

Summary
Background:
A portion of individuals with hemophilia A develop neutralizing antibodies called inhibitors to glycoprotein factor VIII (FVIII). There are multiple risk factors that contribute to the risk of inhibitor formation. However, knowledge of the role of FVIII asparagine (N)-linked glycosylation in FVIII immunity is limited.
Objective:
To evaluate the effect of site-specific N-linked glycan removal on FVIII biochemical properties, endocytosis by murine bone marrow-derived dendritic cells (BMDCs), and antibody responses.
Methods:
Four recombinant B domain deleted (BDD) FVIII variants with single-site amino acid substitutions to remove N-linked glycans were produced for experimental assays.
Results:
BDD FVIII-N41G, FVIII-N239A, FVIII-N1810A, and FVIII-N2118A with confirmed removal of N-linked glycans and similar glycosylation profiles to BDD FVIII were produced. There were no differences in thrombin activation or von Willebrand factor binding of FVIII variants compared to BDD FVIII, however, reduced FVIII expression, activity, and specific activity was observed with all variants. BDD FVIII-N41G and FVIII-N1810A had reduced uptake by BMDCs, but there were no differences in antibody development in immunized hemophilia A mice compared to BDD FVIII. Half of a repertoire of 12 domain-specific FVIII MAbs had significantly reduced binding to ≥1 FVIII variant with a 50% decrease in A1 domain MAb 2-116 binding to FVIII-N239A.
Conclusions:
Modifications of FVIII N-linked glycans reduced FVIII endocytosis by BMDCs and binding of domain-specific FVIII MAbs, but did not alter de novo antibody production in hemophilia A mice suggesting that N-glycans do not significantly contribute to inhibitor formation.
Keywords: dendritic cells, factor VIII, hemophilia A, inhibitors, N-glycosylation
Introduction
Hemophilia A is an inherited X-linked bleeding disorder characterized by a deficiency of clotting glycoprotein factor VIII (FVIII). Individuals with hemophilia A require intravenous infusions of plasma-derived or recombinant FVIII to treat spontaneous and trauma-induced bleeding events such as hemarthrosis, muscle hematomas, and intracranial hemorrhage [1]. A challenging complication of FVIII replacement is the formation of neutralizing antibodies, called inhibitors, against the FVIII protein. Approximately 30% of individuals with severe hemophilia A develop inhibitors [2]. Numerous genetic and environmental risk factors have been investigated for their potential effect on inhibitor development [3–5]. However, an understanding of the mechanisms of inhibitor development is limited.
FVIII is a heavily glycosylated protein consisting of 19 potential asparagine (Asn, N)-linked glycosylation sites within the B domain and 4 N-linked glycan sites outside of the B domain at residues N41 (A1 domain), N239 (A1 domain), N1810 (A3 domain), and N2118 (C1 domain) [6, 7]. N-linked glycosylation is critical for glycoprotein folding, stability, intracellular trafficking, secretion, and clearance by the asialoglycoprotein receptor (ASGPR) [8, 9]. Commercially available recombinant FVIII (rFVIII) products are produced using one of three mammalian cell lines – baby hamster kidney (BHK) cells, Chinese hamster ovary (CHO) cells, or human embryonic kidney (HEK) cells [10]. The primary difference in FVIII produced using these cell lines are the potential glycoforms transferred to the FVIII protein during post-translational modification in the endoplasmic reticulum and Golgi apparatus [11]. The non-human mammalian BHK and CHO cell lines produce N-linked glycoforms N-glycolylneuraminic acid (Neu5Gc) and Gal-α1,3-Gal-β1,4-GlcNAc-R (α-Gal), respectively, that are antigenic epitopes in humans and are not found on FVIII expressed by HEK cell lines [12, 13]. Moreover, rFVIII products expressed by HEK cell lines have similar glycosylation and sulfation patterns to plasma-derived FVIII [13].
Although BHK and CHO cells produce glycoforms that are antigenic to humans, it is unclear whether N-linked glycans affect the immune response to FVIII. Lai et al found that exon 16 (E16) knockout (KO) hemophilia A (FVIII−/−) mice immunized intravenously with BHK-derived rFVIII had a higher incidence of inhibitor positive mice compared to mice immunized with a CHO-derived rFVIII, but 100% of mice in both groups developed anti-FVIII IgG antibodies [14]. In contrast, there were no differences in anti-FVIII IgG antibody positive mice or inhibitor incidence between E16 KO FVIII−/− mice with a R593C point mutation immunized with BHK-derived rFVIII or CHO-derived rFVIII. Another study demonstrated similar anti-FVIII IgG antibody and inhibitor responses in FVIII−/− mice and FVIII and von Willebrand factor (VWF) double KO mice following immunization with B domain deleted (BDD) FVIII or a rFVIII protein lacking the mannose glycoform at N2118 [15]. In this study, we describe the effect of site-specific removal of N-linked glycans outside of the B domain on FVIII biochemical properties, endocytosis, and antibody responses and recognition.
Materials and Methods
Materials
Reagents.
Murine anti-human FVIII monoclonal antibodies (MAbs) were purified from hybridomas as previously described [16–20]. Fluorophore-conjugated antibodies, Pac Blue anti-mouse CD11c (N418) and Live/dead fixable near-IR (NIR) dead cell stain kit, were purchased from BD Biosciences. FVIII C2 domain MAb 2-117 was labeled with DyLight 650 (DyL650) dye using an NHS ester kit from Thermo Fisher Scientific (Waltham, MA). VWF was purified from commercially available human plasma-derived VWF concentrate as previously described [21]. CHO-derived full length FVIII (FL-FVIII) product Advate (Takeda, Lexington, MA) was donated to the lab from the Pediatric Hemophilia Treatment Center at Emory University and Children’s Healthcare of Atlanta. Citrated pooled normal plasma (FACT) and FVIII deficient plasma were purchased from George King Biomedical (Overland Park, KS). All other materials were reagent grade or are described in the cited literature.
Mice.
E16 KO 8-12 week old FVIII−/− male and female mice on a mixed C57BL/6 and 129S4 background were originally obtained from Leon Hoyer (American Red Cross, Holland Laboratory) and used for experiments. Approval for the use of animals and study methods was granted by the Emory University Institutional Animal Care and Use Committee.
Production of FVIII variants with single-site N-linked glycan modifications
A cDNA encoding human BDD FVIII containing a 14 amino acid segment consisting of a SQ linker (sequence: SFSQNPPVLKRHQR) in place of the B domain was cloned into an IRES-Puro or ReNeo mammalian expression plasmid [22–24]. Four cDNAs with alanine (Ala, A) substitutions at N239 (BDD FVIII-N239A), N1810 (BDD FVIII-N1810A), and N2118 (BDD FVIII-N2118A) and glycine (Gly, G) substitution at N41 (BDD FVIII-N41G) for site-specific N-linked glycan removal were designed. Sequences were analyzed and confirmed by Sanger sequencing then ligated into the expression plasmid. XL-1 blue cells were transformed with plasmids and selected colonies were used to transfect BHK-M cells. Puromycin-resistant colonies were selected and screened for FVIII antigen by enzyme-linked immunosorbent assay (ELISA) and FVIII activity by the activated partial thromboplastin time (aPTT) one-stage clotting assay [25]. Approximately 30-100 colonies per variant were screened and the highest expressing colonies were grown to 70% confluence in triple flasks then switch to serum-free AIMV medium. The medium was harvested daily over 5 days, pooled, and centrifuged for 15 minutes at 2400g. Harvested supernatant was frozen at −20°C. The FVIII variants were purified by affinity chromatography on a VIIISelect column followed by cation exchange chromatography with a HiTrap Capto SP ImpRes column. FVIII activity and specific activity of purified FVIII variants were determined by the aPTT one-stage clotting assay and chromogenic assay, respectively, at an absorbance of 280 nm with molar extinction coefficient of 256,300 M−1 cm−1 and molecular weight of 165,300 g/mole. Activation quotients (AQ) of FVIII-containing fractions and purified proteins were determined using one-stage and two-stage activity assays [26, 27].
N-glycopeptide analysis
To confirm removal of site-specific N-linked glycans and ensure the absence of alternative glycoforms with protein modification [28], N-glycopeptide mapping of the five BDD FVIII proteins was performed using LC-MS/MS and multiple fragmentation methods (e.g. CID and HCD) at the University of Georgia Complex Carbohydrate Research Center. Protein samples were reduced, alkylated with iodoacetamide, and digested with 0.5 μg/μl sequencing-grade trypsin at 37ºC for 12 hours. The peptides were analyzed on an Orbitrap Fusion mass spectrometer equipped with a nanospray ion source and connected to an Ultimate 3000 RSLCnano. Pre-packed Nano-LC columns of 15 cm length with 75 μm internal diameter, filled with 2 μm C18 reverse phase material were used for chromatographic separation of the samples. The separation conditions were low-high acetonitrile in solution containing 0.1% formic acid, and the separation time was 3 hours with a 30-minute equilibration time between runs. The precursor ion scan was acquired at 120,000 resolution in an Orbitrap analyzer and precursors at a time frame of 3 seconds were selected for fragmentation using HCD or CID. The threshold for triggering an MS/MS event on the ion-trap was set to 500 counts. Charge state screening was enabled, and precursors with unknown charge state or a charge state of +1 were excluded. Dynamic exclusion was enabled. The fragment ions were analyzed on an Orbitrap for HCD and CID at 30,000 resolution. The proteomic data were processed with Byonic (v2.3.5) and searched against the protein sequences and a catalogue of 300+ mammalian N-glycans. The precursor mass and fragment mass tolerances were wet to 5 ppm and 20 ppm, respectively. Assignments were made using Byonic software. The relative percentages of each glycoform were determined by deconvolution of the LC-MS data at the full MS level, then determining the area under the curve for each full MH+.
Biochemical characterization of FVIII variants
The purity of BDD FVIII and FVIII variants was assessed in the presence and absence of thrombin by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Briefly for SDS-PAGE, 2 μg of FVIII proteins were prepared. Thrombin activated samples were incubated with 4-6 U/mL human thrombin for 5 minutes at 37°C. FVIII proteins were reduced with 10% SDS w/v, 60% glycerol v/v, 0.01% bromophenol blue w/v, 0.6 M Tris, 270 mM DTT reducing buffer at pH 6.8. Samples were heated at 95°C for 5 minutes then loaded onto 4-15% SDS-PAGE precast gels. Gels were run at 30 minutes at 200 volts, washed, and stained with GelCode Blue. The gels were photographed with a Li-Cor Odyssey imaging system.
FVIII binding to VWF was analyzed by ELISA as previously described [20]. Briefly, Immulon-1B plates were coated with 6 μg/ml human VWF in 20 mM HEPES/0.15 M NaCl (HBS)/5 mM CaCl2/0.05% NaN3 and blocked overnight at 4°C. FVIII variants serially diluted twofold between 0.02-1 nM were incubated at room temperature for one hour. FVIII binding was detected by biotinylated anti-C2 domain MAb 2-117, alkaline phosphatase (AP)-conjugated streptavidin, and p-nitrophenyl-phosphate (pnpp) and measured at A405.
Generation of bone marrow derived dendritic cells and endocytosis of FVIII
Murine bone marrow dendritic cells (BMDCs) were derived as previously described [29, 30]. Briefly, the femurs and tibias of euthanized 8-10 week old FVIII−/− mice were harvested and bone marrows flushed with Hank’s balanced salt solution (HBSS). Bone marrow cells were resuspended in red cell lysis buffer and washed twice prior to resuspension in RPMI-1640 medium containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine. Bone marrow cells were seeded at 2 × 106 cells in 100 mm petri dishes with 20 ng/ml GM-CSF and incubated at 37°C at 5% carbon dioxide. Bone marrow cells were fed with medium on day 3 then harvested, washed, and counted on day 6.
FVIII endocytosis experiments were performed as previously described [20, 29] with a few modifications. BMDCs were washed with serum free Iscove’s modified Dulbecco’s medium (IMDM). Approximately 2.5 × 105 BMDCs were incubated with 10 nM BDD FVIII, FVIII variant, or positive control full-length FVIII (FL-FVIII) in IMDM for 30 minutes at 37°C. Cells were washed with phosphate-buffered saline (PBS)/0.5% bovine serum albumin (BSA), stained with fluorophore-conjugated antibodies CD11c and Live/dead NIR, and fixed with 1% ultrapure methanol-free paraformaldehyde. Following additional washes with PBS/0.5% BSA, cells were permeabilized with PBS/0.5% BSA/0.1% saponin and incubated with 1.5 μg/ml of DyL650-conjugated C2 domain MAb 2-117 for 30 minutes at 4°C in the dark. Lastly, cells were washed once more with PBS/0.5% BSA/0.1% saponin and PBS/0.5% BSA. FVIII endocytosis was analyzed by flow cytometry using an Aurora analyzer in the Emory University Pediatrics/Winship Flow Cytometry Core.
Immunization of FVIII−/− mice with BDD FVIII variants
FVIII−/− mice were immunized with 4 weekly retro-orbital injections of 1 μg BDD FVIII or FVIII variant followed by a boost 2 μg dose one week later. One week after the boost dose, mice were euthanized for plasma collection by cardiac puncture. Plasma anti-FVIII IgG antibody titers were determined by ELISA [25]. The inhibitor titer was determined by the Nijmegen Bethesda assay [31].
Anti-FVIII MAb binding to BDD FVIII variants
A repertoire of 12 FVIII MAbs against each FVIII domain were utilized to evaluate binding of FVIII variants compared to BDD FVIII and FL-FVIII by ELISA (Table 1). ELISA plates were coated with FVIII at 1.5 μg/ml in 20 mM Bicine/2 mM CaCl2 and incubated overnight at 4°C. Plates were washed with HBS/2 mM CaCl2/0.05% Tween-20/0.05% NaN3/2% BSA and blocked overnight at 4°C. Anti-FVIII MAbs were serially diluted two-fold starting at 1 μg/ml and incubated for one hour at room temperature. FVIII MAb binding was captured by AP-conjugated goat anti-mouse IgG and detected with substrate pnpp. The ELISA titer was determined by the A405 at 0.3 following a 15-minute incubation.
Table 1.
Summary of FVIII MAbs
| Anti-FVIII MAb | FVIII Domain | IgG Subclass | MAb Binding Epitope [17–20, 56, 59] | Inhibitory Titer (BU/mg IgG) | VWF Binding IC50 (μg/ml) | PL Binding IC50 (μg/ml) |
|---|---|---|---|---|---|---|
| 2-116 | A1 | IgG2aκ | E11-D15, E53-A78 | <1 | >10 | >10 |
| 2-76 | A2 | IgG2aκ | R484-I508 | 38,000 | >10 | >10 |
| 4A4 | A2 | IgG2aκ | D403-H444 | 40,000 | >10 | >10 |
| 4F4 | A2 | IgG2aκ | Indeterminate | 330 | >10 | >10 |
| 2-54 | A2 | IgG1κ | E604-R740 | 34,000 | >10 | >10 |
| 1D4 | A2 | IgG2aκ | E604-R740 | 7,000 | >10 | >10 |
| 2-113 | A3 | IgG1κ | K1818-Y1916 | 156 | >10 | >10 |
| 2A9 | C1 | IgG2aκ | S2063-I2071, N2129-K2136 | 23 | 1.1 | 0.9 |
| B136 | C1 | IgG2aκ | A2077-I2084 | 700 | 0.4 | 0.04 |
| I54 | C2 | IgG2bκ | E2181, D2187, S2206, K2207, H2211, L2212, & Q2213 | 1,300 | 0.02 | 0.02 |
| 1B5 | C2 | IgG2aκ | F2196, T2197, N2198, F2200, T2202, R2220, Q2222, N2225, E2228, K2239, L2252, S2254, H2315, & Q2316 | 930 | 0.05 | 0.03 |
| 2-117 | C2 | IgG2aκ | H2269, Q2270, T2272, L2273, V2282, R2307, & H2309 | >0.4 | >10 | >10 |
BU, Bethesda units; IC50, 50% inhibitory concentration; PL, phospholipid; VWF, von Willebrand factor.
Epitope mapping of A1 domain MAb 2-116 by hydrogen-deuterium exchange mass spectrometry (HDX-MS)
FL-FVIII in the presence or absence of A1 domain MAb 2-116 was used for HDX-MS studies as previously described [20]. Briefly, FVIII (~2 μM) with equimolar equivalent of MAb 2-116 were incubated at room temperature for 15 minutes. FVIII samples were prepared with an autosampler (LEAP Technologies, Carrboro, NC) in 1:7 v/v exchange buffer (10 mM HEPES/5mM CaCl2, pD 7.0) in 99.9% deuterium at 20°C for 10-10,000 seconds in addition to unexchanged controls in sample buffer (10 mM HEPES/5mM CaCl2, pH 7.0). Each time point was repeated three times. Following exchange, samples were quenched at 1°C in 1:1 v/v pre-cooled quenching buffer (2 M guanidine hydrochloride/250 mM Tris(2-carboxyethyl)phosphine hydrochloride, pH 2.5). Samples were injected into a nanoAcquity UPLC with a temperature controlled HDX Manager (Waters, Milford, MA) in which protein digestion and subsequent chromatography are maintained at 0.1°C. Proteins were digested on a 2.1 × 30 mm ProDx Protease column, trapped, and desalted using a BEH C18 2.1 × 5 mm2 column for three minutes with a 100-200 μL/min flow gradient and then separated over a BEH C18 1.0 × 100 mm2 column in eight minutes with a 95-5% water:acetonitrile gradient with 0.1% formic acid at a 40 μL/min flow rate. Peptide carryover was minimized via injection of a digestion column wash solution (1.5 M guanidine hydrochloride/0.1% formic acid) in addition to injections of water with 0.1% formic acid between protein sample injections. Mass spectra were acquired with a Waters Synapt G2-Si HDMS mass spectrometer in positive resolution mode with ion mobility separation. Mass accuracy was determined by simultaneous infusion of reference lock-mass compound leucine enkephalin. FVIII peptides were identified with PLGS 3.0.2, and peptides from autodigested pepsin were excluded. Data were imported into DynamX 3.0 software and peptides were manually inspected to exclude assignments with low signal-to-noise intensity.
Statistical Analysis
Data are presented as mean ± standard deviations for in vitro studies or median with interquartile ranges for immunization studies. Differences in expressed FVIII activity (at 24 and 48 hours) and FVIII internalization by BMDCs between FVIII variants was determined by one-way ANOVA with Dunnett’s correction for multiple comparisons. Differences in ELISA and Bethesda titers were determined by the non-parametric Mann Whitney U test. Differences between binding of MAbs to FVIII variants compared to BDD FVIII and FL-FVIII were determined by two-way ANOVA with Dunnett’s correction for multiple comparisons. A P value <0.05 was considered statistically significant. Statistical analyses were performed with Prism 6.0 (GraphPad Software, La Jolla, CA).
Results
Generation of FVIII variants with single-site N-glycan modifications
To evaluate the effect of single-site removal of N-glycans, we produced four BDD FVIII variants with alanine or glycine substitution at the asparagine site using site-directed mutagenesis. These FVIII variants are designated BDD FVIII-N41G, BDD FVIII-N239A, BDD FVIII-N1810A, and BDD FVIII-N2118A (Fig. 1A). N-glycopeptide analysis was performed to confirm the removal of site-specific N-glycans and characterize N-glycosylation profiles of the FVIII variants including wildtype BDD FVIII. From this analysis, we confirmed the absence of any glycans at each of the intended sites of the four FVIII variants (Fig. 1B).
Figure 1. Design and N-glycopeptide analysis of FVIII variants.
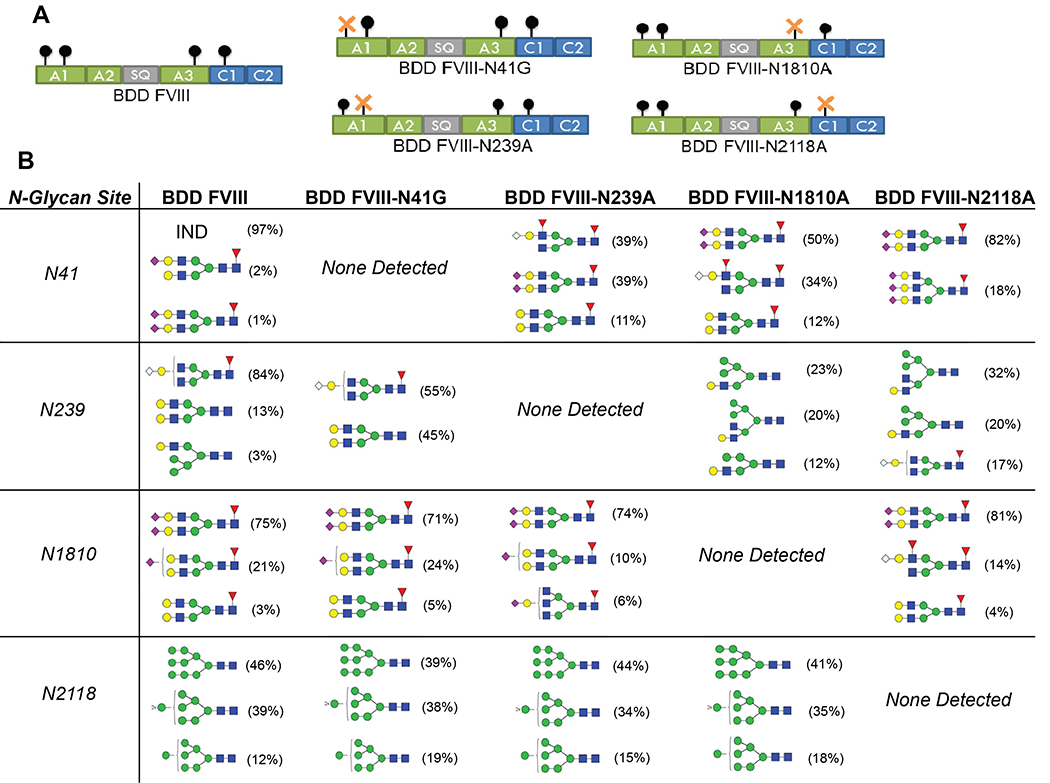
Schematic of BDD FVIII and the four FVIII variants with single-site N-glycan removal designated BDD FVIII-N41G, BDD FVIII-N239A, BDD FVIII-N1810A, and BDD FVIII-N2118A (A). N-glycopeptide analysis of all five FVIII proteins by LC-MS/MS (B). The frequency of the three most abundant N-glycoforms are displayed and were calculated by deconvolution of the full MS spectra then determining the area under the curve of an extraction ion chromatograph. IND, indeterminate.
N-glycopeptide analysis revealed a mixture of high-mannose, complex, and hybrid type N-glycans at N239 for BDD FVIII, BDD FVIII-N41G, BDD FVIII-N1810A, and BDD FVIII-N2118A. At N1810, there was a predominance of complex biantennary, terminally-fucosylated N-glycans with 71-81% of glycans consisting of N-acetylhexosamine (HexNAc) species, specifically HexNAc(4)-Hex(5)-Fuc(1)-NeuAc(2). Additional fucosylated species were observed in all the FVIII variants at residues N41, N239, and N1810. We identified an almost exclusive presence of high-mannose type carbohydrates at N2118, which is consistent with prior analyses of plasma-derived and rFVIII proteins [10, 32]. There was a predominance of the HexNAc(2)-Hex(9) glycan (relative abundance 39-46%) in all FVIII variants with the exception of BDD FVIII-N2118A, which is similar to the predominant glycoform observed at N2118 with commercially available CHO-derived BDD FVIII and BHK-derived FL-FVIII [32]. This is in contrast to a predominance of glycoform HexNAc(2)-Hex(8) at N2118 in CHO-derived B domain-truncated FVIII, HEK-derived BDD FVIII, and plasma-derived FVIII [10, 12]. Sialylated glycoforms N-acetylneuraminic acid (NeuAc) and Neu5Gc were detected in all five FVIII proteins and were primarily located at N41 and N1810. Potential N-glycosylation sites at N582 (A2 domain) and N1685 (A3 domain) were not glycosylated in the FVIII proteins, consistent with prior studies [10, 32].
Characterization of the FVIII variants
All of the FVIII variants had significantly reduced FVIII activity by BHK-M cells compared to BDD FVIII at 24 hours and/or 48 hours of expression (Fig. 2A and 2B). For pooled supernatants, there was a reduction in FVIII activity by 84% (BDD FVIII-N41G), 87% (BDD FVIII-N239A), 80% (BDD FVIII-N1810A), and 54% (BDD FVIII-2118A) compared to BDD FVIII. Following purification of pooled supernatants, specific activities of the purified FVIII variants were determined as 3,000-6,700 units/milligram (IU/mg) compared to 8,300 IU/mg for BDD FVIII (Table 2). FVIII activity and specific activity of purified FVIII variants were reduced by 90% & 64% (BDD FVIII-N41G), 87% & 28% (BDD FVIII-N239A), 86% & 33% (BDD FVIII-N1810A), and 57% & 19% (BDD FVIII-N2118A) compared to BDD FVIII. The AQs of each variant were similar to BDD FVIII with the exception of a 44% reduction for FVIII-N41G suggesting a less functional FVIIIa molecule (Table 2). The importance of N-glycosylation sites outside of the B domain on FVIII expression and activity was apparent with site-directed mutagenesis at all residues, but most notably at residue N41 (Fig. 2A and 2B, Table 2). Initial attempts to produce BDD FVIII-N41A and FVIII-N41Q (glutamine, Gln) variants abolished protein secretion by BHK and CHO cell lines as determined by ELISA and aPTT one-stage clotting assay of supernatant and cell lysates. Subsequent attempts sought to remove the N41 glycan through Ala substitution of the serine (Ser, S) 43 residue (BDD FVIII-S43A), taking into account that the transfer of pre-assembled glycoforms to Asn residues targets a consensus sequence of Asn-X-Ser/threonine where X consists of any amino acid except proline [33]. However, BDD FVIII-S43A was not detected within or expressed by BHK or CHO cell lines. Ultimately, we produced BDD FVIII-N41G through substitution of N41 with the Gly residue, which is also found in the porcine FVIII sequence [34]. Despite this, there was no difference in thrombin activation or VWF binding between the FVIII variants. These results suggest that all of the N-linked glycans outside of the B domain contribute to FVIII expression, in addition to an important role for the N41 residue in FVIII activity and activation.
Figure 2. Biochemical characterization of FVIII variants.
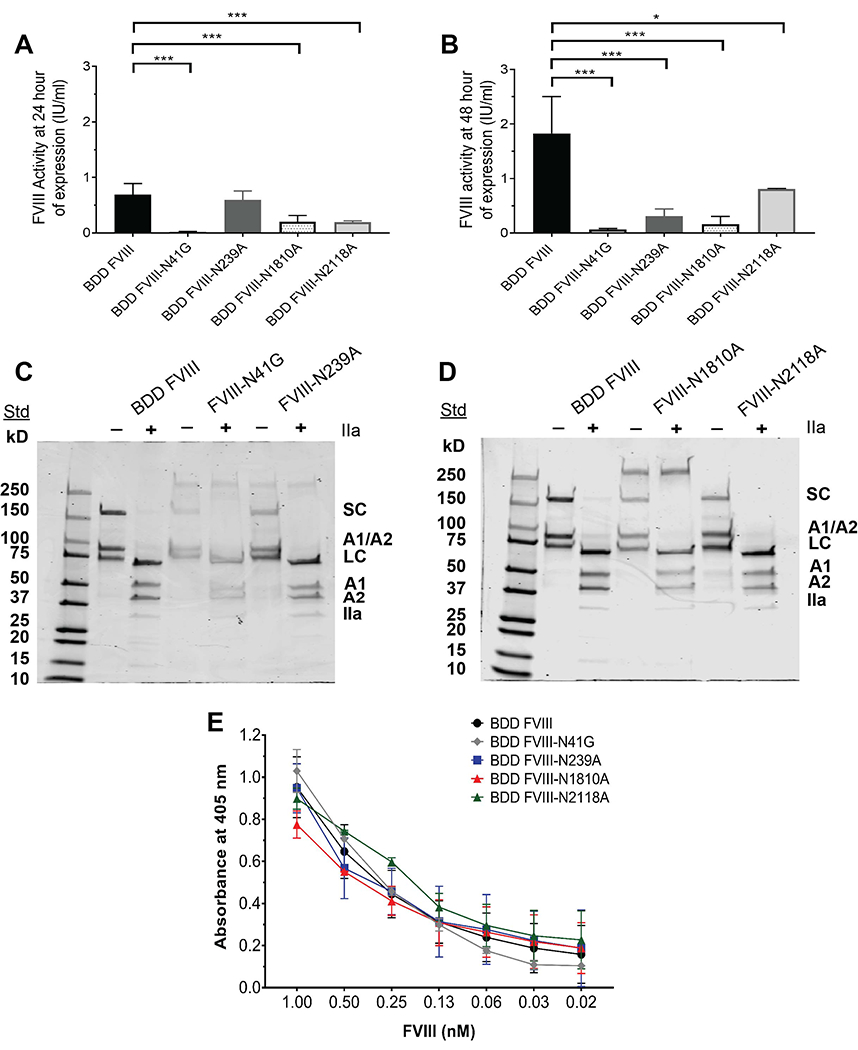
FVIII activity of expressed FVIII supernatant by transfected BHK-M cells at ~70% confluency at ~1 × 106 BHK-M cells for BDD FVIII and FVIII variants determined by the aPTT-based one-stage clotting assay at 24 hours (A) and 48 hours (B). FVIII activity (A and B) are presented as means ± SD of 2-5 measurements per FVIII variant. BDD FVIII activity measurements reflect 4 representative and separate preparations of the protein and the modified FVIII variant reflect one preparation of the proteins. SDS-PAGE of BDD FVIII (C) and FVIII variants (D) are shown. SDS-PAGE was performed in the presence and absence of human thrombin (IIa). A1, FVIII A1 domain; A2, FVIII A2 domain; A1/A2, non-covalently linked A1/A2 domains; LC, FVIII light chain comprised of the A3, C1, and C2 domains; SC, FVIII single chain comprised of the A1, A2, A3, C1, and C2 domains. Binding of BDD FVIII and FVIII variants to human-derived VWF by ELISA is shown (E). Differences in the mean FVIII activity of each of the FVIII variants were compared to the mean activity of BDD FVIII by one-way ANOVA with Dunnett’s correction for multiple comparison. *P <0.05 and ***P <0.001.
Table 2.
FVIII Activities and Activation Quotients
| Pooled Supernatant | Purified Protein | ||||
|---|---|---|---|---|---|
| FVIII Variant | Activity (IU/ml) | AQ† | Activity (IU/ml) | Specific Activity (IU/mg) | AQ† |
| BDD FVIII | 1.18 | 41 | 2800 | 8300 | 78 |
| BDD FVIII-N41G | 0.19 | 35 | 280 | 3000 | 44 |
| BDD FVIII-N239A | 0.15 | 42 | 370 | 6000 | 76 |
| BDD FVIII-N1810A | 0.24 | 37 | 380 | 5600 | 76 |
| BDD FVIII-N2118A | 0.54 | 76 | 1200 | 6700 | 76 |
AQ, activation quotient, is defined as the FVIII activity in the two-stage assay divided by the one-stage assay activity expressed in units per ml. An AQ <20 were excluded due to an increased risk of contaminating activated FVIII in the purified preparation [27].
Protein purity and integrity of thrombin cleavage sites of the FVIII variants were evaluated using SDS-PAGE (Fig. 2C and 2D). Similar to BDD FVIII, the four FVIII variants had bands corresponding to a FVIII single chain, A1/A2 domains, and light chain prior to thrombin cleavage. Following thrombin cleavage, the FVIII light chain, A1 domain, and A2 domain bands were present for each of the FVIII variants comparable to BDD FVIII. This demonstrates that the thrombin cleavage sites at arginine (Arg, R) 372, R740, and R1689 [35] remained intact with site-specific removal of N-glycans permitting FVIII activation.
Lastly, we evaluated the effect of single-site N-glycan removal on the binding of FVIII variants to human-derived VWF. There were no differences in binding of the four FVIII variants to VWF compared to BDD FVIII at FVIII concentrations of 0.02-1 nM (Fig. 2E). Residue N1810 is located within a region of the A3 domain that has been implicated in VWF binding [36], however, there were no differences in the VWF binding capacity of BDD FVIII-N1810A that lacked a N-glycan at this site. This suggests that the loss of this residue and its associated carbohydrate structure does not significantly impact VWF binding.
Removal of N-glycans at N41 and N1810 alters FVIII internalization by BMDCs
While the endocytic receptor responsible for FVIII internalization by antigen presenting cells (APCs) is unknown, oligosaccharides can interact with C-type lectin receptors on the surface of APCs which may influence antigen recognition [37, 38]. Moreover, residues within the C1 and C2 domains have been described as regions that modulate FVIII uptake by APCs [39]. We evaluated whether single-site removal of N-linked glycans would affect FVIII internalization by murine BMDCs. BDD FVIII-N41G and BDD FVIII-N1810A had significantly reduced internalization by 59% (BDD FVIII-N41G mean FVIII uptake 26 ± 10%, P = 0.0049) and 74% (BDD FVIII-N1810A mean FVIII uptake 17 ± 9%, P = 0.0005), respectively, compared to internalization of BDD FVIII (mean FVIII uptake 61 ± 16%) (Fig. 3A and B). There were no differences in internalized BDD FVIII-N239A, BDD FVIII-N2118A, and positive control FL-FVIII compared to BDD FVIII. These results suggest that the amino acid residue or the complex biantennary fucosylated N-glycans at N41 and N1810 may modulate FVIII recognition by dendritic cells by a yet to be determined receptor.
Figure 3. Endocytosis of FVIII variants by murine bone marrow-derived dendritic cells.
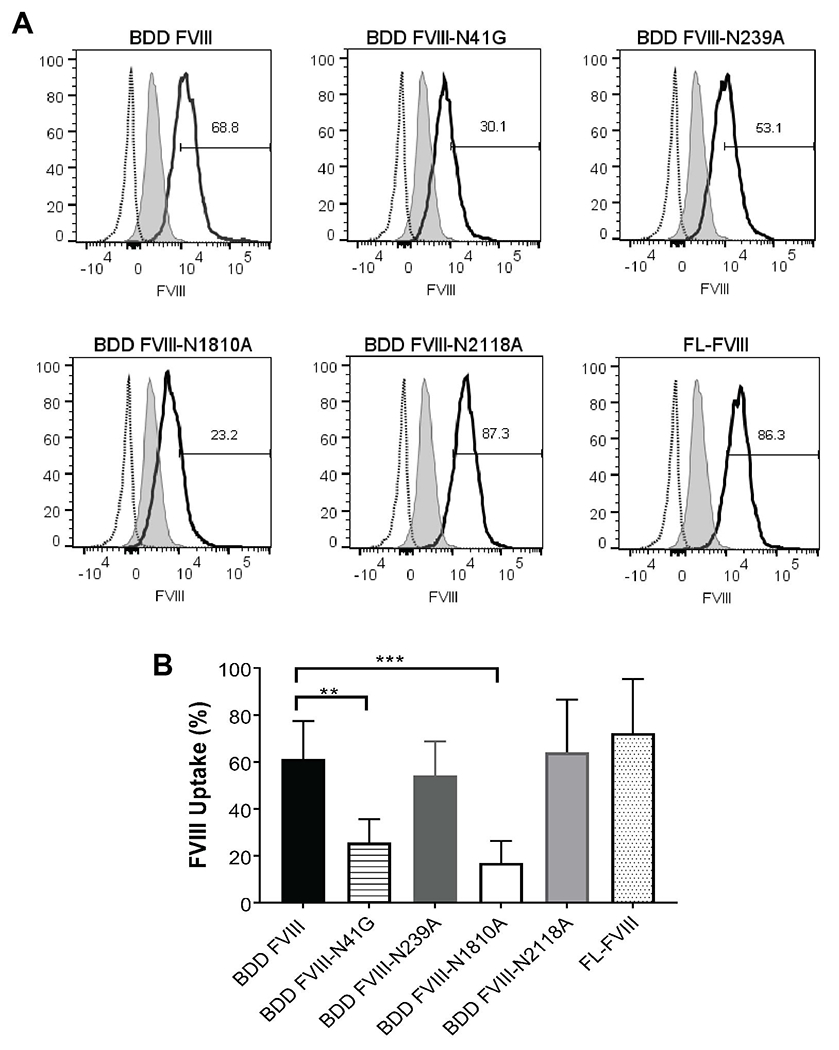
Representative histograms of endocytosis of BDD FVIII and FVIII variants by murine BMDCs detected by DyLight 650-conjugated C2 domain MAb 2-117 (A). Full length FVIII (FL-FVIII) served as positive control. The histogram with the dotted line represents CD11c+ BMDCs treated with serum free medium, which was used to define the starting cut-off point for percent FVIII uptake (i.e. 2 SD above the end of this histogram). The gray shaded histogram represents CD11c+ BMDCs treated with serum free medium and incubated with DyLight 650-MAb 2-117 after permeabilization of BMDCs. The curve with a solid black line represents the histogram of BDD FVIII, FVIII variant, or FL-FVIII. FVIII uptake by FVIII variants are summarized (B). Internalized FVIII percentages were normalized to unstained BMDCs treated with serum-free medium. Differences in uptake of the FVIII variants of 6 replicate measurements (2 replicates on 3 separate days) were compared to BDD FVIII by one-way ANOVA with Dunnett’s correction for multiple comparison. **P <0.01 and ***P <0.001.
Immunization of FVIII−/− mice with FVIII variants induces antibody responses
Next, we evaluated whether differences in internalization of the FVIII variants by BMDCs would translate to altered antibody responses in vivo. FVIII−/− mice were immunized with 1 μg of BDD FVIII variants for four weekly retro-orbital injections followed by a boost 2 μg injection one week later (Fig. 4A). There were no differences in anti-FVIII IgG ELISA or Bethesda titers between FVIII−/− mice immunized with BDD FVIII or the FVIII variants (Fig. 4B and C). Although removal of the predominant complex biantennary type glycans at N41 resulted in reduced uptake by BMDCs, BDD FVIII-N41G showed a trend towards increased anti-FVIII IgG and Bethesda titers compared to BDD FVIII (BDD FVIII-N41G median ELISA and Bethesda titers, 1100 & 97 BU/mL; BDD FVIII median ELISA and Bethesda titers, 675 & 46 BU/mL). However, this difference was not statistically significant. This may indicate presentation of alternate FVIII epitopes or a sufficient number of FVIII variant internalized to activate CD4+ T cells despite reduced uptake by BMDCs.
Figure 4. Immunization of FVIII−/− mice with BDD FVIII or FVIII variants demonstrate similar antibody responses.
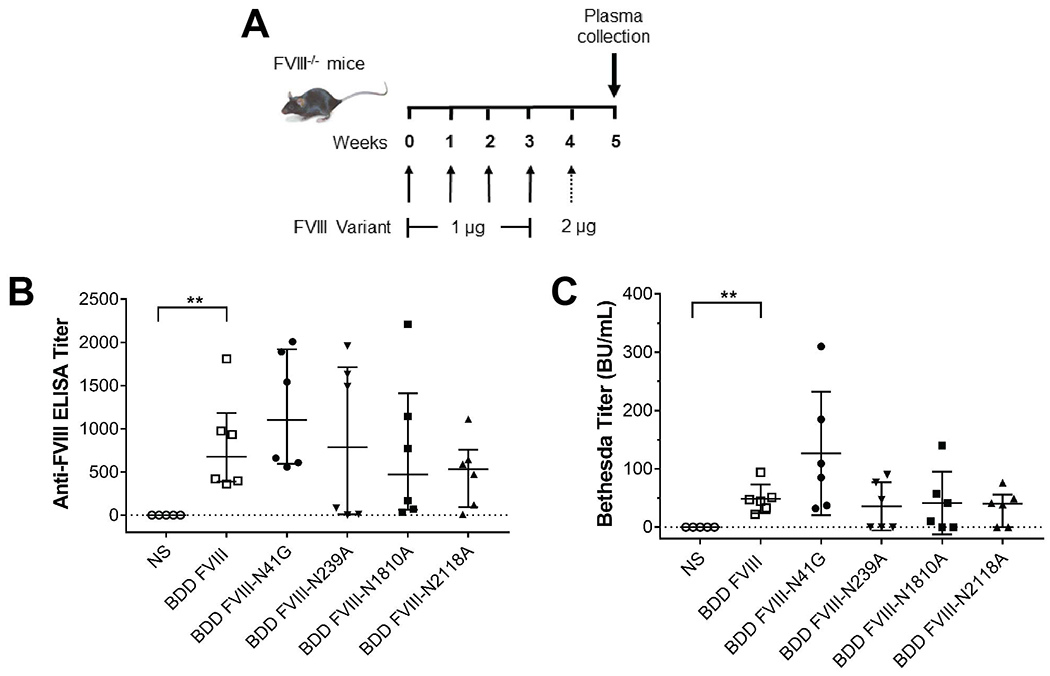
Schematic of the immunization regimen by retro-orbital injection in 8-12 week old FVIII−/− mice with the BDD FVIII proteins (A). Anti-FVIII IgG antibody titers by ELISA (B) and inhibitory antibodies by Bethesda assay (C) in FVIII−/− mice are shown. Mice injected with normal saline (NS) served as a negative control. Differences in the median ELISA and Bethesda titers of each of the FVIII variants were compared to median titers of BDD FVIII mice by the non-parametric Mann Whitney U test. **P <0.01.
N-glycan modifications alter binding of domain-specific FVIII MAbs
We evaluated the effect of single-site N-glycan removal on binding of a repertoire of murine anti-human FVIII MAbs directed against the A1, A2, A3, C1, and C2 domains (Table 1). Six of the 12 (50%) MAbs had significantly reduced binding to ≥1 of the FVIII variants when compared to BDD FVIII binding (Fig. 5A). With the exception of A2 MAb 1D4, N-linked glycan removal at N41 did not reduce binding of the majority of FVIII MAbs. For BDD FVIII-N239A (A1 domain N-glycan removed), there was a significant reduction in binding by A1 MAb 2-116 (Percent reduction in MAb binding: 50 ± 33%), A2 MAb 1D4 (34 ± 12%), and C1 MAb 2A9 (22 ± 12%) compared to BDD FVIII and FL-FVIII (Fig. 5A and 5B). There was a 17-32% reduction in binding of A2 MAb 1D4, A3 MAb 2-113, and both C1 MAbs B136 and 2A9 to BDD FVIII-N1810A with the N-glycan in the A3 domain removed. Lastly, there was decreased binding of A2 MAbs 4F4 and 1D4 by 18% and 27%, respectively, in addition to C1 MAbs B136 and 2A9 by 16% and 18%, respectively, to BDD FVIII-N2118A (C1 domain N-glycan removed). There was increased binding of A1 MAb 2-116 to FVIII-N41G (20 ± 3%) (Fig. 5A), however the remainder of FVIII MAbs did not show increased binding to the FVIII-N239A, FVIII-N1810A, and FVIII-N2118A variants. There was no increased binding of MAbs to the FVIII variants when compared to FL-FVIII binding (Fig. 5B).
Figure 5. Effect of FVIII variants on binding of domain-specific FVIII MAbs.
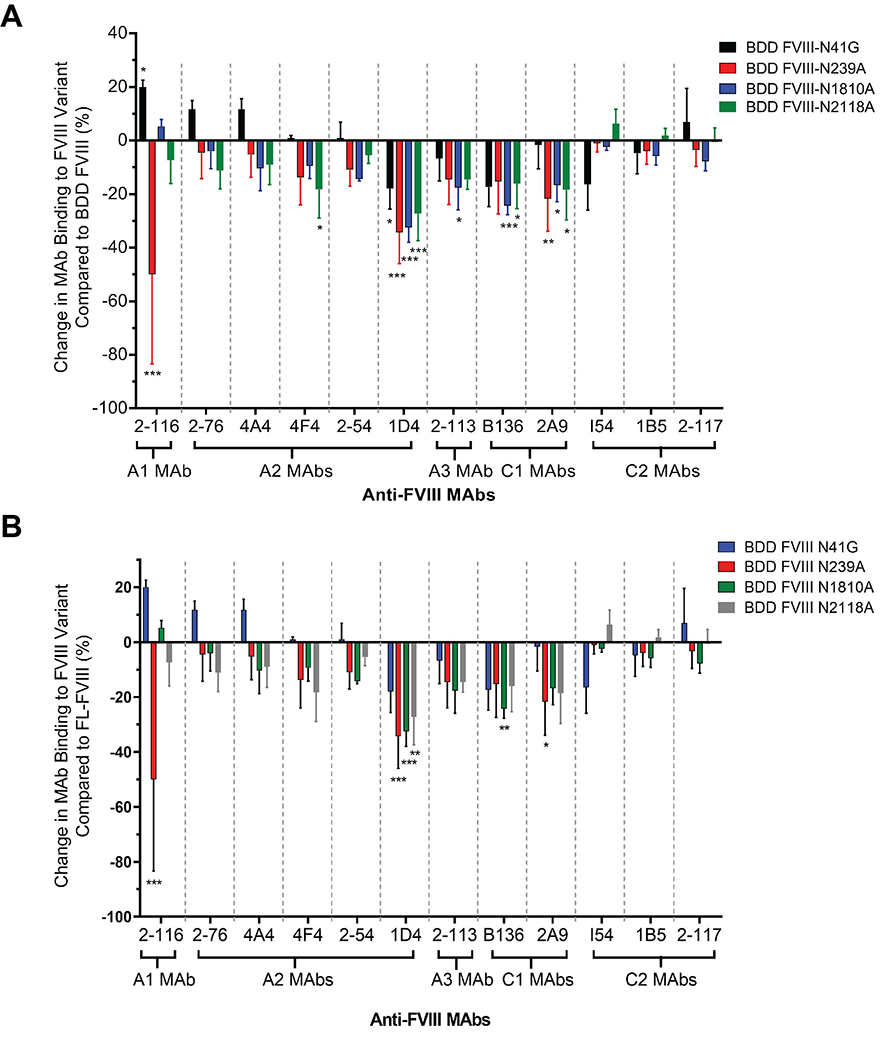
Percent change in FVIII MAb binding to FVIII variants compared to BDD FVIII (A) and FL-FVIII (B) are shown. Negative percent change in MAb binding represents reduced MAb binding to FVIII variants compared to BDD FVIII/FL-FVIII and positive percent change in MAb binding represents increased MAb binding. Differences in binding for each MAb and FVIII variants by ELISA performed on 3 separate days were compared to BDD FVIII and FL-FVIII using an ordinary two-way ANOVA with Dunnett’s correction for multiple comparisons. For BDD FVIII comparison: The main effect of MAbs yielded an F ratio of F(11, 108) = 9.156, P <0.0001, demonstrating a significant difference between FVIII MAbs. The main effect of FVIII variants yielded an F ratio of F(4, 108) = 23.56, P <0.0001, demonstrating a significant difference between FVIII variants. For FL-FVIII comparison: The main effect of MAbs yielded an F ratio of F(11, 96) = 7.461, P <0.0001, demonstrating a significant difference between FVIII MAbs. The main effect of FVIII variants yielded an F ratio of F(4, 96) = 17.98, P <0.0001, demonstrating a significant difference between FVIII variants. The interaction between FVIII MAb and FVIII variants was significant for the BDD FVIII comparison at F(44, 108) = 3.874, (P <0.0001) and FL-FVIII comparison at F(44, 96) = 3.284 (P <0.0001). *P <0.05, **P <0.01 and ***P <0.001.
The IgG subclass of FVIII MAbs did not appear to contribute to MAb binding to FVIII variants. The 6 MAbs with reduced binding to at least one FVIII variant were of the IgG1 (1 MAb) and IgG2a (5 MAbs) subclass. Of the 6 MAbs with unchanged binding to FVIII variants, there were 1 IgG1, 4 IgG2a, and 1 IgG2b MAbs. It is unclear whether reductions in MAb binding to the FVIII variants is a result of the loss of the N-glycan, an allosteric effect of site-specific mutagenesis, or a combination of both. However, these findings suggest that the FVIII variants could serve as a tool to investigate FVIII recognition by affinity-matured FVIII antibodies in memory B cell responses to FVIII upon FVIII re-exposure, as in the case of immune tolerance induction therapy [40].
Mapping the A1 domain MAb 2-116 epitope
A1 MAb 2-116 had the most significant reduction in binding to BDD FVIII-N239A of all the FVIII variants and MAbs tested (Fig. 5). We used HDX-MS to identify the binding epitope for MAb 2-116 and assess the effect of antibody binding on FVIII (Fig. 6A). Analysis of hydrogen-deuterium exchange data revealed a decrease in deuterium uptake at residues Glu11-Asp15 and Glu53-Ala78 of FL-FVIII in complex with MAb 2-116 compared to FL-FVIII alone (Fig. 6B–E). When mapped onto a 3D structure of the A1/A2 domains, these residues clustered to a solvent exposed region of the A1 domain (Fig. 6F), providing evidence that these residues comprise the epitope of MAb 2-116. No other significant differences in deuterium uptake between FL-FVIII and FL-FVIII/MAb 2-116 were observed for the rest of FVIII sequences (Fig. 6G), suggesting that this MAb does not alter native FVIII dynamics beyond its epitope region.
Figure 6. B cell epitope mapping of A1 domain MAb 2-116.
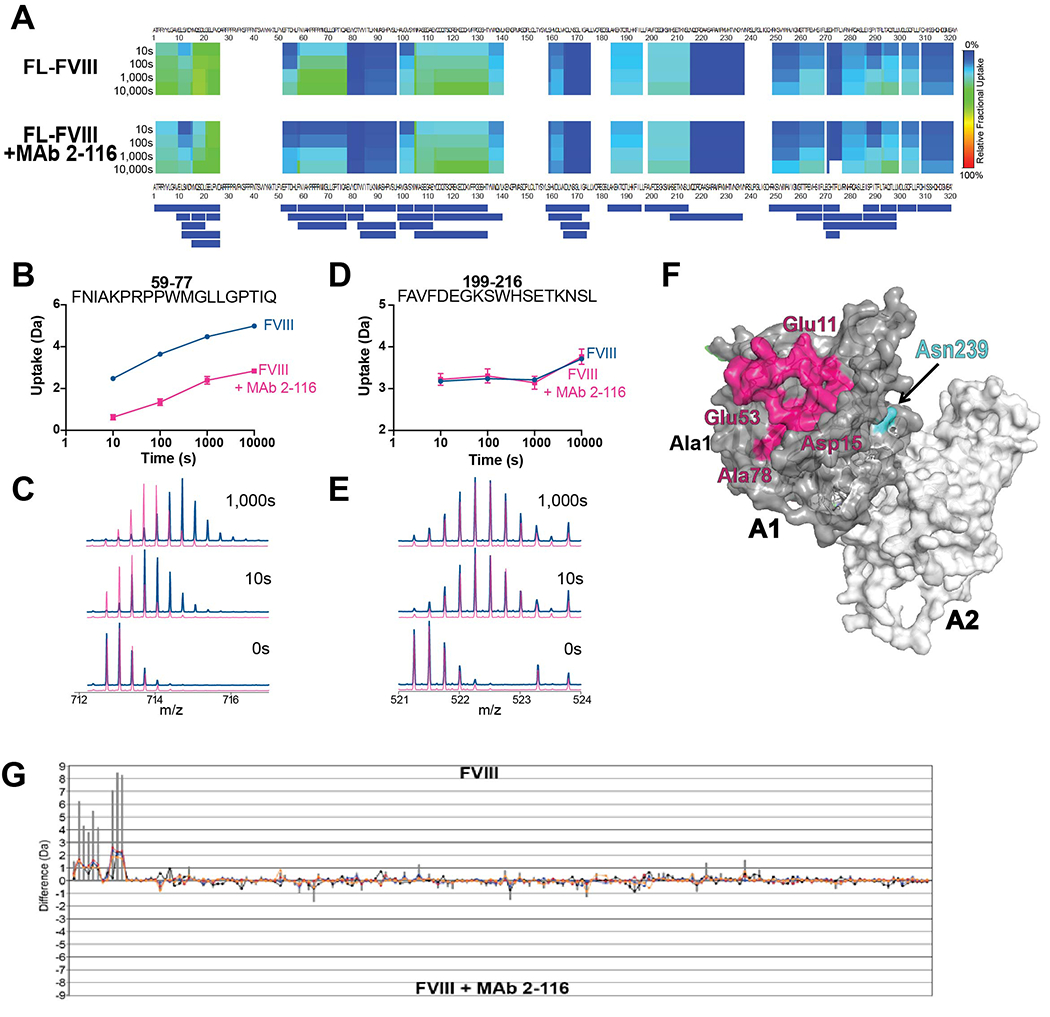
Relative fractional deuterium uptake heat map between residues Ala1-Glu322 over time between FL-FVIII or FL-FVIII/2-116 complex (A). The peptide coverage map is shown below the heat map. Uptake plots of selected peptides at measured time points with corresponding overlaid spectra depicting deuterium uptake patterns of FL-FVIII alone (blue) or FL-FVIII/2-116 complex (magenta) is shown (B-E). Three-dimensional structure of FVIII A1 domain (gray) and A2 domain (white) showing the putative MAb 2-116 binding site spanning residues Glu11-Asp15 and Glu53-Ala78 (magenta) with residue Asn239 (cyan) is shown (PDB: 2R7E) (F). The difference index of deuterium uptake between FL-FVIII and FL-FVIII/MAb 2-116 of all identified peptides in FL-FVIII is shown (G).
Discussion
N-glycosylation is a post-translational modification that contributes to FVIII stability, folding, intracellular trafficking, and secretion [41, 42]. N-glycans within the B domain also mediate the interaction of FVIII with the C-type lectin receptor ASGPR, which may help regulate FVIII clearance [8]. Despite the majority of N-glycans occurring in the B domain, removal of the B domain does not negatively impact FVIII procoagulant function or immunogenicity [43–47]. For this reason, we focused on the effect of single-site removal of N-glycans outside of the B domain for this study. N-glycan structures consist of high-mannose type, complex type, or hybrid type structures [9], which are all found in plasma-derived and rFVIII products [12]. Canis et al. described a predominance of sialylated complex biantennary core-fucosylated type N-glycans at N41, N1810, and glycans within the B domain, while the high mannose type N-glycans predominate at N239 and N2118 [10, 32]. Overall, the wildtype BDD FVIII and FVIII variants with single-site N-glycan removal produced in this study had similar N-glycan profiles to commercially available BDD FVIII products [32].
Selvaraj et al. characterized the effect of single-site N-glycan removal derived from African green monkey kidney fibroblast-like cell line COS-1 [48] and CHO cells on BDD FVIII biochemical properties and secretion [49]. They reported a significant reduction in protein secretion and function with FVIII Asn239Gln [49]. Asn41Gln and Asn1810Gln had modest reductions of protein secretion and function, but there were no differences with Asn2118Gln compared to wildtype BDD FVIII. The authors concluded that N239 was the most critical N-glycan site for proper FVIII folding and secretion. Interestingly, A1 domain residues 227-336 have been implicated in the interaction of FVIII derived from COS-1 cells with the molecular chaperone immunoglobulin-binding protein (BiP) in which stable interaction mediates shuttling of glycoproteins to degradation pathways inhibiting secretion and substitution of this FVIII region with the homologous factor V region increased FVIII expression [50, 51]. Other studies evaluating N239 and/or N2118 variants did not include a N41 variant for comparison of FVIII expression or activity [15, 52]. In this study, all FVIII variants had significantly reduced expression by BHK-M cells compared to BDD FVIII. However, the N41 residue appeared to be a critical site for FVIII expression as well as FVIII activity and co-factor function. This may be due to differences in the FVIII production process and reagents used between the studies such as the expression plasmids, BDD FVIII cDNA, or mammalian cell lines. However, it is possible that the effect of glycine on α-helical backbone of polypeptides may have contributed to reduced N41G expression [53]. Similar to our study, there were no differences in FVIII binding to VWF between BDD FVIII and the FVIII variants. This indicates that multiple variables may affect FVIII biosynthesis, folding, and expression but ultimately spare key FVIII biochemical properties.
The role of N-linked glycans on FVIII recognition and endocytosis by APCs has been described. Prior studies showed that N-glycans modulate FVIII uptake by human monocyte-derived dendritic cells through the macrophage mannose receptor (MMR), which can affect FVIII presentation to CD4+ T cells [15, 38]. However, the role of MMR of FVIII uptake utilizing human monocyte-derived DCs have been conflicting with studies demonstrating differing outcomes with MMR blockade or siRNA-mediated knockdown [29, 38]. Mannose-containing oligosaccharides linked to N239 and N2118 are thought to mediate the interaction of FVIII with the MMR by murine BMDCs [7]. However, removal of the high-mannose type glycans at N239 and N2118 in this study did not reduce FVIII endocytosis by BMDCs. This is in line with prior studies demonstrating that the mechanism of FVIII internalization by murine BMDCs is independent of the MMR [54]. It is less likely that changes in protein confirmation contributed to reductions in internalization of FVIII variants as significant alterations in protein confirmation with altered N-linked glycosylation has not been observed [55]. Although there was reduced uptake of FVIII-N41G and FVIII-N1810A by murine BMDCs, this finding did not translate to reduced antibody responses in vivo in immunized FVIII−/− mice. In fact, there was a trend towards increased anti-FVIII IgG ELISA and Bethesda titers in FVIII−/− mice immunized with BDD FVIII-N41G, albeit this was not statistically significant. Site-directed mutagenesis of N2118 did not affect FVIII uptake by BMDCs, similar to a study that also showed reduced uptake of a modified FVIII2118Q protein by human-derived dendritic cells but not murine BMDCs [15]. Although the primary receptor and mechanism responsible for FVIII recognition and uptake by APCs remains elusive, it is possible that multiple receptors and/or mechanisms for FVIII recognition and uptake exist that differ by species.
Single-site removal of N-glycans reduced binding of various affinity-matured domain-specific FVIII MAbs representing each MAb grouping of each FVIII domain [17–20, 56]. Many of these MAbs are clinically relevant as they compete for FVIII binding with antibodies present in hemophilia A patient plasmas with inhibitors [20, 57, 58]. Interestingly, there was reduced binding of A2 MAb 1D4 to all FVIII variants compared to BDD FVIII and 75% of variants compared to FL-FVIII. MAb 1D4 binds the Glu604-Arg740 epitope at the C-terminus of the A2 domain. There were no differences in A2 MAb 2-54 binding, which also recognizes the Glu604-Arg740 epitope, to the FVIII variants. This suggests that reduced binding of A2 MAb 1D4 to the FVIII variants is independent of the B cell epitope and removal of the large glycan footprint at a single site, but the exact mechanism is unclear. Other factors such as reduced FVIII variant/MAb binding affinity due to alterations in FVIII variant folding may contribute to this finding.
In evaluating the effect of the FVIII variants on FVIII MAb binding, A1 domain MAb 2-116 had a significant ~50% mean reduction in binding to BDD FVIII-N239A. MAb 2-116 is a non-inhibitory MAb that is the sole A1 MAb in our repertoire and the critical binding epitope was previously unknown. To determine whether N239 epitope is a critical binding site, we performed HDX-MS of FL-FVIII with and without MAb 2-116. HDX-MS mapping revealed Glu11-Asp15 and Glu53-Ala78 as the likely A1 domain binding regions for MAb 2-116. Although these regions do not include the N239 epitope (Fig. 6F), N-linked glycans are large covalently-linked molecules that can lie across the FVIII protein surface and mask B cell epitopes [13]. The removal of the N239-linked glycan footprint likely contributed to altered binding of MAb 2-116 to FVIII-N239A resulting in differing MAb 2-116 binding regions identified on FVIII in the HDX-MS analysis. Additionally, HDX-MS analysis of MAb 2-116 binding to FVIII utilized FL-FVIII to ensure appropriate coverage of the entire FVIII protein to examine FVIII dynamics outside of the A1 domain. FVIII MAb binding to FVIII variants were performed by ELISA using FVIII proteins that lacked the B domain. Slight differences in FL-FVIII and BDD FVIII protein folding and 3-D structure may have further contributed to differences in the surface interaction of MAb 2-116 at the FVIII region that encompasses epitope 239, which was further impaired by the absence of the N239-linked glycan with the FVIII-N239A variant. Alternatively, it is possible that removal of the N-glycan at 239 resulted in an allosteric effect that affected MAb 2-116 binding affinity in the ELISA. Nevertheless, the absence of N-glycan at A1 domain residue 239 significantly affected binding of A1 MAb 2-116.
The development and management of FVIII inhibitors remains a challenge in modern hemophilia A care. While the N-glycosylation profile of FVIII is complex, unanswered questions about the role of N-linked carbohydrate structures on FVIII immunity warrant further investigation. Utilizing a variety of multi-disciplinary and novel tools, reagents, and approaches, including rFVIII proteins, may help to illuminate mechanisms of FVIII recognition, processing, and presentation by APCs. In this study, we describe the effect of single-site N-glycan removal of BDD FVIII on FVIII biochemical properties, dendritic cell uptake, and antibody formation in immunized FVIII−/− mice. We demonstrate that single-site N-glycan removal reduced binding of FVIII by half of domain-specific FVIII MAbs that recognize clinically relevant B cell epitopes but do not significantly alter antibody development.
Essentials.
Factor VIII (FVIII) is a glycosylated protein with 25 potential asparagine (N)-linked glycans
N-linked glycan removal at N41 & N1810 reduces FVIII endocytosis but not de novo antibody formation
Site-specific N-glycan removal reduces binding of multiple domain-specific monoclonal antibodies
The role of N-linked glycans on FVIII innate & adaptive immunity warrants further investigation
Acknowledgements
This work was supported by NIH NHLBI K99/R00 grant HL150595 (G.B.), Hemophilia of Georgia Clinical Scientist Development Award (G.B.), Aflac Cancer and Blood Disorders Center Pilot Grant (G.B.), Hemophilia of Georgia Center for Bleeding & Clotting Disorders of Emory Hemostasis Research Award (G.B.), NIH F31 fellowship grant HL149357 (E.R.L), and NIH grant HL141981 and HL143794 (R.L). We thank Dr. Parastoo Azadi and analyst Stephanie Archer-Hartmann at the Complex Carbohydrate Research Center at the University of Georgia, Athens, GA for their N-glycopeptide mapping of the FVIII proteins (NIH grant GM137782).
Footnotes
Conflicts of Interest
G.B. has received honoraria for advisory board participation from Bayer, Genentech, and Octapharma. The remaining authors declare no interests that might be perceived as posing a conflict or bias.
References
- 1.Srivastava A, Santagostino E, Dougall A, Kitchen S, Sutherland M, Pipe SW, Carcao M, Mahlangu J, Ragni MV, Windyga J, Llinás A, Goddard NJ, Mohan R, Poonnoose PM, Feldman BM, Lewis SZ, van den Berg HM, Pierce GF. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020; 26 Suppl 6: 1–158. 10.1111/hae.14046. [DOI] [PubMed] [Google Scholar]
- 2.Dimichele D Inhibitors: resolving diagnostic and therapeutic dilemmas. Haemophilia. 2002; 8: 280–7. [DOI] [PubMed] [Google Scholar]
- 3.Collins PW, Chalmers E, Hart DP, Liesner R, Rangarajan S, Talks K, Williams M, Hay CR. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: (4th edition). UK Haemophilia Centre Doctors Organization. Br J Haematol. 2013; 160: 153–70. 10.1111/bjh.12091. [DOI] [PubMed] [Google Scholar]
- 4.Astermark J, Lacroix-Desmazes S, Reding MT. Inhibitor development. Haemophilia. 2008; 14 Suppl 3: 36–42. 10.1111/j.1365-2516.2008.01711.x. [DOI] [PubMed] [Google Scholar]
- 5.Gouw SC, van den Berg HM. The multifactorial etiology of inhibitor development in hemophilia: genetics and environment. Semin Thromb Hemost. 2009; 35: 723–34. 10.1055/s-0029-1245105. [DOI] [PubMed] [Google Scholar]
- 6.Vehar GA, Keyt B, Eaton D, Rodriguez H, O’Brien DP, Rotblat F, Oppermann H, Keck R, Wood WI, Harkins RN, Tuddenham EG, Lawn RM, Capon DJ. Structure of human factor VIII. Nature. 1984; 312: 337–42. 10.1038/312337a0. [DOI] [PubMed] [Google Scholar]
- 7.Medzihradszky KF, Besman MJ, Burlingame AL. Structural characterization of site-specific N-glycosylation of recombinant human factor VIII by reversed-phase high-performance liquid chromatography-electrospray ionization mass spectrometry. Anal Chem. 1997; 69: 3986–94. 10.1021/ac970372z. [DOI] [PubMed] [Google Scholar]
- 8.Bovenschen N, Rijken DC, Havekes LM, van Vlijmen BJ, Mertens K. The B domain of coagulation factor VIII interacts with the asialoglycoprotein receptor. Journal of thrombosis and haemostasis : JTH. 2005; 3: 1257–65. 10.1111/j.1538-7836.2005.01389.x. [DOI] [PubMed] [Google Scholar]
- 9.Preston RJ, Rawley O, Gleeson EM, O’Donnell JS. Elucidating the role of carbohydrate determinants in regulating hemostasis: insights and opportunities. Blood. 2013; 121: 3801–10. 10.1182/blood-2012-10-415000. [DOI] [PubMed] [Google Scholar]
- 10.Qu J, Ma C, Xu XQ, Xiao M, Zhang J, Li D, Liu D, Konkle BA, Miao CH, Li L, Xiao W. Comparative glycosylation mapping of plasma-derived and recombinant human factor VIII. PloS one. 2020; 15: e0233576. 10.1371/journal.pone.0233576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai J, Hough C, Tarrant J, Lillicrap D. Biological considerations of plasma-derived and recombinant factor VIII immunogenicity. Blood. 2017; 129: 3147–54. 10.1182/blood-2016-11-750885. [DOI] [PubMed] [Google Scholar]
- 12.Hironaka T, Furukawa K, Esmon PC, Fournel MA, Sawada S, Kato M, Minaga T, Kobata A. Comparative study of the sugar chains of factor VIII purified from human plasma and from the culture media of recombinant baby hamster kidney cells. The Journal of biological chemistry. 1992; 267: 8012–20. [PubMed] [Google Scholar]
- 13.Kannicht C, Ramstrom M, Kohla G, Tiemeyer M, Casademunt E, Walter O, Sandberg H. Characterisation of the post-translational modifications of a novel, human cell line-derived recombinant human factor VIII. Thrombosis research. 2013; 131: 78–88. 10.1016/j.thromres.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Lai JD, Swystun LL, Cartier D, Nesbitt K, Zhang C, Hough C, Dennis JW, Lillicrap D. N-linked glycosylation modulates the immunogenicity of recombinant human factor VIII in hemophilia A mice. Haematologica. 2018; 103: 1925–36. 10.3324/haematol.2018.188219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delignat S, Rayes J, Dasgupta S, Gangadharan B, Denis CV, Christophe OD, Bayry J, Kaveri SV, Lacroix-Desmazes S. Removal of Mannose-Ending Glycan at Asn(2118) Abrogates FVIII Presentation by Human Monocyte-Derived Dendritic Cells. Front Immunol. 2020; 11: 393. 10.3389/fimmu.2020.00393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Healey JF, Parker ET, Barrow RT, Langley TJ, Church WR, Lollar P. The humoral response to human factor VIII in hemophilia A mice. Journal of thrombosis and haemostasis : JTH. 2007; 5: 512–9. 10.1111/j.1538-7836.2007.02373.x. [DOI] [PubMed] [Google Scholar]
- 17.Markovitz RC, Healey JF, Parker ET, Meeks SL, Lollar P. The diversity of the immune response to the A2 domain of human factor VIII. Blood. 2013; 121: 2785–95. 10.1182/blood-2012-09-456582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meeks SL, Healey JF, Parker ET, Barrow RT, Lollar P. Antihuman factor VIII C2 domain antibodies in hemophilia A mice recognize a functionally complex continuous spectrum of epitopes dominated by inhibitors of factor VIII activation. Blood. 2007; 110: 4234–42. 10.1182/blood-2007-06-096842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Summers RJ, Meeks SL, Healey JF, Brown HC, Parker ET, Kempton CL, Doering CB, Lollar P. Factor VIII A3 domain substitution N1922S results in hemophilia A due to domain-specific misfolding and hyposecretion of functional protein. Blood. 2011; 117: 3190–8. 10.1182/blood-2010-09-307074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Batsuli G, Deng W, Healey JF, Parker ET, Baldwin WH, Cox C, Nguyen B, Kahle J, Konigs C, Li R, Lollar P, Meeks SL. High-affinity, non-inhibitory pathogenic C1 domain antibodies are present in patients with hemophilia A and inhibitors. Blood. 2016; 128: 2055–67. 10.1182/blood-2016-02-701805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parker ET, Lollar P. Conformation of the von Willebrand factor/factor VIII complex in quasi-static flow. The Journal of biological chemistry. 2021; 296: 100420. 10.1016/j.jbc.2021.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrow RT, Healey JF, Jacquemin MG, Saint-Remy JM, Lollar P. Antigenicity of putative phospholipid membrane-binding residues in factor VIII. Blood. 2001; 97: 169–74. [DOI] [PubMed] [Google Scholar]
- 23.Horton RM, Ho SN, Pullen JK, Hunt HD, Cai Z, Pease LR. Gene splicing by overlap extension. Methods in enzymology. 1993; 217: 270–9. [DOI] [PubMed] [Google Scholar]
- 24.Lind P, Larsson K, Spira J, Sydow-Backman M, Almstedt A, Gray E, Sandberg H. Novel forms of B-domain-deleted recombinant factor VIII molecules. Construction and biochemical characterization. Eur J Biochem. 1995; 232: 19–27. [DOI] [PubMed] [Google Scholar]
- 25.Meeks SL, Cox CL, Healey JF, Parker ET, Doshi BS, Gangadharan B, Barrow RT, Lollar P. A major determinant of the immunogenicity of factor VIII in a murine model is independent of its procoagulant function. Blood. 2012; 120: 2512–20. 10.1182/blood-2012-02-412361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lollar P, Fay PJ, Fass DN. Factor VIII and factor VIIIa. Methods in enzymology. 1993; 222: 128–43. 10.1016/0076-6879(93)22010-d. [DOI] [PubMed] [Google Scholar]
- 27.Doering C, Parker ET, Healey JF, Craddock HN, Barrow RT, Lollar P. Expression and characterization of recombinant murine factor VIII. Thromb Haemost. 2002; 88: 450–8. [PubMed] [Google Scholar]
- 28.Valliere-Douglass JF, Eakin CM, Wallace A, Ketchem RR, Wang W, Treuheit MJ, Balland A. Glutamine-linked and non-consensus asparagine-linked oligosaccharides present in human recombinant antibodies define novel protein glycosylation motifs. The Journal of biological chemistry. 2010; 285: 16012–22. 10.1074/jbc.M109.096412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herczenik E, van Haren SD, Wroblewska A, Kaijen P, van den Biggelaar M, Meijer AB, Martinez-Pomares L, ten Brinke A, Voorberg J. Uptake of blood coagulation factor VIII by dendritic cells is mediated via its C1 domain. The Journal of allergy and clinical immunology. 2012; 129: 501–9, 9.e1–5. 10.1016/j.jaci.2011.08.029. [DOI] [PubMed] [Google Scholar]
- 30.Madaan A, Verma R, Singh AT, Jain SK, Jaggi M. A stepwise procedure for isolation of murine bone marrow and generation of dendritic cells. Journal of Biological Methods. 2014; 1: e1. 10.14440/jbm.2014.12. [DOI] [Google Scholar]
- 31.Barrow RT, Lollar P. Neutralization of antifactor VIII inhibitors by recombinant porcine factor VIII. Journal of thrombosis and haemostasis : JTH. 2006; 4: 2223–9. 10.1111/j.1538-7836.2006.02135.x. [DOI] [PubMed] [Google Scholar]
- 32.Canis K, Anzengruber J, Garenaux E, Feichtinger M, Benamara K, Scheiflinger F, Savoy LA, Reipert BM, Malisauskas M. In-depth comparison of N-glycosylation of human plasma-derived factor VIII and different recombinant products: from structure to clinical implications. Journal of thrombosis and haemostasis : JTH. 2018. 10.1111/jth.14204. [DOI] [PubMed] [Google Scholar]
- 33.Kelleher DJ, Kreibich G, Gilmore R. Oligosaccharyltransferase activity is associated with a protein complex composed of ribophorins I and II and a 48 kd protein. Cell. 1992; 69: 55–65. 10.1016/0092-8674(92)90118-v. [DOI] [PubMed] [Google Scholar]
- 34.Healey JF, Lubin IM, Lollar P. The cDNA and derived amino acid sequence of porcine factor VIII. Blood. 1996; 88: 4209–14. [PubMed] [Google Scholar]
- 35.Graw J, Brackmann HH, Oldenburg J, Schneppenheim R, Spannagl M, Schwaab R. Haemophilia A: from mutation analysis to new therapies. Nat Rev Genet. 2005; 6: 488–501. 10.1038/nrg1617. [DOI] [PubMed] [Google Scholar]
- 36.Shen BW, Spiegel PC, Chang CH, Huh JW, Lee JS, Kim J, Kim YH, Stoddard BL. The tertiary structure and domain organization of coagulation factor VIII. Blood. 2008; 111: 1240–7. 10.1182/blood-2007-08-109918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.García-Vallejo JJ, van Kooyk Y. Endogenous ligands for C-type lectin receptors: the true regulators of immune homeostasis. Immunol Rev. 2009; 230: 22–37. 10.1111/j.1600-065X.2009.00786.x. [DOI] [PubMed] [Google Scholar]
- 38.Dasgupta S, Navarrete AM, Bayry J, Delignat S, Wootla B, Andre S, Christophe O, Nascimbeni M, Jacquemin M, Martinez-Pomares L, Geijtenbeek TB, Moris A, Saint-Remy JM, Kazatchkine MD, Kaveri SV, Lacroix-Desmazes S. A role for exposed mannosylations in presentation of human therapeutic self-proteins to CD4+ T lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104: 8965–70. 10.1073/pnas.0702120104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gangadharan B, Ing M, Delignat S, Peyron I, Teyssandier M, Kaveri SV, Lacroix-Desmazes S. The C1 and C2 domains of blood coagulation factor VIII mediate its endocytosis by dendritic cells. Haematologica. 2017; 102: 271–81. 10.3324/haematol.2016.148502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Georgescu MT, Lai JD, Hough C, Lillicrap D. War and peace: Factor VIII and the adaptive immune response. Cell Immunol. 2016; 301: 2–7. 10.1016/j.cellimm.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 41.Pittman DD, Tomkinson KN, Kaufman RJ. Post-translational requirements for functional factor V and factor VIII secretion in mammalian cells. The Journal of biological chemistry. 1994; 269: 17329–37. [PubMed] [Google Scholar]
- 42.Pipe SW, Morris JA, Shah J, Kaufman RJ. Differential interaction of coagulation factor VIII and factor V with protein chaperones calnexin and calreticulin. The Journal of biological chemistry. 1998; 273: 8537–44. 10.1074/jbc.273.14.8537. [DOI] [PubMed] [Google Scholar]
- 43.Toole JJ, Pittman DD, Orr EC, Murtha P, Wasley LC, Kaufman RJ. A large region (approximately equal to 95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proceedings of the National Academy of Sciences of the United States of America. 1986; 83: 5939–42. 10.1073/pnas.83.16.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pittman DD, Alderman EM, Tomkinson KN, Wang JH, Giles AR, Kaufman RJ. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted factor VIII. Blood. 1993; 81: 2925–35. [PubMed] [Google Scholar]
- 45.Pittman DD, Marquette KA, Kaufman RJ. Role of the B domain for factor VIII and factor V expression and function. Blood. 1994; 84: 4214–25. [PubMed] [Google Scholar]
- 46.Xi M, Makris M, Marcucci M, Santagostino E, Mannucci PM, Iorio A. Inhibitor development in previously treated hemophilia A patients: a systematic review, meta-analysis, and meta-regression. Journal of thrombosis and haemostasis : JTH. 2013; 11: 1655–62. 10.1111/jth.12335. [DOI] [PubMed] [Google Scholar]
- 47.Franchini M, Coppola A, Rocino A, Santagostino E, Tagliaferri A, Zanon E, Morfini M. Systematic review of the role of FVIII concentrates in inhibitor development in previously untreated patients with severe hemophilia a: a 2013 update. Semin Thromb Hemost. 2013; 39: 752–66. 10.1055/s-0033-1356715. [DOI] [PubMed] [Google Scholar]
- 48.Aruffo A, Seed B. Molecular cloning of a CD28 cDNA by a high-efficiency COS cell expression system. Proceedings of the National Academy of Sciences of the United States of America. 1987; 84: 8573–7. 10.1073/pnas.84.23.8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Selvaraj SR, Miao H, Pipe S. Elucidation of the Roles of Individual Asparagine-Linked Glycans Outside of the B Domain on Factor VIII Secretion. Blood. 2011; 118: 2238-. 10.1182/blood.V118.21.2238.2238. [DOI] [Google Scholar]
- 50.Marquette KA, Pittman DD, Kaufman RJ. A 110-amino acid region within the A1-domain of coagulation factor VIII inhibits secretion from mammalian cells. The Journal of biological chemistry. 1995; 270: 10297–303. 10.1074/jbc.270.17.10297. [DOI] [PubMed] [Google Scholar]
- 51.Morris JA, Dorner AJ, Edwards CA, Hendershot LM, Kaufman RJ. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. The Journal of biological chemistry. 1997; 272: 4327–34. 10.1074/jbc.272.7.4327. [DOI] [PubMed] [Google Scholar]
- 52.Wei W, Misra S, Cannon MV, Yang R, Zhu X, Gilmore R, Zhu M, Zhang B. Molecular mechanisms of missense mutations that generate ectopic N-glycosylation sites in coagulation factor VIII. Biochem J. 2018; 475: 873–86. 10.1042/bcj20170884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jacob J, Duclohier H, Cafiso DS. The role of proline and glycine in determining the backbone flexibility of a channel-forming peptide. Biophys J. 1999; 76: 1367–76. 10.1016/s0006-3495(99)77298-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Delignat S, Repessé Y, Navarrete AM, Meslier Y, Gupta N, Christophe OD, Kaveri SV, Lacroix-Desmazes S. Immunoprotective effect of von Willebrand factor towards therapeutic factor VIII in experimental haemophilia A. Haemophilia. 2012; 18: 248–54. 10.1111/j.1365-2516.2011.02679.x. [DOI] [PubMed] [Google Scholar]
- 55.Lee HS, Qi Y, Im W. Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Scientific Reports. 2015; 5: 8926. 10.1038/srep08926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen PC, Lewis KB, Ettinger RA, Schuman JT, Lin JC, Healey JF, Meeks SL, Lollar P, Pratt KP. High-resolution mapping of epitopes on the C2 domain of factor VIII by analysis of point mutants using surface plasmon resonance. Blood. 2014; 123: 2732–9. 10.1182/blood-2013-09-527275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meeks SL, Healey JF, Parker ET, Barrow RT, Lollar P. Nonclassical anti-C2 domain antibodies are present in patients with factor VIII inhibitors. Blood. 2008; 112: 1151–3. 10.1182/blood-2008-01-132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eubanks J, Baldwin WH, Markovitz R, Parker ET, Cox C, Kempton CL, Meeks SL. A subset of high titer anti-factor VIII A2 domain antibodies are responsive to treatment with factor VIII. Blood. 2016; 127: 2028–34. 10.1182/blood-2015-09-670034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gish JS, Jarvis L, Childers KC, Peters SC, Garrels CS, Smith IW, Spencer HT, Doering CB, Lollar P, Spiegel PC. Structure of blood coagulation factor VIII in complex with an anti-C1 domain pathogenic antibody inhibitor. Blood. 2021; 137: 2981–6. 10.1182/blood.2020008940. [DOI] [PMC free article] [PubMed] [Google Scholar]