

Abstract
Efficient and reproducible measurements of multiple polycyclic aromatic hydrocarbon (PAH) metabolites in urinary samples are required to evaluate the complex health effects of PAH exposure. Here, we demonstrate a highly practical, automated off-line solid-phase extraction (SPE) of deconjugated hydroxylated PAHs followed by LC-MS/MS to simultaneously measure eight mono-hydroxylated PAH compounds: 1-hydroxynaphthalene, 2-hydroxynaphthalene, 2-hydroxyfluorene, 1-hydroxyphenanthrene, 2&3-hydroxyphenanthrene, 4-hydroxyphenanthrene and 1-hydroxypyrene. Initially, we observed low recovery rates were observed (e.g., 16% for 1-hydroxypyrene) when using previously published methods. We optimized the procedure by choosing polymeric absorbent-based cartridges, automating the sample loading step by diluting samples with 15% methanol/sodium acetate, and most importantly, replacing acetonitrile with methanol as eluting solvent. Optimized sample preparation has improved the recovery rates to more than 69% for analytes of interest. This improvement led to higher method sensitivity and detection frequency, especially for 1-hydroxypyrene, detected in all of 100 urine samples collected in the New York site of the Legacy Girls Study. The limits of detection ranged from 7.6 pg/mL to 20.3 pg/mL using 1 mL of urine, compared to the 2 mL required in CDC, method 09-OD. The average coefficients of variance of quality control samples (n=60) ranged between 7-21%; variance of repeated measurements (n=45) was <10%. This efficient and reliable method for measuring PAH metabolites will greatly benefit epidemiology studies and biomonitoring programs.
Keywords: PAH metabolites, Urine, Solid-phase extraction, LC-MS/MS, Recovery, Reproducibility
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a group of organic compounds with aromatic ring structures formed from incomplete combustion. Certain PAHs are known for their carcinogenic and mutagenic potential, raising public health concerns[1]. These ubiquitous contaminants can enter the human body through inhalation, ingestion, and dermal absorption [2]. Upon entry, most PAHs are metabolized to mono-hydroxylated compounds in glucuronide conjugated forms, which can then be excreted through urine and feces. Key metabolites include 1-hydroxynaphthalene (1-OH-NAP), 2-hydroxynaphthalene (2-OH-NAP), 2 and 3-hydroxyfluorene (2&3-OH-FLU),1-hydroxyphenanthrene (1-OH-PHEN), 2 and 3-hydroxyphenanthrene (2&3-OH-PHEN), 4-hydroxyphenanthrene (4-OH-PHEN), and 1-hydroxypyrene (1-OH-PYR).
Analyzing these PAH metabolites in urine has been a common approach to characterizing PAH exposure in order to assess its association with adverse health outcomes, such as increased infertility risk in males [3], lower birth weight following prenatal exposure[4], and lung cancer mortality [5]. Furthermore, metabolite concentrations in urine shed light on potential exposure routes and overall body burden of PAHs. For example, by measuring urinary 1-OH-PYR, dermal absorption appeared to be the major exposure pathway responsible for high urinary 1-OH-PYR, while inhaled particulate PAH was found to be resistant to uptake, possibly due to low solubility [6]. However, by measuring multiple PAHs metabolites, heavy traffic exposure was found to be associated with high concentration of urinary OH-PAHs [4]. High dietary PAH was linked with elevated levels of OH-PAHs in children aged 6-11 years [7]. A complete profile of PAH metabolites is necessary to disentangle the complicated health risks of being exposed to unspecific sources of PAHs in the environment and to inform prevention programs based on the source of the metabolites. Thus, effective biomonitoring research with urinary samples that can be used to measure PAH metabolites demands simultaneous measurements of several OH-PAHs in one single analysis run.
There are a number of published methods based on automated sample extraction and gas chromatography/ mass spectrometry (GC/MS)[8],[9],[10]. Automation of sample extractions substantially increases sample throughput and improves recovery in published methods including automated liquid-liquid extraction [8]; automated solid phase extraction [9], and micro phase solid phase extraction [10]. These methods are effective, but generally (a) involve labor-intensive derivatization, (b) demand slow separations and long analysis times per sample, and (c) require large volumes of urine.
Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) has been increasingly used for PAH metabolite analysis as it eliminates the need for a tedious derivatization step and provides high sensitivity. Micro online solid phase extraction combined with LC-MS/MS analysis could significantly improve throughput [11]. However, because urine matrices contain salts and other compounds, online extraction could broaden peaks and block LC columns, leading to lengthened downtime, failed analyses, and costly maintenance. To date, few methods have been published that combine off-line automation solid phase extraction (SPE) and LC-MS/MS. Barbeau et al [12] proposed off-line automated SPE coupled with LC-fluorescent detector but limited the process to only determining one single analyte of interest 3-hydroxybenzo(a)pyrene, while using up to 10ml of urine. Automated procedure offered high productivity and reproducibility which are important to biomonitoring research. This study explored the optimization of automated SPE with LC MS-MS, aiming to achieve high through put and reproducible results.
Furthemore, low recovery, less than 50%, has long been a challenge in PAH analysis due to preferential loss of certain compounds during sample processing. For example, the 2-ringed 1-OH-NAP and 2-OH-NAP, which have high vapor pressure, typically have low recovery rates due to loss in the evaporation step [8],[13]. Significant low extraction recovery has also been observed for 1-OH-PYR, in some cases less than 20% [9],[10],[14], which cannot be explained by evaporative loss. Low recovery rates can lead to low detection frequency and low accuracy if not corrected by internal standards.
For large epidemiology studies which require analysis of hundreds of urine samples, sensitivity, reproducibility, and high throughput are important considerations when choosing an appropriate approach for measuring biomarkers. In this study, we developed a robust, reliable, reproducible method for the aforementioned eight urinary mono hydroxylated metabolites including: 1-OH-NAP, 2-OH-NAP, 2-OH-FLU, 1-OH-PHEN, 2&3-OH-PHEN, 4-OH-PHEN and 1-OH-PYR.
2. Method
2.1. Chemicals and materials:
Standard reference materials.
Standard compounds and mixtures were obtained from high-purity sources in which the identity and composition of the material were independently verified. Chemical standards of 10 hydroxylated PAHs were obtained from Cambridge Isotope Laboratory, Andover, MA: 1-hydroxynaphthalene (1-OH-NAP), 2-hydroxynaphthalene (2-OH-NAP), 2-hydroxyfluorene (2-OH-FLU), 3-hydroxyfluorene (3-OH-FLU), 9-hydroxyfluorene (9-OH-FLU), 1-hydroxyphenanthrene (1-OH-PHEN), 2-hydroxyphenanthrene (2-OH-PHEN), 3-hydroxyphenanthrene (3-OH-PHEN), 4-hydroxyphenanthrene (4-OH-PHEN) and 1-hydroxypyrene (1-OH-PYR). Labeled isotopic compounds including 13C6 1-NAP (99%, 50μg/mL in toluene), 13C6 2-NAP (purity 99%, 50μg/mL in toluene), 13C6 3-PHEN (98%, 50μg/mL in toluene), and 13C6 1-PYR (98%, 50ug/mL in toluene) were purchased from Cambridge Isotope Laboratory. D9-2-FLU (50μg/mL in acetonitrile) was purchased from Santa Cruz Biotech (Santa Cruz, CA).
Other Reagents and Chemicals.
Beta glucuronidase/aryl sulfatase type H1 enzyme from Helix pomatia was purchased from Roche Diagnostic (Indianapolis, IN). We used optima grade methanol, acetonitrile, and water which were supplied by Fisher Scientific (Waltham, MA). Toluene and formic acid (98%-100% were purchased from Merck and Millipore Sigma (Darmstadt, Germany). Anhydrous sodium acetate (for molecular biology >99%), and acetic acid (ACS reagent, 99.7% were purchased from Sigma Aldrich (St. Louis, MO). Bond Elut C18 500mg, 6mL and Bond Elut Focus 60mg, 3mL cartridges were obtained from Agilent (). The liquid chromatography column was a Kinetex 1.7μm Biphenyl 100 Å 50x 2.1mm purchased from Phenomenex, Torrance, CA.
Equipment.
Extractions were performed using a SPE-03 system purchased from PromoChrom Technologies Ltd (Richmond, BC, Canada). Turbo-Vap LV evaporator (Biotage, Charlotte, NC) was used for concentration step. Sterile surfactant-free cellulose acetate (SFCA) membrane syringe filters (0.22um pore size) were obtained from Cole Parmer (Vernon Hill, IL). A Shimadzu ultra-high performance liquid chromatograph (Nexera) was coupled to a Qtrap 6500+ (Sciex, Framingham, MA) for chemical identification and quantification.
2.2. Preparation of standards, synthetic urine, and pooled urine
Stock solution of native standards were diluted to lower concentrations in methanol, ranging from 0.039 ng/mL to 10ng/mL for 2-OH-FLU, 3-OH-FLU, 9-OH-FLU, 1-OH-PHEN, 2-OH-PHEN, 3-OH-PHEN, 4-OH-PHEN and from 0.156 ng/mL to 40 ng/mL for 1-OH-NAP and 2-OH-NAP. A mixture of isotopically-labeled internal standards was prepared in toluene containing 500 ng/mL of D9-2-OH-FLU, 13C6 3-OH-PHEN, 13C6 -1-OH-PYR and 2000 ng/mL of 13C6 1-OH-NAP and 13C6 2-OH-NAP. Calibration standards consisted of mixtures of both native standards and internal standards in methanol. A 9-point-calibration curve was established with the y-axis representing the area ratios of the native standards and corresponding internal standard and the x-axis representing their concentration ratios.
The calibration curves were based on 9-point calibration curves spanning zero to well above levels expected in urine samples. Quantitation was based on peak area ratios relative to their corresponding isotopically-labeled internal standards. For instance, 1-OH-NAP and 2-OH-NAP were quantified using 13C6 1-OH-NAP and 13C6 2-OH-NAP internal standards, respectively; 2-OH-FLU was quantified using deuterated d9-2-OH-FLU; and 1-OH-PYR was quantified using 13C6-1-OH-PYR. 1-OH-PHEN, 2&3-OH-PHEN and 4-OH-PHEN were quantified using 13C6-3-OH-PHEN. Two compounds 2-OH-PHEN and 3-OH-PHEN were co-eluted and quantified as one total concentration of 2&3-OH-PHEN. All native standards, internal standards and sub-standards were stored at 4°C.
Synthetic urine, used as a method blank, was made using the recipe described in a CDC method [15]. The chemicals were weighed and dissolved in 200mL of optima- grade water. Urine samples collected from anonymous volunteers were pooled for use as quality control (QC) samples. These urine samples were filter-sterilized using 0.22mm SFCA membrane filters (Cole Parmer and pipetted in 2mL cryogenic vials. The synthetic urine samples and pooled urine samples were stored at −80°C and thawed prior to processing concurrent with actual samples.
2.3. Sample processing procedure
Immediately prior to analysis, samples and method blanks and QC samples (1 per group) were thawed at room temperature. Internal standard mixtures were added to 1mL of urine samples. To deconjugate the glucuronidated metabolites in urine, we used the β-glucuronidase/aryl sulfatase H1 enzyme (10mg/mL in 0.1M sodium acetate, pH 5.5), which was prepared daily. To do so, 1 mL of the enzyme solution was added to the sample/internal standard mixture and incubated for 17-18 hours in a water bath at 37°C. The hydroxylated compounds were then extracted using solid phase extraction (SPE) using Bond Elut Focus cartridges.
The extraction procedure included 4 steps, conditioning-equilibrating, sample loading, rinsing, and elution. The cartridges were conditioned with 1mL of optima grade water, and equilibrated with 1mL of optima grade methanol. Samples were diluted with 15% methanol in sodium acetate 0.1M, pH 5.5 solution prior to loading at a flow rate of 1mL/min. The cartridges were then rinsed with 1mL of water followed by 3mL of 30% methanol in sodium acetate mixture. Following rinsing, the cartridges were air purged and blown dry with nitrogen gas at 2mL/min flow rate for 10 min. Finally, the PAHs were eluted from the cartridge using 6mL of optima grade methanol. The samples were then concentrated by evaporation with a gentle flow of nitrogen gas (2-4 psi) at 40°C. The resulting concentrate was then reconstituted in methanol to a final volume of 150-300μl for instrumental analysis. Samples were stored in −20°C prior to LC-MS/MS analysis.
2.4. Optimization experiments
A set of 1mL synthetic urine samples and a set of 1mL anonymous pooled urine samples were spiked with different concentrations of OH-PAHs standards mixture. All experiments were conducted in triplicate.
Experiment 1:
Selection of SPE columns. The performance and recoveries of two SPE columns types were evaluated: (a) large-volume Bond Elut C18 (500 mg and 6mL), and (b) small-volume Bond Elut Focus (50mg and 3mL).
Experiment 2:
Comparison of sample loading methods. We experimented with different loading sample conditions to minimize the loss of analytes during transfer from glass vials to the SPE system sample loops, column tubing and pump system. The three methods are (a) direct loading by hand, an option provided by SPE-03 for small sample volume (1mL); (b) automated loading without organic solvents such as methanol being added; and (c) automated loading of diluted samples with 15% methanol in sodium acetate solution.
Experiment 3:
Recovery rate comparison between acetonitrile and methanol. Acetonitrile and methanol are both potentially viable solvents for elution and subsequent analysis by LC-MS/MS. We compared the recovery of each target analyte when using acetonitrile and methanol for extraction, elution and evaporation steps of the SPE procedure.
The extraction efficiencies were determined based on the ratios of internal standards that were spiked before and after sample processing. The relative recoveries were calculated based on the calculated analyte mass of the spiked samples in comparison with the supposed spiked analyte mass.
2.5. LEGACY Cohort
First-void morning urine samples were collected from randomly selected 100 participants in New York Site of the LEGACY Girl Study cohort which is a multicenter prospective study of 1,040 girls and included 5 sites (New York City, NY; Philadelphia PA; Salt Lake City, UT; San Francisco Bay Area, CA; and Toronto, Ontario); for detail see [16],[17]). The percentage of participants who give urine sample is 98%, much higher than that of blood, 49%, [16] emphasized the highly feasibility of urinary metabolite monitoring. Urine samples were collected from girls aged 6-13 years and stored at −80°C before analysis.
2.6. Quality assurance and quality control (QA/QC)
Strict QA/QC protocols were followed in this study. Internal batches included 10% synthetic urine blanks and 10% urine pools whose results had to conform to a stipulated QC range, or else the batch was rerun. Every three samples, a methanol blank was injected to confirm that there was no carry-over between analyses. We also ran 10% duplicate samples to determine the reliability of the instrumental analysis. Precision was assessed by comparing results obtained in these QA/QC samples. Pooled urine and synthetic urine samples were processed and measured in different batches along with actual urine samples.
2.7. Instrumental parameters
High performance liquid chromatography analysis was controlled and coupled with Qtrap 6500+ by Analyst software (version 1.6.2, Sciex, Foster City, CA), and data processing was done on the Multiquant (version 3.0.3, Sciex). The chromatography separation was done using the column Kinetex 1.7Um Biphenyl 100 Å 50x 2.1mm at 40°C. The mobile phase A consisted of 95% water: 5% acetonitrile: 0.005% formic acid, while mobile phase B contained 5% water: 95% acetonitrile: 0.005% formic acid. The gradient program was set up to initially deliver 95% mobile phase A and 5% mobile phase B, and a linear gradient increased the fraction of phase B to 100% over 10 min, held at 100% for two min and decreased back to 5% B at 13 min. The system was equilibrated for 3 min before each run.
Mass spectral analysis involved optimizing factors that influenced ionization and fragmentation. The de-clustering potential, entrance potential, and collision energy were optimized for each compound using manual infusion with mobile phases. Electrospray ionization (ESI) was used in negative ion mode with a set curtain gas pressure of 35 psi, nebulizer gas pressure of 65 psi, turbo gas pressure of 65 psi, source temperature of 650°C, and collision gas pressure of 9 psi.
3. Results
3.1. Compound separation and quantification
Figure 1 shows the chromatogram of a sample in which concentrations of analytes were: 11.14 ng/mL for 2-OH-NAP; 0.153 ng/mL for 2-OH-FLU; 0.091 ng/mL 2&3-OH-PHEN; 0.109 ng/mL for 1-OH-PHEN; 0.045 ng/mL for 4 OH-PHEN; and 0.187 ng/mL for 1-OH-PYR. The concentrations of the isotopic internal standards were 100 ng/mL for 13C6-1-OH-NAP and 13C6-2-OH-NAP; and 25 ng/mL for the others including D9-2-OH-FLU, 13C6-3-OH-PHEN and 13C6-1-OH-PYR.
Figure 1:
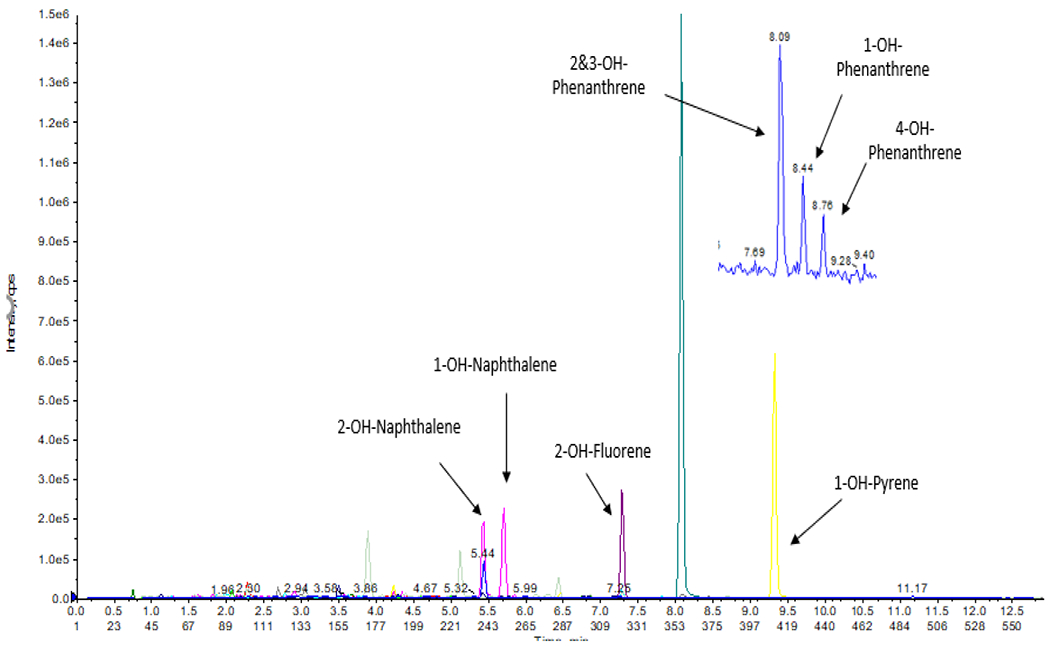
LC- MS/MS Chromatogram of 7 mono-hydroxylated PAHs and the corresponding internal standards in a pooled urine sample.
Separation of 1-OH-NAP and 2-OH-NAP was achieved by adjusting the gradient of the mobile phase. Similarly, 1-OH-PHEN, 3-OH-PHEN, and 4-OH-PHEN were clearly separated. However, 2-OH-PHEN and 3-OH-PHEN could not be eluted separately which was also observed in other previous liquid chromatography methods [18],[19]. Thus, these two isomers were quantified together as 2&3- OH-PHEN. 2-OH-FLU and 3-OH-FLU were also quantified together as 2&3-OH-FLU.
The calibration curves were established using weighted linear regression and a weight factor of 1/x where x is the concentration ratio between the standard and its corresponding internal standard. All correlation coefficients were greater than 0.995 over the entire concentration range. Detection limits of the instrument were calculated based on the signal to noise being equal or greater than 3 from the lowest standard concentration prepared in methanol. The instrumental detection limits are 8 pg/mL for 1-OH-NAP, 4 pg/mL for 2-OH-NAP, 8 pg/mL for 2-OH-FLU,8 pg/mL for 1-OH-PHEN, 2 pg/mL for 2&3-OH-PHEN, 4 pg/mL for 4-OH-PHEN, and 8 pg/mL for 1-OH-PYR.
The method detection limits were calculated from linear regression for each analyte.
where b is the slope of the calibration function, and Sd is the standard deviation of the intercept.
Table 2 shows the detection limits: 18.33 pg/mL for 1-OH-NAP, 12.75 pg/mL for 2-OH-NAP, 7.64 pg/mL for 2-OH-FLU, 20.29 pg/mL for 1-OH-PHEN, 10.74 pg/mL for 2&3-OH-PHEN, 11.75pg/mL for 4-OH-PHEN, and 18.41 pg/mL for 1-OH-PYR. Most published analytical methods reported instrumental detection limits solely based on visual inspection of the signal-to-noise ratio, and found similar detection limits to our rapid throughput method [18],[20].
Table 2:
Method recoveries, limits of detection, limits of quantification and coefficients of variation.
| Analytes | Recovery (%) | LOD (pg/mL) | LOQ (pg/mL) | CV(%) for QC samples (n=60) | CV(%) for actual samples (n=45) |
|---|---|---|---|---|---|
| 1-OH-NAP | 101 | 18.33 | 55.53 | N/A * | 6% |
| 1-OH-PHEN | 97 | 20.29 | 61.49 | 10% | 4% |
| 1-OH-PYR | 93 | 18.41 | 55.78 | 12% | 3% |
| 2-OH-FLU | 98 | 7.64 | 23.15 | 21% | 10% |
| 2-OH-NAP | 69 | 12.75 | 38.65 | 7% | 4% |
| 2&3-OH-PHEN | 77 | 10.74 | 32.56 | 10% | 4% |
| 4-OH-PHEN | 111 | 11.75 | 35.61 | 12% | 9% |
N/A denotes not applicable since there was no detected 1-OH-NAP in actual pooled urine sample.
3.2. Extraction efficiency of two SPE cartridges
Table 3 lists parameters programed in the SPE procedure. Two SPE cartridges were tested with the same manual sample loading and analytes were eluted with acetonitrile. The extraction efficiency using Bond Elut C18 ranged from 40-70% for all analytes except 13C6-1-OH-PYR, which was 16%. Higher extraction efficiencies of OH-PAHs were achieved with Bond Elut Focus columns for 13C6-1-OH-PYR. Importantly, the extraction efficiency of 13C6-1-OH-PYR increased up to 45%, while only minor changes in other analytes were observed. Extraction efficiency are 40% for 13C6-2-OH-NAP and 13C6-1-OH-NAP, 70% for 13C6-3-OH-PHEN and 81% for d9-2-OH-FLU.
Table 3:
Parameters programed for automated solid phase extraction
| Action | Inlet | Flow rate (mL/min) | Volume (mL) |
|---|---|---|---|
| Elute W1 | Methanol | 2 | 1 |
| Elute W2 | Water | 2 | 1 |
| Sample dilution | Sodium acetate buffer + 15% methanol (pH 5.5) | 20 | 8 |
| Add sample | Sample | 0.5 | 4.5 |
| Add sample | Sample | 0.5 | 4.5 |
| Add sample | Sample | 1 | 2 |
| Rinse W1 | Water | 5 | 1 |
| Rinse W1 | Sodium acetate buffer + 30% methanol (pH 5.5) | 5 | 3 |
| Air purge | Air | 10 | 15 |
| Blow N2 | 10 min | 2mL/min | |
| Collect | Methanol | 0 | 6 |
| Air Purge | Air | 5 | 5 |
3.3. Optimizing the SPE sample loading method
Figure 4 shows the comparison in recovery rate among three sample loading methods. Significant improvement of 1-OH-PYR recovery (from 28% to 47%) was observed in samples diluted with sodium acetate solution containing 15% methanol at pH 5.5, compared to the non-dilution sample loading method. Even though manual loading had a good recovery rate because it required less contact with tubing, it was time consuming and labor intensive. Optimized automated loading shortened the sample processing time, which substantially increased the analytical throughput.
Figure 4:
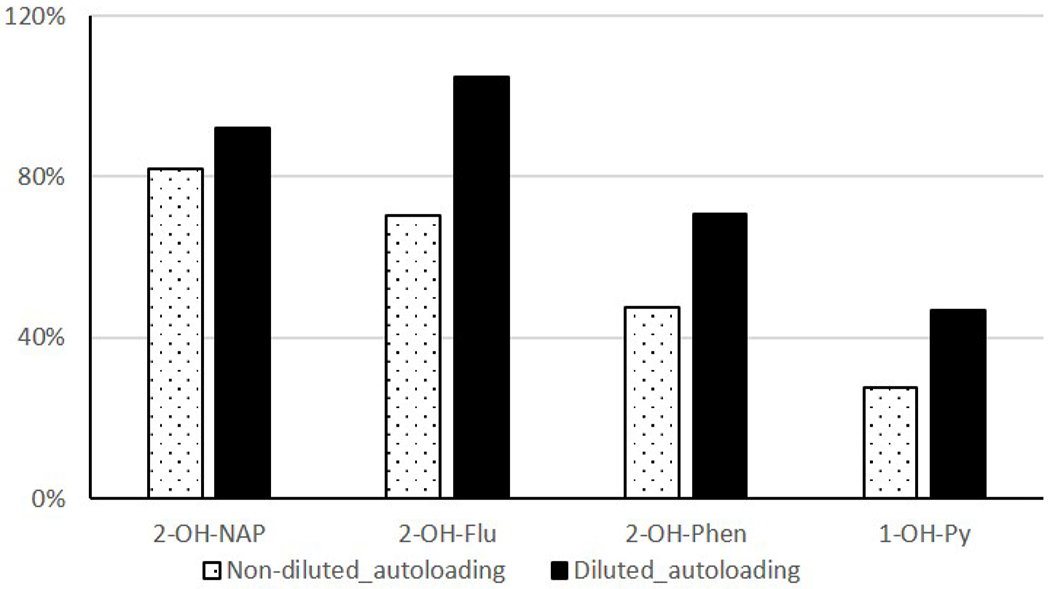
Relative recovery rates of non-diluted autoloading and methanol-diluted autoloading methods compared the manual loading method.
3.4. Selection of eluting solvent
With acetonitrile being used as the eluting solvent, concentration time was lengthy (~2 hrs at 40°C, 5 psi) which led to the loss of 1-OH-NAP. Relative recovery for 1-OH-NAP in acetonitrile elution solution was not even determinable because of the loss. Recovery rates for 2-OH-NAP, 2&3-OH-FLU, 1-OH-PHEN, 4-OH-PHEN, 3-OH-PHEN were 91%, 96%, 82%, 57%, and 97%, respectively. These compounds had adequate recoveries, but the recovery of 1-OH-PYR was very low, 16%.
Using methanol as the eluting solvent, the relative recoveries ranged from 69-111%, comparable with other studies [18],[8] . Specifically, relative recoveries were 101% for 1-OH-NAP, 69 % for 2-OH-NAP, 98% for 2&3 OH-FLU, 77% for 2&3 OH-PHEN, 97% for 1-OH-PHEN, 111% for 4-OH-PHEN, and 93% for 1-OH-PYR (Table 2).
3.5. Detection frequency and reproducibility
The presented method has been applied to measure simultaneously urinary OH-PAHs in about 958 samples from LEGACY Girl Study cohort. For 100 randomly selected samples, the detection frequencies were 85% for 1-OH-NAP, 99 % for 2-OH-NAP, 84% for 2&3 OH-FLU, 100% for 2&3 OH-PHEN, 100% for 1-OH-PHEN, 79% for 4-OH-PHEN, and 100% for 1-OH-PYR. Using only 1mL urine, we achieved up to 90% detectable measurements of most of the analytes (Table 4).
Table 4:
Urinary levels of OH-PAHs (μg/L) in 100 samples randomly selected from participants in the New York City site of the LEGACY Girls Study
| Compounds | New York City LEGACY cohort, aged 6-13 years, n=100, GM(μg/L) | NHANES aged 6-11 years, GM (μg/L)[7] | SD | Range | Detection frequency (%) |
|---|---|---|---|---|---|
| 1-OH-NAP | 0.769 | 1.43 | 18.94 | BDL- 36.9 | 85 |
| 2-OH-NAP | 3.78 | 1.69 | 8.82 | BDL- 28.7 | 99 |
| 2&3-OH-FLU | 0.318 | 0.352 | 0.447 | BDL- 22.26 | 84 |
| 1-OH-PHEN | 0.205 | 0.119 | 0.174 | BDL-3.64 | 100 |
| 2&3-OH-PHEN | 0.180 | 0.146 | 0.153 | BDL-6.65 | 100 |
| 4-OH-PHEN | 0.032 | 0.042 | 0.029 | BDL- 0.56 | 79 |
| 1-OH-PYR | 0.484 | 0.06 | 0.622 | 0.051- 34.6 | 100 |
BDL: Below detection limit
GM: geometric means
The coefficient of variation (CV) values represents the reproducibility of the established method. The averaged CVs of detected OH-PAHs ranged from 7-21%. The CVs for 45 duplicate runs were in the range of 3-10%, with higher relative variance for samples with low concentrations of OH-PAHs.
4. Discussion
Initially, we observed low recovery rates for certain PAH metabolites such as 1-OH-PYR. Similar result was reported in [14] where the recoveries of 1-OH-PYR varied in different matrices including water and synthetic and diluted urine. They suggested that the matrix effect was responsible for the low recovery of this compound [14]. However, in our experiment using C18 SPE column, the recovery of 1-OH-PYR remained low in both synthetic and actual urine matrices. To uncover the reasons behind the low recovery rates, we conducted tests in triplicate for each individual step, including enzyme hydrolysis, evaporation, SPE extraction and calculated the recovery rate in these steps. We found that the selection of solvents is critical for obtaining higher recovery rates. Acetonitrile, used as an elution solution, led to low recoveries for both 1-OH-PYR and 1-OH-NAP (< 30%). Evaporation of acetonitrile took much longer than that of methanol; even under a gentle nitrogen stream (less than 5 psi) for only 60 min, the concentrations of 1-OH-PYR and 13C6-1-OH-PYR were reduced by more than 50%.
Previous methods have reported lower recoveries for 1-OH-NAP which likely is due to the loss of two-ringed analytes during the evaporation process [8],[20],[13]. The evaporation loss is not the main reason for the low recovery rate of 1-OH-PYR, which has low vapor pressure. We speculated that the loss to tubing and vials in SPE could be the reason and an organic solvent that would increase 1-OH-PYR solubility would overcome this limitation. The higher recovery rate using methanol confirmed our speculation. The addition of methanol prior to loading the sample onto the SPE system could help to disassociate the analytes from the culture tube walls and tubing or break down lipid micelles that could contain PAHs. Similar observations of low recoveries due to the tendency of OH-PAH metabolites to stick to glassware were reported previously [21]. The buffer pH of 5.5 is critical to the enzyme hydrolysis step [8] and maintaining this pH throughout loading sample steps ensures that the analytes remain dissolved after enzyme hydrolysis. Thus, we diluted samples with 15% methanol in 0.1M sodium acetate: methanol, pH 5.5.
For the 100 random samples, detection frequencies for 1-OH-PHEN and 2&3-OH-PHEN, and 1-OH-PYR were 100%. This is higher than in previously published methods where detection frequencies for these analytes were reported to be 80%, 85% and 47%, respectively [22]. The detection frequency of 4-OH-PHEN was 79%, similar to [23], but higher than the 74% reported in the National Health and Nutrition Examination Survey (NHANES) data set [7] and the 59% in [14]. Detection frequencies of 1-OH-NAP and 2&3-OH-FLU were similar to those in other published studies at 85% and 84%, respectively [23],[14].
The recovery also varied with the sample matrices. We examined the matrix effects in water, synthetic urine, pooled urine and diluted pooled urine. The pooled urine matrix was identical to real urine samples and most of the analytes were detected (except 1-OH-NAP). Therefore, the pooled urines were used as QC samples for the different analysis batches.
Bond Elut Focus cartridges performed better than C18 columns. Previous methods have shown the insufficient removal of endogenous compounds in urine matrices using Bond Elut C18 cartridges procedure leading to high matrix effects for 1-OH-PYR measurements [14], [24]. Romanoff et al. recommended Bond Elut Focus since a higher percentage of the methanol/water mixture can be used to remove the unwanted chemicals in the collected solution [9]. However, they reported significant loss of all analytes of interest in the process of evaporating 3mL dichloromethane, a GC amenable solvent, unless dodecane was added as the keeper. Due to the incompatibility of dodecane with ESI in LC-MS/MS, instead of dichloromethane, we selected methanol which is compatible with LC-MS/MS hardware. Cleaner extracts were obtained with the usage of Bond Elut Focus which allows the rinse solution up to 30% of methanol to remove the endogenous material in urine matrices. This lengthened the column’s life and reduces the potential downtime of the instruments. Additional benefits of using Bond Elut Focus cartridges include the smaller volume of solvents required for washing and rinsing.
QA and QC measures are critical for obtaining reliable results. Synthetic urine and pooled urine samples were used to test recoveries of each step and the overall method. Synthetic urine results in a reference material with relatively well-constrained concentrations of each analyte, while the pooled samples are matrix-matched and can be studied repeatedly to establish the recovery of each analyte during a given step in the extraction or analysis. Isotopic internal standards were chosen based on their similar structures with their metabolites which are the products of the deconjugation process. Based on [8], the incubation time for efficient deconjugation is dependent on the analyte. In details, the concentration of 2-OH-NAP reached its maximum after 1 hr of hydrolysis and fell over time, while 1-OH-NAP needed more than 12 hr to complete the hydrolysis process [8]. This means the loss of analytes during hydrolysis step will not be accounted for if internal standards are added after enzymatic hydrolysis. We added internal standards prior to the hydrolysis step which allowed us to account for the effect of the hydrolysis on extraction and recovery.
We have optimized the procedure to quantify simultaneously multiple OH-PAHs metabolites in human urine samples using 1ml of urine, relatively less volume compared to other studies [12],[14],[18],[24]. Our method achieved satisfactory recovery and reproducible results with minimal cost of chemical usage, instrument maintenance and reduced laborious laboratory tasks. Automated SPE and relatively short LC run time allowed us to achieve high throughput compared to previously published protocols [18], [24]. We applied this method to quantify the OH-PAHs metabolites for LEGACY cohort.
Table 4 shows higher geometric means of 2-OH-NAP, 1-OH-PHEN, 2&3- OH-PHEN and 1-OH-PYR for the 100 random samples from the New York site of the LEGACY cohort compared to NHANES values for age group 6-11 years [7]. The concentrations of 2-OH-NAP and 1-OH-PYR in our study were notably 3-fold and 7-fold higher, respectively, than those national reference values. This is not unexpected since heavy traffic, building emissions from space heating, as well as thousands of restaurants contribute to PAHs in New York City air.
5. Conclusions
We have developed a fast and sensitive method with an automated sample extraction step followed by a 13-min run on a LC-MS/MS system to simultaneously measure 7 metabolites of PAHs using only 1mL of urine. Using methanol rather than acetonitrile as an elution solvent substantially improved the extraction efficiency. The combination of using the polymeric absorbent SPE cartridge, diluting samples prior to the loading step and using methanol as the eluting solvent improved the recoveries of 1-OH-PYR significantly from 16% to 93%, and overall recovery ranged from 69-111% for target analytes. The inter-day precision varied between 3-10% and the intra-day precision fluctuated in the range of 7-21%, demonstrating good reproducibility in measurements and consistency of sample treatment procedures. Future work could be the inclusion of more target metabolites/analytes for analysis. The presented method effectively and sensitively measures the PAH metabolites with high throughput, allowing for the analysis of large numbers of samples in epidemiology and biomonitoring studies.
Figure 2:
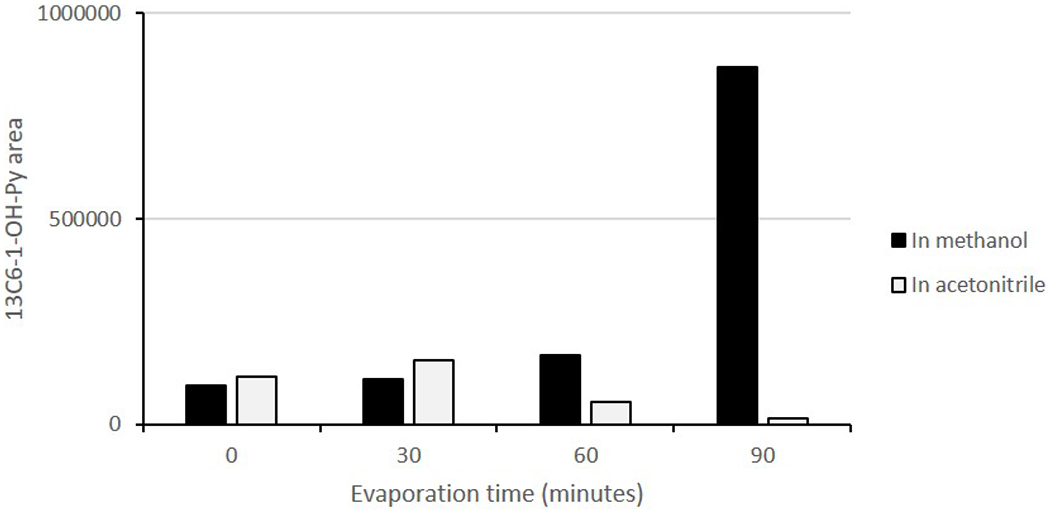
Peak area of 13C6 1-OH-PYR by LC-MS/MS in different solutions after concentration under a stream of nitrogen. As the evaporation time increased, the concentrations of isotopic 13C6-1-OH-PYR increased correspondingly in methanol, but not in acetonitrile.
Figure 3:
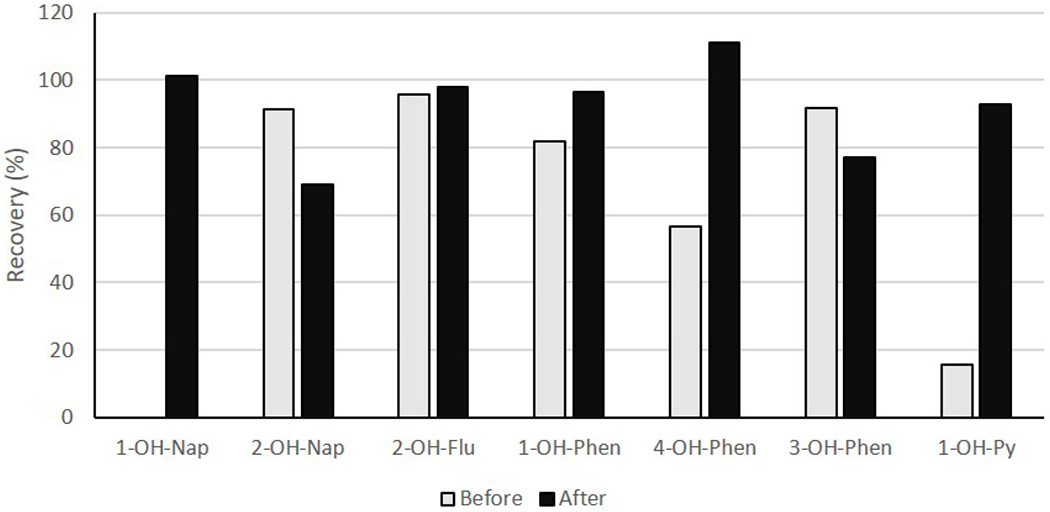
Comparison of the recovery rate of analytes before and after method optimization.
Table 1:
LC-MS/MS Multiple Reaction Monitoring (MRM) parameters used for quantification method of 8 targeted mono-hydroxylated PAHs and their corresponding isotopic internal standards.
| Analytes | Retention time (min) | Precursor ion (m/z) | Product ion (m/z) | Collision energy (eV) | Retention time (min) |
|---|---|---|---|---|---|
| 1-OH-NAP | 5.55 | 142.8 | 115.0 | 34 | 5.59 |
| 2-OH-NAP | 5.28 | 142.8 | 115.0 | 34 | 5.32 |
| 2&3-OH-FLU | 7.32 | 180.9 | 180.0 | 34 | 7.35 |
| 1-OH-PHEN | 8.39 | 192.9 | 165.0 | 42 | 8.44 |
| 2&3-OH-PHEN | 8.04 | 192.9 | 165.0 | 42 | 8.09 |
| 4-OH-PHEN | 8.71 | 192.9 | 165.0 | 42 | 8.76 |
| 1-OH-PYR | 9.27 | 217.0 | 189.1 | 45 | 9.31 |
| 13C6-1-OH-NAP | 5.55 | 148.9 | 121.0 | 34 | 5.58 |
| 13C6-2-OH-NAP | 5.28 | 148.9 | 121.0 | 34 | 5.31 |
| d9-2-OH-FLU | 7.21 | 190.0 | 188.0 | 34 | 7.24 |
| 13C6-3-OH-PHEN | 8.04 | 199.0 | 171.0 | 43 | 8.05 |
| 13C6-1-OH-PYR | 9.27 | 222.8 | 195.0 | 46 | 9.30 |
Highlights.
Simultaneously measured seven urinary OH-PAHs using only 1mL urine aliquot.
Efficient automated off-line solid phase extraction workflow followed by LC-MS/MS.
Improved recovery of 1-hydroxypyrene by selecting methanol as eluting solvent.
Increased detection frequency of 1-hydroxypyrene to 100% in 100 samples.
Satisfactory recoveries of target analytes (69-111%).
Funding
This work was supported by National Institute of Environmental Health Sciences (NIEHS) grants P30ES009089 and 1S10OD020058 and grant 22UB-2308 from the California Breast Cancer, Research Program, grant R01CA138822 from National Cancer Institute and the Breast Cancer Research Foundation. The content is solely the responsibility of the authors and does not, necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Agency for Toxic Substances and Disease Registry, Toxicological profile for polycyclic aromatic carbons, (1995). Agency for Toxic Substances and Disease Registry. [PubMed] [Google Scholar]
- [2].INTERNATIONAL AGENCY FOR RESEARCH ON CANCER, IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, VOLUME 92 Some Non-heterocyclic Polycyclic Aromatic Hydrocarbons and Some Related Exposures, (2010). [PMC free article] [PubMed] [Google Scholar]
- [3].Xia Y, Zhu P, Han Y, Lu C, Wang S, Gu A, Fu G, Zhao R, Song L, Wang X, Urinary metabolites of polycyclic aromatic hydrocarbons in relation to idiopathic male infertility, Hum. Reprod 24 (2009) 1067–1074. 10.1093/humrep/dep006. [DOI] [PubMed] [Google Scholar]
- [4].Nie J, Li J, Cheng L, Li Y, Deng Y, Yan Z, Duan L, Niu Q, Perera F, Tang D, Maternal urinary 2-hydroxynaphthalene and birth outcomes in Taiyuan, China, Environ. Health. 17 (2018) 91. 10.1186/s12940-018-0436-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mumford JL, Li X, Hu F, Lu XB, Chuang JC, Human exposure and dosimetry of polycyclic aromatic hydrocarbons in urine from Xuan Wei, China with high lung cancer mortality associated with exposure to unvented coal smoke, Carcinogenesis. 16 (1995) 3031–3036. 10.1093/carcin/16.12.3031. [DOI] [PubMed] [Google Scholar]
- [6].VanRooij JG, Bodelier-Bade MM, Jongeneelen FJ, Estimation of individual dermal and respiratory uptake of polycyclic aromatic hydrocarbons in 12 coke oven workers, Br. J. Ind. Med 50 (1993) 623–632. 10.1136/oem.50.7.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li Z, Sandau CD, Romanoff LC, Caudill SP, Sjodin A, Needham LL, Patterson DG, Concentration and profile of 22 urinary polycyclic aromatic hydrocarbon metabolites in the US population, Environ. Res 107 (2008) 320–331. 10.1016/j.envres.2008.01.013. [DOI] [PubMed] [Google Scholar]
- [8].Li Z, Romanoff LC, Trinidad DA, Hussain N, Jones RS, Porter EN, Patterson DG, Sjödin A, Measurement of Urinary Monohydroxy Polycyclic Aromatic Hydrocarbons Using Automated Liquid–Liquid Extraction and Gas Chromatography/Isotope Dilution High-Resolution Mass Spectrometry, Anal. Chem 78 (2006) 5744–5751. 10.1021/ac0606094. [DOI] [PubMed] [Google Scholar]
- [9].Romanoff LC, Li Z, Young KJ, Blakely NC, Patterson DG, Sandau CD, Automated solid-phase extraction method for measuring urinary polycyclic aromatic hydrocarbon metabolites in human biomonitoring using isotope-dilution gas chromatography high-resolution mass spectrometry, J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci 835 (2006) 47–54. 10.1016/j.jchromb.2006.03.004. [DOI] [PubMed] [Google Scholar]
- [10].Smith CJ, Walcott CJ, Huang W, Maggio V, Grainger J, Patterson DG, Determination of selected monohydroxy metabolites of 2-, 3- and 4-ring polycyclic aromatic hydrocarbons in urine by solid-phase microextraction and isotope dilution gas chromatography–mass spectrometry, J. Chromatogr. B 778 (2002) 157–164. 10.1016/S0378-4347(01)00456-X. [DOI] [PubMed] [Google Scholar]
- [11].Wang Y, Meng L, Pittman EN, Etheredge A, Hubbard K, Trinidad DA, Kato K, Ye X, Calafat AM, Quantification of urinary mono-hydroxylated metabolites of polycyclic aromatic hydrocarbons by on-line solid phase extraction-high performance liquid chromatography-tandem mass spectrometry, Anal. Bioanal. Chem 409 (2017) 931–937. 10.1007/s00216-016-9933-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Barbeau D, Maître A, Marques M, Highly sensitive routine method for urinary 3-hydroxybenzo[ a ]pyrene quantitation using liquid chromatography-fluorescence detection and automated off-line solid phase extraction, Analyst. 136 (2011) 1183–1191. 10.1039/C0AN00428F. [DOI] [PubMed] [Google Scholar]
- [13].Chetiyanukornkul T, Toriba A, Kameda T, Tang N, Hayakawa K, Simultaneous determination of urinary hydroxylated metabolites of naphthalene, fluorene, phenanthrene, fluoranthene and pyrene as multiple biomarkers of exposure to polycyclic aromatic hydrocarbons, Anal. Bioanal. Chem 386 (2006) 712–718. 10.1007/s00216-006-0628-6. [DOI] [PubMed] [Google Scholar]
- [14].Fan R, Wang D, Ramage R, She J, Fast and Simultaneous Determination of Urinary 8-Hydroxy-2’-deoxyguanosine and Ten Monohydroxylated Polycyclic Aromatic Hydrocarbons by Liquid Chromatography/Tandem Mass Spectrometry, Chem. Res. Toxicol 25 (2012) 491–499. 10.1021/tx200517h. [DOI] [PubMed] [Google Scholar]
- [15].Centers for Disease Control and Prevention, Laboratory Procedure Manual, Monohydroxy-Polycyclic Aromatic Hydrocarbons (OH-PAHs) in Urine, Method 09-OD, (2006). [Google Scholar]
- [16].John EM, Terry MB, Keegan THM, Bradbury AR, Knight JA, Chung WK, Frost CJ, Lilge L, Patrick-Miller L, Schwartz LA, Whittemore AS, Buys SS, Daly MB, Andrulis IL, The LEGACY Girls Study: Growth and development in the context of breast cancer family history, Epidemiol. Camb. Mass 27 (2016) 438–448. 10.1097/EDE.0000000000000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Terry MB, Keegan THM, Houghton LC, Goldberg M, Andrulis IL, Daly MB, Buys SS, Wei Y, Whittemore AS, Protacio A, Bradbury AR, Chung WK, Knight JA, John EM, Pubertal development in girls by breast cancer family history: the LEGACY girls cohort, Breast Cancer Res. 19 (2017) 69. 10.1186/s13058-017-0849-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Onyemauwa F, Rappaport SM, Sobus JR, Gajdošová D, Wu R, Waidyanatha S, Using liquid chromatography–tandem mass spectrometry to quantify monohydroxylated metabolites of polycyclic aromatic hydrocarbons in urine, J. Chromatogr. B 877 (2009) 1117–1125. 10.1016/j.jchromb.2009.02.067. [DOI] [PubMed] [Google Scholar]
- [19].Lu S, Li Y, Zhang J, Zhang T, Liu G, Huang M, Li X, Ruan J, Kannan K, Qiu R, Associations between polycyclic aromatic hydrocarbon (PAH) exposure and oxidative stress in people living near e-waste recycling facilities in China, Environ. Int 94 (2016) 161–169. 10.1016/j.envint.2016.05.021. [DOI] [PubMed] [Google Scholar]
- [20].Fan R, Ramage R, Wang D, Zhou J, She J, Determination of ten monohydroxylated polycyclic aromatic hydrocarbons by liquid–liquid extraction and liquid chromatography/tandem mass spectrometry, Talanta. 93 (2012) 383–391. 10.1016/j.talanta.2012.02.059. [DOI] [PubMed] [Google Scholar]
- [21].Campo L, Rossella F, Fustinoni S, Development of a gas chromatography/mass spectrometry method to quantify several urinary monohydroxy metabolites of polycyclic aromatic hydrocarbons in occupationally exposed subjects, J. Chromatogr. B 875 (2008) 531–540. 10.1016/j.jchromb.2008.10.017. [DOI] [PubMed] [Google Scholar]
- [22].Fan R, Dong Y, Zhang W, Wang Y, Yu Z, Sheng G, Fu J, Fast simultaneous determination of urinary 1-hydroxypyrene and 3-hydroxybenzo[a]pyrene by liquid chromatography–tandem mass spectrometry, J. Chromatogr. B 836 (2006) 92–97. 10.1016/j.jchromb.2006.03.044. [DOI] [PubMed] [Google Scholar]
- [23].Guo Y, Senthilkumar K, Alomirah H, Moon H-B, Minh TB, Mohd MA, Nakata H, Kannan K, Concentrations and Profiles of Urinary Polycyclic Aromatic Hydrocarbon Metabolites (OH-PAHs) in Several Asian Countries, Environ. Sci. Technol 47 (2013) 2932–2938. 10.1021/es3052262. [DOI] [PubMed] [Google Scholar]
- [24].Ramsauer B, Sterz K, Hagedorn H-W, Engl J, Scherer G, McEwan M, Errington G, Shepperd J, Cheung F, A liquid chromatography/tandem mass spectrometry (LC-MS/MS) method for the determination of phenolic polycyclic aromatic hydrocarbons (OH-PAH) in urine of non-smokers and smokers, Anal. Bioanal. Chem 399 (2011) 877–889. 10.1007/s00216-010-4355-7. [DOI] [PubMed] [Google Scholar]