

STROKE STRUCTURED ABSTRACT
Background and Purpose
Clonal hematopoiesis of indeterminate potential (CHIP) is a novel age-related risk factor for cardiovascular disease-related morbidity and mortality. The association of CHIP with risk of incident ischemic stroke was reported previously in an exploratory analysis including a small number of incident stroke cases without replication and lack of stroke sub-phenotyping. The purpose of this study was to discover whether CHIP is a risk factor for ischemic or hemorrhagic stroke.
Methods
We utilized plasma genome sequence data of blood DNA to identify CHIP in 78,752 individuals from 8 prospective cohorts and biobanks. We then assessed the association of CHIP and commonly mutated individual CHIP driver genes (DNMT3A, TET2, ASXL1) with any stroke, ischemic stroke, and hemorrhagic stroke.
Results
CHIP was associated with an increased risk of total stroke (HR= 1.14, 95% CI 1.03–1.27; P=0.01) after adjustment for age, sex, and race. We observed associations with CHIP with risk of hemorrhagic stroke (HR= 1.24, 95% CI 1.01–1.51; P=0.04) and with small vessel ischemic stroke subtypes. In gene-specific association results, TET2 showed the strongest association with total stroke and ischemic stroke, whereas DMNT3A and TET2 were each associated with increased risk of hemorrhagic stroke.
Conclusions
CHIP is associated with an increased risk of stroke, particularly with hemorrhagic and small vessel ischemic stroke. Future studies clarifying the relationship between CHIP and subtypes of stroke are needed.
INTRODUCTION
Clonal hematopoiesis of indeterminate potential (CHIP) is a recently recognized age-related condition defined by clonal expansion of hematopoietic stem cells (HSC) from acquired leukemogenic mutations (typically in DNMT3A, TET2, ASXL1, JAK2, etc) among asymptomatic adults1–5. CHIP can be detected in at least 10–20% of individuals over the age of 70 years using next-generation sequencing of blood DNA. While CHIP is associated with increased risk of hematologic malignancies, CHIP has also been robustly associated with increased coronary heart disease (CHD) risk as well as all-cause and CVD-related mortality3, 6. The association of CHIP with CHD is independent of traditional CVD risk factors such as high blood cholesterol, hypertension, smoking, and diabetes7. Consistently, atherogenic murine models have shown that hematopoietic stem cell deficiency of CHIP genes promotes atherogenesis3, 8, 9. In in vitro systems, monocytes carrying CHIP mutation appear to contribute to acceleration of atherosclerosis through pro-inflammatory mechanisms6 validated through RNA sequencing and biomarker studies in humans6, 10. Murine and human germline genetic analyses aligned with these observations6, 8, 9.
The association of CHIP with risk of incident ischemic stroke was first reported by Jaiswal et al (2014) in an analysis conducted within two cohorts comprising 3,190 persons with 84/3,071 (2.7%) without CHIP developing incident stroke and 12/122 (9.8%) with CHIP developing incident stroke yielded a 2.6-fold age-independent affect2. The ischemic stroke risk appeared to be somewhat greater among persons who had a variant allele fraction of >10%, or at least 10% of circulating blood DNA with a CHIP mutation11–13. This initial report was limited by the relatively small number of incident stroke cases and lack of stroke sub-phenotyping. Moreover, whether CHIP is additionally a risk factor for hemorrhagic stroke, another common type of stroke, is unknown.
Here, we utilized genome sequence data to define CHIP in individuals from 8 prospective cohorts or biobanks with stroke follow-up and sub-phenotyping comprising 86,178 individuals (7,426 stroke cases). We assessed the role of CHIP as a risk factor for all stroke, ischemic stroke, and hemorrhagic stroke. We further assessed CHIP-stroke associations according to commonly mutated CHIP genes (i.e., DNMT3A, TET2, and ASXL1).
MATERIALS AND METHODS
Data Availability
The data that support the findings of this study are available from the corresponding studies and biobanks upon reasonable request. Genomic TOPMed data are available on dbGAP (ARIC, phs001211.v3.p2; CHS, phs001368; FHS, phs000974; JHS, phs000964; MESA, phs001416; WHI, phs001237). UKBB data are available from ukbiobank.ac.uk with an application for qualified researchers. MGBB data are available to qualified researchers by application.
Study participants
The current analysis includes participants from six cohort studies [the Atherosclerosis Risk in Communities (ARIC) study, the Cardiovascular Health Study (CHS), the Framingham Heart Study (FHS), the Jackson Heart Study (JHS), the Multi-Ethnic Study of Atherosclerosis (MESA), the Women’s Health Initiative (WHI)] and two electronic health record-based biobanks [UK Biobank (UKBB) and Mass General Brigham Biobank (MGBB)]. Detailed descriptions of each cohort are provided in the Supplemental Methods. All studies were approved by local Institutional Review Boards and written informed consent was obtained from each participant. This study adheres to the Strengthening the Reporting of Genetic Association Studies (STREGA) extension of the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) checklist for observational studies 14.
We excluded participants who had a known history of stroke at enrollment, incident stroke prior to blood draw, or insufficient CHIP data quality or missing covariates from each participating study. We excluded individuals with a known history of hematologic malignancy or other non-neoplastic clonal disease. We also excluded one of each pair of first-, second-, or third-degree relatives at random from each study. After exclusions, there were a total of 7,426 incident stroke cases and 78,752 controls for analysis. Based on self-reported ancestry, these include White (n=71,820), Black (n=8,667), and Other Race (n=5,715) (Table 1).
Table 1. Baseline Characteristics.
Baseline characteristics of the study population. For continuous variables mean (SD, standard deviation) are displayed. For categorical variables N (%) are displayed. (WHI, Women’s Health Initiative; MESA, Multi-Ethnic Study of Atherosclerosis; JHS, Jackson Heart Study; FHS, Framingham Heart Study; CHS, Cardiovascular Health Study; ARIC, Atherosclerosis Risk In Communities study; MGBB, Mass General Brigham Biobank; UKBB, United Kingdom BioBank; CAD, Coronary Artery Disease; BMI, Body Mass Index; SBP, Systolic Blood Pressure).
| WHI | MESA | JHS | FHS | CHS | ARIC | MGBB | UKBB | |
|---|---|---|---|---|---|---|---|---|
| N | 9683 | 3963 | 1764 | 994 | 2315 | 10355 | 11962 | 45186 |
| Age | 68.9 (6.8) | 61.1 (9.8) | 56.8 (11.4) | 66.4 (12.6) | 73.9 (5.6) | 57.81 (6.0) | 46.5 (14.8) | 56.5 (8.0) |
| Female | 9683 (100) | 2018 (50.9) | 1077 (61.1) | 539 (54.2) | 1297 (56.0) | 5890 (56.9) | 6968 (58.3) | 24656 (54.6) |
| Race | ||||||||
| White | 7988 (82.5) | 1692 (42.7) | 0 (0.0) | 994 (100) | 1889 (81.6) | 7552 (72.9) | 9595 (80.2) | 42110 (93.2) |
| Black | 1195 (12.3) | 875 (22.1) | 1764 (100) | 0 (0.0) | 397 (17.2) | 2783 (26.9) | 717 (6.0) | 936 (2.1) |
| Other | 500 (5.2) | 1396 (35.2) | 0 (0.0) | 0 (0.0) | 29 (1.3) | 0 (0.0) | 1650 (13.8) | 2140 (4.7) |
| Hypertension | 4446 (45.9) | 1531 (41.9) | 1047 (60.6) | 217 (21.9) | 1523 (65.9) | 3765 (36.4) | 1905 (15.9) | 13442 (29.7) |
| Prior Stroke | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Incident Stroke | 4607 (47.6) | 160 (4.0) | 122 (6.9) | 156 (15.7) | 576 (24.9) | 995 (9.6) | 130 (1.1) | 680 (1.5) |
| Current Smoker | 719 (7.4) | 446 (12.2) | 231 (13.2) | 338 (34.1) | 279 (12.1) | 2266 (21.9) | 290 (2.4) | 4050 (9.0) |
| BMI | 28.6 (6.2) | 28.1 (5.2) | 31.6 (7.1) | 25.7 (4.7) | 26.5 (4.5) | 28.19 (5.6) | 28.3 (10.8) | 27.4 (4.78) |
| Follow Up Years | 10.8 (6.4) | 13.5 (2.5) | 12.6 (3.6) | 7.6 (3.5) | 11.3 (7.0) | 20.4 (8.0) | 3.0 (2.0) | 9.9 (2.7) |
CHIP Exposure Definition
For CHS, JHS, MESA, FHS, and WHI, CHIP genotypes were previously determined at the Broad Institute (Cambridge, MA) via deep-coverage whole genome sequencing (WGS) of blood DNA using GATK MuTect215 and hematopathology manual confirmation through the NHLBI TOPMed project (freeze 6) on the basis of pre-specified driver mutations in 74 genes known to promote clonal expansion of hematopoietic stem cells with variant allele frequency (VAF) of >2% as previously described 3, 7. For ARIC, UKBB and MGBB, CHIP was determined at the Broad Institute (Cambridge, MA) via whole exome sequencing (WES) using same genotyping algorithm described above as previously described 6, 7, 16.
CHIP was defined by the presence of pathogenic somatic variants in genes previously implicated in hematologic cancers with a VAF >2% in persons without a known diagnosis of hematologic cancer or other non-neoplastic clonal disease (e.g. myelofibrosis, myelodysplasia). For secondary analyses, VAF >10% was used to define large CHIP and variants grouped by the top genes (i.e., DNMT3A, TET2, and ASXL1) were considered.
Stroke Outcome Definitions
In the six cohort studies, all stroke cases were adjudicated by trained physician adjudicators during follow-up using surveillance, hospitalization records, death certificates and/or International Classification of Disease (ICD) codes to verify stroke cases and time to event or last follow-up using standard algorithms and criteria. Incident stroke was defined as a focal neurological deficit of presumed vascular etiology with a sudden onset and lasting 24 hours or resulting in death. Ischemic stroke was distinguished from hemorrhagic stroke among confirmed strokes when assessment of computed tomographic or magnetic resonance imaging brain scans indicated no sign of cerebral hemorrhage. Ischemic stroke cases in CHS, MESA, and WHI studies were further divided into cardioembolic stroke (CES), large artery stroke (LAS), and small vessel stroke (SVS) according to the Trial of Org 10172 in Acute Stroke Treatment (TOAST) criteria17. Hemorrhagic stroke cases were further divided into intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH). Details are provided in the Supplemental Methods. In UKBB and MGBB a history of prevalent stroke prior to study recruitment was ascertained through a mix of ICD codes and self-report. In both UKBB and MGBB, after study enrollment the identification of stroke cases was based on the 9th and 10th revision of the International Statistical Classification of Diseases and Related Health Problems (ICD) (Supplemental Table I)18. Detailed information on UKBB stroke phenotype curation is available at https://biobank.ndph.ox.ac.uk/showcase/ukb/docs/alg_outcome_stroke.pdf.
Covariates
In the primary analysis, age, sex, and principal components (PCs) 1–10 were used as covariates, and additional covariates were used in sensitivity analyses. Covariates ascertained at the time of blood draw included age, sex, smoking status, prevalent diabetes mellitus, body mass index (BMI), systolic blood pressure (SBP), and self-reported race. Cigarette smoking status was categorized as never, past, and current. Hypertension and diabetes mellitus were either defined by self-reported history of physician diagnosis prior to CHIP determination. BMI (kg/m2) was based upon clinic exams of measured height and weight at the baseline study visit. SBP (mmHg) was measured using standard procedures during baseline clinical exams. In UKBB, history of type 2 diabetes mellitus, were identified by a combination of self-report and ICD codes (ICD-9: 250; ICD-10: E10-E14). SBP was adjusted by adding 15 mmHg for antihypertensive medication users as previously done16, 19.
Statistical methods
Cox proportional hazards model were fitted with adjustment for age, sex and the first 10 principal components of genetic ancestry in the primary model. Schoenfeld residual plots were generated to assess the proportional hazards assumption. We did not observe any pattern with time from the graphical inspection, indicating no violation of proportionality. In WHI, the TOPMed sub-cohort was oversampled for cases of venous thromboembolic events (deep vein thrombosis/pulmonary embolism) and stroke; therefore, inverse probability weighting was used to account for sampling bias and Cox regression was performed using robust standard errors. Using summary results from each study, inverse variance-weighted, fix-effects meta-analysis was used to estimate effect sizes for total stroke, ischemic stroke, hemorrhagic stroke and subtypes. Forest plots were used to summarize effect estimates and confidence intervals of individual study along with pooled results. Sensitivity analyses were performed in the WHI cohort which comprised the majority of cases to adjust for additional covariates including age, smoking status, history of diabetes, history of hypertension and the first 10 principal components of genetic ancestry. Moreover, in WHI we tested the association of CHIP with ICH stratified by age ≤80 years versus >80 years, in a model adjusted for age, race, hypertension, smoking, type 2 diabetes mellitus, systolic blood pressure, and BMI. All statistics were performed using SAS and R version 4.0.2 (https://www.r-project.org). Two-sided p value <0.05 was considered statistically significant.
RESULTS
A total of 86,178 participants with 7,426 (8.6%) incident stroke cases from 8 studies were included in the primary meta-analysis of CHIP and stroke. The mean age of each study ranged from 46.5 to 73.9 (SD between 5.6 and 14.8) years. The overall prevalence of CHIP at baseline was 6.0%. The most common CHIP genes were DNMT3A, TET2, ASXL1 and JAK2 consistent with prior reports. Table 1 compares baseline characteristics for participants with CHIP to those without CHIP. While prevalent stroke cases prior to enrollment were excluded, the total number of incident stroke cases during follow up was 7,426, where WHI contributed 4,607 cases and 5,076 controls; MESA contributed 160 cases and 3,586 controls; JHS contributed 122 cases and 1,642 controls; FHS contributed 156 cases and 838 controls; CHS contributed 576 cases and 1,675 controls; ARIC contributed 995 cases and 9,597 controls; UKBB contributed 680 cases and 44,506 controls; and MGBB contributed 130 cases and 11,832 controls.
CHIP is associated with stroke independently of traditional risk factors
In the fixed-effect meta-analysis, we observed that any CHIP mutation was associated with an increased risk of total stroke (HR= 1.14, 95% CI 1.03–1.27; P=0.01), adjusting for age, race, and sex (Figure 1A). There was no evidence of significant heterogeneity in results across studies. We next performed the CHIP association separately for ischemic and hemorrhagic stroke types (Figure 1B, 1C). Unexpectedly, the risk estimate for CHIP association was numerically greater for hemorrhagic (HR= 1.24, 95% CI 1.01–1.51; P=0.04) than ischemic stroke (HR= 1.11, 95% CI 0.98–1.25; P=0.10), though tests for heterogeneity in these results were negative (p-heterogeneity=0.34). In the main analysis, restricting the definition of CHIP to only individuals with a variant allele fraction >10% (“large CHIP”) did not appreciably alter any of the associations with total stroke (HR 1.18, 95% CI 1.05–1.33; P<0.01), ischemic stroke (HR 1.14, 95% CI 0.99–1.30; P=0.07), or hemorrhagic stroke (HR 1.28, 95% CI 1.03–1.61; P=0.03) (Supplemental Figures IA, IB, IC).
Figure 1: Forest plot of meta-analyzed hazard ratio for the association between CHIP and Stroke.
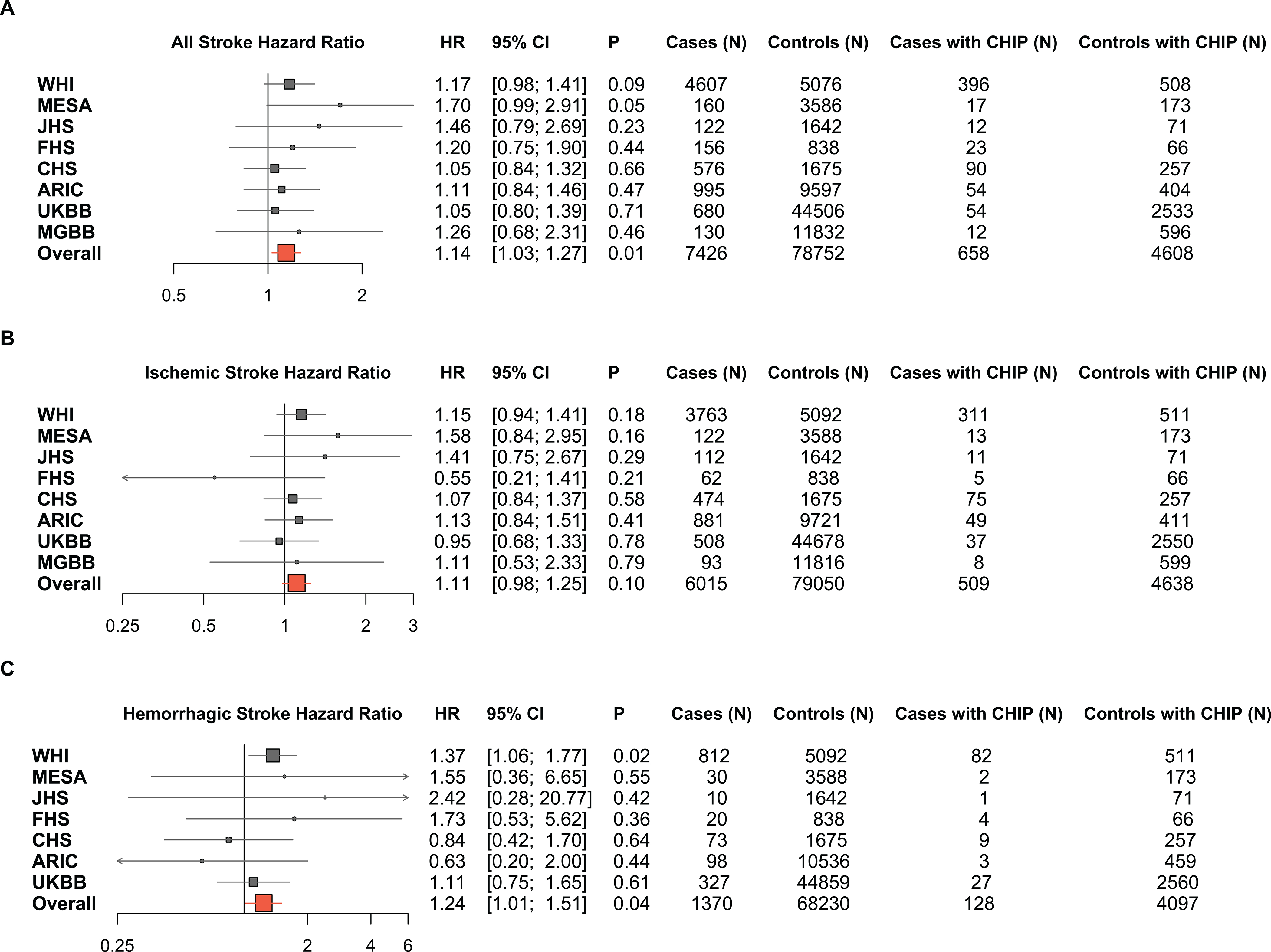
Cox proportional hazards models were fitted, adjusted for age, sex, and the first 10 principal components of genetic ancestry. Here forest plots are used to show the HR, 95% CI and numerical events for each study. (CHIP, Clonal Hematopoiesis of Indeterminate Potential; HR, hazard ratio; CI, confidence interval; WHI, Women’s Health Initiative; MESA, Multi-Ethnic Study of Atherosclerosis; JHS, Jackson Heart Study; FHS, Framingham Heart Study; CHS, Cardiovascular Health Study; ARIC, Atherosclerosis Risk In Communities study; MGBB, Mass General Brigham Biobank; UKBB, United Kingdom Biobank).
We performed sensitivity analyses for any CHIP and “large CHIP” using the WHI cohort, with the largest fraction of cases (i.e., 4,607 of 7,426) to additionally adjust for age, sex, smoking, history of diabetes, history of hypertension and the first 10 principal components of genetic ancestry. In the any CHIP analysis there was no appreciable change in the associations with all stroke (HR 1.23, 95% CI 1.02–1.48; P=0.03), ischemic stroke (HR 1.21, 95% CI 0.98–1.49; P=0.08), or hemorrhagic stroke (HR 1.41, 95% CI 1.09–1.83; P<0.01) with the aforementioned full covariate adjustment, indicating the observed CHIP-stroke associations are independent of traditional stroke risk factors (Table 2).
Table 2: Sensitivity analysis of association between CHIP and Stroke in WHI adjusted for additional covariates.
In the primary analysis age, sex and PC 1–10 were included as covariates. Sensitivity analysis presented here was performed additionally adjusting for diabetes, smoking history and hypertension, which did not explain the association, and demonstrated a stronger association with additional adjustment. (CHIP, Clonal Hematopoiesis of Indeterminate Potential; HR, Hazard Ratio; SE, Standard Error; VAF, Variant Allele; HTN, Hypertension)
| Outcome | CHIP Clone Size | Covariates | HR | beta | SE | P-value |
|---|---|---|---|---|---|---|
|
| ||||||
| Stroke | VAF ≥2% | age, sex, PC1–10 | 1.17 | 0.16 | 0.09 | 0.09 |
| Stroke | VAF ≥2% | age, sex, PC1–10, Diabetes, smoking, HTN | 1.23 | 0.20 | 0.09 | 0.03 |
|
| ||||||
| Stroke | VAF ≥ 10% | age, sex, PC1–10 | 1.12 | 0.11 | 0.10 | 0.25 |
| Stroke | VAF ≥ 10% | age, sex, PC1–10, Diabetes, smoking, HTN | 1.15 | 0.14 | 0.10 | 0.15 |
|
| ||||||
| Ischemic Stroke | VAF ≥2% | age, sex, PC1–10 | 1.15 | 0.14 | 0.11 | 0.18 |
| Ischemic Stroke | VAF ≥2% | age, sex, PC1–10, Diabetes, smoking, HTN | 1.21 | 0.19 | 0.11 | 0.08 |
|
| ||||||
| Ischemic Stroke | VAF ≥ 10% | age, sex, PC1–10 | 1.10 | 0.09 | 0.11 | 0.40 |
| Ischemic Stroke | VAF ≥ 10% | age, sex, PC1–10, Diabetes, smoking, HTN | 1.13 | 0.12 | 0.11 | 0.26 |
|
| ||||||
| Hemorrhagic Stroke | VAF ≥2% | age, sex, PC1–10 | 1.37 | 0.31 | 0.13 | 0.02 |
| Hemorrhagic Stroke | VAF ≥2% | age, sex, PC1–10, Diabetes, smoking, HTN | 1.41 | 0.35 | 0.13 | 0.01 |
|
| ||||||
| Hemorrhagic Stroke | VAF ≥ 10% | age, sex, PC1–10 | 1.30 | 0.26 | 0.14 | 0.07 |
| Hemorrhagic Stroke | VAF ≥ 10% | age, sex, PC1–10, Diabetes, smoking, HTN | 1.32 | 0.28 | 0.14 | 0.05 |
Individual CHIP gene analyses suggest TET2 may be selectively associated with ischemic stroke
Using the WHI cohort sample, we further assessed the association of the most common individual CHIP genes with total, ischemic, or hemorrhagic stroke. CHIP driver mutations were identified in DNMT3A (535 of 45,091; 1.2%), TET2 (212 of 45,091; 0.5%), ASXL1 (64 of 45,091; 0.1%), JAK2 (39 of 45,091; <0.1%), and TP53 (16 of 45,091; <0.1%). In comparing the gene-specific association results, only TET2 had a significant association with total stroke (HR 1.85, p=0.004) (Figure 2; Supplemental Table II). When ischemic and hemorrhagic stroke were analyzed separately, TET2 was associated with increased risk for ischemic stroke (HR 1.93, p=0.006), and the effect sizes for the association of TET2 (HR=1.50, p=0.15) and DMNT3A (HR 1.44, p=0.03) with hemorrhagic stroke were similar.
Figure 2: Forest plot of gene-specific hazard ratios for the association between CHIP and Stroke, amongst the WHI cohort.
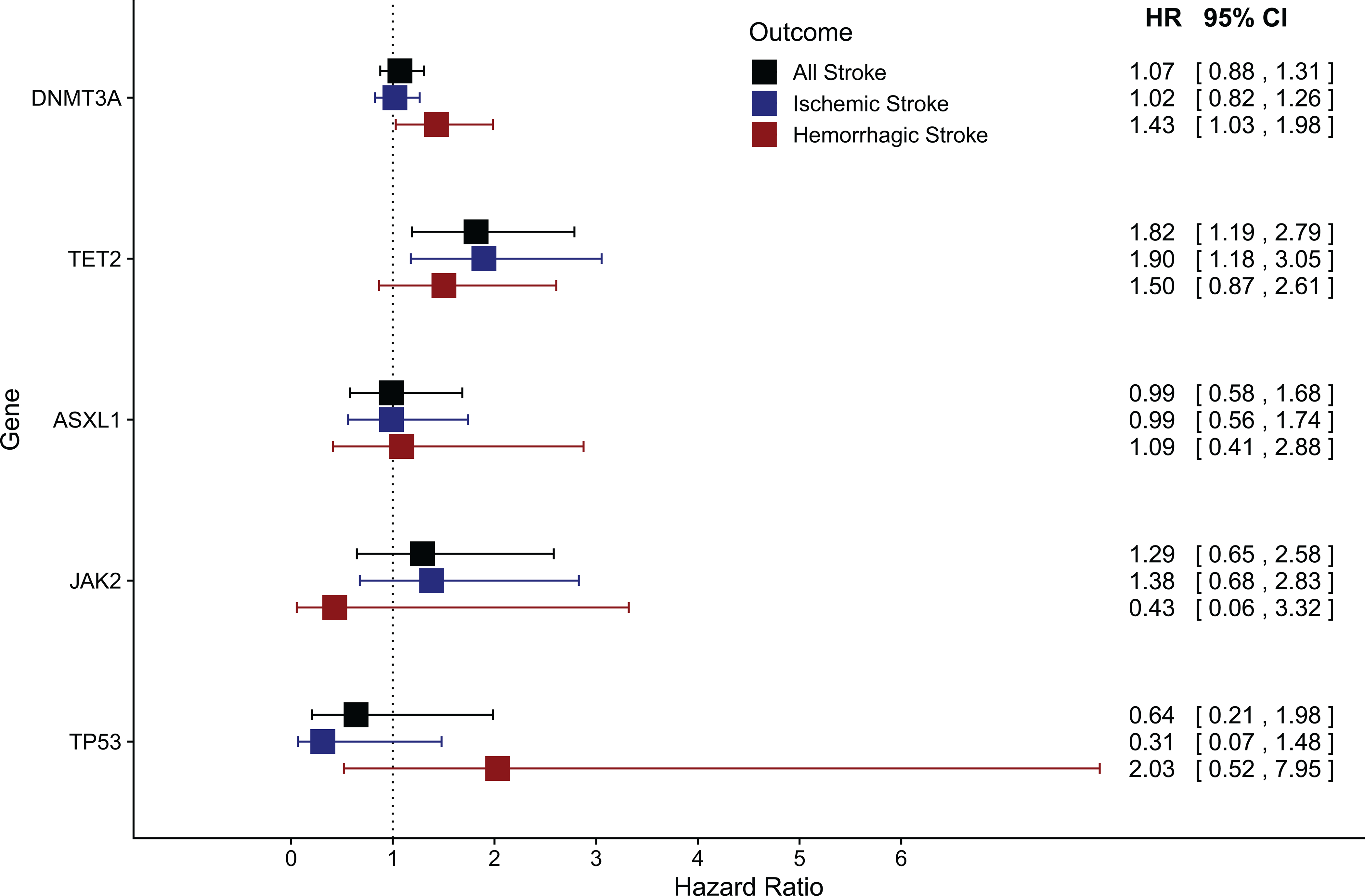
Cox proportional hazards models were fitted, adjusted for age, type 2 diabetes, smoking history, and the first 10 principal components of genetic ancestry. Here forest plots are used to show the HR, 95% CI for each CHIP gene’s association with All Stroke, Ischemic Stroke and Hemorrhagic Stroke. TET2 is found to have a significant association with both overall stroke and ischemic stroke. (CHIP, Clonal Hematopoiesis of Indeterminate Potential; HR, hazard ratio; CI, confidence interval; WHI, Women’s Health Initiative; MESA, Multi Ethnic Study of Atherosclerosis; JHS, Jackson Heart Study; FHS, Framingham Heart Study; CHS, Cardiovascular Health Study; ARIC, Atherosclerosis Risk In Communities study; MGBB, Mass General Brigham Biobank; UKBB, United Kingdom BioBank).
Associations of CHIP according to ischemic and hemorrhagic stroke subtypes
We next assessed the associations of any CHIP with ischemic and hemorrhagic stroke subtypes using information from WHI and MGBB. When ischemic strokes were classified in WHI according to TOAST subtype, CHIP was significantly associated with SVS (HR 1.55, p=0.001) and not with LAS (HR 1.12, p=0.62) or CES (HR 1.05, p=0.68) subtypes (Table 3, Supplemental Figure II). Among hemorrhagic subtypes in WHI, there was a stronger association between CHIP and SAH (HR 1.98, p=0.004) than with ICH (HR 1.31, p=0.063). In WHI, further sensitivity analysis of ICH stratified by age indicated significantly stronger associations among age>80 years (HR 1.84, p=0.01) compared to age≤80 years (HR 1.14, P=0.49), independent of age, race, smoking status, type 2 diabetes mellitus, SBP, and BMI (Supplemental Table III). In the MGBB cohort, there was a higher prevalence of cerebral aneurysms in those with CHIP (19/657 (2.9%)) than those without CHIP (193/11808 (1.6%)) (Chi Sq p=0.001). There was also a higher prevalence of non-traumatic SAH among those with CHIP (15/657 (2.3%)) than those without CHIP (116/11808 (1.0%)) (p=0.003), and a higher incidence of SAH among those with CHIP (5/657 (0.76%)) than those without CHIP (27/11807 (2.3%)) (p=0.02).
Table 3: Association of CHIP with subtypes of stroke within the WHI.
The Trial of Org 10172 in Acute Stroke Treatment (TOAST) defined subtypes of ischemic and hemorrhagic stroke, which are used in standard practice. Here, additional analysis was done within the WHI cohort to investigate the relationship between CHIP and stroke subtypes. CHIP was not associated with increased risk of LAS or CES. CHIP was, however, significantly associated with an increased hazard ratio for SVS Ischemic Stroke (P=0.001) and SAH Hemorrhagic Stroke (P=0.004), as well as a relationship with ICH Hemorrhagic Stroke approaching significance (P=0.06). (CHIP, Clonal Hematopoiesis of Indeterminate Potential; WHI, Women’s Health Initiative; TOAST, Trial of Org 10172 in Acute Stroke Treatment; LAS, Large Artery Stroke; CES, Cardioembolic Stroke; SVS, Small Vessel Stroke; SAH, Subarachnoid Hemorrhage; ICH, intracerebral hemorrhage; SE, standard error).
| Outcome | Total (n) | Event (n) | Hazard Ratio | Beta | SE | P value |
|---|---|---|---|---|---|---|
| All Stroke | 9711 | 4588 | 1.23 | 0.21 | 0.07 | 0.006 |
| Ischemic Stroke | 8875 | 3752 | 1.19 | 0.17 | 0.08 | 0.033 |
| Hemorrhagic Stroke | 5935 | 812 | 1.44 | 0.37 | 0.13 | 0.004 |
| LAS Ischemic Stroke | 5396 | 273 | 1.12 | 0.11 | 0.23 | 0.618 |
| CES Ischemic Stroke | 6282 | 1159 | 1.05 | 0.05 | 0.12 | 0.684 |
| SVS Ischemic Stroke | 5827 | 704 | 1.55 | 0.44 | 0.13 | 0.001 |
| SAH Hemorrhagic Stroke | 5302 | 179 | 1.98 | 0.68 | 0.24 | 0.004 |
| ICH Hemorrhagic Stroke | 5737 | 614 | 1.31 | 0.27 | 0.15 | 0.063 |
DISCUSSION:
CHIP, a recently identified risk factor for cardiovascular disease, is associated with a 14% increased odds of incident stroke when analyzed across 8 cohorts and meta-analyzed after adjustment for age, sex, and ancestry. This relationship was primarily driven by a 24% increased odds of hemorrhagic stroke, particularly SAH. Unselected subtypes of ischemic stroke were not associated with CHIP, and in further analyses of ischemic stroke subtypes in the WHI cohort, CHIP was more strongly associated with small vessel stroke (SVS) than with large artery stroke (LAS) or cardioembolic stroke (CES) subtypes. To contextualize these findings, some discussion of the vascular mechanisms of stroke is warranted.
Hemorrhagic stroke is classified as either ICH or SAH. The most common cause of ICH is small vessel hypertensive disease which leads to lipohyalinosis followed by the formation of small microaneurysms (Charcot-Bouchard aneurysms) that subsequently rupture12, 20. In sensitivity analyses in the WHI, the addition of hypertension as a covariate did not account for the association between CHIP and hemorrhagic stroke, and indeed strengthened the association. An important cause of ICH is cerebral small vessel disease (characterized by subcortical lacunar infarction, white matter lesions, and cerebral microbleeds), which is pathogenetically related to the ischemic stroke subtype SVS. Indeed, CHIP was found to have similar effect sizes for ICH and SVS in the WHI, indicating that CHIP may contribute to ICH by way of SVS21. Furthermore, age-stratified analyses in WHI for ICH identified that the association was stronger among older individuals age>80 years, independent of systolic blood pressure and other cardiovascular risk factors (Supplemental Table III). In older age groups, cerebral amyloid angiopathy is an increasingly common cause of ICH22. Possible mechanisms linking CHIP to ICH include inflammatory signaling pathways linked to aneurysm formation, accelerated arteriosclerosis contributing to vessel fragility in individuals with preexisting age-related risk factors including cerebral amyloid angiopathy13, 22–29.
Non-traumatic SAH is most closely related to saccular intracerebral aneurysm (IA) formation and rupture. The formation of IAs has been linked to inflammatory cytokine activation, recruitment of immune cells, and macrophage activation particularly via matrix metalloproteinases (MMPs) 13, 27. Neuroinflammation also contributes to brain injury and cerebral vasospasm following IA rupture. 13 Macrophage infiltration has been identified as one of the most prominent features of unstable, rupture-prone IAs after pathological analysis using resected aneurysm ruptured and unruptured aneurysm samples 23. The association between CHIP and aneurysm formation has not yet been reported to our knowledge, but CHIP is known to be closely tied to dysregulated inflammation, macrophage activation and infiltration, and is thought to exert its adverse cardiovascular effects primarily through IL1B pathway and NLRP3 inflammasome4, 6, 30, 31. The pathogenesis of an analogous disease state, abdominal aortic aneurysms (AAA), has been shown to involve mast cells in mice which release the proinflammatory cytokines interleukin-6 (IL-6) and interferon-γ (IFN-γ), which may induce aortic SMC apoptosis, matrix-degrading protease expression, and vascular wall remodeling 27. Similar mast cell activation with upregulation of NFKB and MMPs has been observed in IAs 24. Exploratory analysis within the MGBB cohort demonstrated a higher frequency of cerebral aneurysm among those with CHIP. Whether CHIP plays a role in IA formation needs further investigation.
The association of CHIP with incident ischemic stroke was weaker than with hemorrhagic stroke. In exploratory analyses within the WHI cohort (the largest cohort with the most events for the present study), CHIP was found to be associated with 19% increased risk of all ischemic stroke, driven by a 55% increased risk of SVS. Ischemic stroke etiologies are typically divided into embolism from the heart, large extracranial or intracranial embolism or hemodynamic failure, and small vessel occlusion (i.e., lacunar infarcts). These align with the TOAST classifications of CES, LAS, and SVS, respectively. CES is typically related to atrial fibrillation and intracardiac thrombus which has not been shown to be associated with CHIP17, 32. On the other hand, the apparent lack of association of CHIP with LAS ischemic subtype was unexpected. The epidemiology and pathophysiology of LAS subtype is most closely related to ischemic heart disease, both of which involve atherosclerosis and thrombosis driven by traditional CVD risk factors and vascular inflammation, a hallmark of CHIP11. However, compared to ischemic heart disease and coronary atherosclerosis, there is greater uncertainty in stroke subtype classification and greater mechanistic heterogeneity (e.g., ischemic strokes with extracranial carotid stenosis can be due to hemodynamic failure). Thus, the extent of atherosclerotic burden (e.g., carotid stenosis) and subsequent stroke risk are not as tightly correlated in large vessel cerebrovascular disease33, 34 compared to coronary atherosclerosis and risk of MI. Finally, as we note above, the apparent association of CHIP with both SVS and ICH is intriguing, given the pathophysiologic relationships between cerebral small vessel disease and hemorrhage. Overall, these data suggest a relationship between CHIP, SVS, and ICH that requires further characterization. Given the age-related prevalence of CHIP, further assessment of the association of CHIP with white matter intensities on brain MRI and dementia or cognitive impairment in older adults may be warranted.
Our study has several limitations. Firstly, the heterogeneity of study protocols, recruitment and adjudication of patients and clinical events is challenging to harmonize. We attempted through collaboration and rigorous attention to outcome definitions to ensure standard treatment of subjects and events but acknowledge some heterogeneity may persist. However, the inclusion of multiple datasets with diverse individuals improves generalizability of the study findings and simultaneously adds to the strength of the study. Secondly, though these data were prospectively ascertained, they are observational data and thus cannot provide strong causal evidence. Additionally, CHIP was ascertained at a single time point. Having CHIP at multiple time points would allow for stronger evidence linking CHIP and risk of stroke. Lastly, our results were unexpected in linking CHIP to both hemorrhagic and ischemic stroke (particularly to small-vessel disease). Mechanistic links have not yet been robustly investigated that explain this finding in full.
Summary and Conclusions
Our findings identify that CHIP is associated with an increased odds of stroke, and the mechanism of stroke appears to be an important modifier of this relationship. Though CHIP was not found to be associated with ischemic stroke overall, it was found to be associated with small vessel ischemic stroke, with stronger effects for TET2 CHIP. The finding that CHIP was consistently and strongly associated with hemorrhagic stroke raises questions that require further investigation regarding the role of CHIP in vascular fragility and the formation of saccular intracranial aneurysms.
Supplementary Material
Acknowledgements & Sources of Funding
Romit Bhattacharya is supported by the John S. LaDue Memorial Fellowship in Cardiovascular Medicine at Harvard Medical School and this work was additionally supported by NIH Training Grant T32HL007208. Seyedeh M Zekavat is supported by the NIH National Heart, Lung, and Blood Institute (1F30HL149180-01) and the NIH Medical Scientist Training Program Training Grant (T32GM136651). Myriam Fornage is supported by NS114045 and NS100605. Laura M Raffield is supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant KL2TR002490, and by T32HL129982. Alexander G. Bick is supported by a Burroughs Wellcome Foundation career award for medical scientists, the EvansMDS Foundation, RUNX1 Research program, and an NIH Director’s Early Independence Award from the National Institute of Health Common Fund (DP5 OD029586). Bing Yu is supported by NIH grant R01 HL148050. Abhishek Niroula is supported by Knut and Alice Wallenberg Foundation (KAW2017.0436). Christopher J. Gibson is supported by the Damon Runyon Cancer Research Foundation. Bruce M. Psaty, Joshua C. Bis, Russel P. Tracy and W.T. Longstreth note their support via contracts for the Cardiovascular Health Study (noted below). Jerome I. Rotter is supported by TOPMed and MESA funding detailed below. Adolfo Correa is supported by HHSN268201800010I, R01 R01 HL143224, R01 HL142599, R01 AG066134, R01 HL143295, R01 HL146636, R01 AG0624, CC10310/SP13991, AWD-000969, R01 AG067513, R01 HL150170-01A, R01 HL150170-01A1. Sudha Seshadri is supported by R01s NS017950, AG059421, RF1AG059421, U01 AG052409 and AG066546. Andrew D. Johnson is supported by NHLBI Intramural Research funding. Kathleen M. Hayden is supported by NIH grant R01 HL148565; R01 AG058969; NHLBI-WH-11-10; and HHSN271201700002C. Christie M. Ballantyne is supported by NIH grant R01 HL148050. Siddhartha Jaiswal is supported by DP2-HL157540, R01-HL148565. Charles Kooperberg is supported by NIH grant R01 HL148565 and R01 HL136574. JoAnn E. Manson is supported by R01 HL034594, R01 AT011729, HHSN268201100001C, and Mars Edge. Eric A. Whitsel is supported by NIH grant R01-HL148565 and contract 75N92019R0031. Benjamin L. Ebert is supported by the Howard Hughes Medical Institute (HHMI). Alexander P. Reiner is supported by NIH grant R01 HL148565 and R01 HL136574. Pradeep Natarajan is supported by NIH grants R01 HL148050, R01 HL151283, R01 HL148565. Pradeep Natarajan is additionally supported by Fondation Leducq TNE-18CVD04.
The UK Biobank analyses were conducted using application 7089. Building on GWAS for NHLBI-diseases: the U.S. CHARGE consortium” was provided by the NIH through the American Recovery and Reinvestment Act of 2009 (ARRA) (5RC2HL102419). Data for “Building on GWAS for NHLBI-diseases: the U.S. CHARGE consortium” was provided by Eric Boerwinkle on behalf of the Atherosclerosis Risk in Communities (ARIC) Study, L. Adrienne Cupples, principal investigator for the Framingham Heart Study, and Bruce Psaty, principal investigator for the Cardiovascular Health Study. Sequencing was carried out at the Baylor College of Medicine Human Genome Sequencing Center and supported by the National Human Genome Research Institute grants U54 HG003273 and UM1 HG008898.
Whole genome sequencing (WGS) for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). WGS for “NHLBI TOPMed: Multi-Ethnic Study of Atherosclerosis (MESA)” (phs001416.v1.p1) was performed at the Broad Institute of MIT and Harvard (3U54HG003067-13S1). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1, contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity QC, and general study coordination, were provided by the TOPMed Data Coordinating Center (3R01HL-120393; U01HL-120393; contract HHSN268180001I). The MESA project is supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420. Also supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. Infrastructure for the CHARGE Consortium is supported in part by the National Heart, Lung, and Blood Institute (NHLBI) grant R01HL105756.
The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts 75N92021D00001, 75N92021D00002, 75N92021D00003, 75N92021D00004, 75N92021D00005.
The Atherosclerosis Risk in Communities study has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services (contract numbers HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I and HSN268201700005I). The authors thank the staff and participants of the ARIC study for their important contributions.
The Framingham Heart Study is conducted and supported by the NHLBI in collaboration with Boston University (Contract Nos. HHSN26820150001I, 75N92019D00031 and HC 25195), and its contract with Affymetrix, Inc., for genome-wide genotyping services (Contract No. N02-HL-6-4278), for quality control by Framingham Heart Study investigators using genotypes in the SNP Health Association Resource (SHARe) project. A portion of this research was conducted using the Linux Cluster for Genetic Analysis (LinGA) computing resources at Boston University Medical Campus. Ascertainment of phenotypic data in the FHS was funded by AG054076, NS 017950, AG049607
Cardiovascular Health Study: This research was supported by contracts HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, 75N92021D00006, and grants U01HL080295 and U01HL130114 from the National Heart, Lung, and Blood Institute (NHLBI), with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org.
The Jackson Heart Study (JHS) is supported and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I) and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I and HHSN268201800012I) contracts from the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute on Minority Health and Health Disparities (NIMHD). The authors also wish to thank the staffs and participants of the JHS.
Conflicts of Interest / Disclosures
R.B. reports consulting fees from Casana Care Inc, unrelated to present work. A.G.P. serves as a scientific advisor to Foresite Labs, reports stock holdings in TenSixteenBio and compensation from TenSixteenBio. A.N. reports grants from Knut och Alice Wallenbergs Stiftelse. B.M.P. serves on the Yale Open Data Access Project funded by Johnson & Johnson. S.J. has received consulting fees from AVRO Bio, Genentech, Novartis, and Foresite Labs. B.L.E. has received research funding from Celgene, Deerfield, and Novartis and consulting fees from GRAIL. He serves on the scientific advisory boards and holds equity in Skyhawk Therapeutics, Exo Therapeutics, and Neomorph. P.N. reports prior grants from Amgen, Apple, AstraZeneca, Boston Scientific, and Novartis, personal fees from Apple, AstraZeneca, Genentech, Blackstone Life Sciences, Foresite Labs, Novartis and a patent issued for helical synthetic peptides that stimulate cellular cholesterol efflux licensed to Artery Therapeutics, and spousal employment at Vertex, unrelated to the present work.
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
Non-standard Abbreviations and Acronyms
- ARIC
Atherosclerosis Risk In Communities Study
- BMI
Body Mass Index
- CAD
Coronary Artery Disease
- CES
Cardioembolic Stroke
- CHD
Coronary Heart Disease
- CHIP
Clonal Hematopoiesis of Indeterminate Potential
- CHS
Cardiovascular Health Study
- CVD
Cardiovascular Disease
- DM
Diabetes Mellitus
- FHS
Framingham Heart Study
- HSC
Hematopoietic Stem Cell
- HTN
Hypertension
- IA
Intracerebral aneurysm
- ICH
Intracerebral hemorrhage
- JHS
Jackson Heart Study
- LAS
Large artery stroke
- MESA
Multi-Ethnic Study of Atherosclerosis
- MGBB
Mass General Brigham Biobank
- SAH
subarachnoid hemorrhage
- SVS
Small vessel stroke
- T2DM
Type 2 Diabetes Mellitus
- TOAST
Trial of Org 10172 in Acute Stroke Treatment
- UKBB
United Kingdom Biobank
- WHI
Women’s Health Initiative
Footnotes
Citations:
- 1.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Natarajan P, Jaiswal S, Kathiresan S. Clonal hematopoiesis: Somatic mutations in blood cells and atherosclerosis. Circ Genom Precis Med. 2018;11:e001926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bick AG, Pirruccello JP, Griffin GK, Gupta N, Gabriel S, Saleheen D, Libby P, Kathiresan S, Natarajan P. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation. 2020;141:124–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, Szeto MD, Liao X, Leventhal MJ, Nasser J, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, Liu W, Thomas DG, Hajebrahimi MA, Pircher J, et al. The aim2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592:296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. Clonal hematopoiesis associated with tet2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abplanalp WT, Mas-Peiro S, Cremer S, John D, Dimmeler S, Zeiher AM. Association of clonal hematopoiesis of indeterminate potential with inflammatory gene expression in patients with severe degenerative aortic valve stenosis or chronic postischemic heart failure. JAMA Cardiol. 2020;5:1170–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: Mechanisms in search of treatments. Neuron. 2010;67:181–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shuaib A, Hachinski VC. Mechanisms and management of stroke in the elderly. Cmaj. 1991;145:433–443 [PMC free article] [PubMed] [Google Scholar]
- 13.Yang SJ, Shao GF, Chen JL, Gong J. The nlrp3 inflammasome: An important driver of neuroinflammation in hemorrhagic stroke. Cell Mol Neurobiol. 2018;38:595–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Little J, Higgins JP, Ioannidis JP, Moher D, Gagnon F, von Elm E, Khoury MJ, Cohen B, Davey-Smith G, Grimshaw J, et al. Strengthening the reporting of genetic association studies (strega): An extension of the strobe statement. PLoS Med. 2009;6:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu B, Roberts MB, Raffield LM, Zekavat SM, Nguyen NQH, Biggs ML, Brown MR, Griffin G, Desai P, Correa A, et al. Supplemental association of clonal hematopoiesis with incident heart failure. J Am Coll Cardiol. 2021;78:42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams HP Jr., Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, Marsh EE 3rd. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. Toast. Trial of org 10172 in acute stroke treatment. Stroke. 1993;24:35–41 [DOI] [PubMed] [Google Scholar]
- 18.Kokotailo RA, Hill MD. Coding of stroke and stroke risk factors using international classification of diseases, revisions 9 and 10. Stroke. 2005;36:1776–1781 [DOI] [PubMed] [Google Scholar]
- 19.Zekavat SM, Aragam K, Emdin C, Khera AV, Klarin D, Zhao H, Natarajan P. Genetic association of finger photoplethysmography-derived arterial stiffness index with blood pressure and coronary artery disease. Arterioscler Thromb Vasc Biol. 2019;39:1253–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sierra C, Coca A, Schiffrin EL. Vascular mechanisms in the pathogenesis of stroke. Curr Hypertens Rep. 2011;13:200–207 [DOI] [PubMed] [Google Scholar]
- 21.Charidimou A, Pantoni L, Love S. The concept of sporadic cerebral small vessel disease: A road map on key definitions and current concepts. Int J Stroke. 2016;11:6–18 [DOI] [PubMed] [Google Scholar]
- 22.Mehndiratta P, Manjila S, Ostergard T, Eisele S, Cohen ML, Sila C, Selman WR. Cerebral amyloid angiopathy-associated intracerebral hemorrhage: Pathology and management. Neurosurg Focus. 2012;32:E7. [DOI] [PubMed] [Google Scholar]
- 23.Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, Jääskeläinen J. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: Histological analysis of 24 unruptured and 42 ruptured cases. Stroke. 2004;35:2287–2293 [DOI] [PubMed] [Google Scholar]
- 24.Ishibashi R, Aoki T, Nishimura M, Hashimoto N, Miyamoto S. Contribution of mast cells to cerebral aneurysm formation. Curr Neurovasc Res. 2010;7:113–124 [DOI] [PubMed] [Google Scholar]
- 25.Jayaraman T, Berenstein V, Li X, Mayer J, Silane M, Shin YS, Niimi Y, Kiliç T, Gunel M, Berenstein A. Tumor necrosis factor alpha is a key modulator of inflammation in cerebral aneurysms. Neurosurgery. 2005;57:558–564; discussion 558–564 [DOI] [PubMed] [Google Scholar]
- 26.Starke RM, Raper DM, Ding D, Chalouhi N, Owens GK, Hasan DM, Medel R, Dumont AS. Tumor necrosis factor-α modulates cerebral aneurysm formation and rupture. Transl Stroke Res. 2014;5:269–277 [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Wei L, Lu H, Zhu Y. Roles of inflammation in the natural history of intracranial saccular aneurysms. J Neurol Sci. 2021;424:117294. [DOI] [PubMed] [Google Scholar]
- 28.Young AM, Karri SK, You W, Ogilvy CS. Specific tnf-alpha inhibition in cerebral aneurysm formation and subarachnoid hemorrhage. Curr Drug Saf. 2012;7:190–196 [DOI] [PubMed] [Google Scholar]
- 29.Carrillo-Jimenez A, Deniz O, Niklison-Chirou MV, Ruiz R, Bezerra-Salomao K, Stratoulias V, Amouroux R, Yip PK, Vilalta A, Cheray M, et al. Tet2 regulates the neuroinflammatory response in microglia. Cell Rep. 2019;29:697–713 e698 [DOI] [PubMed] [Google Scholar]
- 30.Khetarpal SA, Qamar A, Bick AG, Fuster JJ, Kathiresan S, Jaiswal S, Natarajan P. Clonal hematopoiesis of indeterminate potential reshapes age-related cvd: Jacc review topic of the week. J Am Coll Cardiol. 2019;74:578–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Libby P, Sidlow R, Lin AE, Gupta D, Jones LW, Moslehi J, Zeiher A, Jaiswal S, Schulz C, Blankstein R, et al. Clonal hematopoiesis: Crossroads of aging, cardiovascular disease, and cancer: Jacc review topic of the week. J Am Coll Cardiol. 2019;74:567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolominsky-Rabas PL, Weber M, Gefeller O, Neundoerfer B, Heuschmann PU. Epidemiology of ischemic stroke subtypes according to toast criteria: Incidence, recurrence, and long-term survival in ischemic stroke subtypes: A population-based study. Stroke. 2001;32:2735–2740 [DOI] [PubMed] [Google Scholar]
- 33.Brott TG, Halperin JL, Abbara S, Bacharach JM, Barr JD, Bush RL, Cates CU, Creager MA, Fowler SB, Friday G, et al. 2011 asa/accf/aha/aann/aans/acr/asnr/cns/saip/scai/sir/snis/svm/svs guideline on the management of patients with extracranial carotid and vertebral artery disease: Executive summary: A report of the american college of cardiology foundation/american heart association task force on practice guidelines, and the american stroke association, american association of neuroscience nurses, american association of neurological surgeons, american college of radiology, american society of neuroradiology, congress of neurological surgeons, society of atherosclerosis imaging and prevention, society for cardiovascular angiography and interventions, society of interventional radiology, society of neurointerventional surgery, society for vascular medicine, and society for vascular surgery. Developed in collaboration with the american academy of neurology and society of cardiovascular computed tomography. Catheter Cardiovasc Interv. 2013;81:E76–123 [DOI] [PubMed] [Google Scholar]
- 34.Meschia JF, Bushnell C, Boden-Albala B, Braun LT, Bravata DM, Chaturvedi S, Creager MA, Eckel RH, Elkind MS, Fornage M, et al. Guidelines for the primary prevention of stroke: A statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2014;45:3754–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding studies and biobanks upon reasonable request. Genomic TOPMed data are available on dbGAP (ARIC, phs001211.v3.p2; CHS, phs001368; FHS, phs000974; JHS, phs000964; MESA, phs001416; WHI, phs001237). UKBB data are available from ukbiobank.ac.uk with an application for qualified researchers. MGBB data are available to qualified researchers by application.