

Abstract
Chromosome missegregation over the course of multiple cell divisions, termed chromosomal instability (CIN), is a hallmark of cancer. Multiple causes of CIN have been identified, including defects in the mitotic checkpoint, altered kinetochore-microtubule dynamics, centrosome amplification, and ionizing radiation. Here we review the types, mechanisms, and cellular implications of CIN. We discuss the evidence that CIN can promote tumors, suppress them, or do neither, depending on the rates of chromosome missegregration and the cellular context. Very high rates of chromosome missegregation lead to cell death due to loss of essential chromosomes; thus elevating CIN above a tolerable threshold provides a mechanistic opportunity to promote cancer cell death. Lethal rates of CIN can be achieved by a single insult or through a combination of insults. Because ionizing radiation induces CIN, additional therapies that increase CIN may serve as useful modulators of radiation sensitivity. Ultimately, quantifying the intrinsic CIN in a tumor and modulating this level pharmacologically as well as with radiation may allow for a more rational, personalized radiation therapy prescription, thereby decreasing side effects and increasing local control.
Introduction
CIN results in aneuploid daughter cells, which contain an abnormal chromosome content that deviates from a multiple of the haploid. Both CIN and aneuploidy can be numerical or structural: numerical CIN describes the missegregation of whole chromosomes, while structural CIN involves gain or loss of large chromosome segments. Though the terms aneuploidy and CIN are sometimes used interchangeably, there is an important distinction: aneuploidy describes the state of having an abnormal chromosome number, while CIN involves a persistent rate of chromosome missegregation. Aneuploid chromosome contents can be faithfully propagated to daughter cells over multiple generations or may result in CIN1,2.
CIN and aneuploidy are well-recognized hallmarks of cancer. Aneuploidy occurs in ~85% of human cancers3, with the prevalence of aneuploidy varying between tumor types4,5. Importantly, aneuploidy is more common in cancer than alteration of any single oncogene or tumor suppressor and affects more of the genome than any other somatic genetic alteration6–8. Approximately 25% of the genome of a typical solid tumor is altered through whole-chromosome or chromosome arm changes, with roughly equivalent levels of amplification and deletion, demonstrating the presence of large scale genomic alterations6. CIN, as a rate of ongoing genomic shuffling due to mitotic defects, is more difficult than aneuploidy to quantify in tumors, particularly since most patient samples are fixed specimens collected at a single timepoint. However, one validated method of measuring CIN, via intercellular variability in chromosome number, finds that CIN occurs in 44% of solid tumors and 14% of hematopoietic cancers9. Single cell sequencing approaches also reveal both CIN and stably maintained aneuploid tumor clones10–12, though for both methods it is difficult to discriminate whether clonal aneuploid populations reproducibly produce genetically identical aneuploid progeny, for instance because these genomes have been optimized for tumor growth in their specific microenvironment, or whether other aneuploid karyotypes are produced but eliminated from the surviving population by selection. Nevertheless, both CIN and aneuploidy are common features of a large majority of human cancers.
Causes of CIN
Mammalian cells employ multiple mechanisms to ensure faithful chromosome segregation to prevent CIN. Foremost among these is the mitotic checkpoint (also known as the spindle assembly checkpoint), which controls mitotic progression. During a normal mitosis the chromosomes, which are replicated during S phase and enter mitosis as pairs of sister chromatids linked by cohesins, are sorted and segregated on a bipolar spindle composed of microtubules (reviewed in13). The centromere of each sister chromatid attaches to the microtubules of the mitotic spindle through a protein-based complex termed the kinetochore. The kinetochores of sister chromatid pairs attach to microtubules emanating from opposite spindle poles, which allows the chromatid pairs to align at the spindle equator and form a metaphase plate. Kinetochores frequently make attachments to microtubules from the inappropriate spindle pole, but these are released through an Aurora B-mediated error-correction pathway. Chromatids with unattached kinetochores are at risk of being missegregated and therefore recruit mitotic checkpoint proteins and convert them into active inhibitors of the Anaphase Promoting Complex/Cyclosome (APC/C), the target of the mitotic checkpoint14. Even a single kinetochore lacking microtubule attachments is sufficient to activate the mitotic checkpoint and prevent mitotic progression15. However, once all kinetochores have made stable microtubule attachments, the mitotic checkpoint is satisfied. APC/C E3 ubiquitin ligase activity leads to cleavage of the cohesins connecting sister chromatid pairs, allowing sister chromatid separation, mitotic progression into anaphase, and the creation of two genetically identical daughter cells.
Successful mitosis requires the coordinated activity of kinases, phosphatases, molecular motors, and microtubule associated proteins as well as the mitotic checkpoint. Given the exquisite complexity of mitosis, there are numerous types of cellular defects that result in chromosome missegregation. Though mutations in mitotic checkpoint proteins are rare in human cancers, changes in mRNA expression are common (Fig. 1) and lead to weakening of the mitotic checkpoint and CIN16–18. Mitotic checkpoint genes are most commonly upregulated in cancers compared to normal tissue (Fig. 1A). In part, this is because many mitotic checkpoint genes are cell cycle regulated and are therefore more highly expressed in tumors since they are more proliferative than corresponding normal tissue. However, even mitotic checkpoint genes that are not cell cycle regulated show increased expression in primary cancers relative to normal tissue (Fig. 1A). When comparing amongst tumors that are or are not diploid for a specific checkpoint gene, both increased and decreased expression of mitotic checkpoint genes are observed, implying differences due to copy number changes and/or transcriptional regulation (Fig. 1B). Thus, alterations in mitotic checkpoint gene expression, which often impairs mitotic checkpoint signaling and causes CIN, are common in human cancers, while mutations in these genes are rare.
Figure 1: Mitotic checkpoint gene expression is commonly altered in cancer.
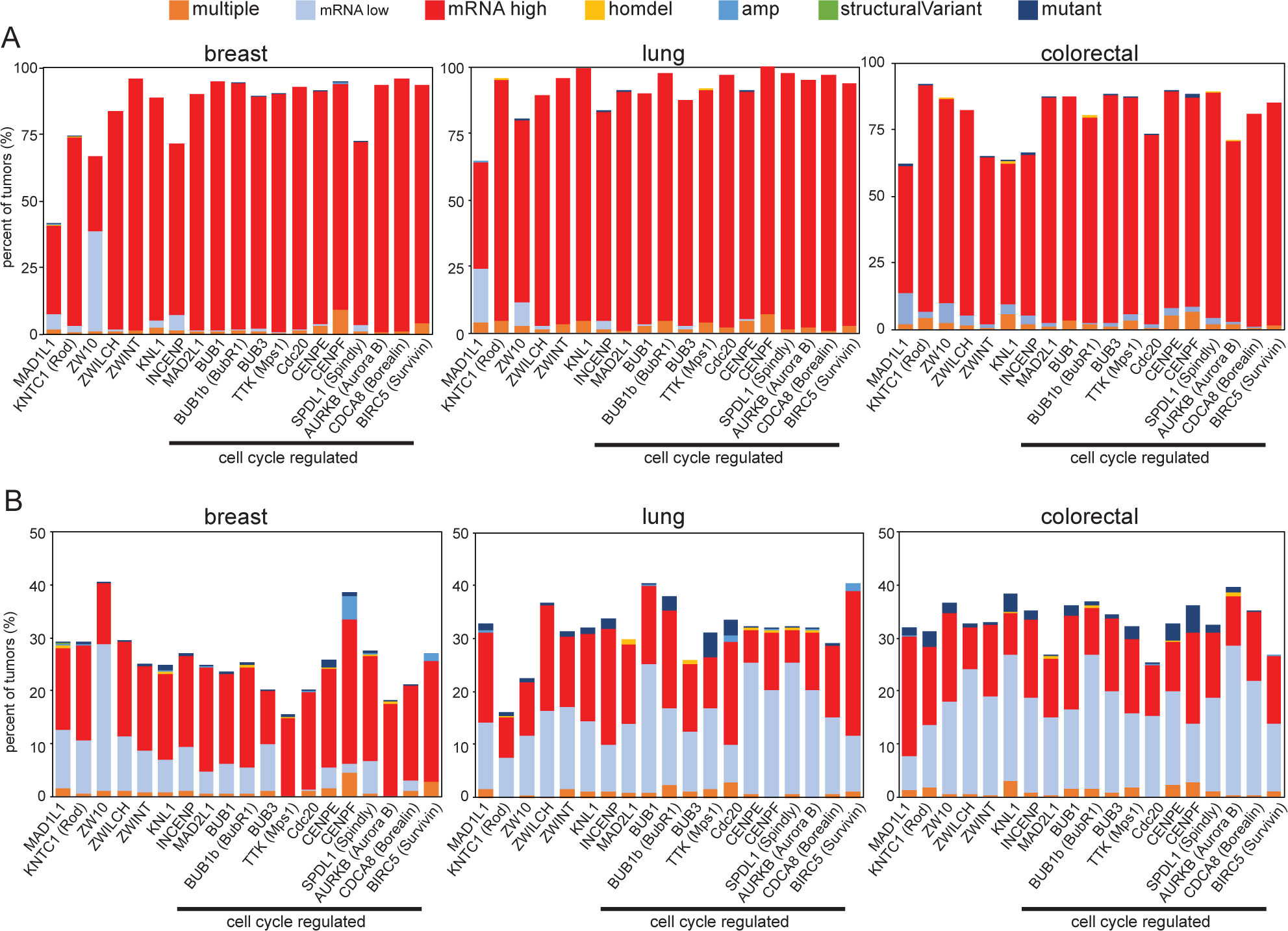
Quantitation of TCGA PanCancer Atlas data111,112 showing the frequency with which mitotic checkpoint genes are mutated, homozygously deleted, amplified, contain a structural variant, exhibit increased or decreased mRNA expression, or have multiple defects in primary cancers as compared to (A) normal samples or (B) tumors that are diploid for each gene, showing that mutations and genomic rearrangements of mitotic checkpoint genes are rare, but altered expression is common. n=994 breast, 466 lung, 524 colorectal samples. Protein names that differ substantially from gene names are indicated in parentheses. Genes that are cell cycle regulated113 are indicated.
Defects in chromosome congression to the metaphase plate result in misaligned chromosomes (Fig. 2A, arrow), some of which are at or near spindle poles (polar chromosomes). Misaligned chromosomes can be whole chromosomes or chromosome fragments (Fig. 2A, arrowhead) and can result from radiation treatment, weakened mitotic checkpoint activity, or decreased expression and/or function of the plus-end directed kinesin CENP-E19. In these circumstances, misaligned chromosomes are often missegregated, leading to CIN.
Figure 2: Causes and examples of CIN.
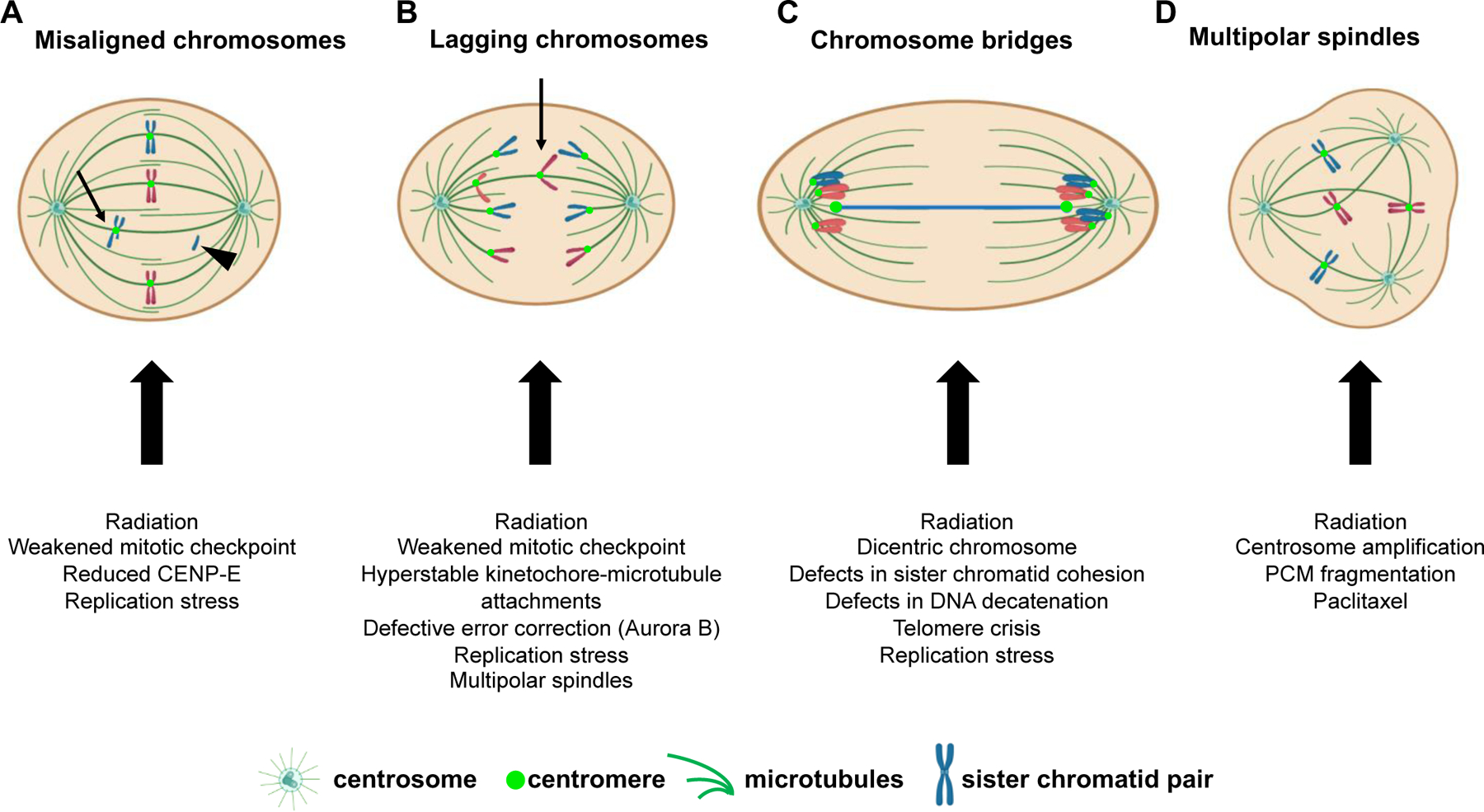
Schematic of hypothetical diploid cell with 4 chromosomes exhibiting various mitotic defects leading to chromosome missegregation. (A) In cells in which most chromosomes have aligned at the spindle equator forming a metaphase plate, one or more chromosomes may remain misaligned. These misaligned chromosomes can be located near the spindle pole (polar chromosomes) or in between the spindle poles and the spindle equator (arrow). Misaligned chromosomes may be intact sister chromatid pairs or a segment of the chromatid pair, including acentric fragments (arrowhead) that lack a centromere and kinetochore. Acentric fragments fail to attach to spindle microtubules and are randomly segregated. (B) Example of a chromosome that lags behind the segregating masses of DNA due to an abnormal merotelic attachment, in which a single kinetochore is attached to microtubules from both spindle poles. Lagging chromosomes can also arise from acentric fragments (not shown). (C) Chromosome bridges, which bridge the segregating chromosome masses, can be caused by dicentric chromosomes in which each centromere is attached to microtubules from opposite spindle poles. (D) Multipolar spindles, which contain greater than two spindle poles, can cause chromosome missegregation in two ways: 1) multipolar spindles that are maintained as cells proceed into anaphase result in division of the two sets of chromosomes in three or more directions. 2) multipolar spindles that are focused into pseudo-bipolar spindles increase the incidence of lagging chromosomes, as in B. PCM, pericentriolar material.
Once chromosomes have begun segregating towards opposite spindle poles during anaphase, additional mitotic errors indicative of chromosome missegregation can become apparent. These include lagging chromosomes (Fig. 2B), which lag behind the segregating masses of DNA, and chromosome bridges (Fig. 2C), which are composed of chromatin stretched between the separating spindle poles. Numerous mechanisms result in lagging chromosomes, including accelerated20 or delayed centrosome separation21, deficits in chromokinesin activity22, and inappropriate merotelic attachments in which a single kinetochore is attached to microtubules from both spindle poles, resulting in a tug-of-war. Though merotelic attachments are not detected by the mitotic checkpoint, errors can be resolved by the error correction pathway, but defects in this pathway substantially increase erroneous attachments leading to lagging chromosomes23,24. Altered kinetochore-microtubule dynamics represent another cause of lagging chromosomes. Certain cancer cell lines with CIN have more stable kinetochore-microtubule attachments than non-CIN cells. These hyperstable kinetochore-microtubule attachments prevent error correction and increase the frequency of lagging chromosomes25,26.
A number of mechanisms also contribute to the formation of chromosome bridges. Bridges can occur due to defects in sister chromatid separation because of incomplete cohesin removal27, defects in DNA decatenation, or lack of topoisomerase activity28. Bridges can also be formed by end-to-end chromosome fusions after double stranded DNA breaks as a consequence of radiation exposure29 or telomere crisis, which both yield dicentric chromosomes produced by rearrangements combining two chromosomes or chromosome fragments that both contain a centromere. Attachment of the two centromeres on a single chromatid to microtubules from opposite spindle poles leads to stretching of the intervening chromatin between the segregating masses of DNA during anaphase, forming a bridge (Fig. 2C). These bridges can be maintained during mitosis30, but subsequently break, resulting in a chromosome breakage-fusion-bridge cycle and further chromosomal rearrangements in subsequent cell cycles. Replication stress during S phase provides an additional, pre-mitotic source of chromosome bridges that can also produce acentric chromosome fragments appearing as misaligned or lagging chromosomes31. Misaligned, lagging, and bridge chromosomes can become isolated from the main nucleus in the subsequent G1 phase of the cell cycle and form their own micronucleus. Chromosomes in micronuclei are often missegregated32 or undergo chromothripsis33, a process in which chromatin from one or two chromosomes is shattered and extensively reorganized, resulting in structural as well as numerical CIN.
An additional mitotic error contributing to aneuploidy and CIN is the formation of multipolar spindles, which can appear in any stage of mitosis (Fig. 2D). Persistence of a multipolar spindle throughout mitosis results in separation of the two sets of replicated chromosomes into three or more daughter cells, most of which die or exhibit cell cycle arrest34. Cells sometimes focus these multipolar spindles into bipolar spindles35, but even the presence of a transient multipolar spindle can promote CIN via merotelic attachments that produce lagging chromosomes34,36. Multipolar spindles are caused by the presence of supernumerary centrosomes, which are readily observed in a variety of cancer types and are induced by the anti-mitotic chemotherapeutic and microtubule stabilizing drug paclitaxel (Taxol™) as well as by radiation. Thus, a wide variety of genetic and cellular alterations contribute to mitotic errors and CIN in cancer.
Ionizing radiation as a cause of CIN
Ionizing radiation is a well-characterized cause of aneuploidy and CIN (Fig. 3). It was established in the 1950s that ionizing radiation damages the “genetic constituents” of cells37 and later that chromosome aberrations from double stranded DNA breaks were a significant source of lethality38. DNA lesions caused by ionizing radiation include base lesions, single strand, and double strand breaks. Repair of double stranded DNA breaks via non-homologous end-joining is error prone and can produce structural aneuploidy in the form of chromosomal translocations, inversions, deletions, and insertions. Radiation-induced structural aneuploidies can be propagated over subsequent generations39, as was observed for multiple rearrangements of chromosome 4 that occurred in a dose dependent fashion after cell recovery and proliferation40. Though some of these rearrangements were stably propagated, most were unstable, indicating CIN. Other types of structural aneuploidy induced by radiation are also associated with CIN. During the mitosis that occurs after radiation, acentric fragments produced by chromosome breaks or rearrangements are unable to attach to the microtubules of the mitotic spindle and are therefore randomly segregated. During cell division, these acentric fragments appear as misaligned or lagging chromosomes, depending on the stage of mitosis (Fig. 2, 3). Radiation also causes dicentric chromosomes, which result in chromosome bridges when the two kinetochores on a single sister chromatid attach to opposite spindle poles (Fig. 2C, 3). Pulling forces may cause breaks in the chromatin, ultimately leading to continued CIN and in some cases chromothripsis, generating further DNA damage. These chromosomal rearrangements are perpetuated with further mitotic cycles and contribute to additional genomic instability41.
Figure 3: Ionizing radiation induces CIN.
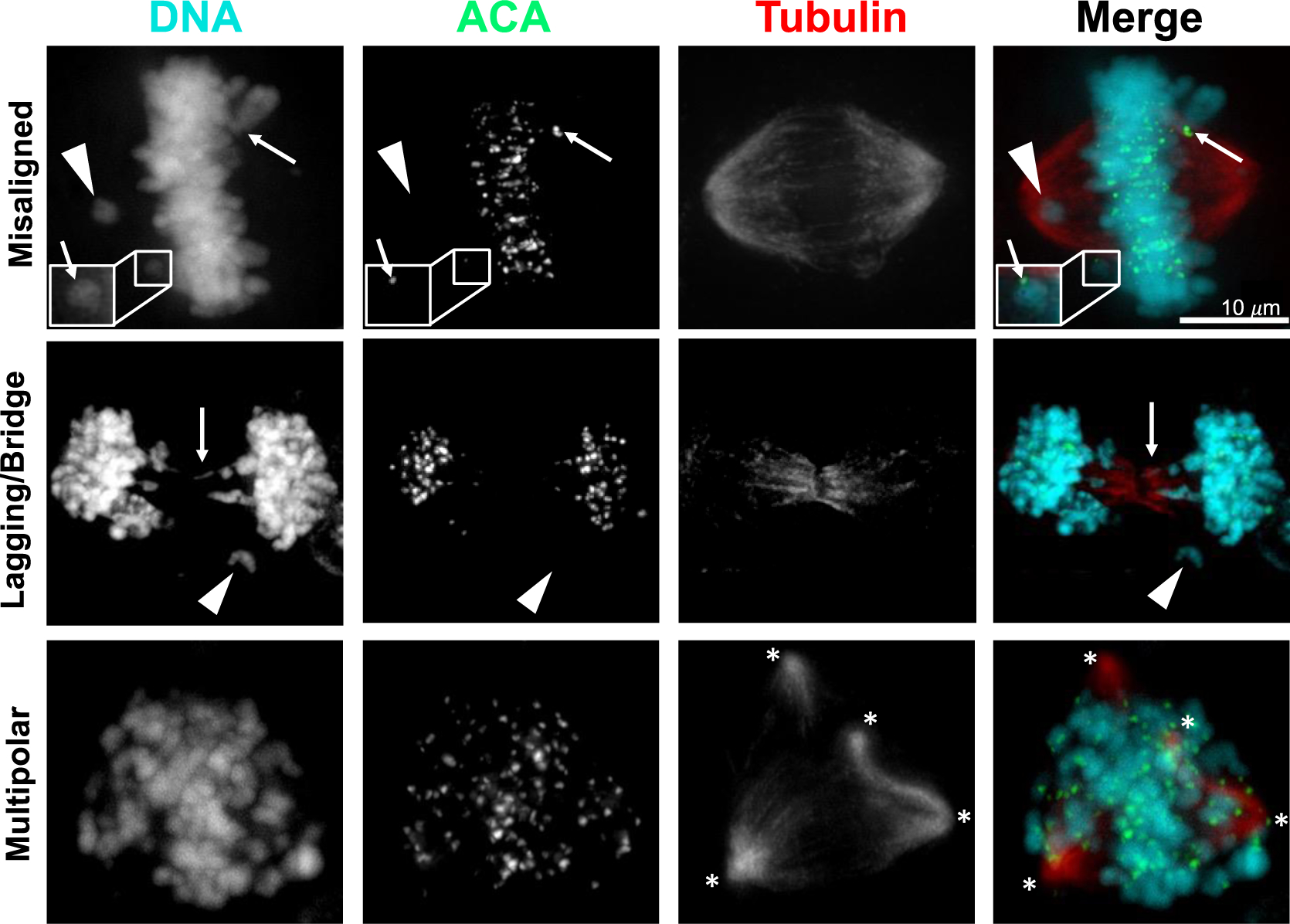
Representative images of HeLa cells 24 hours after 4 Gy radiation. ACA, anti-centromere antibody. Upper row, misaligned chromosomes including acentric (arrowhead) and centric chromosomes (arrows). Middle row, lagging (acentric, arrowhead) and bridge (arrow) chromosomes. Bottom row, multipolar spindles. Asterisks denote spindle poles.
In addition to structural aneuploidy, radiation has also long been recognized to cause numerical aneuploidy and polyploidy42,43. Recent evidence has provided insight into the mechanism through which radiation induces missegregation of whole chromosomes during mitosis. Radiation increases stability of kinetochore-microtubule attachments and thereby prevents correction of erroneous attachments, leading to the formation of lagging chromosomes during anaphase44,45 (Fig. 2B, 3), which is mediated in part by the DNA Damage Response. Interestingly, experimentally destabilizing kinetochore-microtubule attachments by overexpressing Kif2b, a microtubule-depolymerizing kinesin-13 protein, significantly decreased the incidence of lagging chromosomes after radiation and thereby decreased whole chromosome missegregation without affecting the extent of DNA damage or repair44.
Radiation also induces multipolar spindles (Fig. 2D, 3) through at least two mechanisms. First, radiation causes erroneous replication of centrosomes during a subsequent G2 arrest in a Chk1 dependent fashion46–48. Second, radiation induces polyploid cells through cytokinesis failure, cell fusion and/or endocycling, which all produce cells containing supernumerary centrosomes43,49. Multipolar divisions themselves are a source of CIN but, even when multipolar spindles are subsequently focused into pseudo-bipolar spindles, they can increase lagging chromosomes, further increasing CIN34,36. Interestingly, inhibiting centrosome amplification in cultured cells reduced radiation induced cell death, further implicating this as a mechanism of radiation-induced cytotoxicity50. Supernumerary centrosomes are not well tolerated, and it has been shown that clonogenic tetraploid cells formed through inhibiting cytokinesis rapidly decrease their centrosome number through asymmetric clustering of centrosomes during mitosis. This produces one daughter cell with supernumerary centrosomes, which proliferates poorly, and one daughter with only a single centrosome, which shows increased fitness51. Similarly, after induction of centrosome overduplication by overexpression of the kinase Plk4, cells that survive to become clonogenic return to normal centrosome numbers52. Whether cells with radiation-induced centrosome amplification return to a normal centrosome number after experiencing a transient period of CIN due to supernumerary centrosomes is unknown, but evidence from other experimental models suggests this is a possibility. Interestingly, long-term timelapse microscopy has revealed that a fraction of radiation-induced polyploid giant cells continue to divide, sometimes several times, though it is unclear whether these cells are clonogenic43,49,53.
In summary, ionizing radiation induces structural and numerical CIN through formation of dicentric and acentric chromosomes, hyper-stabilization of kinetochore-microtubule attachments, and centrosome amplification. This results in a variety of mitotic defects including misaligned and lagging chromosomes, chromosome bridges, and multipolar spindles, which can all contribute to cell death if occurring with sufficiently high frequency. Conversely, reducing CIN in the form of lagging chromosomes or centrosome amplification leads to radiation resistance and increased cell survival. This implies a mechanistic opportunity to alter CIN levels to enhance radiation induced cell death.
CIN can both promote and suppress tumors
Aneuploidy is clearly well tolerated in cancer cells, suggesting it offers a growth and survival advantage. However, when induced experimentally, aneuploidy is often associated with a significant fitness cost. This apparent contradiction is known as the ‘aneuploidy paradox’54. Induction of aneuploidy experimentally, predominantly through gain of a single chromosome, leads to metabolic alterations, proteotoxic stress, cell cycle delay, and in some cases, CIN55–58. How aneuploid human tumors are able to proliferate and thrive given the general growth disadvantage observed in trisomic cell lines is unclear, although the karyotypes observed in cancer are typically more complex than single chromosome gains. These complex karyotypes could mitigate the fitness costs of trisomy of a single chromosome, which substantially reduces fitness during embryonic development as well as in cultured cells.
It is important to note that, though aneuploid cell lines often show a growth disadvantage under conditions optimized for euploid cells, certain karyotypes exhibit growth advantage under selective conditions, highlighting the importance of chromosome specific as well as global effects of aneuploidy59–62. In cell culture studies, CIN has been associated with therapeutic resistance63, potentially because the shuffling of chromosomes generates a large variety of karyotypes, one or more of which may confer resistance. Consistent with this idea, two recent studies demonstrated that transient induction of CIN in cell lines produced a variety of aneuploid karyotypes that conferred resistance to cytotoxic drugs64,65. Sequencing of these resistant clones revealed outgrowth of specific karyotypes, however, the recurrent chromosome gains and losses that conferred resistance differed in distinct cell lines, suggesting that specific aneuploidies are unlikely to uniformly confer drug resistance. Additionally, the persistent genomic shuffling associated with CIN can increase genomic heterogeneity to such an extent that it can lead to oncogene independence and facilitate further tumor growth66. These findings offer further support for the conclusion that low rates of CIN generate a range of aneuploid karyotypes that permit a subset of cells to survive and proliferate in adverse and changing environments.
Accordingly, mouse models of aneuploidy and CIN have demonstrated tumor promotion67–70, as well as tumor suppression66,71–73, or neither74–78, depending on the specific genetic alteration and the tissue context (reviewed in 9,18,79). Many of the altered genes in these murine models have cellular functions independent of chromosome missegregation (reviewed in16) that complicate interpretation. However, one unifying conclusion is that the effect of aneuploidy on tumors depends on the rate of CIN. Low rates of CIN can be tumor promoting through gain of oncogenes or loss of tumor suppressors, although most aneuploid cells are not transformed. In contrast, higher rates of chromosome missegregation lead to cell death and tumor suppression due to loss of both copies of one or more essential chromosomes80,81. Importantly, higher levels of CIN can drive tumor suppression even in the case of oncogene driven tumors66,71,72. Thus there appears to be a range of CIN that allows for optimal tumor cell fitness; however, further increasing CIN above a maximally tolerated threshold, which appears to vary by tissue context82,83, leads to cell death (Fig. 4). This suggests that tumors with underlying CIN are more sensitive to therapies that further increase CIN, such as radiation.
Figure 4: Cells tolerate only a limited amount of CIN.
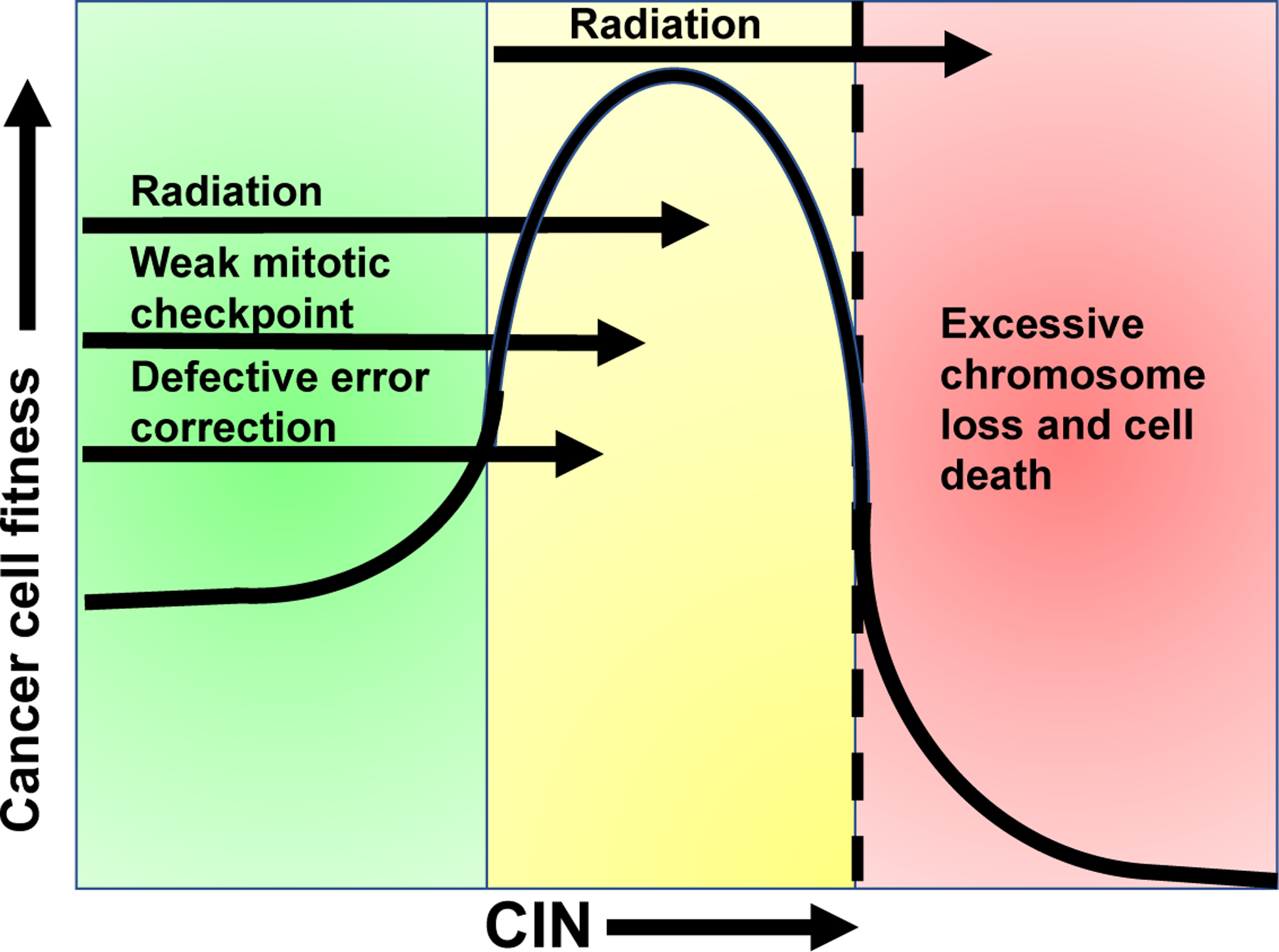
Cells have evolved multiple mechanisms for accurately maintaining their genomes during mitosis (green zone). Modestly increased rates of CIN (yellow zone), due to defective error correction or weakened mitotic checkpoint signaling, can be tumor promoting due to loss of tumor suppressor genes, for instance. However, further increasing the rate of CIN (into the red zone) results in cell death due to loss of both copies of one or more essential chromosomes. Tumors exhibit variable rates of CIN that fall in either the green or yellow zones prior to treatment. Given that radiation induces CIN, irradiation of tumors in the yellow zone is predicted to increase CIN over a maximally tolerated threshold (dashed line), leading to cell death and tumor regression. However, tumors in the green zone are expected to be more tolerant of radiation induced increases in CIN, since it elevates them into the yellow zone. Increasing CIN above a tolerable threshold in these tumors with a CIN-inducing drug provides a mechanistic opportunity to enhance cancer cell death and radiation response.
Combining two sources of CIN results in cell death and tumor suppression
Lethal rates of CIN can be achieved through a single mechanism, for instance by complete loss (rather than weakening) of mitotic checkpoint signaling9,18. However, combining two insults that each cause a low, tolerable rate of CIN can also increase CIN above a maximally tolerated threshold, resulting in cell death and tumor suppression. Since approximately half of solid tumors exhibit CIN prior to treatment, this suggests that increasing CIN could be a useful therapeutic strategy.
Consistent with that hypothesis, both genetic and pharmacologic induction of CIN have been shown to be effective in causing cell death and tumor suppression when combined 84,85. Genetic alteration of a variety of genes results in a low, tolerable rate of CIN coupled with a modest increase in spontaneous tumorigenesis; however, combining two different genetic sources of CIN produces cells with increased rates of CIN ultimately leading to cell death and tumor suppression66,71–73,86,87. Importantly, high CIN is capable of suppressing the growth and progression of tumors that have already formed72. The converse is also true. Decreasing the levels of CIN by overexpressing the microtubule depolymerase Kif2b in a mouse model of K-Ras-induced lung cancer promoted tumor growth88, consistent with the model that there is an optimal rate of CIN for tumor growth, and that increasing CIN above that level suppresses tumor growth. Importantly, p53 is not necessary for cell death and tumor suppression caused by high CIN81. Given the frequency with which the p53 pathway is impaired in cancer, and its role in conferring radioresistance, the continued sensitivity of tumors to high rates of CIN in the absence of p53 suggests that modulating CIN could be a mechanism for conferring radiosensitivity that is applicable to most cancers.
Pharmacologic treatments that induce CIN can cooperate with genetic insults to increase CIN over a maximally tolerated threshold, resulting in tumor suppression (Fig. 4). The best studied examples of pharmacologically induced CIN involve use of taxanes, which are among the most widely used chemotherapeutics and are known to stabilize microtubules. High doses of these drugs arrest cells in mitosis89. However, clinically relevant concentrations of paclitaxel in breast cancer are insufficient to cause mitotic arrest. Instead, these lower doses of paclitaxel induce abnormal multipolar spindles, which result in CIN due to segregation of the two replicated sets of chromosomes in three or more directions90. CIN induced genetically or pharmacologically sensitizes cells and animal tumor models to low dose taxane, as combining these two sources of CIN results in higher rates of CIN, cell death and tumor shrinkage84,85,91. Another example of this phenomenon is shown with the tumor suppressor p38α, which responds to DNA damage and replication stress during S-phase. Loss of this protein results in increased CIN and aneuploidy. Inhibitors of p38α that cause CIN were found to potentiate the effects of CIN-inducing drugs, including taxanes, such that much lower doses of taxanes were necessary to induce cell death92. Similarly, reduction of the MTUS1 gene product ATIP3 increased multipolar spindles and sensitivity to low dose taxane in cell culture and correlated with response to chemotherapy that includes taxane in three cohorts of breast cancer patients93. These studies support the conclusion that tumor cells with a low level of pre-existing CIN are more sensitive to therapies that also cause CIN, such as taxanes and radiation, which is further discussed below.
Clinical studies also support the model that excessive levels of CIN lead to tumor suppression and increased survival in cancer patients, though currently used methods to measure CIN clinically are admittedly imperfect. In general, tumors with CIN exhibit worse patient prognosis than diploid, chromosomally stable tumors94–98. However, tumors with the highest quartile of CIN, as dictated by bioinformatic analysis of gene expression or copy number alterations, correlated with improved recurrence free survival for patients with a variety of cancers, including those of the breast, lung, ovaries and stomach99,100. Improved outcome in patients with tumors with extreme CIN was also shown in two additional cohorts of ER-negative breast cancers using an alternative method of measuring CIN, fluorescence in situ hybridization based detection of intercellular variability in the copy number of two chromosomes101,102. Taken together, the existing data support a model in which a certain range of CIN is weakly tumor promoting, but excessive rates of CIN cause cell death and tumor suppression (Fig. 4).
Evidence for CIN as a radiosensitizer
Since ionizing radiation provides a significant source of both numeric and structural CIN, tumors that exhibit CIN prior to treatment – and are therefore closer to their maximally tolerated threshold – are expected to be more radiosensitive than chromosomally stable (non-CIN) tumors. Historical studies have shown that the increased genomic plasticity induced by radiation could modulate sensitivity by allowing for emergence of resistant clones103. Early studies revealed that more chromosomally unstable clones had greater variation in radiation sensitivity in general than stable clones, but CIN here was defined as clones having rearrangements in 1 chromosome, and is therefore actually more representative of structural aneuploidy rather than CIN104. Thus, these experiments did not test whether pre-existing CIN – or a simultaneous CIN-inducing therapy – sensitizes to radiation. More recent studies that have tested this directly have found that pre-treatment CIN correlates with radiation response. Rectal adenocarcinomas with higher pre-treatment rates of CIN had significantly better pathological response rates to neoadjuvant chemoradiation than more chromosomally stable cancers105. While tumor cells with higher pre-existing CIN are more sensitive to radiation, the converse is also true: reducing CIN by destabilizing hyperstable kinetochore-microtubule attachments decreased radiation sensitivity in an orthotopic model of glioma44, likely by maintaining tolerable rates of CIN.
There is evidence that pharmacologic methods of inducing CIN also increase radiosensitivity. It is well known that taxanes sensitize cancer cells and patient tumors to radiation106–109. Though the sensitization effects of taxanes have previously been assumed to be due to mitotic arrest, the fact that paclitaxel does not reach sufficient concentrations in primary breast cancer to arrest cells in mitosis and causes CIN on multipolar spindles instead90, suggests that taxanes sensitize to radiation via increasing CIN. Chemical inhibition of the mitotic checkpoint kinase Mps1 (gene name TTK), which induces CIN84,85,91, also increases radiation sensitivity in cell and animal breast cancer models110, which was attributed to persistent DNA damage and decreased homologous recombination-mediated DNA damage repair. Though CIN was not evaluated in this study, given that Mps1 inhibition causes CIN84,85,91, it is likely that increased CIN played a role in the enhanced radiosensitivity observed. Similarly, CIN combined with a low level of Mre11, a component of the MRN complex involved in DNA repair, highly correlated with response to chemoradiation in rectal adenocarcinoma105, implying that CIN is particularly detrimental in tumors with DNA repair defects. Together, these studies imply that pre-treatment CIN sensitizes to ionizing radiation and that synergism with CIN-inducing drugs or DNA repair defects may allow for reduced radiation doses.
Conclusions
Manipulating levels of CIN to promote cell death provides a promising mechanistic opportunity in oncology. Tumors contain varying, tolerable rates of CIN prior to treatment, which can be increased to lethal levels pharmacologically and/or with radiation. Given the evidence that tumors with higher CIN are more sensitive to radiation therapy, CIN may serve as a potential biomarker of radiation response that is applicable to all cancer types. Further, targeted manipulation of CIN may allow for enhanced radiation induced cell death, which could have significant clinical implications. Though there remain many unanswered questions, further research on the mechanisms underlying radiation induced CIN, the role of DNA damage repair, and study of the interaction of radiation with CIN-inducing drugs will be essential moving forward. Ultimately, we foresee CIN as a principle determinant of radiation response that can aid in developing a more personalized approach to radiation therapy for complete eradication of tumor with decreased toxicity.
Financial support and disclosures:
Supported in part by an ASCO Young Investigator (PFC), RSNA Fellow Research Grant RF1904 (PFC), and National Institutes of Health grants R01CA234904 (BAW), T32 CA009135 (JBT), T32 GM008688 (SEC), and University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520.
References
- 1.Passerini V et al. The presence of extra chromosomes leads to genomic instability. Nat. Commun. 7, 10754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu J, Pavelka N, Bradford WD, Rancati G & Li R Karyotypic determinants of chromosome instability in aneuploid budding yeast. PLoS Genet. 8, e1002719 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weaver BAA & Cleveland DW Does aneuploidy cause cancer? Curr. Opin. Cell Biol. 18, 658–667 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Taylor AM et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 33, 676–689.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denomy C et al. Banding Together: A Systematic Comparison of The Cancer Genome Atlas and the Mitelman Databases. Cancer Res. 79, 5181–5190 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Beroukhim R et al. The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitelman F Recurrent chromosome aberrations in cancer. Mutat. Res. 462, 247–253 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Sivakumar S et al. Pan cancer patterns of allelic imbalance from chromosomal alterations in 33 tumor types. Genetics 217, 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zasadil LM, Britigan EMC & Weaver BA 2n or not 2n: Aneuploidy, polyploidy and chromosomal instability in primary and tumor cells. Semin. Cell Dev. Biol. 24, 370–379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolhaqueiro ACF et al. Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat. Genet. 51, 824–834 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Gao R et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 48, 1119–1130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson L et al. A living biobank of ovarian cancer ex vivo models reveals profound mitotic heterogeneity. Nat. Commun. 11, 822 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McIntosh JR Mitosis. Cold Spring Harb. Perspect. Biol. 8, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Musacchio A The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr. Biol. 25, R1002–1018 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Rieder CL, Cole RW, Khodjakov A & Sluder G The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 130, 941–948 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Funk LC, Zasadil LM & Weaver BA Living in CIN: Mitotic Infidelity and Its Consequences for Tumor Promotion and Suppression. Dev. Cell 39, 638–652 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan SD et al. Up-regulation of the mitotic checkpoint component Mad1 causes chromosomal instability and resistance to microtubule poisons. Proc. Natl. Acad. Sci. 109, E2205–2214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ricke RM, van Ree JH & van Deursen JM Whole chromosome instability and cancer: a complex relationship. Trends Genet. 24, 457–466 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weaver BAA et al. Centromere-associated protein-E is essential for the mammalian mitotic checkpoint to prevent aneuploidy due to single chromosome loss. J. Cell Biol. 162, 551–563 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nam H-J & van Deursen JM Cyclin B2 and p53 control proper timing of centrosome separation. Nat. Cell Biol. 16, 538–549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silkworth WT, Nardi IK, Paul R, Mogilner A & Cimini D Timing of centrosome separation is important for accurate chromosome segregation. Mol. Biol. Cell 23, 401–411 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almeida AC & Maiato H Chromokinesins. Curr. Biol. 28, R1131–R1135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cimini D et al. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 153, 517–527 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cimini D, Moree B, Canman JC & Salmon ED Merotelic kinetochore orientation occurs frequently during early mitosis in mammalian tissue cells and error correction is achieved by two different mechanisms. J. Cell Sci. 116, 4213–4225 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Bakhoum SF, Genovese G & Compton DA Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr. Biol. 19, 1937–1942 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ertych N et al. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer cells. Nat. Cell Biol. 16, 779–791 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chestukhin A, Pfeffer C, Milligan S, DeCaprio JA & Pellman D Processing, localization, and requirement of human separase for normal anaphase progression. Proc. Natl. Acad. Sci. 100, 4574–4579 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dykhuizen EC et al. BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature 497, 624–627 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bender MA & Gooch PC Types and rates of x-ray-induced chromosome aberrations in human blood irradiated in vitro. Proc. Natl. Acad. Sci. 48, 522–532 (1962). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pampalona J et al. Chromosome Bridges Maintain Kinetochore-Microtubule Attachment throughout Mitosis and Rarely Break during Anaphase. PloS One 11, e0147420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burrell RA et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He B et al. Chromosomes missegregated into micronuclei contribute to chromosomal instability by missegregating at the next division. Oncotarget 10, 2660–2674 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C-Z et al. Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ganem NJ, Godinho SA & Pellman D A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwon M et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 22, 2189–2203 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silkworth WT, Nardi IK, Scholl LM & Cimini D Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PloS One 4, e6564 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puck TT & Marcus PI Action of x-rays on mammalian cells. J. Exp. Med. 103, 653–666 (1956). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carrano AV Chromosome aberrations and radiation-induced cell death. I. Transmission and survival parameters of aberrations. Mutat. Res. 17, 341–353 (1973). [DOI] [PubMed] [Google Scholar]
- 39.Little JB Radiation-induced genomic instability. Int. J. Radiat. Biol. 74, 663–671 (1998). [DOI] [PubMed] [Google Scholar]
- 40.Marder BA & Morgan WF Delayed chromosomal instability induced by DNA damage. Mol. Cell. Biol. 13, 6667–6677 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Umbreit NT et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 368, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berry RJ Quantitative Studies of Relationships between Tumor Cell Ploidy and Dose Response to Ionizing Radiation in Vivo: Modification of Radiation Response in a Previously Irradiated Tumor. Radiat. Res. 18, 236 (1963). [Google Scholar]
- 43.Chu K, Teele N, Dewey MW, Albright N & Dewey WC Computerized video time lapse study of cell cycle delay and arrest, mitotic catastrophe, apoptosis and clonogenic survival in irradiated 14–3-3sigma and CDKN1A (p21) knockout cell lines. Radiat. Res. 162, 270–286 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Bakhoum SF et al. Numerical chromosomal instability mediates susceptibility to radiation treatment. Nat. Commun. 6, 5990 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bakhoum SF, Kabeche L, Murnane JP, Zaki BI & Compton DA DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov. 4, 1281–1289 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dodson H, Wheatley SP & Morrison CG Involvement of centrosome amplification in radiation-induced mitotic catastrophe. Cell Cycle 6, 364–370 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Dodson H et al. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J. 23, 3864–3873 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bourke E et al. DNA damage induces Chk1-dependent centrosome amplification. EMBO Rep. 8, 603–609 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Illidge TM, Cragg MS, Fringes B, Olive P & Erenpreisa JA Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol. Int. 24, 621–633 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Sato N et al. A possible role for centrosome overduplication in radiation-induced cell death. Oncogene 19, 5281–5290 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Baudoin NC et al. Asymmetric clustering of centrosomes defines the early evolution of tetraploid cells. eLife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sala R, Farrell KC & Stearns T Growth disadvantage associated with centrosome amplification drives population-level centriole number homeostasis. Mol. Biol. Cell 31, 2646–2656 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ianzini F et al. Activation of meiosis-specific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Res. 69, 2296–2304 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weaver BA & Cleveland DW The aneuploidy paradox in cell growth and tumorigenesis. Cancer Cell 14, 431–433 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sheltzer JM, Torres EM, Dunham MJ & Amon A Transcriptional consequences of aneuploidy. Proc. Natl. Acad. Sci. 109, 12644–12649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hwang S et al. Consequences of aneuploidy in human fibroblasts with trisomy 21. Proc. Natl. Acad. Sci. 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vasudevan A et al. Aneuploidy as a promoter and suppressor of malignant growth. Nat. Rev. Cancer 21, 89–103 (2021). [DOI] [PubMed] [Google Scholar]
- 58.Stingele S et al. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 8, 608 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rutledge SD et al. Selective advantage of trisomic human cells cultured in non-standard conditions. Sci. Rep. 6, 22828 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scopel EFC, Hose J, Bensasson D & Gasch AP Genetic variation in aneuploidy prevalence and tolerance across Saccharomyces cerevisiae lineages. Genetics 217, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang F et al. The fitness costs and benefits of trisomy of each Candida albicans chromosome. Genetics (2021) doi: 10.1093/genetics/iyab056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Selmecki AM, Dulmage K, Cowen LE, Anderson JB & Berman J Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet. 5, e1000705 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee AJX et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 71, 1858–1870 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lukow D et al. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Prepr. BioRxiv (2020) doi: 10.1101/2020.09.25.314229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ippolito M et al. Aneuploidy-driven genome instability triggers resistance to chemotherapy. Prepr. BioRxiv (2020) doi: 10.1101/2020.09.25.313924. [DOI] [Google Scholar]
- 66.Rowald K et al. Negative Selection and Chromosome Instability Induced by Mad2 Overexpression Delay Breast Cancer but Facilitate Oncogene-Independent Outgrowth. Cell Rep. 15, 2679–2691 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iwanaga Y et al. Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res. 67, 160–166 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Jeganathan K, Malureanu L, Baker DJ, Abraham SC & van Deursen JM Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J. Cell Biol. 179, 255–267 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li M, Fang X, Wei Z, York JP & Zhang P Loss of spindle assembly checkpoint-mediated inhibition of Cdc20 promotes tumorigenesis in mice. J. Cell Biol. 185, 983–994 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sotillo R et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 11, 9–23 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Silk AD et al. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. 110, E4134–4141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zasadil LM et al. High rates of chromosome missegregation suppress tumor progression but do not inhibit tumor initiation. Mol. Biol. Cell 27, 1981–1989 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weaver BAA, Silk AD, Montagna C, Verdier-Pinard P & Cleveland DW Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11, 25–36 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Baker DJ et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 36, 744–749 (2004). [DOI] [PubMed] [Google Scholar]
- 75.Kalitsis P et al. Increased chromosome instability but not cancer predisposition in haploinsufficient Bub3 mice. Genes. Chromosomes Cancer 44, 29–36 (2005). [DOI] [PubMed] [Google Scholar]
- 76.Malureanu L et al. Cdc20 hypomorphic mice fail to counteract de novo synthesis of cyclin B1 in mitosis. J. Cell Biol. 191, 313–329 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cowley DO, Muse GW & Van Dyke T A dominant interfering Bub1 mutant is insufficient to induce or alter thymic tumorigenesis in vivo, even in a sensitized genetic background. Mol. Cell. Biol. 25, 7796–7802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ricke RM, Jeganathan KB, Malureanu L, Harrison AM & van Deursen JM Bub1 kinase activity drives error correction and mitotic checkpoint control but not tumor suppression. J. Cell Biol. 199, 931–949 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holland AJ & Cleveland DW Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep. 13, 501–514 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kops GJPL, Foltz DR & Cleveland DW Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. 101, 8699–8704 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Funk LC et al. p53 Is Not Required for High CIN to Induce Tumor Suppression. Mol. Cancer Res. 19, 112–123 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Foijer F et al. Deletion of the MAD2L1 spindle assembly checkpoint gene is tolerated in mouse models of acute T-cell lymphoma and hepatocellular carcinoma. eLife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoevenaar WHM et al. Degree and site of chromosomal instability define its oncogenic potential. Nat. Commun. 11, 1501 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Janssen A, Kops GJPL & Medema RH Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc. Natl. Acad. Sci. 106, 19108–19113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maia ARR et al. Mps1 inhibitors synergise with low doses of taxanes in promoting tumour cell death by enhancement of errors in cell division. Br. J. Cancer (2018) doi: 10.1038/s41416-018-0081-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.de Cárcer G et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat. Commun. 9, 3012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Godek KM et al. Chromosomal Instability Affects the Tumorigenicity of Glioblastoma Tumor-Initiating Cells. Cancer Discov. 6, 532–545 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Laucius CD, Orr B & Compton DA Chromosomal instability suppresses the growth of K-Ras-induced lung adenomas. Cell Cycle 18, 1702–1713 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schiff PB & Horwitz SB Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. 77, 1561–1565 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zasadil LM et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 6, 229ra43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maia ARR et al. Inhibition of the spindle assembly checkpoint kinase TTK enhances the efficacy of docetaxel in a triple-negative breast cancer model. Ann. Oncol. 26, 2180–2192 (2015). [DOI] [PubMed] [Google Scholar]
- 92.Cánovas B et al. Targeting p38α Increases DNA Damage, Chromosome Instability, and the Anti-tumoral Response to Taxanes in Breast Cancer Cells. Cancer Cell 33, 1094–1110.e8 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Rodrigues-Ferreira S et al. Improving breast cancer sensitivity to paclitaxel by increasing aneuploidy. Proc. Natl. Acad. Sci. 116, 23691–23697 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Carter SL, Eklund AC, Kohane IS, Harris LN & Szallasi Z A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 38, 1043–1048 (2006). [DOI] [PubMed] [Google Scholar]
- 95.Goh JY et al. Chromosome 1q21.3 amplification is a trackable biomarker and actionable target for breast cancer recurrence. Nat. Med. 23, 1319–1330 (2017). [DOI] [PubMed] [Google Scholar]
- 96.Turajlic S et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 173, 581–594.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shukla A et al. Chromosome arm aneuploidies shape tumour evolution and drug response. Nat. Commun. 11, 449 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kheir SM et al. Prognostic significance of DNA aneuploidy in stage I cutaneous melanoma. Ann. Surg. 207, 455–461 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Birkbak NJ et al. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 71, 3447–3452 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Andor N et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 22, 105–113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jamal-Hanjani M et al. Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: a prospective validation cohort study from the TACT trial. Ann. Oncol. 26, 1340–1346 (2015). [DOI] [PubMed] [Google Scholar]
- 102.Roylance R et al. Relationship of extreme chromosomal instability with long-term survival in a retrospective analysis of primary breast cancer. Cancer Epidemiol. Biomark. Prev. 20, 2183–2194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morgan WF & Murnane JP A role for genomic instability in cellular radioresistance? Cancer Metastasis Rev. 14, 49–58 (1995). [DOI] [PubMed] [Google Scholar]
- 104.Limoli CL, Corcoran JJ, Jordan R, Morgan WF & Schwartz JL A role for chromosomal instability in the development of and selection for radioresistant cell variants. Br. J. Cancer 84, 489–492 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zaki BI, Suriawinata AA, Eastman AR, Garner KM & Bakhoum SF Chromosomal instability portends superior response of rectal adenocarcinoma to chemoradiation therapy. Cancer 120, 1733–1742 (2014). [DOI] [PubMed] [Google Scholar]
- 106.Dey S et al. Low-dose fractionated radiation potentiates the effects of Paclitaxel in wild-type and mutant p53 head and neck tumor cell lines. Clin. Cancer Res. 9, 1557–1565 (2003). [PubMed] [Google Scholar]
- 107.Formenti SC et al. Concurrent paclitaxel and radiation therapy for breast cancer. Semin. Radiat. Oncol. 9, 34–42 (1999). [PubMed] [Google Scholar]
- 108.Choy H et al. Phase II trial of weekly paclitaxel and concurrent radiation therapy for locally advanced non-small cell lung cancer. Clin. Cancer Res. 4, 1931–1936 (1998). [PubMed] [Google Scholar]
- 109.Tishler RB et al. A Phase I/II trial of concurrent docetaxel and radiation after induction chemotherapy in patients with poor prognosis squamous cell carcinoma of the head and neck. Cancer 95, 1472–1481 (2002). [DOI] [PubMed] [Google Scholar]
- 110.Chandler BC et al. TTK inhibition radiosensitizes basal-like breast cancer through impaired homologous recombination. J. Clin. Invest. 130, 958–973 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grant GD et al. Identification of cell cycle-regulated genes periodically expressed in U2OS cells and their regulation by FOXM1 and E2F transcription factors. Mol. Biol. Cell 24, 3634–3650 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]