PITPNC1, phosphatidylinositol transfer protein cytoplasmic 1) and no further improvement as compared to the reference model (IBS: reference model 0.092; fixed model 0.092) (Figure 1A).
In contrast, using the “equally penalized model” on all variables from the reference model and GEP data resulted in selection of only three confirmed prognostic markers (FCR, β2-m, del(17p)) along with ten GEP variables comprising the genes CLEC2B, RGS1, LDOC1, L3MBTL4, PRKCA, FHL1, SGCE, DCLK2, VSIG1, CD72 (Online Supplementary Table S3A). When assessing the prediction accuracy, this model performed similarly as the reference model (IBS: reference model 0.092; equally penalized model 0.096) (Figure 1A). When analyzing PFS by prediction models including a signature of selected GEP variables for PFS (Online Supplementary Table S2B) with the same approach, the “fixed model” did not lead to selection of GEP variables besides the confirmed prognostic variables. Conversely, only four confirmed prognostic markers (FCR, del(11q), del(17p), SF3B1 mutation) were selected in the “equally penalized model”, together with 11 GEP variables including the genes RGS1, EIF1AY, LDOC1, L3MBTL4, DCAF12, PLD5, GTSF1L, NIPAL2, CYBRD1, ANXA1 (Online Supplementary Table S3B). Again, variables selected in the “equally penalized model” performed similar to the “reference model” as demonstrated by prediction error estimates (IBS: reference model 0.160; equally penalized model 0.166; fixed model 0.160) (Figure 1B). Of note, strong prognostic markers like TP53 and IGHV mutation status (Online Supplementary Table S1) were substituted in both models by prognostic GEP variables (Online Supplementary Table S3).
For the prognostication of PFS, inclusion of GEP data alone or in addition to non-genetic variables (β2-m, TK, WBC, ECOG, study medication, sex and age) compensated for missing genetic information in patients with late disease progression (Figure 1B). In such models, GEP reliably increased prediction accuracy for patients over time as prediction error curves converged with those of the reference model. Prediction accuracy was comparable with the reference model at 60 months.
The overall number of prognostic variables remained similar for either model (“reference model”: OS/PFS 15 variables vs. “equally penalized”: OS 13 and PFS 15 variables) and although chromosomal gains or losses covered multiple genes, these variables were substituted by the expression of a few genes only. Furthermore, expression variables selected along with clinical variables in the penalized models for OS and PFS were not derived from genes localized in the recurrently deleted or amplified chromosomal regions (Online Supplementary Table S3A and B).
Gene expression profiling signatures refine prognostic estimation and retain strong prognostic value in an independent cohort of unselected patients
In order to illustrate the distribution for OS and PFS within the different prediction models, conditional Kaplan- Meier estimates were generated and survival curve estimates are shown for lowest, median, and highest values of the prognostic variable combinations (Figure 2A to F).
GEP variables are especially suitable to predict cases with late progression, while established prognostic factors compensate in the remaining cases with early progression (Figure 1A and B). Specifically, patients with long-term PFS were more accurately identified with models using prognostic GEP signatures (Figure 2D and F) when compared with models using established prognostic variables only (Figure 2B) or single genetic characteristics. This aspect was further exemplified in a subgroup analysis for patients <60 years and those receiving FCR (Online Supplementary Figure S1A and B).
In order to validate the results we tested our prognostic gene signature in an independent cohort.22 This cohort was selected to be most heterogeneous from CLL8 to confirm the strength and independence of our prognostic score for OS (Online Supplementary Table S2A; Figure 2E and F). While the CLL8 cohort consisted of treatment-naive patients receiving FC/FCR and GEP was derived from CD19+ purified tumor cells, the validation cohort contained samples with heterogeneous tumor cell purity from both treatmentnaive and pretreated patients. The CLL8-based signature was estimated on the validation cohort and evaluated for individual performance. For comparison, we used the gene signature established for the validation cohort with respective weights as provided.22 Notably, the CLL8-derived gene signature performed highly similar to the gene signature originally established for this dataset (Online Supplementary Figure S2).22
Gene expression profiling variables balance prognostic inaccuracy of established markers
GEP variables selected both for OS and PFS contained the genes RGS1 (regulator of G protein signaling 1), LDOC1 (LDOC1 regulator of NF-κB signaling) and L3MBTL4 (L3MBTL histone methyl-lysine binding protein 4). While RGS1 was homogeneously distributed across the expression range, LDOC1 and L3MBTL4 expression showed a bimodal distribution (Online Supplementary Figure S3). When evaluating expression level distributions of RGS1, LDOC1 and L3MBTL4 in relation to genetic variables, we could not identify an exclusive association with known prognostic factors (Figure 3; Online Supplementary Table S4A to D).
Figure 1.

Prediction error estimates for prognostic model combinations. Prediction error curves for combinations of prognostic variables in models are shown for overall survival (OS) (A) and progression-free survival (PFS) (B). Combinations of prognostic variables contain the confirmed prognostic variables, as used in the reference model (age, sex, study medication, Eastern Cooperative Oncology Group [ECOG], log white blood cells [WBC], β2-microglobulin [β2- m], log thymidine kinase [TK], IGHV mutation status, del(11q), del(13q), del(17p), trisomy 12, TP53 mutation, NOTCH1 mutation, SF3B1 mutation) and gene expression profiling (GEP) variables. Prognostic GEP variables were selected in addition to (fixed model) or instead of (equally penalized model) the confirmed prognostic variables. In a separate approach prognostic GEP variables were selected in addition to (fixed model) or instead of (equally penalized model) non-genetic prognostic variables (only age, sex, study medication, ECOG, log WBC, log TK, β2-m). GEP variables selected in the fixed or equally penalized model largely overlap with the full prognostic gene signature (Online Supplementary Table S2), which is separately used in the “GEP data only” prediction error curve. Combination of prognostic variables selected in the equally penalized model performed highly similar to the model containing only confirmed prognostic variables. Strong overlap was found for prediction error curves represented by the red and blue solid lines.
Figure 2.

Conditional Kaplan-Meier survival estimates illustrate the distribution for overall survival and progression-free survival within the different prediction models. Kaplan-Meier estimates were generated for the lowest, the median, and the highest observed values of the prognostic variable combinations. Kaplan-Meier estimates illustrate overall survival (OS) (A, C and E) and progression-free survival (PFS) (B, D and F) with regard to the “reference model” (confirmed prognostic variables only, A and B), the “equally penalized model” (confirmed prognostic variables and GEP equally penalized, C and D) and prognostic GEP signatures only (as represented in the Online Supplementary Table S2A and B) (E and F).
In order to elucidate the biologic context from which the prognostic impact of these three genes may derive, we dichotomized patient samples regarding the upper and lower quartile of RGS1, LDOC1 and L3MBTL4 expression and assessed the differential expression of associated genes. Differentially expressed genes with a false discovery rate (FDR) of <0.01 and a fold-change (FC) of >1.5 were assessed for overlaps of the respective expression signatures (Figure 4A). Only 12 genes were overlapping between all three gene-specific comparisons (Figure 4A). Expression signatures associated with RGS1 were highly distinct from the other profiles and showed only nine of 341 genes exclusively overlapping with the LDOC1 specific signature. Conversely, 51 of 69 genes contained in the L3MBTL4 signature exclusively overlapped with the LDOC1 signature and therefore support a similar biologic context. Genes contained in different signatures showed highly correlated expression profiles (Figure 4B). LDOC123 and other genes overlapping for the L3MBTL4 and LDOC1 signature, such as LPL or CRY1, were previously reported as surrogate markers for the IGHV mutation status. 24,25 We specifically investigated ZAP70 in this context, since it has also been identified as a surrogate marker for the IGHV mutation status.25,26,27 While ZAP70 had a foldchange lower than the previously set cut-off (FC>1.5), we found a highly significant (q<1x10-7) association with LDOC1 and L3MBTL4 (Figure 4C). Provided that LDOC1 and L3MBTL4 expression levels did not show an exclusive association with the IGHV mutation status (Figure 3; Online Supplementary Table S4A to D), we wondered if the combined status of these two genes may explain the observed similarities. Notably, expression of LDOC1 and L3MBTL4 was highly correlated with each other and the combination of both variables reliably identified the majority of cases with IGHV homology <98% (Figure 5). However, we observed several “discordant” cases with mutated IGHV and high expression levels of LDOC1 and L3MBTL4 or IGHV unmutated cases with low expression levels (Figure 3; Figure 5). Provided the fact that these continuous variables were selected due to the higher prognostic accuracy instead of the categorical IGHV mutation status, these markers therefore better mirror prognostic effects and the related biology of a variable sequence homology, especially in “discordant” cases.
Discussion
In the presented study, we evaluated the significance of GEP as a means for prognostic modeling in CLL. The CLL8 study cohort provides a valid basis for this as it was designed as a large international, multi-center phase III study defining current standard treatment, with full genetic characterization and long follow-up. Importantly, CD19+ purified tumor cells were procured at enrollment allowing valid GEP analysis.
While GEP was unable to improve prediction when used in addition to confirmed prognostic variables, GEP substituted for many of these variables when tested in direct comparison in the equally penalized model and reliably predicted OS and PFS, similar to models integrating only confirmed prognostic variables. Furthermore, for the prognostication of PFS, GEP was able to compensate for missing genetic information in the subgroup with late progression events.
High prediction accuracy for late progression and confirmation of the independent prognostic value for previously reported high-risk markers,4,5,28 which were selected in the equally penalized model, implies that GEP-based prognostication can primarily substitute for intermediate and lowrisk prognostic variables. However, GEP-based prognostic modeling was also able to substitute for “unmutated IGHV”, one of the most important variables with negative prognostic impact on OS and PFS.1,6,7,28
GEP variables selected for PFS and OS in the equally penalized models were largely heterogeneous, a finding that may reflect both methodological and biological differences when modeling these endpoints. Conversely, we identified RGS1, LDOC1 and L3MBTL4 to have prognostic value both for PFS and OS. While the combined expression of LDOC1 and L3MBTL4 was highly associated with IGHV homology and therefore may be viewed as surrogate marker of the IGHV mutation status at first, one has to consider that both genes were selected in the prognostic model instead of the IGHV mutation status. This indicates that these genes and the associated biology have a considerable impact on the prognosis and not merely substitute for the IGHV mutation status.
This study further demonstrates the potential of GEP to reduce biologic dimensionality. As such, chromosomal aberrations affecting a multitude of genes, also if minimally deleted regions only are considered, can be replaced by less than a dozen genes. The fact that the genes contained in the prognostic GEP scores were not located on recurrently affected chromosomal regions indicates that the deregulated expression does not derive from a mere gene dosage effect but represents a convergence of various biologic traits. Genes of the identified signatures likely constitute important elements in overactive signaling cascades impacting on the clinical course. In addition, GEP variables represent continuous variables and therefore may hold more potential to fine-tune prognostic modeling in contrast to categorical variables such as aberrations and mutations.
The efficacy resulting from the addition of rituximab to FC treatment and substantial benefit for patients with distinct genetic features leading to long-term disease control and OS has been confirmed recently in a long-term followup analysis.1 Notably, prognostic variables selected in the equally penalized model or the GEP signature estimated the clinical course of long-term PFS within this cohort better compared to the model using only genetic factors or parameters previously identified to characterize such patients.1
Future studies will provide insight, if prognostic models including GEP also hold advantage over recently reported prognostic models using epigenetic subgrouping.29,30,31 Patients with DNA methylation profiles reflecting memory B-cell-like CLL were reported to strongly benefit from treatment with chemoimmunotherapy on two phase II trials.31 A major strength of our study was the possibility to exclusively use CD19+ sorted patient samples from a randomized phase III trial and extensive characterization for established prognostic variables, including availability of the TP53, SF3B1 and NOTCH1 mutation status in >95% of cases. Future comparative studies assessing the prognostic impact of methylation markers need to include a comprehensive genetic characterization since SF3B1 and NOTCH1 mutations were found to have independent prognostic and predictive impact for chemoimmunotherapy5 and show a heterogeneous distribution within epigenetic subgroups.29,31 In addition, the CLL8 trial design provided an ideal basis to differentiate between the prognostic and predictive value of markers and therefore to specifically assess for the prognostic strength of established and GEP variables. Notably, GEP variables selected in our model also reliably substituted for IGHV mutation status and showed strong prognostic impact irrespective of treatment for both PFS and OS in contrast to the epigenetic subgrouping.31
Figure 3.

Association of RGS1, LDOC1 and L3MBTL4 with genetic variables. Boxplots showing distribution for log2 expression of genes selected for both overall survival (OS) and progressionfree survival (PFS), namely RGS1, LDOC1 and L3MBTL4. LDOC1 and L3MBTL4 show a bimodal distribution. Distribution of the three genes was not exclusively associated with distinct genetic variables.
Figure 4.

Assessment of genes showing concordant or discordant expression with RGS1, LDOC1 and L3MBTL4. (A) Venn diagram illustrating overlaps for differentially expressed genes (fold-change [FC] >1.5; false discovery rate [FDR] <0.01) between patient samples with either high or low expression (upper vs. lower quartile) for RGS1, LDOC1 and L3MBTL4. (B) Heatmap showing clustered expression pattern (Pearson correlation and average linkage) of 12 genes found in all three gene specific signatures and heatmap showing expression pattern of 51 genes found in gene specific signatures of LDOC1 and L3MBTL4. (C) Scatter plots for ZAP70 expression with regard to groups showing high and low LDOC1 and L3MBTL4 expression (upper vs. lower quartile).
Figure 5.

Combined status of LDOC1 and L3MBTL4 is correlated with IGHV sequence homology and identifies cases with “discordant” clinical course. The figure highlights the correlation between expression levels of LDOC1 (x-axis), L3MBTL4 (y-axis) and the immunoglobulin heavy chain variable (IGHV) gene sequence homology (color coded). Cases with IGHV sequence homology <98% are indicated in blue, cases with IGHV sequence homology ≥98% are indicated in red. LDOC1 and L3MBTL4 expression identifies “discordant” cases with mutated IGHV but poor clinical course (high expression of LDOC1 and/or L3MBTL4) and vice versa.
While storage and workup conditions were found to change expression levels of multiple transcripts in an RNA sequencing-based study on healthy donor samples, prognostic GEP variables selected in our study largely represented transcripts with low reported variability.32 Stable expression of our prognostic GEP variables selected for the respective clinical endpoints is further supported since prognostic markers unaffected by surrounding conditions (e.g., chromosomal aberrations, gene mutation status) were reliably substituted in the multivariate analysis. Validation of the prognostic impact of selected GEP variables was achieved in an independent data set differing with regard to storage conditions, workup and sorting of samples from a patient cohort with heterogeneous treatment, 22 further demonstrating the prognostic robustness of selected GEP variables.
While novel compounds have revolutionized the landscape of CLL treatment in particular for high-risk patients,10,11,12,13 the long-term benefit and treatment related toxicities still remain to be evaluated. Further, the significant economic burden may limit the access in some healthcare systems.33 In this study, we were able to confirm that GEP variables can achieve a higher prognostic accuracy, better reflect IGHV sequence homology and reliably identify “discordant” patients with mutated IGHV but poor clinical course and vice versa. This is especially promising since treatment with BTK inhibitors and FCR was reported with similar PFS in patients with mutated IGHV.14
Although the depth of biological characterization has reached a new dimension with the use of RNA sequencing, both array and RNA sequencing-based prognostic modeling were found to perform equally well for the prediction of major clinical endpoints.34 Studies evaluating FCR and BTK inhibitor treatment in a randomized fashion14 would provide an ideal basis for marker validation using RNA sequencing and easy to apply quantitative real-time polymerase chain reaction based approaches in parallel. Prognostic models used here may therefore hold promise for future selection, substitution and harmonization of prognostic markers, which show variable prognostic value within the respective treatment context.
Supplementary Material
Acknowledgements
The authors thank all patients and their physicians for trial participation and donation of samples; the DCLLSG; Sabrina Schrell and Christina Galler for their excellent technical assistance; and Myriam Mendila, Nancy Valente, Stephan Zurfluh, and Jamie Wingate for their support in conception and conduct of the trial.
Funding Statement
Funding: This work was supported by grants from BMBF (PRECISE), European Commission / BMBF (“FIRE CLL”, 01KT160), Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 1074 project B1 and B2), DJCLS R 11/01, and F. Hoffmann-La Roche.
References
- 1.Fischer K, Bahlo J, Fink AM, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127(2):208-215. [DOI] [PubMed] [Google Scholar]
- 2.Keating MJ, O`Brien S, Albitar M, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol. 2005;23(18):4079-4088. [DOI] [PubMed] [Google Scholar]
- 3.Rossi D, Rasi S, Fabbri G, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119(2):521-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910-1916. [DOI] [PubMed] [Google Scholar]
- 5.Stilgenbauer S, Schnaiter A, Paschka P, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123(21):3247-3254. [DOI] [PubMed] [Google Scholar]
- 6.Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840-1847. [PubMed] [Google Scholar]
- 7.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848-1854. [PubMed] [Google Scholar]
- 8.Woyach JA, Ruppert AS, Heerema NA, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379(26):2517-2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moreno C, Greil R, Demirkan F, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(1):43-56. [DOI] [PubMed] [Google Scholar]
- 10.Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with Venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17(6):768-778. [DOI] [PubMed] [Google Scholar]
- 12.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and Rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shanafelt TD, Wang V, Kay NE, et al. A randomized phase III study of ibrutinib (PCI- 32765)-based therapy vs. standard fludarabine, cyclophosphamide, and rituximab (FCR) chemoimmunotherapy in untreated younger patients with chronic lymphocytic leukemia (CLL): a trial of the ECOG-ACRIN cancer research group (E1912). Blood. 2018;132(Supplement 1):LBA-4. [Google Scholar]
- 15.Bengtsson H, Simpson K, Bullard J, Hansen K. aroma.affymetrix: A generic framework in R for analyzing small to very large Affymetrix data sets in bounded memory. Tech Reports. 2008;745:1-9. [Google Scholar]
- 16.van Buuren S, Groothuis-Oudshoorn K. MICE: multivariate imputation by chained equations in R. J Stat Software. 2011;45(3). [Google Scholar]
- 17.Fan J, Li R. Variable selection via nonconcave penalized likelihood and its oracle properties. J Am Stat Assoc. 2001;96:1348-1360. [Google Scholar]
- 18.Willi Sauerbrei, Buchholz A, Boulesteix AL, Binder H. On stability issues in deriving multivariable regression models. Biom J. 2015;57(4):531-555. [DOI] [PubMed] [Google Scholar]
- 19.Mogensen UB, Ishwaran H, Gerds TA. Evaluating random forests for survival analysis using prediction error curves. J Stat Softw. 2012;50(11):1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beran R. Nonparametric regression with randomly censored survival data. Tech Report. 1981 University of California, Berkeley. [Google Scholar]
- 21.Gerds TA. Prodlim: Product-limit estimation for censored event history analysis 2014. URL https//CRAN.R-project.org/package=prodlim. R Packag. version 1, 460 (2016). [Google Scholar]
- 22.Herold T, Jurinovic V, Metzeler KH, et al. An eight-gene expression signature for the prediction of survival and time to treatment in chronic lymphocytic leukemia. Leukemia. 2011;25(10):1639-1645. [DOI] [PubMed] [Google Scholar]
- 23.Duzkale H, Schweighofer CD, Coombes KR, et al. LDOC1 mRNA is differentially expressed in chronic lymphocytic leukemia and predicts overall survival in untreated patients. Blood. 2011;117(15):4076-4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morabito F, Cutrona G, Mosca L, et al. Surrogate molecular markers for IGHV mutational status in chronic lymphocytic leukemia for predicting time to first treatment. Leuk Res. 2015;39(8):840-845. [DOI] [PubMed] [Google Scholar]
- 25.Rosenwald A, Alizadeh AA, Widhopf G, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001;194(11):1639-1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rassenti LZ, Huynh L, Toy TL, et al. ZAP-70 compared with immunoglobulin heavychain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med. 2004;351(9):893-901. [DOI] [PubMed] [Google Scholar]
- 27.Klein U, Tu Y, Stolovitzky GA, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194(11):1625-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.International CLL-IPI working group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLLIPI): a meta-analysis of individual patient data. Lancet Oncol. 2016;17(6):779-790. [DOI] [PubMed] [Google Scholar]
- 29.Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44(11):1236-1242. [DOI] [PubMed] [Google Scholar]
- 30.Oakes CC, Seifert M, Assenov Y, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet. 2016;48(3):253-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wojdacz TK, Amarasinghe HE, Kadalayil L, et al. Clinical significance of DNA methylation in chronic lymphocytic leukemia patients: results from 3 UK clinical trials. Blood Adv. 2019;3(16):2474-2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dvinge H, Ries RE, Ilagan JO, Stirewalt DL, Meshinchi S, Bradley RK. Sample processing obscures cancer-specific alterations in leukemic transcriptomes. Proc Natl Acad Sc. USA. 2014;111(47):16802-16807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Q, Jain N, Ayer T, et al. Economic burden of chronic lymphocytic leukemia in the era of oral targeted therapies in the United States. J Clin Oncol. 2017;35(2):166-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang W, Yu Y, Hertwig F, et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015;16(1):133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.