

Abstract
Queuosine is a structurally unique and functionally important tRNA modification, widely distributed in eukaryotes and bacteria. The final step of queuosine biosynthesis is the reduction/deoxygenation of epoxyqueuosine to form the cyclopentene motif of the nucleobase. The chemistry is performed by the structurally and functionally characterized cobalamin-dependent QueG. However, the queG gene is absent from several bacteria that otherwise retain queuosine biosynthesis machinery. Members of the IPR003828 family (previously known as DUF208) have been recently identified as non-orthologous replacements of QueG and this family was renamed QueH. Here we present the structural characterization of QueH from Thermotoga maritima. The structure reveals an unusual active site architecture with a [4Fe-4S] metallocluster along with an adjacent coordinated iron metal. The juxtaposition of cofactor and coordinated metal ion predicts a unique mechanism for a two electron reduction/deoxygenation of epoxyqueuosine. To support the structural characterization, in vitro biochemical and genomic analyses are presented. Overall, this work reveals new diversity in the chemistry of iron/sulfur dependent enzymes and novel insight into the last step of this widely conserved tRNA modification.
Keywords: tRNA modification, queuosine biosynthesis, iron/sulfur cofactor, reductase
INTRODUCTION
Post-transcriptional modifications of transfer RNAs (tRNAs) are common and essential steps, providing functional and structural elements for key players in protein translation.(1,2) The modifications range from, relatively simple, nucleoside methylation to de novo production of hypermodified bases. Modifications contribute to tRNA stability, folding and fidelity of decoding.(3–5) tRNAs lacking modifications have an accelerated decay rate, accompanied by loss of aminoacylation function.(6,7) tRNA modifications can also affect the function of the overall translation machinery, such as aminoacyl-tRNA synthetases, in addition to playing a key role in decoding capacity of the machinery.(6)
The hypermodified nucleoside queuosine (Q) is common at the wobble position of the anticodon stem loop (ASL) of tRNAs that encode for Asn, Asp, His or Tyr (G/Q-U-N anticodon motif).(1,8–11) Q was discovered half century ago, yet its molecular mechanism of function remains vaguely defined. Although Q is not required for cell growth under ordinary conditions, the absence of Q reduces fitness under stress as well as translational accuracy.(4,12,13) In eukaryotes, Q-tRNA levels control the translational speed of the Q-decoded codon and suppresses second-position misreading.(14,15) Q is broadly distributed in bacteria and eukaryotes, but only bacteria are capable of de novo biosynthesis.(16) The corresponding free base queuine (q) is salvaged by eukaryotes and considered a micronutrient for humans.(17) The biosynthesis of Q starts from GTP requiring GTP cyclohydrolase I, along with the QueDECF enzymes, to produce the intermediate PreQ1 (Figure S1).(1,16,18) tRNA guanine transglycosylase (TGT) then inserts PreQ1 into a tRNA.(12,19) The final two enzymatic steps are completed by the bacteria-specific tailoring enzymes QueA and QueG/QueH (Figures 1a, S1).(20–22) Salvage pathways have been described where YhhQ specifically transports PreQ0 and PreQ1 and a representative example is in E. coli.(23) In addition, two Q salvage mechanisms have been characterized in some pathogenic and commensal bacteria which includes transporters of Q and/or biosynthetic precursors, a specific TGT enzyme, along with queuine lyase.(24)
Figure 1.
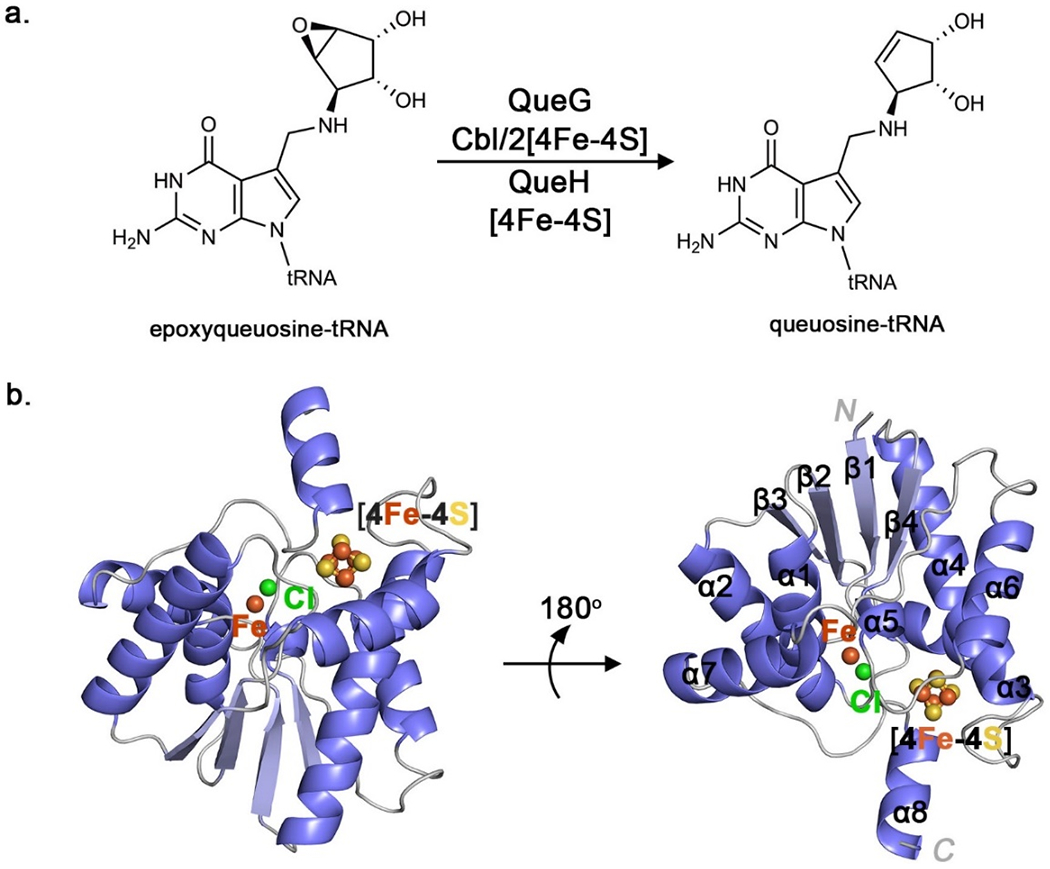
The epoxyqueuosine reductase, QueH. A. Reduction and deoxygenation of epoxyqueuosine catalyzed by QueG (colbalmin-dependent) or QueH. B. The overall structure of T. maritima showing iron (orange), chloride (green) and sulfur (yellow) atoms.
The final enzyme in the Q biosynthetic pathway, epoxyqueuosine reductase (EC 1.17.99.6 QueG), catalyzes the reduction/deoxygenation of epoxyqueuosine (oQ) to yield the Q incorporated tRNA (Figure 1a).(21,25) QueG is a member of the cobalamin/[4Fe-4S] enzyme family and activity was assigned to the yjeS gene by systematic screening of E. coli genes.(25) Protein structures of QueG from Streptococcus thermophilus and Bacillus subtilis confirm a cobalamin/[4Fe-4S] redox chain, along with a HEAT-like protein/RNA interaction domain, and the enzymatic mechanism has been proposed to involve reduction via a cobalt-carbon bound intermediate.(26,27) QueG orthologs could only be identified in 694 out of 1273 eubacteria genomes examined that also harbor tgt and queA homologs. (22) Using a comparative genomics approach on bacterial genomes lacking queG, epoxyqueuosine reductase activity was assigned to the DUF208 gene family, in some instances located proximal to tgt and queA. An opposite distribution pattern between duf208 genes and queG suggests duf208 can fill the gap in genomes missing queG. Indeed, in an E. coli mutant, heterologous expression of duf208 from different species restore the functional conversion of oQ to Q, establishing DUF208 as a non-orthologous replacement of QueG in vivo, and the family was renamed QueH (Figure 1a). (22) The QueH family (IPR003828) was previously annotated as a member of the ATPase, adenine nucleotide α-hydrolase (AANH) superfamily. AANH enzymes are known to be involved in tRNA modifications for example QueC and the 4-thiouridine biosynthesis enzyme, ThiI.(28,29) Present knowledge of the function and mechanism of the QueH family is very limited, a ortholog from Helicobacter pylori (Hp0100) has been identified as a component of a tRNA-independent transamidosome enzyme complex, and gene expression accelerates the catalysis of transamidation.(30)
In the presented study, QueH from Thermotoga maritima is structurally and biochemically characterized. The structure reveals a unique protein architecture with a [4Fe-4S] metallocluster, along with an adjacent coordinated metal ion (Fe) in the active site. Biochemical analyses support catalytic roles for the iron centers in reductase chemistry, and preliminary analysis suggests a novel active site environment and metal function. Overall, a unique structure and proposed enzyme chemistry is described for the QueH protein family.
MATERIALS AND METHODS
Protein purification and crystallization experiments were performed under anaerobic conditions in a strictly argon atmosphere. Chemicals and reagents were purchased from Sigma-Aldrich or Fisher Scientific. All solutions used for QueH purification were exhaustively degassed and purged with argon gas prior to use. Protein structure surface analysis was executed using CAVER(31) and protein cavities depicted using POCASA software along with the CASTp server.(32,33) Graphical illustrations of protein structure were prepared using PyMOL and electrostatic surface representations using the APBS plugin.(34,35)
Expression and purification of QueH
A plasmid construct of affinity tag-free T. maritima queH in the pET-28b vector was utilized for expression and purification. The coding sequence was inserted into the NcoI and NotI sites of pET28b, after PCR amplification with the following primers:
5’-CATGCCATGGGCACTGTGCTGATTCATGTTT-3’
5’-ATAAGAATGCGGCCGCTTACATATGACCACGTTTTCTGG-3’
The resulting plasmid was transformed into E. coli BL21(DE3) cells and grown at 37°C in LB media to an OD600 of 0.6. Overexpression was initiated by adding IPTG (0.25 mM final concentration). Growth continued for 20 hr at 18 °C and cells were harvested by centrifugation. The cell pellet was resuspended in 20 mM Tris-HCl pH 7.5, 500 mM NaCl, 2 mM 2-mercaptoethanol (60 mL) and lysed at 14,000 psi through a microfluidizer cell (M-110L Pneumatic). The lysate was clarified by centrifugation at 14,000 g for 20 min at 4 °C, transferred to a sealed bottle, heated for 15 min at 68 °C, then cooled for 5 min on ice. The lysate was further clarified by centrifugation at 18,000 g at 4 °C for 30 min. Solid ammonium sulfate was added to the clarified lysate to 35% saturation and stirred at 4 °C. The sample was decanted and ammonium sulfate was added to a final concentration of 70% saturation. Precipitated protein was collected by centrifugation at 18,000 g for 30 min at 4 °C then resuspended in 10 mL of 20 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM 2-mercaptoethanol (β-ME). The solution was then dialyzed against 1 L of 20 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM β-ME overnight. QueH was further purified by cation exchange chromatography (HiTrap SP HP 1 mL, GE Healthcare) with a linear gradient of 0 to 500 mM NaCl over 30 min followed by size-exclusion chromatography (HiLoad 16/600 Superdex 75, GE Healthcare) with 20 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM β-ME as the running buffer. Protein fractions were pooled and dialyzed against 1 L of 20 mM Tris-HCl pH 7.5, 100 mM KCl, 10% glycerol (storage buffer), concentrated to 12 mg/mL and moved into an anaerobic chamber for further reconstitution.
In vitro reconstitution of holo [4Fe-4S] QueH
Enzyme reconstitution was performed in an anaerobic chamber. Stock solutions of 1.0 M dithiothreitol (DTT), 500 mM iron(III)chloride and 500 mM sodium sulfide were freshly prepared in the storage buffer. Holo-QueH was reconstituted by rapidly mixing of DTT, FeCl3 and sodium sulfide solutions (10 μM each). The reaction was incubated at room temperature for 4 h then desalted using a PD-10 column equilibrated with storage buffer containing 10 mM DTT.
TmQueH crystallization
Initial crystallization conditions were obtained in an anaerobic chamber using the hanging drop method at 22 °C. 2 μL TmQueH solution (storage buffer with 10 mM DTT) and 2 μL of 100 mM bis-TrisHCl pH 5.5, 200 mM lithium sulfate monohydrate and 25% PEG-3350 were mixed and equilibrated against 500 μL reservoir solution. Crystal optimization using an additive screen (Hampton Research), gave hexagonal crystals over 12 days in 100 mM bis-TrisHCl pH 5.5, 200 mM lithium sulfate monohydrate, 25% PEG-3350 and 10 mM CaCl2
Data collection, structure solution and refinement
Diffraction data for TmQueH were collected on beamline 23-ID-D of the GM/CA CAT facility, Argonne National Laboratory Advanced Photon Source (APS-ANL) at a wavelength of 1.0332 Å. Data were collected at 100 K using a Pilatus 6M detector, integrated, merged and scaled using the XDS package(36) to a resolution of 1.50 Å with one monomer per asymmetric unit in space group C2221. As no apparent homology model was available for molecular replacement, the hybrid substructure search protocol (HySS) (37) was used to locate heavy metals and an experimental phase solution was determined using AutoSol in PHENIX (38,39) with a figure of merit of 0.52 to 1.45 Å and CC-BAYES of 59.9. The R-work/R-free after density modification was 0.26/0.27. Experimental density maps enabled automated building of 149 residues and the remaining residues were built manually using COOT.(40) Refinement was performed with PHENIX and REFMAC5.(38,41) A iron-sulfur cluster parameter file was generated using the cofactor structure of QueG.(27,42) Waters and metals were added in the later rounds of refinement. Sigma-A weighted, simulated annealing composite omit maps were used to judge and verify structures throughout refinement. Model geometry was validated through MolProbity,(43) with 98.3% and 97.7% of protein residues in TmQueH are in the favored region of Ramachandran analysis.
Electron Paramagnetic Resonance Spectroscopy
Anaerobically reconstituted TmQueH (as described above, 150 μL, 10 mg/mL) was mixed with 1 mM sodium dithionite and transferred into a sealed EPR tube in an anaerobic glovebox. Samples were removed from the glovebox and flash frozen in liquid nitrogen before EPR analysis. EPR spectra were acquired with a Bruker Elexsys E500 system utilizing an Oxford ESR900 cryostat at a frequency of 9.644 GHz, in the perpendicular setting of a Bruker ER4116-DM dual mode resonator. The field modulation frequency was set at 100 kHz, modulation amplitude at 10 G, and the microwave power at 2 mW at 10 K. All spectra were baseline-corrected by subtraction of the EPR signal of a blank. For the oxidized sample, the tube was thawed and exposed to air for 30 minutes before being frozen in liquid nitrogen.
UV-vis spectroscopy
Anaerobically reconstituted QueH (as described above, 100 μL, 1.1 mg/mL) was opened to ambient air and the UV-vis spectra was recorded at 10 and 30 minutes. A second sample was mixed with 1 mM sodium dithionite and transferred to an anaerobic cuvette before measuring UV-vis spectra.
Expression and purification of C-terminal hexahistidine-tagged TmQueH
The TmQueH coding sequence was cloned into the plasmid pET-28b at the restriction sites NcoI and NotI, after PCR amplification with the following primer sequences:
5’-CATGCCATGGGCACTGTGCTGATTCATGTTT-3’
5’-ATAAGAATGCGGCCGCCATATGACCACGTTTTCTGGAA-3’
The construct, along with a plasmid containing the E. coli ISC operon, was co-transformed into E. coli BL21(DE3) cells. A starter culture (100 mL LB media with 50 μg/mL kanamycin, 50 μg/mL spectinomycin) was inoculated at 37 °C and grown to an OD600=0.3. 4 L of M9 media was inoculated with the starter culture and incubated at 37 °C to an OD600 =0.3. L-arabinose (final concentration 2 g/L) was then added to induce expression of the ISC operon and growth continued to an OD600 of 0.6. FeCl3 (final concentration 50 μM) and L-cysteine (final concentration 150 μM) were added along with IPTG (0.25 mM final) and growth continued for 20 h at 18 °C. The cell pellet was resuspended in HEPES-KOH pH 8.0, 300 mM KCl, 10% glycerol and lysed using B-PER (ThermoFisher Scientific) after introduction into an anaerobic chamber. The lysate was incubated at 65 °C for 15 minutes before being clarified by centrifugation at 39,000 g for 30 min at 4 °C. The soluble fraction was purified by NTA-affinity chromatography, brown colored fractions were pooled, concentrated, flash frozen in liquid nitrogen and stored at −80 °C.
Expression and purification of N-terminally hexa-His-tagged SpQueH
The S. pyogenes QueH coding sequence was cloned into the plasmid pET-28b at the restriction sites NdeI and NotI, after being amplified with the primers:
5’-GGGAATTCCATATGATTGATTTACAAGAGATTCTCG-3
5’-ATAAGAATGCGGCCGCTTACATCTCATCCCCACGAT-3
The expression protocol for SpQueH follows that described for TmQueH and was carried out aerobically. In short, cell pellets were resuspended in HEPES-KOH pH 8.0, 300 mM KCl, 10% glycerol and lysed by sonication before being clarified at 39,000 g for 30 minutes at 4 °C. The supernatant was then purified by NTA-affinity chromatography. Brown fractions were pooled, concentrated, flash frozen in liquid nitrogen and stored at −80 °C for further use.
Reconstitution of TmQueH for in vitro analysis
TmQueH (50 μM) was reconstituted in buffer containing 100 mM HEPES-KOH pH 8.0, 300 mM KCl, 10% glycerol. The protein was first incubated with 10 mM DTT for 2 h, then 10 molar equivalents of iron (III) chloride was slowly added and incubation continued for 30 min. 10 molar equivalents of sodium sulfide was then added and incubated overnight at 4 °C. Reconstituted TmQueH was purified by size-exclusion chromatography (HiLoad 16/600 Superdex 75, GE Healthcare). tRNA substrate for QueH was purified from the E. coli strain (ΔqueG::KanR JW4124, abbreviated ΔqueG) along with the corresponding wild type strain BW25113 (KEIO collection).(44,45) QueH reactions (24 mM HEPES-NaOH/pH 8.0, 200 μg/mL tRNA, 100 μM sodium dithionate, 1 or 10 μM QueH) were incubated at 37°C for 20 hours in an anaerobic chamber. Reaction mixtures were filtered with a 10 kDa cutoff and digested with benzonase (0.2 U/μL), phosphodiesterase (0.002 U/μL) and alkaline phosphatase (0.02 U/μL) for 3 hours at 37°C before passing again through a 10 kDa cutoff filter.
Mass analysis of QueH reactions
5 µL of the reaction mixture was analyzed on a Waters Synapt G2-Si instrument/CORTECS UPLC C18 column with 5 mM NH4OAc (A) and 40% acetonitrile/water (B) as running buffers. Protocol: 100% of A for 1 min, gradient to 88% A over 16 min, 50% A over 6 min and 0% A to 25 min followed by isocratic 0% A to 39 min. Positive electrospray ionization mode was used, 50 Da to 2000 Da range, 0.5 s scan duration, 3.5 kV and source temperature of 120 °C. The queuosine nucleoside was analyzed at 410 Da.
RESULTS
X-ray crystal structure of TmQueH
The queH gene (TM0731) of T. maritima was previously shown to complement a ΔqueG strain of E. coli by restoring functionally production of Q.(22) This gene was previously characterized as a probe to the misfolded protein response in E. coli as a representative misfolded/insoluble gene product.(46) To favor soluble expression of QueH, an affinity tag-free, recombinant TM0731 was constructed in the pET-28b vector, heterologously expressed in E. coli followed by purification and anaerobic reconstitution of a predicted [4Fe-4S] cluster in the presence of iron(III)chloride and sodium sulfide (Figure S2). The reconstituted protein was crystallized and diffraction data collected to 1.50 Å resolution in the space group C2221 (Table 1). As no apparent structural homologs were available, the phase solution was determined using SAD phasing approaches based on anomalous diffraction of native iron atoms.
Table 1.
Data collection and refinement statistics
| QueH_Apo (PDB: 7LC5) | |
|---|---|
| Data collection | |
| Space group | C2221 |
| Cell dimensions | |
| a, b, c (Å) | 53.22, 105.18, 73.90 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 42.85-1.50(1.55-1.50)* |
| Rmerge (%) | 4.4 (70.8) |
| I/σI | 36.83(4.85) |
| CC 1/2 | 1(0.929) |
| Completeness (%) | 99.33(98.05) |
| Redundancy | 13.0(12.4) |
| Refinement | |
| Resolution (Å) | 42.85-1.50 |
| No. reflections | 33562(3275) |
| Rwork / Rfree | 19.54/20.96 |
| No. atoms | |
| Protein | 1472 |
| Ligand/ion | 10 |
| Water | 98 |
| B-factors | |
| Protein | 30.01 |
| Ligand/ion | 26.68 |
| Water | 36.47 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 0.81 |
Values in parentheses are for highest-resolution shell.
Overall architecture of T. maritima QueH
The crystal structure of T. maritima QueH contains one protein monomer per asymmetric unit, consistent with size exclusion chromatography suggesting a monomeric, soluble protein (Figure 1a). A structural homology search revealed 4-thiouridine (s4U) synthetase (ThiI) as most similar to QueH(47) (Table S1). Although their substrates and chemistries are significantly different, QueH and ThiI both are members of the AANH superfamily,(22) exemplifying a minimal structural core motif for this family with diverse chemistries.
QueH comprises a single protein domain with a mixed N-terminal extension β/α fold (β1-4, α5) flanked by three α-helices of each side of the core along with an extended α-helix (α8) at the C-terminus (Figure 1b). The overall architecture is similar to a ‘thumbs-up’ shape with three adjacent α-helices (α1, α2 and α7) and β-strands as fingers, and α8 as thumb. The N-terminal extension begins at β1, leading to a tight-turn with α1. The core fold includes a loop, following strand β1, containing two conserved cysteines Cys9, Cys10 and an aspartate residue (Asp13) as an unpredicted, metal binding center (Figures 1b, 2c). A canonical [4Fe-4S] cluster is coordinated with a four cysteine motif: Cys87-X2-Cys90 (α4) and Cys169-X-Cys171 (located in a loop between α7, α8) with helix α3 helping to enclose the active site (Figures 1b, 2a). The unusual arrangement does resemble a limited number of catalytic metalloclusters with ~9 Å separating an [4Fe-4S] cluster and a single coordinated metal ion.(48,49)
Figure 2.
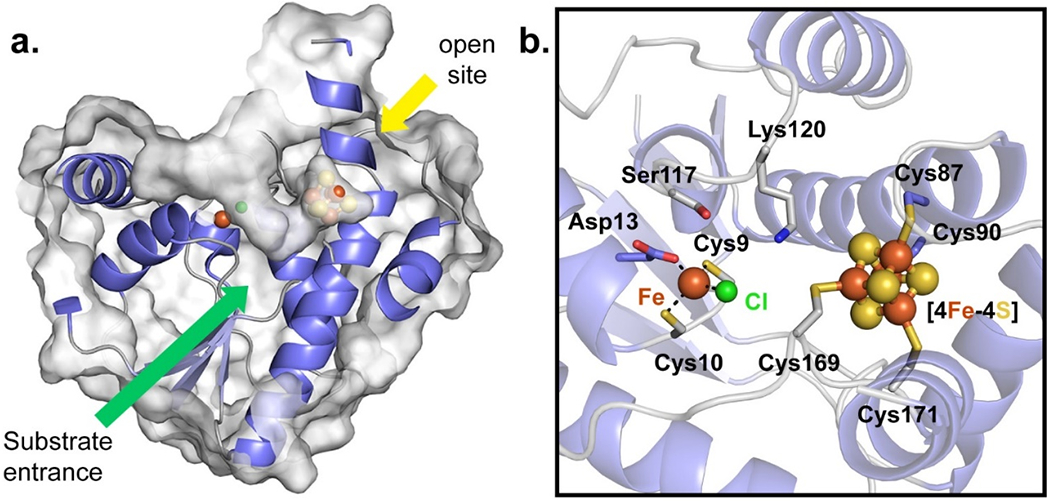
Substrate binding pocket and active site of TmQueH. A. Surface representation of TmQueH showing a large “V-shape” binding pocket leading to the [4Fe-4S] cluster/Fe active site. An orthogonal opening (yellow arrow) allows access to external redox machinery. B. Detailed view of the active site of TmQueH showing enzyme bound metal, a ligated chloride ion and iron/sulfur cluster as spheres.
Structural features outside of the metallocluster core are likely involved in substrate binding and redox chemistry. Notably, two concurrent loops (α5-loop-α6 and α7-loop-α8, Figure S3) define a funnel shaped pocket large enough to accommodate a tRNA substrate localized adjacent to the metallocluster core (Figure 2a). The active site has a second opening, opposite the large pocket, linking the iron/sulfur cluster to bulk solvent (Figure 2b). Unlike QueG, no common RNA binding motif or ferredoxin-like domain is readily identifiable in the structure of QueH, although positive charged patches of residues are present at the C-terminal extension of the protein suggesting a favorable protein-RNA interface (Figure S4).
Unusual metal architecture in the QueH active site
Sequence analysis shows conservation of the six cysteine residues among QueH family members; corresponding to four cysteines coordinating to the [4Fe-4S] cluster, along with two others. The two cysteines not coordinating the metal cluster were previously predicted to undergo disulfide bond chemistry through a redox cycle, similar to the mechanism of vitamin K epoxide reductase.(50) In contrast to this prediction, a single metal ion was found coordinated with the two cysteines (Cys9, Cys10) along with Asp13 and a non-proteinogenic ligand, consistent with a chloride ion based on the crystallization conditions, to complete a tetrahedral geometry (Figures 2b, 3a). Based on the anomalous diffraction data, along with the observed coordination geometry, the single metal is consistent with an iron atom (Figure 3a), however this result can be influenced by the presence of FeCl3 during in vitro [4Fe-4S] cluster reconstitution used to prepare the enzyme. Sequence alignment shows that Asp13 is commonly replaced in some bacteria species by a cysteine residue, suggesting possible alternative bonding modes and/or coordinating metals. Prediction software tools (IonCom server) suggested Zn+2 as a favorable ligand based on the architecture of the protein metal binding site.(51) The stand-alone metal could function as a cofactor providing structure stability and/or directly participating in the enzyme chemistry during catalysis.(52)
Figure 3.
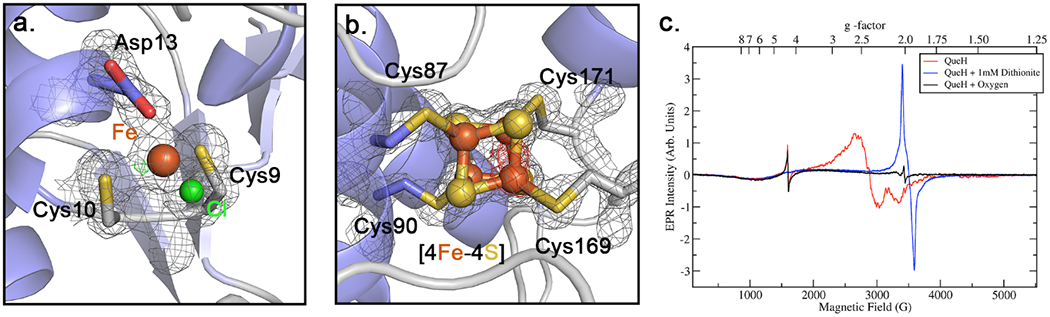
Metal binding and conserved [4Fe-4S] cluster sites. a. Coordination of the mononuclear metal-binding site, electron density map (2Fo-Fc, 2σ) is shown along with a non-proteinogenic ligand (chloride). mFo-DFc difference map contoured at ±4σ and shown as green and red, respectively. b. Four cysteine coordinated [4Fe-4S] with an electron density map (2Fo-Fc, 2σ) and mFo-DFc diffence map (green/red, ±4σ). c. EPR spectra of the QueH. red line: anaerobically prepared protein, blue line: dithionite reduced, black line: air oxidized. For acquisition parameters see Materials and Methods.
Conserved [4Fe-4S] cluster binding motif
The observed [4Fe-4S] cluster is buried and located opposite to the mononuclear metal-binding site in the active site cavity(Figures 2b, 3b). Clear electron density is present for a complete [4Fe-4S] cluster with four iron atoms coordinated by sulfhydryls of four conserved cysteine residues from Cys87-X2-Cys90 and Cys169-X-Cys171 (Figure 3b). The position of the [4Fe-4S] cluster at the surface of the active site cavity suggests that it is in a position to directly interact with a bound substrate epoxide. This orientation is also confirmed by an open active site where the, relatively large, tRNA nucleoside substrate can bind and participate in the established reduction chemistry. To complete a catalytic cycle, QueH requires a two electrons, presumably trafficked from the [4Fe-4S] cluster, typically supplied by ferredoxin/ferredoxin reductase machinery.(53,54) Three tunnels are calculated leading from the [4Fe-4S] cluster to surrounding solvent (Figures 2a, S5) suggesting potential binding modes for protein partners, and the largest tunnel, with an average length 15 Å, can reasonable accommodate a tRNA substrate.
Spectroscopic support for a redox active [4Fe-4S] cluster in QueH
EPR spectral analyses were recorded to investigate the redox property of the single [4Fe-4S] cluster (Figure 3c, S9). Both the anaerobically prepared (red) and the oxidized preparations (black) show a relatively sharp signal at 1600 G (g ≈ 4.3) which is typical for high-spin Fe(III) in a rhombic environment and consistent with the assigned metal based on the structure. Dithionite reduction removes this signal entirely since high-spin Fe(II) is usually EPR-silent in the X-band and the low-spin state is diamagnetic.
At higher fields, the QueH sample shows an unusually broad and apparently axial spectrum between 2000 and 3500 G (g‖≈2.58, g⊥≈2.06) which is demonstrated to be sensitive to both reduction and oxidation (red trace in Figure 3c). Typical EPR spectra of [4Fe-4S] complexes show axial or rhombic spectra within a much more narrow effective g-factor range between 1.8 and 2.1.(55) This broad spectrum may be rationalized as due to the [4Fe-4S], but broadened by a strong magnetic coupling between with the neighboring lone Fe center. As corroboration of the EPR analysis, UV-vis spectrometer of the anaerobically prepared/reduced and air oxidized protein display characteristic spectra of [4Fe-4S] clusters (Figure S8)
In vitro analysis of recombinant QueH
Recombinant TmQueH was evaluated for in vitro activity, using a variety of constructs and conditions. The substrate used was bulk tRNA extracted from ΔqueG E. coli, harboring epoxyqueuosine at position 34 (Table S2). tRNAs were then digested to nucleosides and the presence of epoxyqueuosine and queuosine was evaluated using LC-MS.(25) Epoxyqueuosine can be identified in negative control reactions and that of queuosine in a positive control (wild-type E. coli). With added TmQueH, the epoxyqueuosine signal disappears, however, a mass corresponding to queuosine was not apparent (Figure S6). This suggests epoxyqueuosine modified tRNAs are being consumed under these conditions and, overall, is consistent with previous in vivo results with TmQueH (22). In an attempt to further characterize enzyme activity, enzyme purification conditions were varied to include the presence or absence of a hexa-histidine affinity tag, aerobic purification/reconstitution and/or inclusion of titanium citrate as an electron donor. However, none of the approaches resulted in apparent masses corresponding to products (Figure S7). Additionally, screening of the QueH homolog from S. pyogenes for in vitro activity also yielded similar negative results as with TmQueH.
Discussion
The enzymatic reduction of oQ is the final step in the biosynthesis of the Q nucleobase, a modification key to the maturation of tRNAs and the integrity of protein translation.(14,15,56) The structure and mechanism of QueG has previously been characterized as a unique member of the cobalamin-dependent family of enzymes, catalyzing epoxide reduction to a cyclopentene product olefin. The chemistry of the cobalamin cofactor is diverse, playing a central role in multiple complex enzyme-catalyzed chemistries.(57–59) Interestingly, cobalamin-independent enzymes commonly coexist with pathways that complement and/or replace functionally homologous cobalamin-dependant enzymes. Examples include the chemistry of methionine synthase, ribonucleotide reductase (RNR) and methylmalonyl-CoA mutase (MCM) . QueG is likewise replaced or supplemented by QueH in a significant number of organisms.(22) Sequence analysis indicated that QueH is a cobalamin-independent enzyme with an active site 4Fe-4S metallocluster but little structural and mechanistic homolog to known metalloenzymes. Our initial proposed mechanism involved reduction of the epoxide by a conserved pair of cysteines with electrons required for turnover mediated by the [4Fe4S] metallocluster, this model was inspired by the similar mechanism of vitamin K epoxide reductase(60). However, the reduction chemistry of oQ to Q would be predicted more challenging both kinetically (a less electrophilic epoxide) and thermodynamically (higher energy product), relative to vitamin K epoxide reductase, justifying alternative enzyme strategies could be necessary.
To structural characterize QueH, we screened various genes for soluble protein expression/crystallization and the homolog from the thermophilic bacteria, T. maritima, provided suitable crystals. In the T. maritima genome, queH is flanked by genes encoding the tRNA repair enzyme, tRNA deacylase and tRNA-associated protein threonine-tRNA ligase, although each are not physically clustered with genes involved in Q biosynthesis or salvage. Using Genome Neighborhood Networks (GNN) analyses,(61) co-occurrence of QueH with TGT and other Q related genes is low and no additional genes are obvious as co-localized with queH. In addition to the functional assignment of the DUF208 family to QueH, a DUF208 homolog from H. pylori (Hp0100, a DUF208 member with a C-terminal domain sequence of unknown function) was previously characterized as a component of asparagine-transamidosome.(30)
The structure of TmQueH shows a unique architecture with an active site containing [4Fe-4S]/Fe metalloclusters available for epoxide ring reduction chemistry. Common structural motifs related to the chemistry of QueH are not present in the structure, including a cobalamin binding domains, RNA binding domains or a ferredoxin-like domain. The enzyme does contain a deep active site cavity of size and shape to accommodate the substrate tRNA oQ nucleoside. Unpredicted, based on sequence analysis and orthologous chemistries, the structure contains a stand-alone, tetrahedrally coordinated metal (identifiable as iron in the protein crystal structure) in addition to a [4Fe-4S] metallocluster. As common, metal cations can play a direct role in redox chemistry or catalysis through stabilization of high energy radical or anion intermediates.(63) [4Fe-4S] clusters in proteins generally participate in redox electron transport, as the metalloclusters have reduction potentials ranging from over 400 mV to below −400 mV, allowing diverse and varied electron transfer chemistries(63,64).
Our in vitro analysis of QueH did not corroborate observed activity in vivo(22), namely QueH-based production of an isolatable product, Q. The reactions did, however, show disappearance of substrate oQ. Possible reasons for not observing a product in vitro could include: a lack of an effective electron donor for complete turnover, unstable intermediates in the in vitro reaction and/or the fact that complete QueH function requires an unknown protein partner present in vivo. As the reconstitution conditions to generate holo-QueH in vitro favor the incorporation of iron, it is also reasonable that the mononuclear metal observed in the native enzyme is not an iron or the site is not bound with any metal ion.
A structural homology search indicates that TmQueH is most similar to tRNA modifying enzymes responsible for thiolation of uridine: with the tRNA s4U8 biosynthesis enzyme ThiI, the s2U synthetase, TtuA and the s2U34 thiouridylase MnmA as the top matches (Table S1). TtuA and MnmA are similar sulfurtransferases of s2U and share similar catalytic domains. The TmQueH structure is most homologous with the pyrophosphatase domain of ThiI, and the catalytic domain of TtuA and MnmA, underscoring functional conservation within tRNA modification family enzymes and evolutionary constrains on protein structure. Based on homology to the co-complex structure of MnmA with tRNA substrate,(65) a model of Q-tRNA interactions with QueH was prepared. The model illustrates that the overall structure and active site can accommodate the substrate placing the tRNA on a distinct positively charge protein surface where the extended aminomethyl–epoxycyclopentenediol moiety of the substrate can bind in the active site adjacent to the two metal clusters observed in the crystal structure (Fig. 4a).
Figure 4.
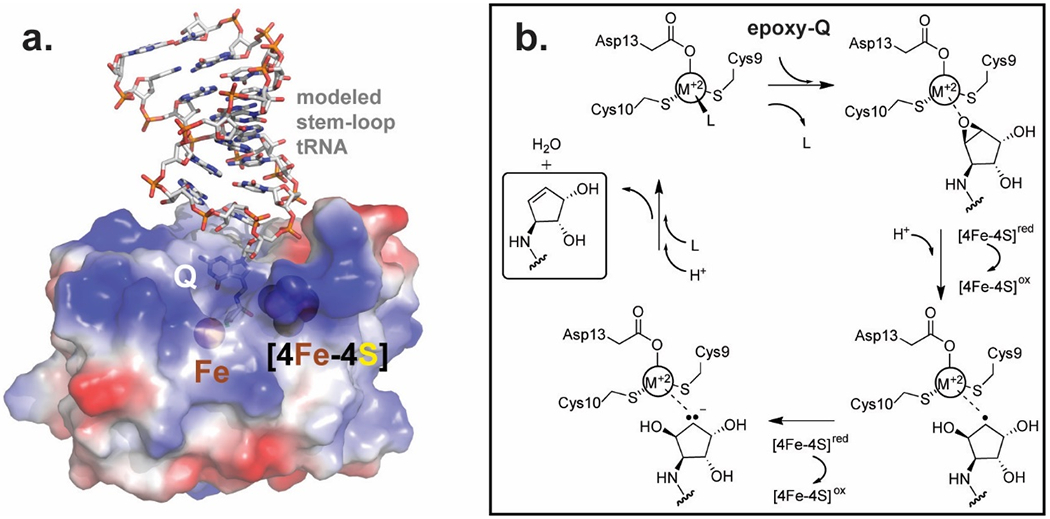
Modeled substrate binding and mechanism a. Proposed interaction oQ tRNA substrate and QueH. Q-tRNA (pdb code:5D0B) bound with QueH was modeled using MnmA/tRNA (pdb code:2DER) as a homologous scaffold. b. Proposed chemical mechanism of QueH catalyzed conversion of oQ to Q.
The reduction of an epoxide to an alkene and water requires two protons/two electrons. Based on the presented structures, a tRNA anticodon stem loop can bind to QueH in a large, electropositive cavity, such that a oQ nucleobase is inserted between the [4Fe-4S] and Fe binding sites, excluding solvent diffusion into the active site. As common with the motif, a [4Fe-4S] cluster could be involved in transfer of the two required electrons, generating radical and anionic intermediates, however the source of the electrons in vivo is not apparent. Based on the identity of the metal (M+2 in Figure 4b), high energy intermediates could be stabilized by forming a covalent carbon/metal (as with QueG) or through ionic-type interactions. EPR spectral analysis provides further insight in the metal environment. The spectra at high fields show differences between QueH and canonical protein [4Fe-4S] clusters most likely due to magnetic coupling with the single nearby iron ion. Upon oxidation in air, the broad EPR spectrum is quenched leaving a small signal near g ~ 2 with a linewidth of the order of 20 G, which may be a residual carbon-based radical. After chemical reduction with dithionite, the broad spectrum sharpens and shows intensity in the effective g-factor range between 1.85 and 2.1, as expected for a [4Fe-4S] signal (blue line, Figure 3c). Besides the main features at 3401 (maximum) and 3589 G (minimum), the spectrum shows shoulders at 3327, 3478, 3517, 3558, and 3696 G (Figure S9) suggesting the possibility that more than one paramagnetic species is present or sample heterogeneity has to be considered. It should be noted that this narrow spectrum cannot be interpreted as an isolated carbon-based radical spectrum due to its linewidth of almost 200 G. Overall, the results show that the [4Fe4S] cluster is spectroscopically active and the spectra are strongly dependent on the redox state of the protein.
In summary, presented is the first structure of the alternative epoxyqueuosine reductase, QueH, providing a structural basis for the final enzyme step of this complex tRNA modification. Additionally, the work expands the general knowledge of [4Fe-4S] clusters and the diverse utility of [4Fe-4S]-dependent enzymes.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by National Institutes of Health (Grant R01 GM70641 to V dC-L and SDB, and P01 GM118303 to J.A.G.). We acknowledge the Advanced Photon Source, particularly the staff and resources at GM/CA CAT. GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We would also like thank Martin McLauglin and Wilfred A. van der Donk for providing the guidance and the plasmid isc-pBADCDF for in vivo maturation of metalloenzymes. The LC-MS analyses were performed by Furong Sun at the Mass Spectrometry Laboratory, a Service Facility from the School of Chemical Sciences at University of Illinois at Urbana–Champaign.
Funding Sources:
This work was supported by NIH grant R01 GM041916
Footnotes
Supporting Information. Supporting material includes: graphics illustrating the queuosine biosynthetic pathway and protein surface representations of QueH, UV-Vis spectra of QueH preparation samples/TmQueH redox activity, additional EPR spectra for TmQueH, LC-MS data for TmQueH in vitro reactions and tables with structural homology details and QueH in vitro reaction conditions.
REFERENCES
- (1).El Yacoubi B, Bailly M, and de Crécy-Lagard V (2012) Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet 46, 69–95. [DOI] [PubMed] [Google Scholar]
- (2).de Crécy-Lagard V, and Jaroch M (2021) Functions of bacterial tRNA modifications: From ubiquity to diversity. Trends Microbiol. 29, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Boccaletto P, Machnicka MA, Purta E, Piątkowski P, Bagiński B, Wirecki TK, de Crécy-Lagard V, Ross R, Limbach PA, Kotter A, Helm M, and Bujnicki JM (2017) MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lorenz C, Lünse CE, and Mörl M (2017) tRNA modifications: impact on structure and thermal adaptation. Biomolecules 7, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Motorin Y, and Helm M (2010) tRNA stabilization by modified nucleotides. Biochemistry 49, 4934–4944. [DOI] [PubMed] [Google Scholar]
- (6).Koh CS, and Sarin LP (2018) Transfer RNA modification and infection – Implications for pathogenicity and host responses. Biochim. Biophys. Acta - Gene Regul. Mech 1861, 419–432. [DOI] [PubMed] [Google Scholar]
- (7).Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, Grayhack EJ, and Phizicky EM (2006) Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell 21, 87–96. [DOI] [PubMed] [Google Scholar]
- (8).Goodman HM, Abelson J, Landy A, Brenner S, and Smith JD (1968) Amber suppression: a nucleotide change in the anticodon of a tyrosine transfer RNA. Nature 217, 1019–1024. [DOI] [PubMed] [Google Scholar]
- (9).Kasai H, Ohashi Z, Harada F, Nishimura S, Oppenheimer NJ, Crain PF, Liehr JG, Von Minden DL, and McCloskey JA (1975) Structure of the modified nucleoside Q isolated from Escherichia coli transfer ribonucleic acid. 7-(4,5-cis-Dihydroxy-1-cyclopenten-3-ylaminomethyl)-7-deazaguanosine. Biochemistry 14, 4198–4208. [DOI] [PubMed] [Google Scholar]
- (10).Harada F, and Nishimura S (1972) Possible anticodon sequences of tRNAHis, tRNAAsn, and tRNAAsp from Escherichia coli. Universal presence of nucleoside O in the first position of the anticodons of these transfer ribonucleic acid. Biochemistry 11, 301–308. [DOI] [PubMed] [Google Scholar]
- (11).Katze, Basile B, and McCloskey J (1982) Queuine, a modified base incorporated posttranscriptionally into eukaryotic transfer RNA: wide distribution in nature. Science 216, 55–56. [DOI] [PubMed] [Google Scholar]
- (12).Noguchi S, Nishimura Y, Hirota Y, and Nishimura S (1982) Isolation and characterization of an Escherichia coli mutant lacking tRNA-guanine transglycosylase. Function and biosynthesis of queuosine in tRNA. J. Biol. Chem 257, 6544–6550. [PubMed] [Google Scholar]
- (13).Manickam N, Joshi K, Bhatt MJ, and Farabaugh PJ (2015) Effects of tRNA modification on translational accuracy depend on intrinsic codon–anticodon strength. Nucleic Acids Res. 44, 1871–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tuorto F, Legrand C, Cirzi C, Federico G, Liebers R, Müller M, Ehrenhofer-Murray AE, Dittmar G, Gröne H-J, and Lyko F (2018) Queuosine-modified tRNAs confer nutritional control of protein translation. EMBO J. 37, e99777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Müller M, Legrand C, Tuorto F, Kelly VP, Atlasi Y, Lyko F, and Ehrenhofer-Murray AE (2019) Queuine links translational control in eukaryotes to a micronutrient from bacteria. Nucleic Acids Res. 47, 3711–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hutinet G, Swarjo MA, and de Crécy-Lagard V (2017) Deazaguanine derivatives, examples of crosstalk between RNA and DNA modification pathways. RNA Biol. 14, 1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zallot R, Brochier-Armanet C, Gaston KW, Forouhar F, Limbach PA, Hunt JF, and de Crécy-Lagard V (2014) Plant, animal, and fungal micronutrient queuosine is salvaged by members of the DUF2419 protein family. ACS Chem. Biol 9, 1812–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).McCarty RM, and Bandarian V (2012) Biosynthesis of pyrrolopyrimidines. Mech. Enzymol. - Part II 43, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Romier C, Reuter K, Suck D, and Ficner R (1996) Crystal structure of tRNA-guanine transglycosylase: RNA modification by base exchange. EMBO J. 15, 2850–2857. [PMC free article] [PubMed] [Google Scholar]
- (20).Slany RK, Boesl M, Crain PF, and Kersten H (1993) A new function of S-adenosylmethionine: The ribosyl moiety of AdoMet is the precursor of the cyclopentenediol moiety of the tRNA wobble base queuine. Biochemistry 32, 7811–7817. [DOI] [PubMed] [Google Scholar]
- (21).Miles ZD, Myers WK, Kincannon WM, Britt RD, and Bandarian V (2015) Biochemical and spectroscopic studies of epoxyqueuosine reductase: a novel iron–sulfur cluster- and cobalamin-containing protein involved in the biosynthesis of queuosine. Biochemistry 54, 4927–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zallot R, Ross R, Chen W-H, Bruner SD, Limbach PA, and de Crécy-Lagard V (2017) Identification of a novel epoxyqueuosine reductase family by comparative genomics. ACS Chem. Biol 12, 844–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zallot R, Yuan Y, and De Crécy-Lagard V (2017) The Escherichia coli COG1738 Member YhhQ Is Involved in 7-Cyanodeazaguanine (preQ0) Transport. Biomolecules 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yuan Y, Zallot R, Grove TL, Payan DJ, Martin-Verstraete I, Šepić S, Balamkundu S, Neelakandan R, Gadi VK, Liu C-F, Swairjo MA, Dedon PC, Almo SC, Gerlt JA, and de Crécy-Lagard V (2019) Discovery of novel bacterial queuine salvage enzymes and pathways in human pathogens. Proc. Natl. Acad. Sci 116, 19126–19135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Miles ZD, McCarty RM, Molnar G, and Bandarian V (2011) Discovery of epoxyqueuosine (oQ) reductase reveals parallels between halorespiration and tRNA modification. Proc. Natl. Acad. Sci 108, 7368–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Payne KAP, Fisher K, Sjuts H, Dunstan MS, Bellina B, Johannissen L, Barran P, Hay S, Rigby SEJ, and Leys D (2015) Epoxyqueuosine reductase structure suggests a mechanism for cobalamin-dependent tRNA modification. J. Biol. Chem 290, 27572–27581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dowling DP, Miles ZD, Köhrer C, Maiocco SJ, Elliott SJ, Bandarian V, and Drennan CL (2016) Molecular basis of cobalamin-dependent RNA modification. Nucleic Acids Res. 44, 9965–9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Gaur R, and Varshney U (2005) Genetic analysis identifies a function for the quec (ybax) gene product at an initial step in the queuosine biosynthetic pathway in Escherichia coli. J. Bacteriol 187, 6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mueller EG, Buck CJ, Palenchar PM, Barnhart LE, and Paulson JL (1998) Identification of a gene involved in the generation of 4-thiouridine in tRNA. Nucleic Acids Res. 26, 2606–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Silva GN, Fatma S, Floyd AM, Fischer F, Chuawong P, Cruz AN, Simari RM, Joshi N, Kern D, and Hendrickson TL (2013) A tRNA-independent mechanism for transamidosome assembly promotes aminoacyl-tRNA transamidation. J. Biol. Chem 288, 3816–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Jurcik A, Bednar D, Byska J, Marques SM, Furmanova K, Daniel L, Kokkonen P, Brezovsky J, Strnad O, Stourac J, Pavelka A, Manak M, Damborsky J, and Kozlikova B (2018) CAVER Analyst 2.0: analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics 34, 3586–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yu J, Zhou Y, Tanaka I, and Yao M (2009) Roll: a new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 26, 46–52. [DOI] [PubMed] [Google Scholar]
- (33).Tian W, Chen C, Lei X, Zhao J, and Liang J (2018) CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 46, W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Dolinsky TJ, Nielsen JE, McCammon JA, and Baker NA (2004) PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA (2001) Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci 98, 10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kabsch W (2010) XDS. Acta Crystallogr. Sect. D 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Grosse-Kunstleve RW, and Adams PD (2003) Substructure search procedures for macromolecular structures. Acta Crystallogr. Sect. D 59, 1966–1973. [DOI] [PubMed] [Google Scholar]
- (38).Adams PD, Grosse-Kunstleve RW, Hung L-W, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, and Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. Sect. D 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- (39).Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart PH, and Hung L-W (2009) Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. Sect. D 65, 582–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (41).Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Moriarty NW, Grosse-Kunstleve RW, and Adams PD (2009) electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. Sect. D 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Phillips G, Swairjo MA, Gaston KW, Bailly M, Limbach PA, Iwata-Reuyl D, and de Crécy-Lagard V (2012) Diversity of archaeosine synthesis in crenarchaeota. ACS Chem. Biol 7, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, and Mori H (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol 2:2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lesley SA, Graziano J, Cho CY, Knuth MW, and Klock HE (2002) Gene expression response to misfolded protein as a screen for soluble recombinant protein. Protein Eng. Des. Sel 15, 153–160. [DOI] [PubMed] [Google Scholar]
- (47).Holm L (2019) Benchmarking fold detection by DaliLite v.5. Bioinformatics 35, 5326–5327. [DOI] [PubMed] [Google Scholar]
- (48).Seiffert GB, Ullmann GM, Messerschmidt A, Schink B, Kroneck PMH, and Einsle O (2007) Structure of the non-redox-active tungsten/[4Fe:4S] enzyme acetylene hydratase. Proc. Natl. Acad. Sci 104, 3073–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).tenBrink F, Schink B, and Kroneck PMH (2011) Exploring the active site of the tungsten, iron-sulfur enzyme acetylene hydratase. J. Bacteriol 193, 1229–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Goodstadt L, and Ponting CP (2004) Vitamin K epoxide reductase: homology, active site and catalytic mechanism. Trends Biochem. Sci 29, 289–292. [DOI] [PubMed] [Google Scholar]
- (51).Hu X, Dong Q, Yang J, and Zhang Y (2016) Recognizing metal and acid radical ion-binding sites by integrating ab initio modeling with template-based transferals. Bioinformatics 32, 3260–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Dutta A, and Bahar I (2010) Metal-binding sites are designed to achieve optimal mechanical and signaling properties. Structure 18, 1140–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Lill R (2009) Function and biogenesis of iron–sulphur proteins. Nature 460, 831–838. [DOI] [PubMed] [Google Scholar]
- (54).Yan R, Adinolfi S, and Pastore A (2015) Ferredoxin, in conjunction with NADPH and ferredoxin-NADP reductase, transfers electrons to the IscS/IscU complex to promote iron–sulfur cluster assembly. Biochim. Biophys. Acta - Proteins Proteomics 1854, 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Guigliarelli B, and Bertrand P (1999) Application of EPR Spectroscopy to the Structural and Functional Study of Iron-Sulfur Proteins, in Advances in Inorganic Chemistry (Sykes AG, Ed.), pp 421–497. Academic Press. [Google Scholar]
- (56).Zaborske JM, Bauer DuMont VL, Wallace EWJ, Pan T, Aquadro CF, and Drummond DA (2014) A nutrient-driven tRNA modification alters translational fidelity and genome-wide protein coding across an animal genus. PLOS Biol. 12, e1002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Brown KL (2005) Chemistry and enzymology of vitamin B12. Chem. Rev 105, 2075–2150. [DOI] [PubMed] [Google Scholar]
- (58).Banerjee R, and Ragsdale SW (2003) The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem 72, 209–247. [DOI] [PubMed] [Google Scholar]
- (59).Shelton AN, Seth EC, Mok KC, Han AW, Jackson SN, Haft DR, and Taga ME (2019) Uneven distribution of cobamide biosynthesis and dependence in bacteria predicted by comparative genomics. ISME J. 13, 789–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Silverman RB (1981) Chemical model studies for the mechanism of vitamin K epoxide reductase J. Am. Chem. Soc 103, 5939–5941. [Google Scholar]
- (61).Zallot R, Oberg N, and Gerlt JA (2019) The EFI Web resource for genomic enzymology tools: leveraging protein, genome, and metagenome databases to discover novel enzymes and metabolic pathways. Biochemistry 58, 4169–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Andreini C, Bertini I, Cavallaro G, Holliday GL, and Thornton JM (2008) Metal ions in biological catalysis: from enzyme databases to general principles. JBIC J. Biol. Inorg. Chem 13, 1205–1218. [DOI] [PubMed] [Google Scholar]
- (63).Rees DC, and Howard JB (2003) The interface between the biological and inorganic worlds: iron-sulfur metalloclusters. Science 300, 929–931. [DOI] [PubMed] [Google Scholar]
- (64).Beinert H (2000) Iron-sulfur proteins: ancient structures, still full of surprises. JBIC J. Biol. Inorg. Chem 5, 2–15. [DOI] [PubMed] [Google Scholar]
- (65).Numata T, Ikeuchi Y, Fukai S, Suzuki T, Nureki O (2006) Snapshots of tRNA sulphuration via an adenylated intermediate. Nature, 442, 419–424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.