

Abstract
Introduction and importance
Myoepithelial tumors are under-recognized neoplasms that could be difficult to identify due to their rarity and limited comprehension. Their diverse morphology, varied cytologic features and heterogenous immunohistochemical characteristics create a significant diagnostic challenge.
Case presentation
We report the case of a 72-year-old-male patient who received conservative treatment during one year for a popliteal mass on the right knee that showed synovial hyperplasia (benign findings) at initial open tissue biopsy. New symptoms of popliteal area enlargement and discomfort required a second incisional biopsy to reach the diagnosis of a soft tissue myoepithelial tumor through tissue analysis and immunohistochemical staining.
Clinical discussion
The myoepithelial tumors represent a medical dilemma due to their heterogenic features requiring high level of suspicion and adequate immunohistochemical markers for their diagnosis.
Conclusion
Orthopaedic surgeons should be aware of the atypical presentation of these rare neoplasms to provide an early diagnosis and adequate management.
Abbreviations: MT, myoepithelial tumors; MES, myoepithelioma of soft tissue; MC, myoepithelial carcinoma; CK, cytokeratin; EMA, epithelial membrane antigen; S100p, S-100 protein; GFAP, glial fibrillary acidic protein; MRI, magnetic resonance imaging; UPR, University of Puerto Rico
Keywords: Popliteal mass, Myoepithelial tumors, Immunohistochemical markers, Open biopsy, Diagnosis
Highlights
-
•
Consider myoepithelial tumor within the differential of a soft tissue mass.
-
•
Immunohistochemical markers are essential for diagnostic confirmation.
-
•
Surgical resection with negative margins remains the mainstay of treatment.
-
•
There is limited evidence regarding radiotherapy or chemotherapy effectiveness.
-
•
This case highlights the deceiving nature and an unusual location of a myoepithelial tumor.
1. Introduction and importance
Myoepithelial neoplasms are rare soft tissue tumors that have been recognized recently and are still being characterized [1], [2], [3]. These myoepithelial tumors (MT) are difficult to diagnose as they demonstrate varied morphology, heterogenous immunohistochemical features and exhibit a wide range of cytologic and architectural features, both within a given lesion and between different tumors [4]. MT can be divided into myoepithelioma of the soft tissue (MES) and myoepithelial carcinoma (MC) based on the presence of cytologic atypia [1]. MES is characterized by having mild cytologic atypia, whereas MC has moderate to severe cytologic atypia [1]. In addition, MES usually ranges in size from 0.7 cm to 12 cm in soft tissues, whereas MC ranges in size from 1.3 cm to 20 cm and may present necrosis and hemorrhage as they enlarge. A set of appropriate immunohistochemical markers, which include a combination of epithelial indicators such as cytokeratin (CK), epithelial membrane antigen (EMA), S-100 Protein (S-100p), and glial fibrillary acidic protein (GFAP) are fundamental for the diagnosis of these neoplasms [1], [5], [6]. Previous articles showed that MT remains a controversial diagnosis due to its heterogenic features [1], [4], [7].
Both MES and MC have potential for local recurrence with higher risk among malignant epithelial tumors [7]. In the literature, around 18% of soft tissue myoepitheliomas have recurred, compared to a 39–42% of myoepithelial carcinomas [1], [8], [9]. Surgical resection with negative margins has been commonly used with debatable results [10]. Currently, there are no well-established guidelines for their management.
MT remain an erratic and non-fully comprehended neoplasm. Its diagnosis could be even more challenging by involving uncommon anatomic sites such as the popliteal area. Early evaluation and tissue biopsy may not be enough to address this diagnosis in early stages due to its abnormal and deceiving presentation. We report the diagnostic dilemma of MT in the popliteal area to increase awareness about the atypical nature of these neoplasms.
This case is reported according to the updated consensus-based surgical case report (SCARE) guidelines [11].
2. Case presentation
A 72-year-old Hispanic male with a past medical history significant for hypertension, atrial fibrillation, congestive heart failure, and diabetes mellitus presented to our musculoskeletal oncology clinic for follow-up due to a right popliteal mass which had been biopsied by our service one year prior to this appointment. At that time, the biopsy reported benign findings including hyperplastic synovium, cellular synovial lining with hypertrophy and hyperplasia, chronic inflammatory cells, hemosiderin pigment and hemorrhage (Fig. 1). These results were confirmed by a national major referral center. Based on the patient's age, multiple comorbidities, good functionality with minimal symptoms and the benign report, he was treated conservatively.
Fig. 1.

H&E slide showing synovial hyperplasia with chronic inflammatory cells, hemosiderin pigment and hemorrhage.
At the one-year follow-up, he complained of increased discomfort and progressive sensation of fullness in the back of his right knee. Constitutional symptoms such as fatigue, night sweats, or weight loss were denied. Physical exam revealed a non-tender popliteal area enlargement causing decreased range of motion, with an unremarkable neurovascular exam. Knee radiographs were not contributory. Magnetic Resonance Imaging (MRI) showed a large lobulated complex mass with heterogeneous signal posterior to the distal femur but anterior to the neurovascular bundle. Multiple nodules were described ranging from 0.7 cm to 3.9 cm in size (Fig. 2). Due to concerns of malignancy, additional tests such as bone scintigraphy and chest computed tomography with intravenous contrast were performed, and these came back negative for distant lesions.
Fig. 2.

Magnetic resonance images with IV contrast. A large lobulated complex mass with a heterogeneous signal on sagittal reconstructions (A) shows contrast enhancement (B). T2 sequences demonstrate no bone involvement of the lesion (C) but there is a close relationship to neurovascular structures on axial slices (D).
A second incisional open biopsy was performed by the senior author, and a 7.0 cm mass on the posterolateral aspect of the knee was found abutting against the common peroneal nerve and popliteal vessels. Intraoperative pathologic evaluation was inconclusive. The case was consulted with the University of Puerto Rico (UPR): School of Medicine - Pathology program and a major national referral center. The UPR Pathology Service reported a mixed tumor/myoepithelioma with atypical features such as high cellularity, tumor necrosis, cytologic atypia and elevated Ki67 index, all of which were considered features of malignancy (Fig. 3). In addition, immunohistochemical staining (Fig. 4) was performed with positive results for Parakeratin (i.e., CK), EMA, Vimentin, Ki67 index (10%), and CD99 antigen. On the other hand, S-100p, GFAP and Desmin were negative. These findings were reviewed by a national reference laboratory, who confirmed our diagnosis.
Fig. 3.

H&E slides showing a discrete area of increased cellularity associated with hemorrhage (A) magnification 10×. Areas of tissue necrosis (B) can be appreciated (*) at a magnification of 10×. Cellular detail can be observed (C) showing increased cell to matrix ratio of round to ovoid cells, uniform dark nuclei but no significant atypia or atypical mitotic figures (20×). (D) shows cells with mild atypia over a chondroid matrix (20×), no mitotic figures seen.
Fig. 4.
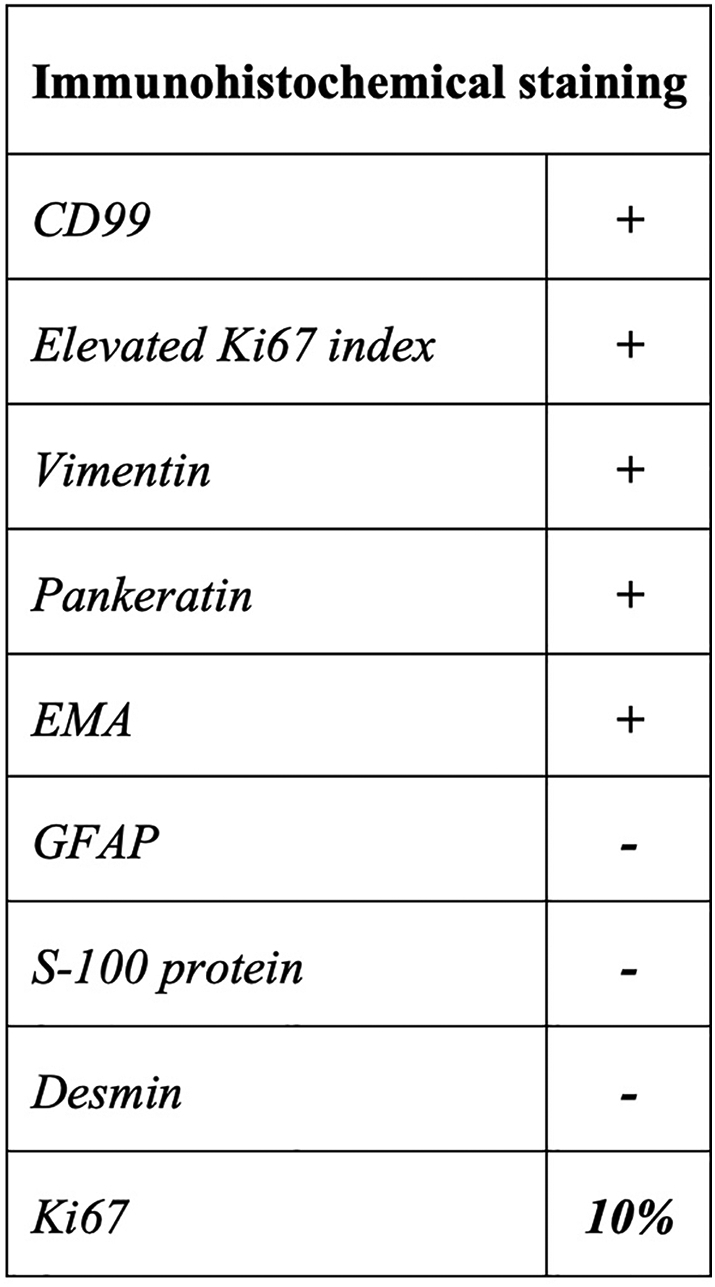
Immunohistochemical staining outcomes.
The treatment approach to this case was challenging due to the tumor's proximity to the neurovascular bundle. Consequently, a trans-femoral amputation was suggested to obtain local control of the disease as recommended by previous reports. The case was discussed with an expert panel (Tumor Board), and a trans-femoral amputation was recommended. Unfortunately, the patient refused our recommendations and was lost to follow-up.
3. Discussion
Myoepithelial tumors (MT) of soft tissue lack any known normal cellular counterpart, which has likely contributed to its under-recognition in the past [4]. Benign and malignant forms have been described (i.e., myoepithelioma of the soft tissue and myoepithelial carcinoma, respectively). MT are equally distributed between genders over a wide age range, mostly presenting between the third and fifth decades of life [5]. These tumors are commonly found in the subcutaneous and deep soft tissues of the upper and lower extremities [1]. Hornick et al. reviewed 101 myoepithelial tumors of soft tissue and described that most were in the buttocks, groin/inguinal area, neck, forearm, and shoulder [1].
Usually, the MT are multinodular or lobular and, despite being well-circumscribed, often show infiltrative growth. Microscopically, they are described as spindled, ovoid, or epithelioid tumor cells arranged in a combination of reticular, trabecular, or nested growth patterns associated with a variably chondromyxoid or hyalinized stroma [9]. Some tumors may show a dominant single growth pattern. Other occasional morphologic appearances include plasmacytoid “hyaline cells” with densely eosinophilic cytoplasm, tumor cells with copious clear vacuolated cytoplasm, and rhabdoid morphology [9]. Up to 15% of cases show heterologous differentiation, including chondro-osseous, adipocytic, and squamous components [1], [2], [8], [12], [13]. Nucleoli are small or inconspicuous, hyperchromatic, and vesicular nuclear changes are absent. Mitotic activity may be observed, but atypical cellular replication should not be present. Rare examples have shown perineural invasion, but tumor necrosis is rare. Cytologic atypia remains the sole criterion for malignancy, as it is the best predictor for malignant behavior [1], [8]. No other validated criteria for distinguishing benign and malignant tumors have been established. Therefore, the absence of comprehensive criteria to define malignancy in MT could explain the benign report after the first biopsy to our patient, which does not include immunohistochemical staining due to the evident benign findings of synovial hyperplasia on histologic examination.
However, to confirm the diagnosis of an MT, appropriate immunohistochemical staining is needed. Studies have reported that MT mostly expresses epithelial antigens (cytokeratin and EMA) and S-100p [14]. This was not the case in our patient, who did not express S-100p. Vickie Jo reviewed the immunohistochemical features in MT and described that broad-spectrum cytokeratin is positive in 93–100% of cases, compared with a lower EMA positivity of 19–66% [14]. S-100p is positive in 72–100% of cases, and GFAP staining is lower, ranging from 27% to 54%. Of the myogenic markers, calponin positivity goes from 86 to 100%, while desmin is less frequently positive in 0–20% of cases [14]. Several studies have agreed that the combination of epithelial markers including CK, EMA, S-100p, and GFAP is fundamental for diagnosing these neoplasms [1], [5], [6], [14].
The differential diagnosis of MT can be extensive and includes extra-skeletal myxoid chondrosarcomas of high grade, malignant melanoma, epithelioid sarcomas, metastatic carcinoma, chordomas, and epithelioid malignant peripheral nerve sheath tumors [4], [7].
MT (i.e., MES and MC) have potential for local recurrence with higher risk among those neoplasms with malignant features [7]. Up to 18% of MES are known to recur, and the risk appears higher with inadequate surgical resection [1], [8], [9]. Some MCs show aggressive behavior with a 39% to 42% recurrence rate and the development of distant metastasis in 32% to 52% of affected patients [1], [8]. The reported disease-related deaths in several extensive series ranges from 13% to 43% [1], [8].
Surgical resection with negative margins has been the recommended treatment [7], [15], [16]. Moreover, Bisogno et al. suggested aggressive local control with resection and radiotherapy systemically applied for these lesions [16]. Some authors argue that it may have a role in the adjuvant setting after removing the primary tumor. However, as a sole treatment, radiotherapy has limited evidence of effectiveness.
Additionally, MT are described as poorly responsive to systemic chemotherapy [2], [17], even though some literature supports chemoradiation for the local control of unresectable metastases [18]. On the other hand, Gleason et al. reported two cases of MT of soft tissue unresectable and solitary (without metastases in transit), who underwent hyperthermic isolated limb perfusion without response leading to amputation of the limb [8]. Nowadays, no well-defined guidelines have been presented in the literature for management of MT.
This case represents a diagnostic and treatment dilemma. An uncommon presentation of a MT with atypical features in the popliteal area was initially diagnosed as synovial hyperplasia after an open tissue biopsy. A high level of suspicion is needed to correctly diagnose these tumors, along with a correct immunohistochemical panel. Due to the high morbidity and significant mortality with MT, orthopaedic surgeons need to be aware of the atypical presentation of these rare neoplasms to provide an early diagnosis and adequate management.
Source of funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Ethical approval
This study is exempt from ethical approval in our institution.
Informed consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
CRediT authorship contribution statement
Lucas De Virgilio-Salgado, MD: Conceptualization, investigation, reviewing, and editing.
Norberto J. Torres-Lugo, MD: Investigation, visualization, writing, reviewing, and editing.
Gerardo Olivella, MD: Conceptualization, investigation, data curation, and writing.
John M. Watson-Pérez, BS: Investigation, data curation, reviewing, and editing.
Norman Ramírez, MD: Supervision, reviewing, and editing.
Juan Bibiloni-Rodríguez, MD: Investigation, supervision, data curation, reviewing, and editing.
Provenance and peer review
Not commissioned, externally peer-reviewed.
Research registration
Not applicable.
Guarantor
Lucas De Virgilio-Salgado, MD and Norberto J. Torres Lugo, MD.
Declaration of competing interest
The authors have no conflict to disclose.
Acknowledgments
We are grateful to Dr. María Marín, pathologist, for giving us insightful advice about the case and its pathologic images.
References
- 1.Hornick J.L., Fletcher C.D. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am. J. Surg. Pathol. 2003;27:1183–1196. doi: 10.1097/00000478-200309000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Kilpatrick S.E., Hitchcock M.G., Kraus M.D., et al. Mixed tumors and myoepitheliomas of soft tissue: a clinicopathologic study of 19 cases with a unifying concept. Am. J. Surg. Pathol. 1997;21:13–22. doi: 10.1097/00000478-199701000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Michal M., Miettinen M. Myoepitheliomas of the skin and soft tissues. Report of 12 cases. Virchows Arch. 1999;434:393–400. doi: 10.1007/s004280050358. [DOI] [PubMed] [Google Scholar]
- 4.Jo V.Y. Myoepithelial tumors: an update. Surg. Pathol. Clin. 2015;8(3):445–466. doi: 10.1016/j.path.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Balraam K.V., Shelly D., Mishra P.S., Sharma I., Sampath K.S., Bharadwaj R. Myoepithelial carcinoma of soft tissue: a report of two cases. J. Cancer Res. Pract. 2019;6:136–139. [Google Scholar]
- 6.Rekhi B., Sable M., Jambhekar N.A. Histopathological, immunohistochemical and molecular spectrum of myoepithelial tumours of soft tissues. Virchows Arch. 2012;461:687–697. doi: 10.1007/s00428-012-1335-7. [DOI] [PubMed] [Google Scholar]
- 7.Gleason B.C., Hornick J.L. Myoepithelial tumors of skin and soft tissue: an update. Diagn. Histopathol. 2008;14:552–562. [Google Scholar]
- 8.Gleason B.C., Fletcher C.D. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am. J. Surg. Pathol. 2007;31:1813–1824. doi: 10.1097/PAS.0b013e31805f6775. [DOI] [PubMed] [Google Scholar]
- 9.Thway K., Bown N., Miah A., et al. Rhabdoid variant of myoepithelial carcinoma, with EWSR1 rearrangement: expanding the spectrum of EWSR1-rearranged myoepithelial tumors. Head Neck Pathol. 2015;9(2):273–279. doi: 10.1007/s12105-014-0556-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rastrelli M., Del Fiore P., Damiani G.B., Mocellin S., Tropea S., Spina R., Costa A., Cavallin F., Rossi C.R. Myoepithelioma of the soft tissue: a systematic review of clinical reports. Eur. J. Surg. Oncol. 2019 Sep;45(9):1520–1526. doi: 10.1016/j.ejso.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Agha R.A., Franchi T., Sohrabi C., Mathew G., for the SCARE Group The SCARE 2020 guideline: updating consensus Surgical CAse REport (SCARE) guidelines. Int. J. Surg. 2020;84:226–230. doi: 10.1016/j.ijsu.2020.10.034. [DOI] [PubMed] [Google Scholar]
- 12.Hornick J.L., Fletcher C.D. Cutaneous myoepithelioma: a clinicopathologic and immunohistochemical study of 14 cases. Hum. Pathol. 2004;35:14–24. doi: 10.1016/j.humpath.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 13.Mentzel T., Requena L., Kaddu S., et al. Cutaneous myoepithelial neoplasms: clinicopathologic and immunohistochemical study of 20 cases suggesting a continuous spectrum ranging from benign mixed tumor of the skin to cutaneous myoepithelioma and myoepithelial carcinoma. J. Cutan. Pathol. 2003;30:294–302. doi: 10.1034/j.1600-0560.2003.00063.x. [DOI] [PubMed] [Google Scholar]
- 14.Jo V.Y. Soft tissue special issue: myoepithelial neoplasms of soft tissue: an updated review with emphasis on diagnostic considerations in the head and neck. Head Neck Pathol. 2020 Mar;14(1):121–131. doi: 10.1007/s12105-019-01109-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee J.R., Georgi D.E., Wang B.Y. Malignant myoepithelial tumor of soft tissue: a report of two cases of the lower extremity and a review of the literature. Ann. Pathol. 2007;11(3):190–198. doi: 10.1016/j.anndiagpath.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 16.Bisogno G., Tagarelli A., Schiavetti A., et al. Myoepithelial carcinoma treatment in children: a report from the TREP project. BMC Neurol. 2013;May 4:13:40. doi: 10.1002/pbc.24818. [DOI] [PubMed] [Google Scholar]
- 17.Puls F., Arbajian E., Magnusson L., et al. Myoepithelioma of bone with a novel FUS-POU5F1 fusion gene. Histopathology. 2014;65:917–922. doi: 10.1111/his.12517. [DOI] [PubMed] [Google Scholar]
- 18.Rastrelli M., Passuello N., Cecchin D., et al. Metastatic malignant soft tissue myoepithelioma: a case report showing complete response after locoregional and systemic therapy. J. Surg. Case Rep. 2013 Dec 16;2013(12) doi: 10.1093/jscr/rjt109. [DOI] [PMC free article] [PubMed] [Google Scholar]