

Abstract
Recessive variants in RARS2, a nuclear gene encoding a mitochondrial protein, were initially reported in pontocerebellar hypoplasia. Subsequently, a recessive RARS2 early‐infantile (<12 weeks) developmental and epileptic encephalopathy was described with hypoglycaemia and lactic acidosis. Here, we describe two unrelated patients with a novel RARS2 phenotype and reanalyse the published RARS2 epilepsy phenotypes and variants. Our novel cases had infantile‐onset myoclonic developmental and epileptic encephalopathy, presenting with a progressive movement disorder from 9 months on a background of normal development. Development plateaued and regressed thereafter, with mild to profound impairment. Multiple drug‐resistant generalized and focal seizures occurred with episodes of non‐convulsive status epilepticus. Seizure types included absence, atonic, myoclonic, and focal seizures. Electroencephalograms showed diffuse slowing, multifocal, and generalised spike‐wave activity, activated by sleep. Both patients had compound heterozygous RARS2 variants with likely impact on splicing and transcription. Remarkably, of the now 52 RARS2 variants reported in 54 patients, our reanalysis found that 44 (85%) have been shown to or are predicted to affect splicing or gene expression leading to protein truncation or nonsense‐mediated decay. We expand the RARS2 phenotypic spectrum to include infantile encephalopathy and suggest this gene is enriched for pathogenic variants that disrupt splicing.
Keywords: developmental and epileptic encephalopathy, epilepsy, infantile, movement disorder, myoclonic, RARS2
1. INTRODUCTION
While RARS2 was first identified as a recessive gene for pontocerebellar hypoplasia (PCH), 1 a 2020 review of 25 cases with reported imaging found PCH in only 48%. 2 There are now 52 published cases with RARS2 variants with clinical information available for 43. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 Seizures are reported in 40 (93%) individuals with onset prior to age 3 months in 37 (93%). RARS2 encodes a mitochondrial aminoacyl‐tRNA synthetase (mtARS) which catalyzes the attachment of arginine, vital for mtRNA–protein translation. 30
The reported 50 pathogenic RARS2 variants (Table S1) occur in families as compound heterozygous mutations in 81% and are most commonly missense (62%. 31/50) (Figure 1B). There are ten recurrent variant positions (p.Met1(Val/Leu) 15 , 18 , 29 ; p.Gln12Arg 4 , 6 , 13 , 28 ; c.110+5A>G 1 , 8 , 13 ; p.Lys158del 3 , 7 , 28 ; p.Gln208* 20 , 26 ; p.Arg258His 5 , 26 ; p.Leu283Gln 7 , 23 , 25 ; p.Met342Ile 12 , 20 ; p.Asp515Gly 9 , 22 ; p.Val522Ile 28 , 29 ); however no clear genotype–phenotype correlation has been identified. 2
FIGURE 1.
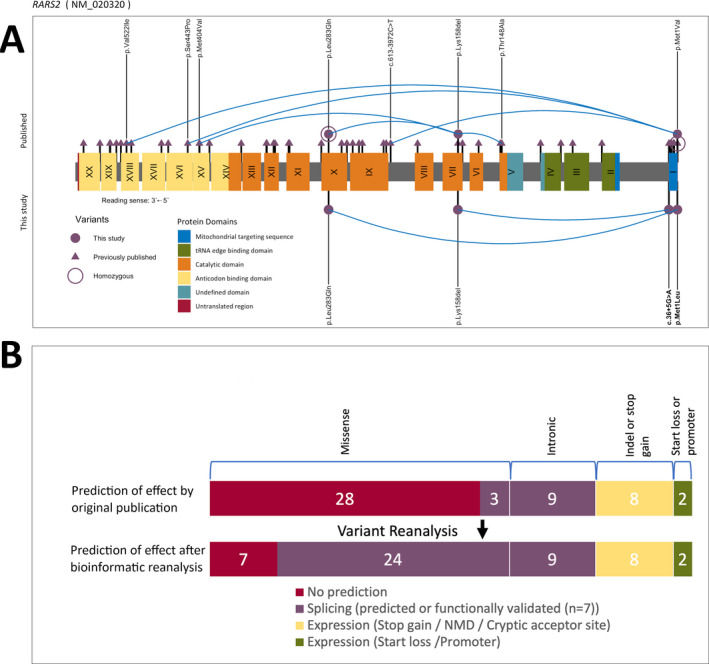
A, RARS2 variants and their position in the RARS2 gene. Previously published variants are shown above the protein. Previously published variants not found in our cases are represented with a triangle. Our cases are shown below the protein and variants found in our cases or affecting the same animoacid are represented by a circle. Blue lines link compound heterozygote variants identified in our cases and published cases who have at least one variant also found in our cases. cDNA changes in bold represent novel variants found in this study. Introns and UTRs are represented in a 1:100 scale. B, Effect on expression and splicing of the published RARS2 variants. The top bar represents represent the effect of the variants as considered by original publication and the bottom bar represents the effect of the same variants after our bioinformatic reanalysis. NMD, nonsense‐mediated decay
We describe a new RARS2 phenotype of infantile‐onset myoclonic developmental and epileptic encephalopathy in two unrelated children and compare this to the epileptology in the previously reported cases. We reanalyze the published variants and suggest there is enrichment for variants that disrupt splicing in this gene.
2. METHODS
Two children had compound heterozygous RARS2 (RefSeq: NM_020320.5) pathogenic variants identified. Trio genome sequencing was performed on Case A and her unaffected parents as part of our epilepsy genetics research cohort of 250 patients with developmental and epileptic encephalopathies (DEEs) who underwent whole genome sequencing (28) or whole exome sequencing (222) analysis. Case B had singleton whole exome sequencing. Variants were called using the standard genome analysis toolkit pipeline and annotated with SnpEff. We filtered coding variants with a frequency <0.0001 in population databases (1000 Genomes phase 3, NHLBI GO Exome Sequencing Project, ExAC, and gnomAD) and an impact severity predicted by SnpEff of ‘HIGH’ or ‘MEDIUM’. Variants were validated and segregation performed with Sanger sequencing. We used Human Splicing Finder (HSF), SpliceAI, and Transcript Inferred Pathogenicity Score (TraP) to predict impact on splicing by our variants and published variants (Table S1).
We reviewed epilepsy and medical history, examination, magnetic resonance imaging (MRI), and electroencephalogram (EEG) findings. The study was approved by Austin Health and New Zealand Health and Disability Ethics Committees. Written informed consent was obtained from the parents.
We reviewed RARS2 publications prior to Aug 2021 and identified 52 novel RARS2 cases (Table S1). 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 We compared our cases with the epileptology reported for RARS2 encephalopathy; adequate information was only available for 15/52 reported cases (Table 1). 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11
TABLE 1.
Symptoms that child presented with are shaded yellow
| CASE | CASE A | CASE B | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 |
|---|---|---|---|---|---|---|---|---|---|
| Publication | Ours | Ours | Glamuzina et al, 2012 | Cassandrini et al, 2013 | Cassandrini et al, 2013 | Cassandrini et al, 2013 | Kastrissianikis et al, 2013 | Kastrissianikis et al, 2013 | Rankin et al, 2010 |
| Patient (sex, age at study) | A (F,6 y ‐died) | B (F, 10 y) | IV‐1.1 (F, 2 y) | A‐01 (M,11 y) | B‐01 (F, 9 y) | B‐02 (F, 3 y) | Sibling 1 (F, 3 y) | Sibling 2 (M, 12 m) | 1 (F, 4 y 6 m) |
| Epilepsy Syndrome | Infantile onset DEE | Infantile onset DEE | Neonatal onset DEE | Neonatal onset DEE | Neonatal onset DEE | Neonatal onset DEE | Neonatal onset DEE | Neonatal onset DEE | Neonatal onset DEE |
| Seizure onset | 2 y 2 m | 3 y | 4 w | 11 d | 20 d | 11 d | 2 w | 1 d | 2 d |
| First seizure type | My; A | A | Motor SE | FM (clonic) | Motor SE | FM (clonic) | TC | FM (clonic) | My |
| Subsequent seizures | FBTC; T; Ab (atypical); Non‐convulsive & motor SE | Ab (atypical); MyA; non‐convulsive SE | UK; TC | FM (multifocal) | FM (multifocal) | FM (multifocal) | TC | FM (multifocal) | My |
| Pharmaco‐resistant | Y | Y | Y | Y | Y | Y | Y | Y | NR |
| Developmental concern: onset age: outcome | 17 m: Profound ID (G‐tube) | 15 m: Mild ID | Birth: Profound DD (G‐tube) | Birth: Severe ID | Birth: Severe ID (G‐tube) | Unclear: Severe DD | Unclear <3 m: Severe DD (G‐tube) | Birth: Severe DD | Birth: Profound DD (G‐tube) |
| Regression (age) | Y (17 m) | Y (5 y) | N | N | N | N | N | N | N |
| Examination (neuro) | Central hypotonia, increased tone, reflexes | Ataxia, dysarthria, café au lait lesions, freckling | Hypotonia | Initial hypotonia developed spastic quadriplegia | Initial hypotonia developed spastic quadriplegia | Spastic quadriplegia | Spastic quadriplegia | Increased tone & reflexes | Hypotonia & increased reflexes |
| Microcephaly | N | N | Y | Y | Y | Y | Y | N | Y |
| Movement disorder: age of onset & type | Yes: 9 m, myoclonus | Yes: 8 m, myoclonus | Yes: 1 m, severe dystonia | Possibly | Possibly | Possibly | N | N | N |
| EEG | 2 y 4 m: MFD; 2 y 5 m & 2 y 9 m: Slow, MFD; 3 y 4 m & 4 y 5 m: Slow, MFD, GSW, PSW | 2 y: Normal; 3 y: GSW; 7 y 9 m: Slow, GSW, 8 y & 10 y: Slow, GSW, MFD, PPR | 4 w: BS; 6 m: Slow, bifrontal discharges | Slow, MFD | Slow, MFD | Slow, MFD | 14 w: Focal slow, MFD; 16 m: CSWS | 1 d: Normal; 3 w: BS in sleep, MFD |
1 w: Discharges, 2 y: GSW |
| MRI | Subtly small pons & cerebellum, mild PVWM hyper‐intensity |
Mild cortical atrophy; Focal lesion right cerebellum |
Pontocerebellar hypoplasia; cortical & optic nerves atrophy | Cerebellar vermis hypoplasia; Cortical & cerebellar atrophy | Progressive cortical & pontocerebellar atrophy | Progressive cortical & pontocerebellar atrophy | Cortical & cerebellar atrophy | Cortical & cerebellar atrophy | Cortical & pontocerebellar atrophy |
| Metabolic abnormalities (peak blood lactate level)a | Normal (blood 1.3 to 2.2 mmol/L; CSF 1.4 mmol/L) | Normal (blood 2.0 mmol/L; CSF 1.4mmol/L) | Neonatal hypoglycaemia & lactic acidosis (14.2 mmol/L) | Neonatal lactic acidosis (6.7 mmol/L) | Neonatal hypoglycaemia & lactic acidosis (3.7 mmol/L) | Neonatal onset of lactic acidosis (2.4 mmol/L) | Mildly elevated CSF lactate (blood lactate normal) | Neonatal hypoglycaemia & lactatic acidosis (3.7 mmol/L) | Neonatal hypoglycaemia & lactic acidosis (16 mmol/L) |
| Variant 1* |
c.848T>A; p.L283Qp |
c.36+5G>Am;intronic ‐ |
c.1211T>A; p.M404Km |
c.25G>A; p.I9Vp |
c.734G>A; p.R245Qm |
c.734G>A; p.R245Qm |
c.773G>A; p.R258H |
c.773G>A; p.R258H |
c.1024A>G; p.M342Vm |
| SIFT | D (0) | ‐ | D (0) | T (0.12) | D (0) | D (0) | D (0) | D (0) | D (0) |
| PolyPhen | B (0.021) | 10.96 | D (0.953) | B (0.03) | D (0.997) | D (0.997) | D (0.992) | D (0.992) | P (0.688) |
| CADD | 22 | ‐4.3 | 29.5 | 15.42 | 32 | 32 | 34 | 34 | 24.7 |
| GERP RS | 3.97 | 0 | 5.9699 | 3.868 | 6.17 | 6.17 | 5.51 | 5.51 | 4.659 |
| gnomADex | 2 | 0 | 0 | 5 | 6 | 6 | 70 | 70 | 0 |
| gnomADgen | 0 | 0 | 0 | 0 | 0 | 14 | 14 | 0 | |
| Variant 2* |
c.1A>T; p.M1L m |
c.472_474delAAA;p.K158delKp ‐ |
c.471_473delCAA;p.K158delKp ‐ |
c.1586+3A>Tm ‐ |
c.1406G>A; p.R469Hp |
c.1406G>A; p.R469Hp |
c.1651‐2A>G ‐ |
c.1651‐2A>G ‐ |
c.35A>G; p.Q12Rp |
| SIFT | D (0) | ‐ | ‐ | ‐ | D (0) | D (0) | ‐ | ‐ | T (0.07) |
| PolyPhen | B (0.0998) | ‐ | ‐ | 23 | D (1) | D (1) | 33 | 33 | B (0) |
| CADD | 27 | 4.4327 | 4.84 | 5.28 | 32 | 32 | 5.1999 | 5.1999 | 23.3 |
| GERP RS | 5.5 | 32 | 0 | 0 | 5.11 | 5.11 | 0 | 0 | 3.97 |
| gnomADex | 2 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 17 |
| gnomADgen | 0 | 0 | 0 | 0 |
| CASE | Case 8 | Case 9 | Case 10 | Case 11 | Case 12 | Case 13 | Case 14 | Case 15 |
|---|---|---|---|---|---|---|---|---|
| Publication | Nevanlinna et al, 2020 | Ngoh et al, 2016 | Ngoh et al, 2016 | Nishri et al, 2016 | Nishri et al, 2016 | Van Dijk et al, 2017 | Zhang et al, 2018 | Xu et al, 2020 |
| Patient (sex, age at study) | 1 (M, 11 y‐died) | Patient 1 (M, 11 y) | Patient 2 (M, 6 y) | II‐3 (F, 3 y‐died) | II‐4 (M, 4 y‐died) | B (F, 3 m‐died) | I (M, 3 y) | (F, 7 m) |
| Epilepsy Syndrome | Neonatal onset DEE | Neonatal onset DEE to Infantile spasms | Early onset DEE to infantile spasms | Early onset DEE | Early onset DEE | Early onset DEE | Early onset DEE | Neonatal onset DEE to Infantile Spasms |
| Seizure onset | 6 w | 5 w | 8 w | 9 w | 12 w | Unclear <12 w | 12 w | 19 d |
| First seizure type | FTBCS | Motor seizures (clonic) | Motor seizures (clonic) | FM (clonic) | FM (clonic) | FM (clonic) | FM (multifocal) | My |
| Subsequent seizures | FM; FIAS; My: non‐convulsive SE | ES; T; GTCS (+SE) | ES; My; ES | FM (multifocal), My | My; T | Motor SE | FM (multifocal) | Convulsive status epilepticus, ES |
| Pharmaco‐resistant | Y | Y | Y | Y | Y | Y | Y | Y |
| Developmental concern: onset age: outcome | 2 m: Profound ID (G‐tube) | 1 m: Profound ID (G‐tube) | 2 m: Severe DD (G‐tube) | Birth: Profound DD (G‐tube) | Unclear <2 m: Profound DD | Unclear<3 m: Severe DD | 5 m: Profound DD (G‐Tube) | Unclear: Severe DD |
| Regression (age) | N | N | N | Y (4m) | Y (9m) | N | N | NR |
| Examination (neuro) | Initial hypotonia developed spastic quadriplegia | Spastic quadriplegia | Axial hypotonia & spastic quadriplegia | Increased reflexes | Axial hypotonia & spastic quadriplegia | Hypotonia | Initial hypotonia, later hypertonia | NR |
| Microcephaly | Y | Y | Y | Y | N | NR | Y | NR |
| Movement disorder: age of onset & type | N | N | N | N | N | N | N | NR |
| EEG | 3 m: Normal; 4 m: Slow, MFD | 6 w: Normal; 8 m: modified Hyps; 11 m: MFD | 5 m: modified Hyps; 2.5 y: MFD | 9 w: Normal; 4 m: Slow, FD, PSW, EDE; 3 y: MFD (migratory) | 3 m: FD; 9 m: MFD, PSW | 3 m: MFD | 6 m: Slow, MFD | 24 d: BS |
| MRI | Cortical & basal ganglia atrophy | Thin corpus callosum, small cerebellum | Thin corpus callosum, small cerebellum | Cortical & cerebellar atrophy | Cortical atrophy | Cortical atrophy | Cortical, basal ganglia & white matter atrophy | Cortical atrophy, hypomylenation |
| Metabolic abnormalities (peak blood lactate level)a | Elevated lactate (5.3 mmol/L) | Neonatal hypoglycaemia; elevated lactate (4.9 mmol/L) | 6 y‐mild elevation of CSF glycine (blood lactate normal) | Normal | Normal | MRS lactate peak (CSF & blood normal) | 4m: Lactic acidosis (9.5 mmol/L) | Neonatal Hypoglycaemia & lactic acidosis (value NR) |
|
Variant 1* |
c.795delA; p.E265Dfsm |
c.848T>A; p.L283Q |
c.848T>A; p.L283Q |
c.110+5A>Gm; intronic ‐ |
c.110+5A>Gm;intronic ‐ |
c.1544A>G; p.D515Gm |
c.1718C>T; p.T573I |
c.282_285delAGAGp ‐ |
| SIFT | ‐ | ‐ | ‐ | ‐ | ‐ | D (0.01) | T (0.06) | ‐ |
| PolyPhen | ‐ | ‐ | ‐ | 16.17 | 16.17 | D (0.901) | B (0.411) | ‐ |
| CADD | ‐ | ‐ | ‐ | 5.4499 | 5.4499 | 25.8 | 23.2 | ‐ |
| GERP RS | 4.2 | 4.84 | 4.84 | 3 | 3 | 5.3699 | 4.65 | 6 (0) |
| gnomADex | 0 | 2 | 2 | 0 | 0 | 7 | 26 | 1 (0) |
| gnomADgen | 0 | 0 | 0 | 1 | 0 | |||
| Variant 2* |
c.961C>T; p.L321Fp |
c.472_474delAAA;p.K158delK D (0) |
c.472_474delAAA;p.K158delK D (0) |
c.878+5G>Tp ‐ |
c.878+5G>Tp ‐ |
c.297+2T>Gp ‐ |
c.991A>G; p.I331V |
c.773G>A p.R258Hm |
| SIFT | D (0.03) | D (0.998) | D (0.998) | ‐ | ‐ | ‐ | T (0.42) | 0 (D) |
| PolyPhen | D (0.874) | 27 | 27 | 18.87 | 18.87 | ‐ | B (0.291) | 0.992 (D) |
| CADD | 31 | 5.5 | 5.5 | 5.51 | 5.51 | 4.29 | 22.1 | 33 |
| GERP RS | 5.599 | 0 | 0 | 0 | 0 | 0 | 5.8299 | 5.51 |
| gnomADex | 1 | 0 | 0 | 0 | 0 | 0 | 8831 + 300 | 70 (0) |
| gnomADgen | 0 | 1071 + 35 | 14 (0) |
Abbreviations: A, atonic seizure; Ab, absence seizure; B, Benign; BS, burst suppression; CSF, cerebrospinal fluid; CSWS, continuous spike and wave in sleep; D, Damaging; d, days; DD, developmental delay; DEE, developmental and epileptic encephalopathy; EDE, electro decremental event; ES, epileptic spasm; F, female; FBTC, focal to bilateral tonic clonic seizure; FIAS, focal impaired awareness; FM, focal motor seizure; GSW, generalized spike and slow wave; GTCS, generalized tonic clonic seizure; Hyps, hypsarrhythmia; ID, intellectual disability; M, male; m, maternal inheritance; m, months; MFD, multifocal discharges; MRS, magnetic resonance spectroscopy; My, myoclonic seizure; MyA, myoclonic atonic seizure; N, no; NR, not recorded; p, paternal inheritance; P, Probably damaging; PPR, photo paroxysmal response; PSW, polyspike and slow wave; PVWM, periventricular white matter; SE, status epilepticus; seizure; T, Tolerated; T, tonic seizure; TC, tonic clonic seizure; UK, unknown seizure type; w, weeks; y, years; Y, yes.
alactate levels are in blood unless otherwise specified.
*In‐silico predictions, GERP conservation values and genetic database frequencies obtained from: CADD, SIFT, PolyPhen, GERP: https://cadd.gs.washington.edu/snv, gnomAD: https://gnomad.broadinstitute.org/gene/ENSG00000146282?dataset=gnomad_r2_1
3. RESULTS
3.1. Clinical phenotyping
Case A, who died at 6 years, was born following a normal pregnancy to unrelated parents of European ancestry. Early development was normal. At 9 months, she developed action myoclonus of her hands which became more prominent over time. By 17 months, she was ataxic and had dysphagia, with developmental regression. At 2 years 2 months, she developed myoclonic and atonic seizures associated with fever. By 2.5 years, she could no longer sit or speak and required gastrostomy‐tube feeding. She had central hypotonia with increased peripheral tone, increased reflexes, and dystonia. She had continuous multifocal myoclonus of her limbs and face. Electroencephalogram showed that her myoclonus did not correlate with epileptiform discharges, despite frequent multifocal and generalised spike wave (GSW) and polyspike wave, together with diffuse background slowing. The GSW was maximal independently in the bifrontal and bioccipital areas and continuous in sleep. She developed focal motor seizures at 2.5 years. Tonic, atypical absence, and definite epileptic myoclonic seizures were apparent by 3 years. By 3.5 years, she had convulsive and non‐convulsive status epilepticus. Her epilepsy was drug‐resistant; oral prednisolone and diazepam resulted in short‐term improvement of her myoclonus and EEG. She was noted to have accelerated growth without pubertal signs from 4 years. At 6 years, she died following convulsive status epilepticus.
Magnetic resonance imaging of the brain showed a thick rostrum/genu of the corpus callosum, mild periventricular white matter hyperintensity, and structurally normal but the small pons and cerebellum (just under the 3rd percentile volume for age). Metabolic investigations including blood lactate, skin, rectal, and muscle biopsies were uninformative.
Case B is a 10‐year‐old girl born to an unrelated Japanese mother and Australian father. She developed myoclonus of her fingers and hands at 8 months; an EEG capturing myoclonus at age 2 years was normal. The myoclonus continued, and at 3 years she developed atonic seizures. At 4 years of age, her myoclonus became more prominent and atypical absence seizures began. She developed episodes of non‐convulsive status epilepticus from 6.5 years that were responsive to steroids. At 8 years, she remains drug‐resistant with daily myoclonic, atypical absence and atonic seizures and the movement disorder (Video S1). She has had over 10 anti‐seizure medicines and the ketogenic diet.
Early development was normal with single words at 1 year and walking at 14 months; however, by 15 months her development slowed, and she remained ataxic. Cognitive development plateaued at 4 years, and her speech regressed from 5 years. By 8 years, her speech was limited to 15‐word sentences in English and Japanese (Video S2).
On examination, she had 30 café‐au‐lait lesions and axillary freckling. She was ataxic and dysarthric and had prominent widespread myoclonus but was otherwise neurologically normal with normal head circumference. Her EEG showed diffuse slowing and very frequent multifocal spikes and 5 seconds bursts of GSW. The GSW was either maximal bifrontal or bioccipital, and she developed photosensitivity by 7 years. In sleep, bursts of GSW evolved to long runs of rhythmic monomorphic high voltage delta lasting up to 30 minutes. Video‐EEG monitoring at 8 years captured atypical absence seizures with eyelid fluttering and gradual loss of tone with paroxysms of GSW. Magnetic resonance imaging shows generalized sulcal prominence and a non‐specific focal lesion in the right cerebellar white matter. Lactate and metabolic markers were unremarkable.
The RARS2 literature included 15 cases with sufficient clinical information to compare with our patients (Table 1). All had drug‐resistant developmental and epileptic encephalopathies. Eight had neonatal onset, and seven had onset from 4 weeks to 3 months. They presented with seizures (13/15), developmental delay (10/15), hypoglycemia (6/15), and lactic acidosis (8/15). All showed delay in development by 5 months and seven in the neonatal period. Five children died aged 3 months to 11 years, and surviving children had severe to profound disability.
3.2. Molecular findings
Our two children had compound heterozygous pathogenic variants in RARS2 (Table 1).
For Case A, the maternally inherited start‐loss variant is at the same position as previously published variants, 15 , 18 , 29 and the paternally inherited missense variant is a recurrent pathogenic variant 7 , 23 , 25 (Table 1). No other plausible pathogenic variants were identified in the trio genome.
For Case B, the paternally inherited in‐frame deletion is a recurrent pathogenic variant reported in three patients. 3 , 7 , 28 The novel maternally inherited splice site variant is predicted by three independent in silico splicing prediction tools (HSF score 90.21 > 80.64 [−10.61%] [alteration of the wild‐type donor site, the most probably affecting splicing], TraP score 0.856 [probably damaging], and SpliceAI score 0.1593 for donor loss [low probability]) to lead to loss of the wild type donor site and disrupt splicing. This patient also has a mosaic pathogenic stop‐gain variant in NF1 (Table S2). Given that only 3.7% of children with NF1 have epilepsy, 31 that NF1 epilepsy is usually focal, 31 and that the six cases with her NF1 variant did not have seizures, 32 , 33 , 34 this mosaic variant is unlikely to be contributing to her epilepsy phenotype.
Reanalysis of the published missense variants predicts that 24/31 (77%) affect splicing, (Figure 1B, Table S1) meaning that 86% (43/50) of all variants likely affect expression.
4. DISCUSSION
Here we describe two children with a new RARS2 phenotype that is quite distinct from the previously described early infantile developmental and epileptic encephalopathy RARS2 phenotype. In the 15 published cases that describe the RARS2 epilepsy phenotype in detail, 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 seizure onset occurred by 3 months in the setting of abnormal development. In contrast, our children presented with a progressive movement disorder at 8 and 9 months on a background of normal development. They subsequently developed a myoclonic developmental and epileptic encephalopathy with developmental slowing between 15 and 17 months and seizure onset after 2 years of age.
Of the previously published cases in Table 1, only Case 1 was recognised to have a movement disorder which was described as near continuous jerks and severe dystonia from 4 weeks. 3 The distinction between a non‐epileptic and epileptic myoclonus can be challenging. Interestingly Case 2, 3, and 4 were described as having frequent upper‐limb myoclonus after the first year of life which may have been an unrecognised movement disorder. 4
Eight families share at least one of the variants or a variant affecting the same amino acid as the variants in our cases (Figure 1A); for six families comparable clinical information is provided. 3 , 7 , 15 , 18 , 23 , 25 Four had early infantile‐onset DEE. 3 , 7 , 15 , 18 The other two families with three affected individuals had homozygous p.Leu283Gln variants. 23 , 25 Although there was limited epilepsy phenotyping information, two were siblings described as having progressive myoclonic epilepsy with myoclonic jerks noted on day one, childhood febrile seizures, and mild to severe intellectual disability. 23 The third had adolescent onset cerebellar ataxia, a single seizure, and delayed development. 25 In addition to the six families with shared variants, there is one family with a homozygous promotor variant (c.‐2A>G variant) likely to have a similar effect to the start‐loss variant in our Case A. This family had two children with slow development, seizure onset at 9 months in one, and developmental regression without the movement disorder. 16
RARS2 is highly tolerant to missense variation (Z = −0.06, gnomAD) which, although not unusual for recessive genes, is out of keeping with the proportion (62%) of pathogenic missense variants reported in this gene. As most of these exonic RARS2 variants occur close to exon boundaries (Figure 1A), we theorised that they may affect splicing. Indeed, our bioinformatic reanalysis predicted that a high proportion (24/31, 77%) of these RARS2 missense variants are predicted to affect expression. Although these predictions are based only on bioinformatic tools, aberrant splicing of RARS2 has been functionally confirmed in the recurrent p.Gln12Arg variant using an exon trap vector in vitro. 6
Overall, our bioinformatic reanalysis suggests that 85% of the now 52 RARS2 variants likely impact on splicing or expression of the gene and that most cases with RARS2 encephalopathy have at least one variant with this effect (46/54 with two variants, 7/54 with one variant) (Table S1). Mitochondrial genes, such as RARS2, are required for adenosine triphosphate production 4 , 30 and so are essential for organs with high energy demand, such as the brain. Abnormal splicing in these genes is particularly problematic in the brain as mitochondrial human aminoacyl‐tRNA synthetases already have low splicing efficiency in neurons compared to other tissues due to naturally occurring weak splice sites. 35 We speculate that in RARS2, these low levels do not reach the threshold for sufficient mitochondrial function and result in the progressive neurological disease. 1 , 30 , 35
CONFLICT OF INTERESTS
Prof Sadleir is funded by the Health Research Council of New Zealand and Cure Kids New Zealand. She is a consultant for the Epilepsy Consortium and has received travel grants from Seqirus and Nutricia. She has received research grants from Zynerba. Prof Scheffer serves/has served on scientific advisory boards/consulted for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia, Rogcon, Xenon Pharmaceuticals, Zynerba Pharmaceuticals, Ovid Therapeutics, Atheneum Partners, Chiesi, Encoded Therapeutics and Knopp Biosciences; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, Biomarin and Eisai; and has served as an investigator for Zogenix, Zynerba, Ultragenyx, GW Pharma, UCB, Eisai, Anavex Life Sciences, Ovid Therapeutics, Epigenyx, Encoded Therapeutics and Marinus. She may accrue future revenue on pending patent WO2009/086591; has a patent for SCN1A testing held by Bionomics Inc and licensed to various diagnostic companies; and has a patent molecular diagnostic/ therapeutic target for benign familial infantile epilepsy (BFIE) [PRRT2] WO/2013/059884 with royalties paid. The remaining authors have no conflicts of interest.
ETHICAL APPROVAL
The authors confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Table S1
Table S2
Video S1
Video S2
ACKNOWLEDGMENTS
The authors thank the children and their families for participating in our research. We gratefully acknowledge support from the Health Research Council of New Zealand, Cure Kids New Zealand, the Ted and Mollie Car Endowment Trust, and the National Health and Medical Research Council of Australia. The authors thank the Epi25 consortium for data generation for some of the patients. They thank the Epi25 principal investigators, local staff from individual cohorts, and all of the patients with epilepsy who participated in the study for making possible this global collaboration and resource to advance epilepsy genetics research. This work is part of the Centers for Common Disease Genomics (CCDG) program, funded by the National Human Genome Research Institute (NHGRI) and the National Heart, Lung, and Blood Institute (NHLBI). Centers for Common Disease Genomics–funded Epi25 research activities at the Broad Institute, including genomic data generation in the Broad Genomics Platform, are supported by NHGRI grant UM1 HG008895 (PIs: Eric Lander, Stacey Gabriel, Mark Daly, Sekar Kathiresan). The Genome Sequencing Program efforts were also supported by NHGRI grant 5U01HG009088‐02. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also made possible through the Victorian State Government Operational Infrastructure Support and Australian Government National Health and Medical Research Council (NHMRC) independent research Institute Infrastructure Support Scheme (IRIISS). MB was supported by an NHMRC Senior Research Fellowship (1102971).
de Valles‐Ibáñez G, Hildebrand MS, Bahlo M, et al. Infantile‐onset myoclonic developmental and epileptic encephalopathy: A new RARS2 phenotype. Epilepsia Open.2022;7:e12553. 10.1002/epi4.12553
Contributor Information
Guillem de Valles‐Ibáñez, Email: guille.devallesibanez@otago.ac.nz.
Lynette G. Sadleir, Email: Lynette.sadleir@otago.ac.nz.
REFERENCES
- 1. Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, et al Deleterious mutation in the mitochondrial arginyl‐transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81(4):857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nevanlinna V, Konovalova S, Ceulemans B, Muona M, Laari A, Hilander T, et al A patient with pontocerebellar hypoplasia type 6: Novel RARS2 mutations, comparison to previously published patients and clinical distinction from PEHO syndrome. Eur J Med Genet. 2020;63(3):103766. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31536827 [DOI] [PubMed] [Google Scholar]
- 3. Glamuzina E, Brown R, Hogarth K, Saunders D, Russell‐Eggitt I, Pitt M, et al Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl‐tRNA synthetase, RARS2 . J Inherit Metab Dis. 2012;35(3):459–67. [DOI] [PubMed] [Google Scholar]
- 4. Cassandrini D, Cilio MR, Bianchi M, Doimo M, Balestri M, Tessa A, et al Pontocerebellar hypoplasia type 6 caused by mutations in RARS2: definition of the clinical spectrum and molecular findings in five patients. J Inherit Metab Dis. 2013;36(1):43–53. [DOI] [PubMed] [Google Scholar]
- 5. Kastrissianakis K, Anand G, Quaghebeur G, Price S, Prabhakar P, Marinova J, et al Subdural effusions and lack of early pontocerebellar hypoplasia in siblings with RARS2 mutations. Arch Dis Child. 2013;98(12):1004–7. [DOI] [PubMed] [Google Scholar]
- 6. Rankin J, Brown R, Dobyns WB, Harington J, Patel J, Quinn M, et al Pontocerebellar hypoplasia type 6: a British case with PEHO‐like features. Am J Med Genet Part A. 2010;152(8):2079–84. [DOI] [PubMed] [Google Scholar]
- 7. Ngoh A, Bras J, Guerreiro R, Meyer E, Mctague A, Dawson E, et al RARS2 mutations in a sibship with infantile spasms. Epilepsia. 2016;57(5):e97–102. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27061686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishri D, Goldberg‐stern H, Noyman I, Blumkin L, Kivity S, Saitsu H RARS2 mutations cause early onset epileptic encephalopathy without ponto‐cerebellar hypoplasia. Eur J Paediatr Neurol. 2016;20(3):412–7. 10.1016/j.ejpn.2016.02.012 [DOI] [PubMed] [Google Scholar]
- 9. van Dijk T, van Ruissen F, Jaeger B, Rodenburg RJ, Tamminga S, van Maarle M, et al RARS2 mutations: is pontocerebellar hypoplasia type 6 a mitochondrial encephalopathy? In: JIMD Rep. 2017;33:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang J, Zhang Z, Zhang Y, Wu Y. Distinct magnetic resonance imaging features in a patient with novel RARS2 mutations: a case report and review of the literature. Exp Ther Med. 2018;15(1):1099–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu Y, Wu B‐B, Wang H‐J, Zhou S‐Z, Cheng G‐Q, Zhou Y‐F. A term neonate with early myoclonic encephalopathy caused by RARS2 gene variants: a case report. Transl Pediatr. 2020;9(5):707–12. Available from: http://tp.amegroups.com/article/view/50764/html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Minardi R, Licchetta L, Baroni MC, Pippucci T, Stipa C, Mostacci B, et al Whole‐exome sequencing in adult patients with developmental and epileptic encephalopathy: it is never too late. Clin Genet. 2020;98(5):477–85. 10.1111/cge.13823 [DOI] [PubMed] [Google Scholar]
- 13. Namavar Y, Barth PG, Kasher PR, van Ruissen F, Brockmann K, Bernert G, et al Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain. 2011;134(1):143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joseph JT, Innes AM, Smith AC, Vanstone MR, Schwartzentruber JA, Bulman DE, et al Neuropathologic features of pontocerebellar hypoplasia type 6. J Neuropathol Exp Neurol. 2014;73(11):1009–25. [DOI] [PubMed] [Google Scholar]
- 15. Lax NZ, Alston CL, Schon K, Park SM, Krishnakumar D, He L, et al Neuropathologic characterization of pontocerebellar hypoplasia type 6 associated with cardiomyopathy and hydrops fetalis and severe multisystem respiratory chain deficiency due to novel RARS2 mutations. J Neuropathol Exp Neurol. 2015;74(7):688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Z, Schonberg R, Guidugli L, Johnson AK, Arnovitz S, Yang S, et al A novel mutation in the promoter of RARS2 causes pontocerebellar hypoplasia in two siblings. J Hum Genet. 2015;60(7):363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alkhateeb AM, Aburahma SK, Habbab W, Thompson IR. Novel mutations in WWOX, RARS2, and C10orf2 genes in consanguineous Arab families with intellectual disability. Metab Brain Dis. 2016;31(4):901–7. 10.1007/s11011-016-9827-9 [DOI] [PubMed] [Google Scholar]
- 18. Legati A, Reyes A, Nasca A, Invernizzi F, Lamantea E, Tiranti V, et al New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochimica et Biophysica Acta (BBA) ‐ Bioenergetics. 2016;1857(8):1326–35. 10.1016/j.bbabio.2016.02.022 [DOI] [PubMed] [Google Scholar]
- 19. Lühl S, Bode H, Schlötzer W, Bartsakoulia M, Horvath R, Abicht A, et al Novel homozygous RARS2 mutation in two siblings without pontocerebellar hypoplasia – further expansion of the phenotypic spectrum. Orphanet J Rare Dis. 2016;11(1):140. 10.1186/s13023-016-0525-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pronicka E, Abramczuk DP, Ciara E, Trubicka J, Rokicki D, Więckowska AK, et al New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole‐exome sequencing at a national paediatric centre. J Transl Med. 2016;14(1):174. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27290639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frésard L, Smail C, Ferraro NM, Teran NA, Li X, Smith KS, et al Identification of rare‐disease genes using blood transcriptome sequencing and large control cohorts. Nat Med. 2019;25(6):911–9. 10.1038/s41591-019-0457-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gieldon L, Mackenroth L, Kahlert AK, Lemke JR, Porrmann J, Schallner J, et al Diagnostic value of partial exome sequencing in developmental disorders. PLoS One. 2018;13(8):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mathew T, Avati A, D'Souza D, Therambil M. Expanding spectrum of RARS2 gene disorders: Myoclonic epilepsy, mental retardation, spasticity, and extrapyramidal features. Epilepsia Open. 2018;3(2):270–275. 10.1002/epi4.12108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Al Balushi A, Matviychuk D, Jobling R, Salomons GS, Blaser S, Mercimek‐Andrews S. Phenotypes and genotypes of mitochondrial aminoacyl‐tRNA synthetase deficiencies from a single neurometabolic clinic. JIMD Rep. 2020;51(1):3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shakya S, Kumari R, Suroliya V, Tyagi N, Joshi A, Garg A, et al Whole exome and targeted gene sequencing to detect pathogenic recessive variants in early onset cerebellar ataxia. Clin Genet. 2019;96(6):566–74. [DOI] [PubMed] [Google Scholar]
- 26. Wu T‐H, Peng J, Zhang C‐L, Wu L‐W, Yang L‐F, Peng P, et al Mutations in aminoacyl‐tRNA synthetase genes: an analysis of 10 cases. Zhongguo Dang Dai Er Ke Za Zhi. 2020;22(6):595–601. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32571458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang HF, Deng J, Fang F, Li H, Wang XH, Dai LF. [Early onset epileptic encephalopathy caused by mitochondrial arginyl‐tRNA synthetase gene deficiency: report of two cases and literature review]. Chinese J Pediatr. 2020;58(11):893–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33120460 [DOI] [PubMed] [Google Scholar]
- 28. Roux C‐J, Barcia G, Schiff M, Sissler M, Levy R, Dangouloff‐Ros V, et al Phenotypic diversity of brain MRI patterns in mitochondrial aminoacyl‐tRNA synthetase mutations. Mol Genet Metab. 2021;133(2):222–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33972171 [DOI] [PubMed] [Google Scholar]
- 29. Weng X, Liu Y, Peng Y, Liang Z, Jin X, Cheng L, et al Analysis of genetic variant in a fetus featuring pontocerebellar hypoplasia type 6. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2021;38(7):667–70. Available from: http://www.ncbi.nlm.nih.gov/pubmed/34247374 [DOI] [PubMed] [Google Scholar]
- 30. Sissler M, González‐Serrano LE, Westhof E. Recent advances in mitochondrial aminoacyl‐tRNA synthetases and disease. Trends Mol Med. 2017;23(8):693–708. [DOI] [PubMed] [Google Scholar]
- 31. Bernardo P, Cinalli G, Santoro C. Epilepsy in NF1: a systematic review of the literature. Childs Nerv Syst. 2020;36(10):2333–50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32613422 [DOI] [PubMed] [Google Scholar]
- 32. Ars E, Serra E, García J, Kruyer H, Gaona A, Lázaro C, et al Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. 2000;9(2):237–47. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10607834 [DOI] [PubMed] [Google Scholar]
- 33. Maruoka R, Takenouchi T, Torii C, Shimizu A, Misu K, Higasa K, et al The use of next‐generation sequencing in molecular diagnosis of neurofibromatosis type 1: a validation study. Genet Test Mol Biomarkers. 2014;18(11):722–35. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25325900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zafar R, Hsiao EY, Botteron KN, McKinstry RC, Gutmann DH. De novo development of gliomas in a child with neurofibromatosis type 1, fragile X and previously normal brain magnetic resonance imaging. Radiol Case Rep. 2016;11(1):33–5. Available from: https://linkinghub.elsevier.com/retrieve/pii/S193004331530580X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tyynismaa H. Mitochondrial aminoacyl‐tRNA synthetases. In: Wong LJC, editor. Mitochondrial disorders caused by nuclear genes. New York: Springer; 2013. p. 263–76. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Video S1
Video S2