

Abstract
Objective
The purpose of this single-center, randomized, open, two-period, two-sequence crossover, single-dose administration, bioequivalence research was to evaluate the bioequivalence and safety of the generic formulations of metformin hydrochloride sustained-release (MH-SR) 500 mg tablets (test preparation [T]: Yuantang® SR) and the original formulation (reference preparation [R]: Glucophage® XR) in 36 healthy Chinese volunteers under postprandial conditions.
Methods
Subjects received 500 mg T/R in each period, with a 7-day washout period. Venous blood samples of 4 mL each were collected from each subject 19 times spanning predose (0 h) to 36 h postdose. The metformin concentration in deproteinized plasma was determined by high-performance liquid chromatography–tandem mass spectrometry. Bioequivalence (80.00–125.00%) was assessed by adjusted geometric mean ratios (GMRs) and two-sided 90% confidence intervals (CIs) of the area under the curve (AUC) and maximum concentration (Cmax) for each component. SAS 9.4 software was used for statistical analysis and Phoenix WinNonlin software v7 was used to analyze the pharmacokinetic parameters.
Results
Thirty-four volunteers completed the clinical study. The 90% CIs (96.12–105.44% for AUC from time zero to the time of the last measurable concentration [AUCt], 96.22–105.54% for AUC extrapolated from time zero to infinity [AUC∞], and 98.42–105.00% for Cmax) of T/R adjusted GMRs were within the bioequivalence acceptance range of 80.00–125.00%, indicating that they are bioequivalent. No serious adverse events occurred in this study, indicating that the two formulations were effective and well tolerated.
Conclusions
Yuantang® SR was confirmed to be a well tolerated and bioequivalent alternative to Glucophage® XR when taken under postprandial conditions in healthy Chinese volunteers. The Clinical Trials Registry Platform used for this study was http://www.chinadrugtrials.org.cn/clinicaltrials.searchlistdetail.dhtml. The trial registration numbers (TRNs) and dates of registrations were CTR20180476 (19 April 2018) for this clinical trial and CTR20171595 (11 January 2018) for the pilot trial.
Key Points
| Metformin hydrochloride (MH) is a first-line drug for the treatment of diabetes mellitus; however, because of the large number of diabetic patients, the original MH-sustained release (SR) formulation cannot meet market demand. For developing countries, original metformin is expensive. |
| After the bioequivalence and safety of the generic MH-SR formulation (Yuantang® SR, 500 mg/tablet) and the brand name (Glucophage® extended release [XR], 500 mg/tablet) were confirmed under fasting conditions, we conducted a single-center, randomized, open, two-period, two-way crossover, bioequivalence trial in 36 Chinese adult healthy volunteers under postprandial conditions. |
| The conclusions that Yuantang® SR (500 mg/tablet), manufactured by Guangdong Sinocorp Pharmaceutical Co., Ltd, China, was bioequivalent and well tolerated to Glucophage® SR (500 mg/tablet), manufactured by Merck Serono limited company, UK, provides a good choice for clinicians and patients. |
| Generic MH-SR reduces the costs for clinical supplies of brand-name MH-SR and alleviates the contradiction between supply and demand due to insufficient supply in the original MH-SR market. |
Introduction
Diabetes mellitus (DM), a group of metabolic disorders characterized by hyperglycemia and associated with major complications, including diabetic neuropathy, retinopathy, and cardiovascular disease, is one of the most serious and common diseases worldwide [1, 2]. Approximately 90% of total DM cases are type 2, which has been treated with metformin hydrochloride (MH) (Fig. 1), a herb-derived biguanide oral antihyperglycemic agent, since the 1950s [3]. MH is currently the first-line pharmacologic treatment for type 2 DM and is prescribed either alone or in combination with insulin or other hypoglycemic therapies [4].
Fig. 1.
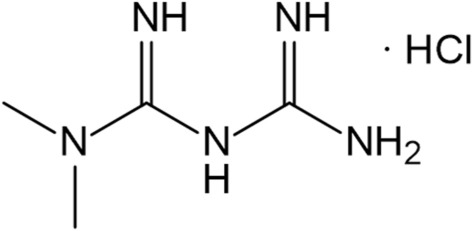
Chemical structure of metformin hydrochloride
Unfortunately, MH tablets have low bioavailability (50–60%) and a short half-life, leading to the development of sustained-release forms of the drug [5–11]. MH sustained-release (MH-SR) tablets overcame the shortcomings of the original tablets and have been widely used in clinical applications. To address clinical shortages and reduce the costs associated with brand-name drugs, generic formulations have been developed.
Owing to the growing market for generic drugs, the Chinese government has strengthened the quality requirements for generic drugs while encouraging their use. At the 2017 International Council for Harmonization (ICH) meeting in Montreal, Canada, the ICH Assembly approved the inclusion of the China Food and Drug Administration (CFDA) as a new regulatory member and the Pharmaceutical Inspection Co-operation Scheme (PIC/S) as a new observer [12]. According to CFDA requirements, any generic drugs that were approved before adopting the new regulatory measures for chemical drugs must be re-evaluated for comparable quality and efficacy to the original drug [13]. Therefore, MH-SR (Yuantang® SR, 500 mg/tablet), produced by Guangdong Sinocorp Pharmaceutical Co., Ltd, China, must be reassessed for quality and efficacy compared with the original drug. According to the CFDA catalog of reference preparations for generic drugs, Glucophage® XR (MH-SR, 500 mg/tablet), manufactured and marketed by Merck Serono Co., Ltd, UK, is designated as the reference preparation for bioequivalence. After the bioequivalence and safety of the generic MH-SR formulations (Yuantang® SR, 500 mg/tablet) and the brand name (Glucophage® XR, 500 mg/tablet) were confirmed under fasting conditions, they were assessed in this study under high-fat meal conditions.
Material and Methods
Study Design
A single-center, randomized, open, two-period, two-sequence crossover, single-dose administration, bioequivalence trial of two MH-SR brands (test preparation [T]: Yuantang® SR; and reference preparation [R]: Glucophage® XR) was conducted on 36 healthy Chinese subjects under postprandial conditions from 17 April to 29 May 2018. The clinical trial [14] was completed at the Phase I Clinical Trial Center of Beijing Shijitan Hospital, affiliated with Capital Medical University, China. The results of this study will be used in an application for a listing qualification certificate from the Chinese Center for Drug Evaluation.
The protocol and all documents were reviewed and approved by the Institutional Review Board of Beijing Shijitan Hospital on 16 October 2017 (registration number: 2017Y123) (http://www.chinadrugtrials.org.cn/clinicaltrials.searchlistdetail.dhtml). The clinical trial was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, Good Laboratory Practice, and Pharmaceutical Administration Law. The nature and purpose of this study were explained to all participants prior to obtaining informed consent.
Study Products
Test Product: MH-SR tablets (generic drug)
Trade name:Yuantang® SR
Specification: 500 mg/tablet
Batch No.: 1171115010, expiry 11/2019
Manufacturer: Guangdong Sinocorp Pharmaceutical Co., Ltd, China
Provider: Guangdong Sinocorp Pharmaceutical Co., Ltd, China
Reference Product: MH extended-release tablets (brand-name drug)
Trade name:Glucophage® XR
Specification: 500 mg/tablet
Batch No.: Y02494, expiry 12/2019
Manufacturer: Merck Serono Co., Ltd, UK
Provider: Guangdong Sinocorp Pharmaceutical Co., Ltd, China
Sample Size Calculation Using Pass Software
The minimum sample size required for equivalence tests for the ratio of two geometric means in a 2 × 2 crossover design was calculated using PASS 15 (Power Analysis and Sample Size System) software (NCSS, LLC, Kaysville, UT, USA). The value of power, the probability of rejecting the null hypothesis when it was false, was set to 80%, and the value of alpha, the probably of a type I error that was rejecting the null hypothesis of non-equivalence when in fact the groups were equivalent, was set to 5%. The lower and upper equivalence limit for the ratio of the two geometric means were set to 80.00% and 125.00%, respectively, and the value of the ratio of the two geometric means at which the power was to be calculated was set to 1.0.
In theory, the coefficient of variation (CV) was the ratio of the standard deviation (SD) and the mean (SD/mean). The CV was defined on the original (not logarithmic) scale from our pilot study, calculated using the relationship between the means and variances of Y and X. Suppose data on a response variable Y were collected. The procedure of PASS assumed that the values of X = Ln(Y) would be analyzed using an appropriate analysis of variance (ANOVA) procedure (PASS 15, help center). In our study, the mean square error (MSE) of the maximum concentration (Cmax), area under the curve from time zero to the time of the last measurable concentration (AUCt), and AUC extrapolated from time zero to infinity (AUC∞) [after logarithmic transformation] was 0.77%, 1.11% and 1.10%, respectively, after excluding the variation among preparations, individuals, periods, and drug sequences by multivariate ANOVA according to our pilot trial (Ethical Approval No.: 2017Y123; Registration number: CTR20171595) (http://www.chinadrugtrials.org.cn/clinicaltrials.searchlistdetail.dhtml). MSE was brought into the following formula estimation of individual in vivo variation: CV(Y) = [Exp{σ(X)2} − 1]1/2 (PASS 15, help center), where CV(Y) was the estimated value of the individual in vivo variation of pharmacokinetic parameters, and σ(X)2 was the MSE after logarithmic transformation of the pharmacokinetic parameters. The estimated values of the intraindividual variation of Cmax, AUCt and AUC∞ were 8.81%, 10.58% and 10.51%, respectively.
However, considering that the CV value reported in the literature was 17–29% [15], we set the CV value to 0.30. We estimate the minimum sample size was 32 subjects. Considering the 10% withdrawal rate, 36 subjects were finally determined.
Study Population
Inclusion criteria: Thirty-six healthy Chinese volunteers who were > 18 years of age, with a body mass index of 19.0–28.0 kg/m2 and weighing > 50 kg (male) or > 45 kg (female) were recruited. Based on medical history, clinical examinations, and laboratory investigations, no subject had a history or evidence of serious diseases or allergies, or contraindications to metformin or its excipients. Women of childbearing age were confirmed not to be pregnant and were required to use effective contraceptives throughout the study and for at least 6 months following the trial.
Exclusion criteria: Subjects who had trouble with venous blood collection and a history of venipuncture syncope or were exposed to any drugs within 2 weeks prior to the trial were excluded from the study. Participants were instructed to abstain from taking any medications that affect liver drug-metabolizing enzymes, including inducers (e.g., barbiturates, carbamazepine, phenytoin sodium, and glucocorticoids) and inhibitors (e.g., cimetidine, diltiazem, macrolides, nitroimidazoles, sedative hypnotics, verapamil, fluoroquinolones, antihistamines, and omeprazole), for a minimum of 3 months prior to and during the study period. Participants were forbidden to consume alcohol and beverages or food containing methylxanthines 48 h before the clinical trial and after drug administration until the final blood sample was collected. Volunteers who donated or lost > 400 mL of blood within 3 months prior to the clinical trial were excluded.
Withdrawal criteria: Researchers withdrew a subject from the trial if (1) they considered it medically unethical to continue the trial; (2) a serious adverse event (SAE) occurred; (3) the subject would benefit from withdrawal; or (4) the subject was noncompliant. Noncompliance included the following situations: (1) the subject did not take the drug or accept examination according to the protocol; or (2) the subject’s behavior would affect the results. Subjects had the right to withdraw from the trial or to refuse medication and testing without formally withdrawing their informed consent. When possible, the reasons for a withdrawal were recorded.
Drug Administration
All subjects were admitted to the hospital the day preceding drug administration (D−1). Examinations included symptom analysis, vital sign measurements, alcohol breath tests, urine drug screening analysis, and blood pregnancy tests for women of childbearing age. The subjects fasted, except for water, for 10 h prior to drug administration. On day 1 of drug administration (D1), subjects were randomly assigned by the principal investigator to one of the two dosing-order subgroups, T/R and R/T, which were equally represented. According to the summary of Glucophage XR characteristics, the recommended starting dose is 500 mg (one tablet) orally once daily with the evening meal; increase the dose in increments of 500 mg weekly based on glycemic control and tolerability, up to a maximum of 2000 mg (four tablets) once daily with the evening meal [16]. According to the protocol, subjects took a single dose (500 mg) of either the T or R MH-SR formulation with 240 mL of water after eating. A statistician performed block randomization using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).
Beginning from the underfed state, subjects ate a prescribed high-fat trial diet 30 min before taking their formulation and were required to finish the meal within 20 min (Table 1). Subjects could not consume water from 1 h before to 1 h after drug administration. Lunch and dinner were served at 4 and 10 h post drug administration, respectively. After meals, subjects remained seated and were observed closely for 4 h. Subjects were ambulatory at other times during the trial but were prohibited from strenuous activity. They remained in hospital for an additional 2 days and were discharged on day 3 (D3) after blood collection and examinations. According to the summary of Glucophage® XR characteristics, the plasma elimination half-life (t½) was approximately 6.2 h and the blood t½ was approximately 17.6 h [16]. The preparation was cross-administered after a 7-day washout (>5 × t½).
Table 1.
Nutritional composition of a high-fat diet
| Materials | Weight (g) |
Food (g) |
Energy (kcal) |
Protein (g) |
Fat (g) |
Carbohydrate (g) |
|---|---|---|---|---|---|---|
| Unified flour | 80 | 80 | 295.12 | 12.56 | 2 | 56.72 |
| Romaine lettuce | 53.19 | 50 | 7.95 | 0.65 | 0.15 | 1 |
| Chicken | 50 | 50 | 58.95 | 12.3 | 0.95 | 0.3 |
| Milk | 250 | 250 | 136 | 7.5 | 8.0 | 8.5 |
| Egg | 56.82 | 50 | 68.8 | 7.2 | 3.2 | 2.8 |
| Salad oil | 35 | 35 | 314.47 | 0 | 34.9 | 0.11 |
| Total | 881.29 | 40.21 | 49.20 | 69.43 | ||
| Caloric ratio (%) | 18.25 | 50.24 | 31.51 |
Blood Sampling
As the plasma t½ of Glucophage® XR was approximately 6.2 h [16], the sample collection period was set for 36 h (>5 × t½ = 31 h). Approximately 19 venous blood samples (4 mL) were collected through an indwelling cannula from each subject to assay metformin from predose (0 h; within 30 min before administration) to 36 h postdose at preset time-points (1, 2, 3, 4, 5, 5.5, 6, 6.5, 7, 7.5, 8, 8.5, 9, 10, 12, 15, 24, and 36 h). Blood samples were collected using vacuum blood collection tubes containing dipotassium ethylene diamine tetraacetate (K2-EDTA) anticoagulant and centrifuged (2500g) at 4°C for 10 min. Evenly separated plasma was frozen at – 20 °C in two tubes, one for testing and the other as a backup. Samples were transferred to – 70 °C freezers for long-term storage within 72 h. After a 7-day washout, the trial was repeated in the same manner to complete the crossover design.
Bioanalytical Methods
Plasma concentrations of metformin were determined by Nanjing Clinical Tech Laboratories Inc. using a validated high-performance liquid chromatography–tandem mass spectrometry (HPLC-MS/MS) method [17]. The HPLC was fitted with a liquid phase pump (LC-20ADXR), column heater (CTO-20AC), controller (CBM-20Alite), degasser (DGU-20A5R), and automatic injector (SIL-30AC) [Shimadzu Co., Kyoto, Japan]. Electrospray ionization mass spectrometry was performed using an API 4000 (Applied Biosystems/Sciex, Foster City, CA, USA).
HPLC was performed using an analytical/elution column (Poroshell 120 EC-CN, 2.1 × 50 mm, 2.7 μm; Agilent Technologies, Santa Clara, CA, USA) fit with a pre-column (Security Guard Cartridges C18, 4 × 2.0 mm; Phenomenex, Torrance, CA, USA). The column and autosampler were maintained at 40 °C and 8 °C, respectively. The mobile phases were an aqueous solution of 0.1% formic acid and 5.0 mM NH4Ac (A) and 100% ethanol (B). The injection volume was 6.00 μL and the retention time was approximately 1.60 min for both metformin and metformin-d6.
Mass spectrometry was performed for 5.00 min using the following parameters: ion spray voltage, 1700 V; curtain gas, 45 psi; atomizing gas, 55 psi; auxiliary gas, 55 psi; temperature, 650 °C; collision-activated dissociation, 10 U; positive ion mode; and multiple reaction monitoring. The ion pair of metformin was 130.1/71.1 m/z with a dwell time of 100 ms. The ion pair of the metformin-d6 internal standard was 136.1/60.1 m/z with a dwell time of 100 ms.
Based on verification, the linear range was 3.00–1200 ng/mL. The lower limit of quantification was 3.00 ng/mL. The overall within-run precision and accuracy for metformin ranged from 1.9 to 6.8% and 98.7 to 107.7%, respectively, while the overall between-run precision and accuracy for metformin ranged from 2.0 to 8.0% and 102.0 to 104.0%, respectively.
Pharmacokinetic Analysis
The primary pharmacokinetic parameters were Cmax, AUCt and AUC∞. Phoenix WinNonlin software v7 (Pharsight®, a Certara™ company, Raleigh, NC, USA) was used to analyze the pharmacokinetic parameters. AUCt was calculated according to the linear trapezoidal rule as the AUC from time zero to the time of the last measurable concentration. The AUC was extrapolated from time zero to infinity (AUC∞), using the formula AUC∞ = AUCt + Ct/λz, where Ct is the final measurable concentration and λz is the elimination rate constant represented by the slope of the terminal segment of the logarithmically transformed drug concentration versus the time linear regression curve. The criteria for estimation of λz was minimum R-squared value, minimum span from the first to last timepoint selected for regression in relation to derived half-life. The ratio of AUCt/AUC∞ should be more than 80%. The time to peak concentration (Tmax) was obtained directly from the concentration-time (c − t) curve, and the t½ was calculated as 0.693/λz. Log-transformed primary pharmacokinetic parameters were tested using ANOVA. The results of the mixed-effect model included subjects, sequence, period, and preparation factors. The bioequivalence of Yuantang® SR and Glucophage® XR was evaluated using the geometric mean ratio (GMR), 90% confidence interval (CI), and two-sided t-test. Yuantang® SR and Glucophage® XR were considered bioequivalent if they were within the equivalent interval (80.00–125.00%).
Safety Assessment
Subjects were monitored for any adverse events (AEs), SAEs, concomitant medications, nonpharmacological treatments, changes in clinical laboratory results (e.g., routine blood and urine tests, blood biochemistry), 12-lead electrocardiogram results, clinical symptoms, vital signs, and physical examination results. All AEs were evaluated using the Common Terminology Criteria of Adverse Events (CTCAE) version 4.03. The correlation between AEs and drug administration was stratified into five levels—definite, probable, possible, unlikely, and no relationship—according to the judgment standard of causality between drugs and AEs. Definite, probable and possible were considered adverse drug reactions.
Statistical Analysis
The normality of data distributions was assessed using the Kolmogorov–Smirnov test. Normally distributed data were expressed as mean ± SD (x̄ ± SD) and non-normally distributed data were expressed as quartiles (25%, 75%). Count data were expressed in cases and percentages (n, %) and analyzed using the Chi-square test or Fisher’s exact test if the theoretical frequency was < 5.
The primary pharmacokinetic parameters were typically log-normally distributed and thus were log-transformed prior to statistical analysis [18]. Differences between parameters were considered statistically significant at p < 0.05. Phoenix WinNonlin software v7 (Pharsight®, a Certara™ company) was used to analyze pharmacokinetic parameters, while SAS version 9.4 software was used for other parameters.
Results
Demographics
Thirty-six healthy Chinese adults were included in the bioequivalence trial and randomized into T/R and R/T subgroups without difference in sex ratio (p = 0.72) [Fig. 2]. A total of 25 (69.44%) participants were male, with 88.89% (32/36) of participants representing Han nationality. The average age was 31.36 ± 8.18 years, ranging from 19 to 46 years, and average body weight was 64.89 ± 7.53 kg, ranging from 47.9 to 82.4 kg. Average height was 165.01 ± 7.63 cm, ranging from 150.8 to 182.0 cm, and median BMI was 23.83 ± 2.30 kg/m2, ranging from 19.58 to 27.99 kg/m2 (Table 2).
Fig. 2.
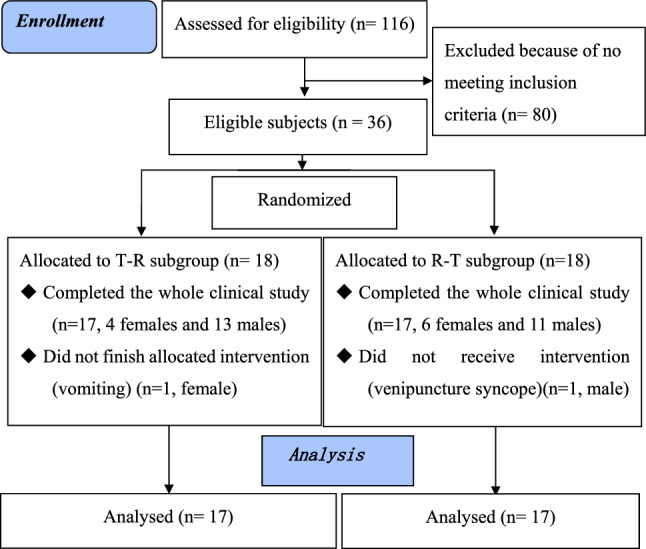
Flow diagram. T test preparation, Yuantang® SR, R reference preparation, Glucophage® XR
Table 2.
The demographic and baseline characteristics of subjects included in the clinical trials
| Characteristics | T/R subgroup | R/T subgroup | Total |
|---|---|---|---|
| No. of subjects | 18 | 18 | 36 |
| Age, years | 32.61 ± 7.29 | 30.11 ± 9.02 | 31.36 ± 8.18 |
| Male sex [n (%)] | 13 (72.22) | 12 (66.67) | 25 (69.44) |
| Han nationality [n (%)] | 16 (88.89) | 16 (88.89) | 32 (88.89) |
| Other nationalities [n (%)] | 2 (11.11) | 2 (11.11) | 4 (11.11) |
| Height, cm | 165.41 ± 6.98 | 164.62 ± 8.41 | 165.01 ± 7.63 |
| Body weight, kg | 65.59 ± 6.57 | 64.18 ± 8.53 | 64.89 ± 7.53 |
| Body mass index, kg/m2 | 24.00 ± 2.26 | 23.65 ± 2.39 | 23.83 ± 2.30 |
Data are expressed as mean ± standard deviation unless otherwise specified
T test preparation, Yuantang® SR, R reference preparation, Glucophage® XR
Pharmacokinetics
Both Yuantang® SR and Glucophage® XR were demonstrated to be readily absorbed from the gastrointestinal (GI) tract as metformin was measurable from the first sampling time (1.00 h) in all 34 subjects (Fig. 3). The concentration increased over time until it reached a maximum at 5.00 h after drug administration and then decreased rapidly, although it was still detectable for up to 36 h (Table 3 and Fig. 3). The t½ of T and R was 4.70 ± 0.55 and 4.81 ± 0.42 h (Table 3), and shorter than that in the summary of the Glucophage® XR characteristics (t½ = 6.2 h) [16]. The 90% CIs (96.12–105.44% for AUCt, 96.22–105.54% for AUC∞, and 98.42–105.00% for Cmax) of the T/R GMR for these pharmacokinetic parameters were within the bioequivalence acceptance range of 80.00–125.00% (Table 4). Additionally, no significant differences between the main parameters of T and R were identified by ANOVA (p > 0.05) [Table 5].
Fig. 3.
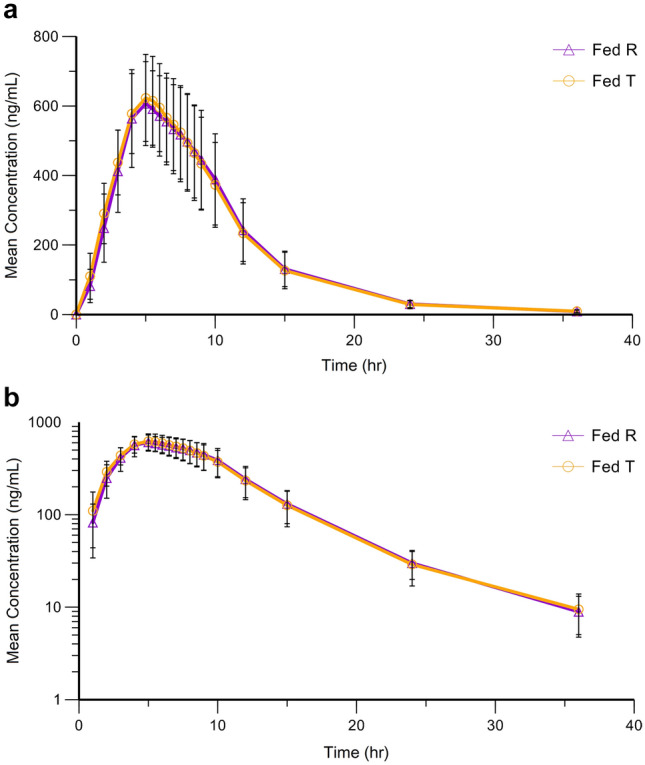
a Mean (n = 34) plasma concentration (± SD)–time curves of Yuantang® SR (T) and Glucophage® XR (R) after a single 500 mg oral dose under postprandial conditions. a Linear plot. b Semilogarithmic plot. T test preparation, Yuantang® SR, R reference preparation, Glucophage® XR, SD standard deviation
Table 3.
Pharmacokinetic parameters after a single dose of 500 mg of Yuantang® SR (T) and Glucophage® XR (R)
| Pharmacokinetic parameter | T [n = 34] | R [n = 34] |
|---|---|---|
| AUCt, h ng/mL | 6366.13 ± 1492.17 (23.44) | 6305.72 ± 1410.58 (22.37) |
| AUC∞, h ng/mL | 6434.14 ± 1508.94 (23.45) | 6367.92 ± 1426.40 (22.40) |
| Cmax, ng/mL | 651.41 ± 123.68 (18.99) | 640.29 ± 116.22 (18.15) |
| Tmax, h (minimum, maximum) | 5.00 (4.00, 7.50) | 5.00 (4.00, 8.00) |
| t½, h | 4.70 ± 0.55 (11.71) | 4.81 ± 0.42 (8.81) |
| λz, 1/h | 0.15 ± 0.02 (11.17) | 0.15 ± 0.01 (8.45) |
Data are expressed as arithmetic mean ± SD (%CV) unless otherwise specified
AUCt area under the curve from time zero to the time of the last measurable concentration, AUC∞ area under the curve extrapolated from time zero to infinity, Cmax maximum plasma concentration, Tmax time to reach maximum concentration, t½ elimination half-life, λz elimination rate constant represented by the slope of the terminal segment of the logarithmically transformed drug concentration versus time linear regression curve, SR sustained release, XR extended release, T test preparation, Yuantang® SR, R reference preparation, Glucophage® XR
Table 4.
Bioequivalence analysis of Yuantang® SR (T) and Glucophage® XR (R) after a single dose of 500 mg T and R
| Pharmacokinetic parameter | Geometric mean value and ratio | 90% CI (%) | CV (%) | Power | ||
|---|---|---|---|---|---|---|
| T | R | T/R (%) | ||||
| AUCt, h ng/mL | 6185.62 ± 0.23 | 6144.37 ± 0.22 | 100.67 | 96.12–105.44 | 11.30 | > 99.99 |
| AUC∞, h ng/mL | 6252.63 ± 0.23 | 6204.80 ± 0.22 | 100.77 | 96.22–105.54 | 11.29 | > 99.99 |
| Cmax, ng/mL | 640.26 ± 0.19 | 629.82 ± 0.18 | 101.66 | 98.42–105.00 | 7.89 | > 99.99 |
SR sustained release, XR extended release, T test preparation, Yuantang® SR, R reference preparation, Glucophage® XR, CI confidence interval, CV coefficient of variation, AUCt area under the curve from time zero to the time of the last measurable concentration, AUC∞ area under the curve extrapolated from time zero to infinity, Cmax maximum plasma concentration
Table 5.
Results (p-values) of ANOVA test of bioequivalence between the two formulations
| Influence factor | lnAUCt | lnAUC∞ | lnCmax |
|---|---|---|---|
| Sequence | 0.13 | 0.12 | 0.30 |
| Periods | 0.18 | 0.19 | 0.96 |
| Formulations | 0.81 | 0.78 | 0.40 |
ANOVA analysis of variance, AUCt area under the curve from time zero to the time of the last measurable concentration, AUC∞ area under the curve extrapolated from time zero to infinity, Cmax maximum plasma concentration
Safety and Tolerability
The T and R formulations of MH-SR were safe and well tolerated in the postprandial trial, with no SAEs or unsolicited adverse reactions observed. One subject from the T/R subgroup was removed from the trial as she vomited 4.25 h after the first administration of medication T, shorter than the plasma t½ (t½ = 6.2 h in the summary of Glucophage® XR characteristics [16]; t½ = 4.70 ± 0.55 h in this study (Table 3). This subject was analyzed in the safety set but excluded from the pharmacokinetic set. A volunteer from the R/T subgroup withdrew prior to drug administration due to venipuncture syncope and was eliminated from the safety and pharmacokinetic sets. The remaining 34 participants completed the clinical study and were analyzed in the safety and pharmacokinetic sets.
The primary adverse reactions to the administration of T and R were symptomatic digestive tract reactions (52.94%, 9/17) and abnormal blood and urine laboratory tests (41.18%, 7/17). Most of the drug-related AEs (88.24%, 15/17) were grade I and of mild severity. All patients recovered from the drug-related AEs without special treatment. There were no significant differences in subjects’ age and sex for the occurring AEs, and the type, severity and prognosis of AEs between the T and R treatment groups (p > 0.05) (Table 6).
Table 6.
Adverse events of Yuantang® SR and Glucophage® XR (T and R)
| Items | Postprandial trial | p-Value | |
|---|---|---|---|
| T | R | ||
| No. of subjects | 36 | 36 | > 0.99a |
| Analyzed | 35 (97.22) | 34 (94.44) | |
| Dropout | 1 (2.78) | 2 (5.56) | |
| Subjects with drug-related adverse events | 3 (8.57) | 12 (35.29) | 0.01 |
| Drug-related (n) | 4 | 13 | 0.371a |
| Dry mouth | 0 (0.00) | 1 (7.69) | |
| Bitter taste of mouth | 0 (0.00) | 1 (7.69) | |
| Nausea | 1 (25.0) | 0 (0.00) | |
| Vomiting | 1 (25.0) | 0 (0.00) | |
| Abdominal pain | 0 (0.00) | 1 (7.69) | |
| Diarrhea | 0 (0.00) | 4 (30.77) | |
| Increased platelet count | 0 (0.00) | 1 (7.69) | |
| Hypertriglyceridemia | 1 (25.0) | 3 (23.08) | |
| Increased white blood cell count in urine | 0 (0.00) | 1 (7.69) | |
| Urine occult blood [+] | 1 (25.0) | 0 (0.00) | |
| Upper respiratory tract infection | 0 (0.00) | 1 (7.70) | |
| Severity of drug-related adverse events | 0.426a | ||
| Grade I | 3 (75.00) | 12 (92.31) | |
| Grade II | 1 (25.00) | 1 (7.69) | |
| Outcomes | > 0.99a | ||
| Recovery | 4 (100) | 13 (100) | |
Data are expressed as n (%) unless otherwise specified
SR sustained release, XR extended release, T test preparation, Yuantang® SR, R reference preparation, Glucophage® XR
aFisher’s exact test
Discussion
Metformin is only approved for the treatment of type 2 DM; however, a growing number of studies have suggested benefits from its off-label use in other areas, such as dementia [19], weight loss [20], glioblastoma [21], inflammation [22], hair growth [23], miscarriage and premature delivery, polycystic ovarian syndrome [24], breast and colorectal cancer [25], and cardiovascular protection [26]. These findings have increased interest in metformin and the development of generic MH-SR drugs.
The most important objective of a bioequivalence trial is to ensure that the two formulations have similar rates and extents of absorption, which would indicate that they are therapeutically equivalent [27, 28]. After a single drug dose, particularly for fast-release formulations, Cmax reflects the absorption rate and AUC reflects the degree of absorption; these represent the primary pharmacokinetic parameters. It is generally accepted that the standard equivalency range of 80.00–125.00% holds for basic pharmacokinetic characteristics [29].
Recently, Zhou, et al. [30] studied the bioequivalence of two matrix-type metformin extended-release tablets under postprandial conditions. The reference preparation was Glucophage® XR. AUC36, AUC∞ and Cmax of generic metformin extended-release under postprandial states were 6563 ± 814 ng h/mL, 6627 ± 817 ng h/mL and 619 ± 60 ng/mL. The 90% CIs of Ln AUC36, Ln AUC∞, and Ln Cmax from the generic formulation versus the branded formulation were 91.4–105.0%, 91.3–104.7%, and 101.2–119.4%, respectively. The results obtained from the study by Zhou et al. suggest that the two drugs were bioequivalent in Chinese subjects. This study confirmed that another generic MH sustained-release tablet is bioequivalent with brand-name drugs and provides a new good choice for clinicians and patients. The new generic MH-SR reduces the costs for clinical supplies of brand-name MH-SR and alleviates the contradiction between supply and demand due to insufficient supply in the original MH-SR market.
The common (> 1.0%) and most common (> 5.0%) solicited adverse reactions detailed in MH-SR medication guides include diarrhea, nausea/vomiting, constipation, abdominal discomfort, flatulence, dyspepsia/heartburn, asthenia, headache, dizziness, hypoglycemia, myalgias, upper respiratory infection, dyspnea, nail disorder, rash, increased sweating, taste disorders, chest discomfort, chills, flushing and palpitation [16]. The drugs in this clinical trial were well tolerated, with GI symptoms and laboratory abnormalities being the primary AEs, most of which did not require special treatment apart from clinical observation until the patient recovered naturally. All AEs were consistent with those listed in the summary of Glucophage® XR characteristics and other clinical trials [15, 30].
The limitations of this study include its small sample size, comprising only healthy Chinese adults; therefore, the safety data may not have enough statistical power. Another limitation is that two subjects withdrew during the trial; however, we fully considered this issue before commencement of the trial and included an additional 10% of subjects.
The AUCt, AUC∞ and Cmax of the two formulations compared in this study did not differ significantly, indicating that the blood profiles generated by Yuantang® SR (500 mg/tablet) were comparable with those produced by Glucophage® XR (500 mg/tablet). The 90% CIs for the GMR of AUCt, AUC∞, and Cmax revealed that these values were within the bioequivalence acceptance range of 80.00–125.00%. Furthermore, ANOVA for these parameters suggested no significant difference (p > 0.05) between the two MH-SR brands, either in sequence, periods or formulations. Based on these results, we can safely conclude that Yuantang® SR (500 mg/tablet), manufactured by Guangdong Sinocorp Pharmaceutical Co., Ltd, China, is bioequivalent to Glucophage® XR (500 mg/tablet), manufactured by Merck Serono Co., Ltd, UK, when taken under postprandial conditions. Additionally, this clinical trial confirmed that both products were safe and well tolerated by healthy Chinese adults.
Conclusions
The generic drug, Yuantang® SR (500 mg/tablet), is a well tolerated, bioequivalent alternative to the original drug, Glucophage® XR (500 mg/tablet), under postprandial conditions after comparing their pharmacokinetic characters in healthy Chinese adults.
Acknowledgements
The authors thank the volunteers for their participation in this trial, and are grateful to the Beijing Giant Med-Tech Development Co., Ltd, acting as the contract research organization; Nanjing Clinical Tech Laboratories, Inc., acting as the testing unit, Beijing Bio-Know Information Technology Co., Ltd, acting as the data management unit; and Peking University Clinical Research Institute, acting as the statistical unit, for their work during this clinical trial.
Declarations
Funding
This trial was funded by Guangdong Sinocorp Pharmaceutical Co., Ltd, China.
Conflicts of Interest/Competing interests
Xiang-Dong Luo, the third author, was a senior engineer at Guangdong Sinocorp Pharmaceutical Co., Ltd, China. Ming-Li Sun, Hui-Juan Liu, Xiang-Dong Luo, Yu Wang, Wei Zhang, Chen Liu and Xinghe Wang have no financial or other substantive conflicts of interest that may be construed to influence the results or interpretations reported in this manuscript.
Ethics approval
The protocol and all documents were reviewed and approved by the Institutional Review Board of Beijing Shijitan Hospital on 16 October 2017 (registration number: 2017Y123).
Consent to participate
All subjects signed the consent form under fully informed conditions prior to commencement of the trial.
Consent for publication
All researchers, participants, institutions, and sponsors consented to the submission of this report to the journal.
Availability of data and material
All data generated or analyzed during this study are included in this published article.
Author contributions
Ming-Li Sun, working as the sub-I of the trial, and first author of this article, wrote the first and successive versions of this manuscript. Hui-Juan Liu and Wei Zhang were principal research nurses for the clinical trial. Yu Wang was quality control (QC). Xinghe Wang was the principal investigator (PI) of the clinical trial and corresponding author of this article. Xiang-Dong Luo was the sponsor. All authors took part in the research design and contributed many constructive opinions and suggestions to the clinical trial and this study article. All authors approved the final version of the manuscript.
Code availability
Not applicable.
References
- 1.Luo Y, Zhu Y, Chen J, Gao X, Yang W, Zou X, et al. A decision-support software to improve the standard care in Chinese type 2 diabetes. J Diabetes Res. 2019 doi: 10.1155/2019/5491743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, IDF Diabetes Atlas Committee et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019;157:107843. doi: 10.1016/j.diabres.2019.107843. [DOI] [PubMed] [Google Scholar]
- 3.Flory J, Lipska K. Metformin in 2019. JAMA. 2019;321:1926–1927. doi: 10.1001/jama.2019.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qaseem A, Barry MJ, Humphrey LL, Forciea MA, Clinical Guidelines Committee of the American College of Physicians Oral pharmacologic treatment of type 2 diabetes mellitus: a clinical practice guideline update from the American College of Physicians. Ann Intern Med. 2017;166:279–290. doi: 10.7326/M16-1860. [DOI] [PubMed] [Google Scholar]
- 5.Beloica S, Cvijić S, Homšek I, Bogataj M, Parojčić J. An in vitro–in silico–in vivo approach in biopharmaceutical drug characterization: metformin hydrochloride IR tablets. Pharmazie. 2015;70:458–465. doi: 10.1691/ph.2015.4168. [DOI] [PubMed] [Google Scholar]
- 6.Cetin M, Sahin S. Microparticulate and nanoparticulate drug delivery systems for metformin hydrochloride. Drug Deliv. 2016;23:2796–2805. doi: 10.3109/10717544.2015.1089957. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, Desai D, Good D, Crison J, Timmins P, Paruchuri S, et al. Mathematical model-based accelerated development of extended-release metformin hydrochloride tablet formulation. AAPS PharmSciTech. 2016;17:1007–1013. doi: 10.1208/s12249-015-0423-9. [DOI] [PubMed] [Google Scholar]
- 8.Mandal U, Gowda V, Ghosh A, Bose A, Bhaumik U, Chatterjee B, et al. Optimization of metformin HCl 500 mg sustained release matrix tablets using Artificial Neural Network (ANN) based on Multilayer Perceptrons (MLP) model. Chem Pharm Bull (Tokyo). 2008;56:150–155. doi: 10.1248/cpb.56.150. [DOI] [PubMed] [Google Scholar]
- 9.Mandal U, Gowda V, Ghosh A, Selvan S, Solomon S, Pal TK. Formulation and optimization of sustained release matrix tablet of metformin HCl 500 mg using response surface methodology. Yakugaku Zasshi. 2007;127:1281–1290. doi: 10.1248/yakushi.127.1281. [DOI] [PubMed] [Google Scholar]
- 10.Wadher KJ, Kakde RB, Umekar MJ. Study on sustained-release metformin hydrochloride from matrix tablet: Influence of hydrophilic polymers and in vitro evaluation. Int J Pharm Investig. 2011;1:157–163. doi: 10.4103/2230-973X.85966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wadher KJ, Kakde RB, Umekar MJ. Formulation and evaluation of a sustained-release tablets of metformin hydrochloride using hydrophilic synthetic and hydrophobic natural polymers. Indian J Pharm Sci. 2011;73:208–215. doi: 10.4103/0250-474x.91579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ICH: press release, ICH Assembly meeting in Montreal, Canada, May/June 2017. Geneva: International Council for Harmonization. https://www.ich.org/news/press-release-ich-assembly-meeting-montreal-canada-mayjune-2017. Accessed 1 Aug 2021
- 13.Huang XM, Wang GZ, He BB, Gao T, Long P, Zhang BK. Bioequivalence and pharmacokinetic evaluation of two metformin hydrochloride tablets under fasting and fed conditions in healthy Chinese volunteers. Clin Pharmacol Drug Dev. 2020;9:910–917. doi: 10.1002/cpdd.849. [DOI] [PubMed] [Google Scholar]
- 14.Center for Drug Evaluation, National Medical Products Administration. Bioequivalence of metformin hydrochloride sustained release tablets. (First registration: CTR20171580, updated registration: CTR20180476). 2018. http://www.chinadrugtrials.org.cn/clinicaltrials.searchlist.dhtml. Accessed 1 Aug 2021
- 15.Batolar LS, Iqbal M, Monif T, Khuroo A, Sharma PL. Bioequivalence and pharmacokinetic comparison of 3 metformin extended/sustained release tablets in healthy Indian male volunteers. Arzneimittelforschung. 2012;62:22–26. doi: 10.1055/s-0031-1295428. [DOI] [PubMed] [Google Scholar]
- 16.Summary of Glucophage, Glucophage® XR characteristics. RxList: Brand Name: Glucophage, Glucophage XR. 2019. https://www.rxlist.com/glucophage-drug.htm. Accessed 1 Aug 2021.
- 17.Marques MA, Ade SS, Pinto OW, Barroso PT, Pinto DP, Ferreira-Filho M, et al. Simple and rapid method determination for metformin in human plasma using high performance liquid chromatography tandem mass spectrometry: application to pharmacokinetic studies. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;852:308–316. doi: 10.1016/j.jchromb.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 18.Ii Y. Investigation of the properties of the “switching decision rule” described in the Japanese guideline for human bioequivalence studies. Pharm Stat. 2019;18:260–273. doi: 10.1002/pst.1922. [DOI] [PubMed] [Google Scholar]
- 19.Samaras K, Makkar S, Crawford JD, Kochan NA, Wen W, Draper B, et al. Metformin use is associated with slowed cognitive decline and reduced incident dementia in older adults with type 2 diabetes: the Sydney memory and ageing study. Diabetes Care. 2020;43:2691–2701. doi: 10.1002/pst.1922. [DOI] [PubMed] [Google Scholar]
- 20.Dludla PV, Nkambule BB, Mazibuko-Mbeje SE, Nyambuya TM, Mxinwa V, Mokgalaboni K, et al. Adipokines as a therapeutic target by metformin to improve metabolic function: a systematic review of randomized controlled trials. Pharmacol Res. 2021;163:105219. doi: 10.1016/j.phrs.2020.105219. [DOI] [PubMed] [Google Scholar]
- 21.Xu X, Shen X, Feng W, Yang D, Jin L, Wang J, et al. d-Galactose induces senescence of glioblastoma cells through YAP-CDK6 pathway. Aging (Albany NY). 2020;12:18501–18521. doi: 10.18632/aging.103819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alwarawrah Y, Nichols AG, Green WD, Eisner W, Kiernan K, Warren J, et al. Targeting T-cell oxidative metabolism to improve influenza survival in a mouse model of obesity. Int J Obes (Lond) 2020;44:2419–2429. doi: 10.1038/s41366-020-00692-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chai M, Jiang M, Vergnes L, Fu X, de Barros SC, Doan NB, et al. Stimulation of hair growth by small molecules that activate autophagy. Cell Rep. 2019;27:3413–3421. doi: 10.1016/j.celrep.2019.05.070. [DOI] [PubMed] [Google Scholar]
- 24.Guan Y, Wang D, Bu H, Zhao T, Wang H. The effect of metformin on polycystic ovary syndrome in overweight women: a systematic review and meta-analysis of randomized controlled trials. Int J Endocrinol. 2020 doi: 10.1155/2020/5150684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rennert G, Rennert HS, Gronich N, Pinchev M, Gruber SB. Use of metformin and risk of breast and colorectal cancer. Diabetes Res Clin Pract. 2020;165:108232. doi: 10.1016/j.diabres.2020.108232. [DOI] [PubMed] [Google Scholar]
- 26.Emelyanova L, Bai X, Yan Y, Bosnjak ZJ, Kress D, Warner C, et al. Biphasic effect of metformin on human cardiac energetics. Transl Res. 2021;229:5–23. doi: 10.1016/j.trsl.2020.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kano EK, Chiann C, Fukuda K, Porta V. Effect of different sampling schedules on results of bioavailability and bioequivalence studies: evaluation by means of Monte Carlo simulations. Drug Res (Stuttg). 2017;67:451–457. doi: 10.1055/s-0043-105797. [DOI] [PubMed] [Google Scholar]
- 28.Najib N, Idkaidek N, Beshtawi M, Bader M, Admour I, Alam SM, et al. Bioequivalence evaluation of two brands of metformin 500 mg tablets (Dialon and Glucophage)—in healthy human volunteers. Biopharm Drug Dispos. 2002;23:301–306. doi: 10.1002/bdd.326. [DOI] [PubMed] [Google Scholar]
- 29.National Medical Products Administration. Circular of NMPA on issuing statistical guidelines for bioequivalence studies (No. 103 of 2018). 2018. https://www.nmpa.gov.cn/yaopin/ypggtg/ypqtgg/20181029173101911.html. Accessed 1 Aug 2021.
- 30.Zhou Z, Wang C, Li M, Lan Q, Yu C, Yu G, et al. In vitro dissolution and in vivo bioequivalence evaluation of two metformin extended-release tablets. Clin Pharmacol Drug Dev. 2021;10(4):414–419. doi: 10.1002/cpdd.857. [DOI] [PubMed] [Google Scholar]