

Abstract
Objective
Among standard treatments for infantile spasms, adrenocorticotropic hormone (ACTH) is reported as the best treatment, but ACTH is ineffective in one‐half of the patients. To establish precision medicine, we examined pharmacoresistance of focal epileptic spasms (ES), generalized ES, and generalized ES combined with focal seizures, diagnosed based on the revised seizure classification of ILAE in 2017.
Methods
We conducted a retrospective nationwide study in Japan on the long‐term seizure outcome of ES. Long‐term seizure outcome was evaluated by seizure‐free rate, seizure‐free period, and Kaplan‐Meier curve. Seizure‐free was defined as seizure control for longer than 2 months.
Results
From the medical history of 501 patients, 325 patients had generalized ES only (GES group) at the start of the first treatment, 125 patients had generalized ES after focal seizure onset (FS‐GES group), seven patients had focal ES after focal seizure onset (FS‐FES group), and 24 patients had generalized ES combined with focal seizures after focal seizure onset (FS‐GES + FS group). Seizure‐free period of ES (generalized ES and focal ES) [mean (95% confidence interval)] was 2.7 (0.0‐5.4) months in GES group, 1.1 (0.1‐2.2) months in FS‐GES group, 1.0 (0.2‐1.9) months in FS‐GES + FS group, and 0.1 (−0.2‐0.5) months in FS‐FES group. Seizure‐free rate, seizure‐free period, and Kaplan‐Meier curve of generalized ES were almost the same in GES group and FS‐GES group, with characteristics of superior response to ACTH. Mean seizure‐free period of generalized ES combined with focal seizures was significantly shorter in FS‐GES + FS group than in GES group. Mean seizure‐free period of focal ES in FS‐FES group was extremely short with exceedingly early relapse.
Significance
Pharmacoresistance was different in generalized ES, focal ES, and generalized ES combined with focal seizures. ES with focal features or with focal seizures may have focal lesions, thus consider surgical options earlier in the course.
Keywords: combined generalized epileptic spasms, focal epileptic spasms, generalized epileptic spasms, infantile spasms, seizure outcome
Key Points.
Patients with epileptic spasms (ES) were subclassified into various groups according to the type of ES and seizure evolution after the onset of epilepsy
Generalized epileptic spasms (GES) had better long‐term seizure outcome and were controlled by adrenocorticotropic hormone irrespective of whether ES were preceded by focal seizures
Coexistence of GES and focal seizures had unfavorable long‐term seizure outcome
Focal ES had extremely unfavorable long‐term seizure outcome with early relapse
1. INTRODUCTION
Infantile spasms (ISs) are an epilepsy syndrome with onset age younger than 2 years and clinical manifestation of epileptic spasms (ES). 1 ES are characterized by sudden flexion, extension, or mixed extension‐flexion of predominantly proximal and truncal muscles. 2 Previous studies have reported that hormonal treatment is the optimal monotherapy, except for patients with tuberous sclerosis complex (TSC), in whom vigabatrin (VGB) appears superior, and that combination therapy (hormone plus VGB) may be more effective than either agent alone. 3 , 4 , 5 Hormone and vigabatrin are the first‐line agents for ISs, but only 55.3% of ISs respond to combination therapy of VGB and prednisolone. 6 Furthermore, in patients with ISs, normal development was seen in only 12%, 7 and the risk of autistic spectrum disorder was 19.9%. 8 These data suggest that current therapeutic strategies are unsatisfactory for seizure and cognitive outcomes in patients with ISs.
Although adrenocorticotropic hormone (ACTH) therapy is considered the most effective among standard treatments for ISs, the response rate of ACTH varied from 36.7% to 87% in previous reports. 3 This diversity may suggest that the effectiveness of ACTH depends on several factors including etiology, lead time, and seizure evolution. As etiology is considered the most important factor that determines the outcome of ISs, establishment of underlying disease‐specific treatment is an ideal goal of treatment for ISs. 3 However, determination of etiologies is not always possible at the start of treatment for ISs. Search for other factors that predict effective treatments for individual patients is needed.
In 2017, ILAE revised the classification of seizure types, and ES are classified into generalized ES and focal ES. 2 Because fundamental pathophysiologic understanding of differing seizure presentations has not been confirmed, this revised classification is derived for practical clinical use from opinion of experts. This revision of seizure types has allowed precise subclassification of ES at the start of treatment for ISs. Other than subclassification of ES, ES may show heterogeneity of seizure evolution and different combinations of seizures at the start of treatment. Many patients with ES present with de novo epileptic seizures, but a small number of patients are in the process of evolution of focal seizures. We studied the long‐term seizure outcome of treatments for ES with respect to the subclassification of ES, evolution of ES, and association of focal seizures.
2. METHODS
2.1. Retrospective registration of patients
The National Hospital Organization (NHO) in Japan is constituted of 140 hospitals nationwide, and expert pediatric neurologists from 11 hospitals of NHO joined this nationwide study, to recruit patients from 2015 to 2018. Patients with ES in their medical history were eligible for this study. ES were diagnosed clinically or electroclinically based on the ILAE classification. 2 ES include single or clustered ES irrespective of etiology, onset age of ES, age at recruitment in this study, interictal EEG findings, severity of cognitive dysfunction, and coexisting seizures other than ES. After written informed consents, patients with ES were registered.
2.2. Retrospective data collection
After registration, the following data were collected retrospectively: etiology, onset ages of epilepsy and ES, semiology of epileptic seizures, interictal and ictal EEG (if possible, with video), MRI findings, history of treatments including ACTH therapy, oral antiseizure medications (ASMs), etc, severity of cognitive and motor dysfunction, seizure frequency before and after treatments, and age at the last examination. Among registered patients, we selected patients with onset age of ES before 2 years old, to restrict patients of ISs.
2.3. Diagnosis of seizure types
Seizure types were determined by expert pediatric neurologists mostly based on video‐EEG monitoring data (Figure 1). Atypical absence was diagnosed only by video‐EEG monitoring data.
FIGURE 1.
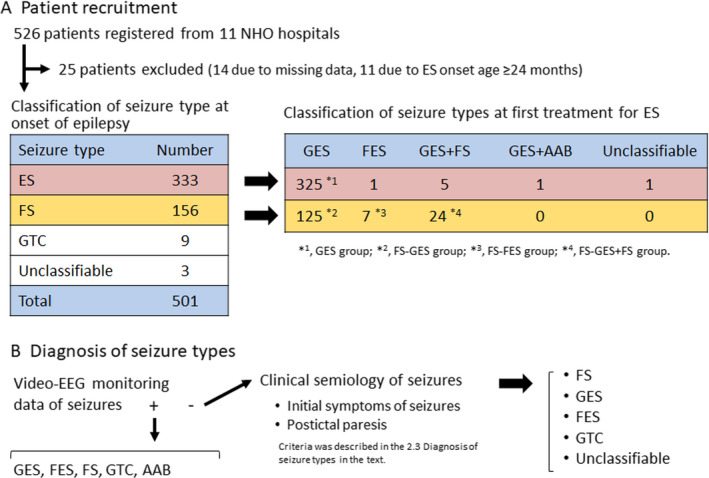
Selection of patients and definition of groups based on seizure evolution and seizure type in this study. A, Patient recruitment. Among patients receiving care in eleven hospitals nationwide, 526 patients with a history of epileptic spasms were registered. B, Diagnosis of seizure types. Seizure types at the start of ES were diagnosed in the majority of patients by video‐EEG monitoring and in a few patients by clinical semiology of seizures. AAB, atypical absence; ES, epileptic spasms; FES, focal epileptic spasms; FS, focal seizure; GES, generalized epileptic spasms; GTC, generalized tonic‐clonic seizure
In patients without video‐EEG monitoring data, seizure types were determined based on clinical semiology of seizures, according to the instruction manual for the ILAE 2017 operational classification of seizure type. 2 Focal seizures were diagnosed by the initial symptoms of seizures, showing focal tonic or clonic convulsion, lateral version of head, lateral clonic eye version, and automatism involving extremities and autonomic signs (cyanosis or salivation) without motor components. Postictal paresis after convulsive seizures supported the diagnosis of focal seizures. Generalized tonic‐clonic seizure was diagnosed by symmetric tonic convulsion flowed by symmetric clonic convulsion. Generalized ES was diagnosed by the symmetric spasms of extremities with upward eye deviation for less than one second. Focal ES was diagnosed by the asymmetric spasms of extremities, spasms with lateral eye version, and spasms seamlessly followed by focal tonic or clonic convulsion. Unclassifiable seizures were diagnosed, when ES, focal seizures, generalized tonic‐clonic seizure, or atypical absence were not confirmed by semiology.
2.4. Treatment protocols for ES in Japan
Treatments for ES available in Japan include oral ASMs, ACTH, and ketogenic diet (KD). Synthetic ACTH (Cortrosyn Z) is used in ACTH therapy, and the dosage of synthetic ACTH was decreased from 0.025 mg (equivalent to 1.0 IU of natural ACTH) to 0.0125 mg/kg/day according to the recommendation by the guideline committee of Japan Epilepsy Society in 2006. 9 A few doctors conducted repeated ACTH therapy in patients with recurred ES after the first ACTH treatment. VGB has been launched since 2016, but strict governmental regulation has restricted prescription of VGB. Subsequently, there are little data on VGB in this study. Protocols and prioritization of treatments for ES were not standardized in Japan during the study period. Therefore, treatment protocols varied among hospitals and era.
2.5. Definition of the initial treatment
The initial treatment was defined as the first therapy immediately after onset of ES, irrespective of oral ASMs prescribed for focal seizures before onset of ES (Figure S1). In patients with focal seizures and started treatment with oral ASM before the onset of ES, if the dose of the prescribed ASM was increased after the onset of ES, the ASM with increased dosage was defined as the first treatment.
2.6. Evaluation of long‐term seizure outcome
Seizure outcome of the first treatment for ES was evaluated qualitatively and quantitatively. Qualitative seizure outcome was classified into three categories: controlled (seizure‐free for longer than 2 months from the initiation of a therapy without modification of treatment), relapse (recurrence of seizure after seizure control for longer than 2 months from the initiation of a therapy), and ineffective (seizure‐free for less than 2 months from the initiation of a therapy). Modification of treatment included addition of novel treatment and dosage increase of concomitant oral ASM prescribed for preceding focal seizures. Recurrence of seizures was usually diagnosed clinically, but in patients with subtle seizures, recurrence was confirmed with video‐EEG recordings. Qualitative seizure outcome was evaluated by seizure‐free rate (SFR).
Quantitative seizure outcome was evaluated by seizure‐free period (SFP) and Kaplan‐Meier curve (KMC) for freedom from treatment failure at the last observation for the ASM. SFP was defined as the duration of seizure control without additional treatment from the initiation of a therapy to the last observation. KMC was used to evaluate the characteristics of relapse of ES and to estimate the final SFR at the longest observation.
2.7. Developmental outcome
Cognitive and motor dysfunctions were evaluated by our original scores on a scale of six grades and a scale of four grades, respectively. 10 Cognitive dysfunction scores depend on intelligent quotient. Motor dysfunction scores depend on mobility capability.
2.8. Statistical analysis
Statistical data are expressed as mean ± SD. Statistical analyses were performed using chi‐square test, log‐rank test, Kruskal‐Wallis test, Fisher's exact test, or Mann‐Whitney test by statisticians at Imepro, Inc. Statistical difference of two groups was evaluated by two‐tailed test. Statistical significance was determined by P < .05.
2.9. Ethical approval
This study was approved by the ethical committee of the NHO in Japan for the registration period from 2015 to 2018.
3. RESULTS
3.1. Characteristics of the patients
From December 2015 to February 2018, 526 patients were registered from 11 NHO hospitals (Figure 1). Fourteen patients were excluded due to missing data around the onset of epilepsy, and eleven patients were excluded due to onset of ES at 24 months of age or above. Among 501 patients included in the study, seizure types diagnosed at the onset of epilepsy were as follows: ES in 333 patients, focal seizures in 156 patients, generalized tonic‐clonic seizures in nine patients, and seizures unclassifiable from clinical semiology in three patients. In 93.6% of the patients, diagnosis of seizure types was determined by video‐EEG monitoring data at the first treatment of ES. However, seizure type was determined only by clinical semiology of seizures without video‐EEG monitoring data in 6.4% of the patients. Among 333 patients with ES at onset of epilepsy, 325 patients had generalized ES (GES group), five patients had generalized ES and focal seizures, one patient had generalized ES and atypical absence, and one patient had focal ES at the first treatment for ES. Among 156 patients with focal seizures at onset of epilepsy, 125 patients evolved to generalized ES only (FS‐GES group), seven patients to focal ES only (FS‐FES group), and 24 patients to generalized ES and focal seizures (FS‐GES + FS group) at the first treatment for ES. We compared seizure outcome among four groups (GES, FS‐GES, FS‐FES, and FS‐GES + FS groups).
The characteristics of patients in the four groups are shown in Table 1. The etiology, mean age at onset of epilepsy, mean age at start of treatment for epilepsy, and EEG findings were significantly different among four groups. Furthermore, at the start of the first treatment for ES, the ratios of patients with concomitant oral ASMs prescribed before onset of ES were significantly different among four groups (P < .0001). Hypsarrhythmia was frequent in GES group. MRI was performed in 410 out of 512 patients, and MRI lesion was found in 52.2% of patients of GES group, 63.7% of patients of FS‐GES group, 68.2% of FS‐GES + FS group, and 33.3% of patients of FS‐FES group. Age at the start of ACTH therapy, starting dose of ACTH, and duration of ACTH therapy for ES were not significantly different among four groups (data not shown). Age at the last observation and duration of observation were not different among four groups. Outcomes of seizure, cognitive function, and motor function were significantly different among four groups. Patients in GES group had better cognitive and motor outcome than FS‐GES group (P < .0001).
TABLE 1.
Characteristics of the patients at the first treatment
| GES group | FS‐GES group | FS‐GES + FS group | FS‐FES group | Statistics | |
|---|---|---|---|---|---|
| Number (Male/Female) | 325 (145/180) | 125 (64/61) | 24 (13/11) | 7 (3/4) | |
| Etiology‐no. | Unknown, 171; Hypoxic encephalopathy, 46; Structural abnormality, 21; Tuberous sclerosis, 27; Chromosome abnormality, 25; Gene mutation, 7; Encephalitis, 7; Intracranial hemorrhage, 4; others, 17. | Unknown, 36; Hypoxic encephalopathy, 26; Structural abnormality, 17; Tuberous sclerosis, 6; Chromosome abnormality, 5; Gene mutation, 9; Encephalitis, 9; Intracranial hemorrhage, 8; others, 9. | Unknown, 8; Hypoxic encephalopathy, 3; Structural abnormality, 4; Tuberous sclerosis, 2; Chromosome abnormality, 2; Gene mutation, 3; Intracranial hemorrhage, 2. | Unknown, 3; Hypoxic encephalopathy, 1; Gene mutation, 3. | P < .0001 a |
| Mutated genes‐no. | STXBP1, 2; WDR45, 2; NR2F1, 1; HERC1, 1; undisclosed, 1 | STXBP1, 4; SCN2A, 2; ARX, 1; SLC35A2, 1; TUBA1A, 1 | STXBP1, 1; CDKL5, 2 | CDKL5, 3 | |
| Age at onset of epilepsy‐months | 6.4 ± 3.6 | 3.5 ± 3.5 | 3.1 ± 4.0 | 2.4 ± 1.9 | P < .0001 b |
| Age at start of treatment of epilepsy‐months | 7.1 ± 4.3 | 4.3 ± 4.3 | 3.9 ± 5.2 | 3.1 ± 1.7 | P < .0001 b |
| ASM before onset of ES‐no. | 4/325 | 67/129 | 21/24 | 3/7 | P < .0001 a |
| Age at onset of ES‐months | 6.4 ± 3.6 | 7.0 ± 4.4 | 7.0 ± 5.3 | 4.1 ± 2.5 | P > .05 b |
| EEG findings‐no. | Hypsarrhythmia, 232;focal, 46;SBP, 7;generalized, 7;wnl, 4;unknown, 32. | Hypsarrhythmia, 65;focal, 38;SBP, 6;generalized, 8;wnl, 2;unknown, 12. | Hypsarrhythmia, 13;focal, 7;SBP, 4;generalized, 0;wnl, 0;unknown, 1. | Hypsarrhythmia, 3;focal, 2;SBP, 0;generalized, 0;wnl, 1;unknown, 2. | P < .0001 a |
| Initial treatment‐no. | B6, 160; VPA, 100; ZNS, 19; ACTH, 16; PB, 14; CBZ, 6: CZP, 5; NZP, 3; TRH, 1; LEV, 1 | B6, 39; VPA, 43; ZNS, 17; ACTH, 6; PB, 5; CBZ, 3: CZP, 5; NZP, 1; LEV, 2; CLB, 2; IVIg, 1; PRM, 1 | B6, 8; VPA, 4; ZNS, 1; ACTH, 6; PB, 2; CLB, 1; DZP, 1; KBr, 1 | B6, 3; ZNS, 1; ACTH, 1; PB, 2 | |
| Age at the last observation‐years | 8.6 ± 7.7 | 8.1 ± 7.6 | 5.5 ± 5.2 | 11.7 ± 7.0 | P > .05 b |
| Duration of observation‐years | 8.0 ± 7.7 | 7.8 ± 7.6 | 5.2 ± 5.2 | 11.9 ± 6.6 | P > .05 b |
| No seizure at the last observation‐no. (rate) | 157 (48.3%) | 38 (30.4%) | 7 (29.2%) | 0 (0%) | P < .02 b |
| ES free at the last observation‐no. (rate) | 208 (64.0%) | 60 (48.0%) | 14 (58.3%) | 4 (57.1%) | P < .03 a |
| Cognitive function score at the last observation‐ 0‐5/5 | 1.7 ± 1.5/5 | 1.1 ± 1.1/5 | 0.8 ± 1.1/5 | 1.1 ± 1.3/5 | P < .0001 b |
| Motor function score at the last observation‐ 0‐3/3 | 1.8 ± 1.3/3 | 1.1 ± 1.2/3 | 0.5 ± 1.0/3 | 1.0 ± 0.2/3 | P < .0001 b |
Data are expressed as mean ± SD or number of patients (percent).
Abbreviations: ACTH, adrenocorticotropic hormone; ASM, antiseizure medication; B6, vitamin B6 (pyridoxal phosphate hydrate); CBZ, carbamazepine; CDKL5, Cyclin‐dependent kinase‐like 5; CLB, clobazam; CZP, clonazepam; DZP, diazepam; FES, focal epileptic spasms; focal, focal interictal discharge; FS, focal seizure; generalized, generalized interictal discharge; GES, generalized epileptic spasms; HERC1, HECT And RLD Domain Containing E3 Ubiquitin Protein Ligase Family Member 1; IVIg, intravenous immunoglobulin therapy; KBr, potassium bromide; LEV, levetiracetam; NR2F1, Nuclear Receptor Subfamily 2 Group F Member 1; NZP, nitrazepam; PB, phenobarbital; PRM, primidone; SBP, suppression burst pattern; SCN2A, Sodium Voltage–Gated Channel Alpha Subunit 2; SLC35A2, Solute Carrier Family 35 Member A2; STXBP1, Syntaxin‐binding protein 1; TRH, thyroid hormone‐releasing hormone; TUBA1A, Tubulin Alpha 1a; VPA, valproate; WDR45, WD repeat domain 45; wnl, within normal limit; ZNS, zonisamide.
Chi‐square test.
Kruskal‐Wallis test.
3.2. Seizure outcome of all patients in four groups
Among all 481 patients in four groups, generalized or focal ES was controlled in three patients by ACTH and in two patients by valproate (VPA), but two of three patients controlled by ACTH had focal seizures as relapse. Therefore, SFR for all seizures was 0.6% (3/481), SFR for generalized ES was 1.1% (5/474), SFR of focal ES was 0% (0/7), and SFR of focal seizures was 8.3% (2/24) in four groups.
Seizure‐free period [mean (lower‐upper 95% CI of mean)] of ES (generalized or focal) was 16.2 (0.7‐31.8) months when treated with ACTH (n = 30), 3.2 (−1.8‐8.1) months with VPA (n = 151), 1.0 (−0.1‐2.1) months with zonisamide (ZNS) (n = 40), 0.5 (−0.1‐1.1) months with phenobarbital (PB) (n = 23), and 0.2 (0.1‐0.3) months with vitamin B6 (n = 211; Figure 2A). SFP was significantly different among drugs as the first treatment for ES (generalized or focal; P < .0001). SFP was better using ACTH than using vitamin B6 (P < .0001), VPA (P < .0001), ZNS (P < .0001), PB (P < .0005), carbamazepine (CBZ; P < .004), or clonazepam (CZP; P < .04).
FIGURE 2.
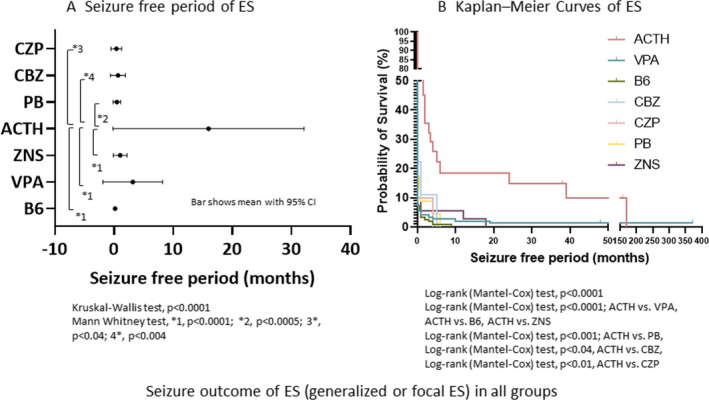
Seizure outcome of ES (generalized or focal ES) in all groups. A, Seizure‐free period of ES. SFP of ES in the first treatment. Bars show mean with 95% confidence interval of SFP after the start of the first treatment for ES. B, Kaplan‐Meier curves of ES. Probability of control of ES by various drugs is shown. ACTH, ACTH therapy; B6, vitamin B6 (pyridoxal phosphate hydrate); CBZ, carbamazepine; CZP, clonazepam; ES, epileptic spasms; PB, phenobarbital; SFP, seizure‐free period; VPA, valproate; ZNS, zonisamide
Kaplan‐Meier curve analysis suggested that the characteristics of relapse of ES (generalized or focal) were different among drugs (P < .0001; Figure 2B). The probability of ES control by ACTH was significantly superior to that by VPA (P < .0001), B6 (P < .0001), CBZ (P < .04), CZP (P < .01), PB (P < .001), or ZNS (P < .0001).
3.3. Comparison of seizure outcome among four groups
In GES group, two of 325 patients were free from ES, but one of the two had focal seizures as relapse. In FS‐GES group, two of 125 patients were free from both ES and focal seizures. In FS‐GES + FS group, one patient was free from ES, but focal seizures were uncontrolled, and two were free from focal seizures, while generalized ES were uncontrolled. In FS‐FES group, no patients were free from ES and focal seizures. SFR of generalized and focal ES by the first treatment was not significantly different among four groups, and those SFRs were extremely poor in all four groups (Figure 3A).
FIGURE 3.
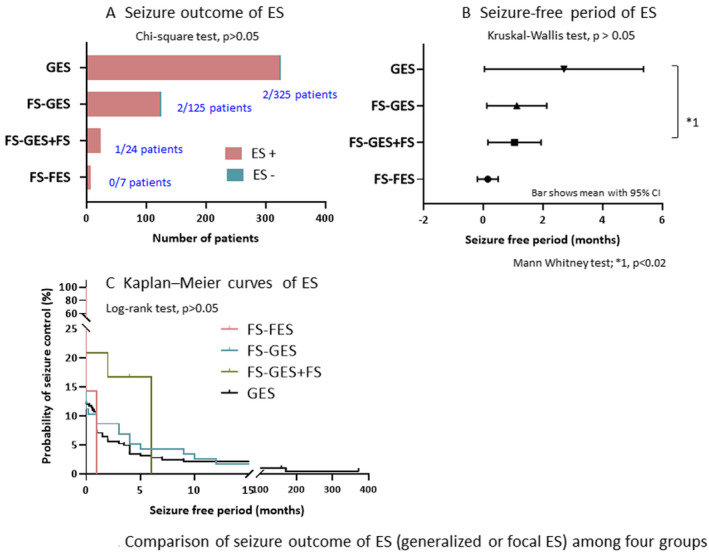
Comparison of seizure outcome of ES (generalized or focal ES) among four groups. A, Seizure outcome of ES. ES + denotes no control of ES and ES‐denotes control of ES. B, Seizure‐free period of ES. Seizure‐free periods (months) of ES are compared between four groups. Bars show mean with 95% confidence interval of seizure‐free period after the start of treatment for ES. C, Kaplan‐Meier curves of ES. Probabilities of control of ES in four groups are shown. ES, epileptic spasms; GES, generalized ES only at epilepsy onset; FS‐GES, focal seizures at epilepsy onset evolving to generalized ES; FS‐FES, focal seizures at epilepsy onset evolving to focal ES; FS‐GES + FS, focal seizures at epilepsy onset evolving to generalized ES and focal seizures
Mean SFP of generalized and focal ES was 2.7 (0.0‐5.4) months in GES group (n = 325), 1.1 (0.1‐2.2) months in FS‐GES group (n = 125), 1.0 (0.2‐1.9) months in FS‐GES + FS group (n = 24), and 0.1 (−0.2‐0.5) months in FS‐FES group (n = 7; Figure 3B). Mean SFP of ES was not significantly different among four groups (P > .05), but SFP of generalized ES in GES group was significantly longer than that in FS‐GES + FS group (P < .02).
Kaplan‐Meier curve analysis suggested that the characteristics of relapse of generalized and focal ES were not different among four groups (P >.05; Figure 3C). KMCs of generalized ES in GES and FS‐GES groups showed similar characteristics, with early rapid relapse followed by few relapses over the long term. KMCs of generalized and focal ES in FS‐FES and FS‐GES + FS groups had similar characteristics with early relapse in all patients.
3.4. Characteristics of pharmacoresistance to drugs in GES group
Mean SFPs of generalized ES were significantly different among drugs (P < .0001; Figure 4A). Mean SFP was 0.2 (0.0‐0.3) months when treated with B6 (n = 160), 4.0 (−3.4‐11.4) months with VPA (n = 100), 1.0 (−0.9‐3.0) months with ZNS (n = 19), 25.8 (−3.6‐55.2) months with ACTH (n = 16), and 0.7 (−0.4‐1.8) months with PB (n = 14). SFP was significantly longer using ACTH than using B6, VPA, ZNS, or PB.
FIGURE 4.
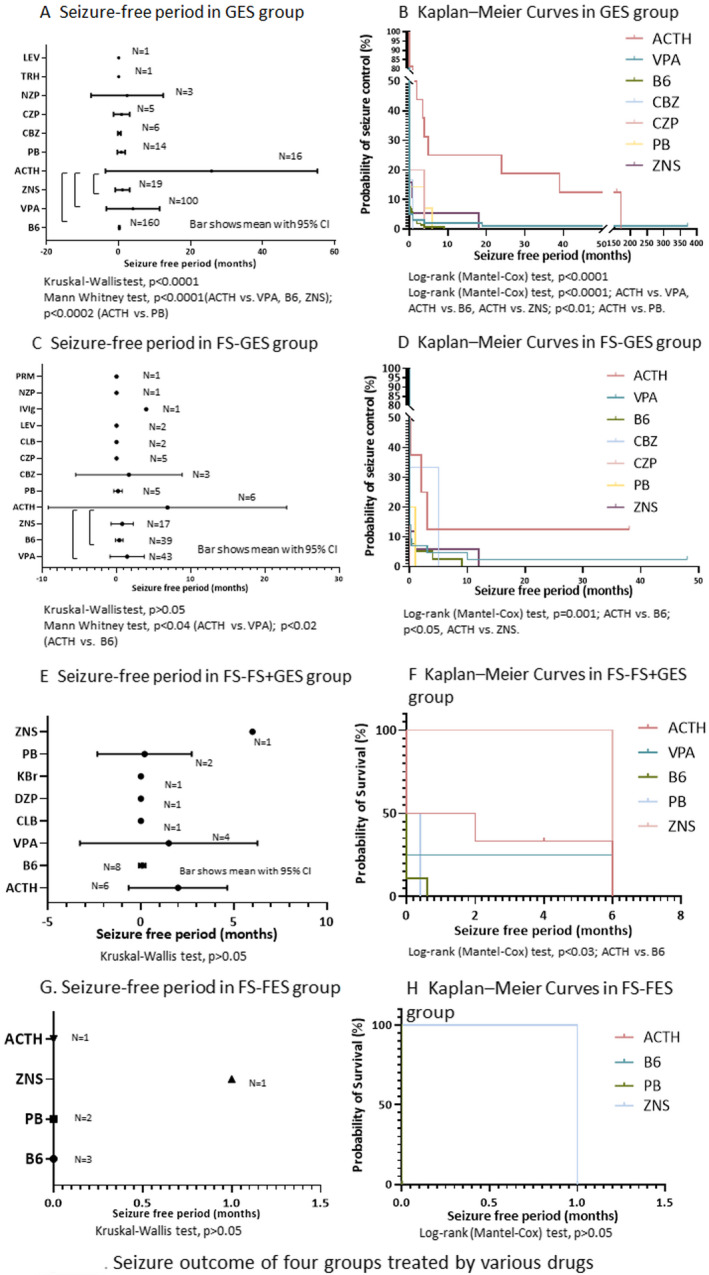
Seizure outcome of four groups treated by various drugs. Bars show mean with 95% confidence interval of seizure‐free period (SFP) after the start of first treatment for ES (A, C, E, and G). Probabilities of control of ES by ASMs in four groups are shown (B, D, F and H). FS‐FES, focal seizures at epilepsy onset evolving to focal ES; FS‐GES, focal seizures at epilepsy onset evolving to generalized ES; FS‐GES + FS, focal seizures at epilepsy onset evolving to generalized ES and focal seizures; GES, generalized ES only at epilepsy onset; ACTH, ACTH therapy; B6, vitamin B6 (pyridoxal phosphate hydrate); CBZ, carbamazepine; CLB, clobazam; CZP, clonazepam; DZP, diazepam; IVIg, intravenous immunoglobulin therapy; KBr, potassium bromide; LEV, levetiracetam; NZP, nitrazepam; PB, phenobarbital; PRM, primidone; TRH, thyroid hormone‐releasing hormone; VPA, valproate; ZNS, zonisamide
Kaplan‐Meier curve analysis suggested that the characteristics of relapse of generalized ES was different among drugs (P < .0001; Figure 4B). KMCs of generalized ES treated with ACTH showed significantly better seizure outcome compared with VPA, B6, ZNS, or PB. KMCs for VPA had characteristics of early rapid relapse with few relapses in the chronic stage. In GES group, ACTH had better long‐term seizure outcome and later relapse of generalized ES compared to oral ASMs.
3.5. Characteristics of pharmacoresistance of drugs in FS‐GES group
Mean SFPs of generalized ES were not significantly different among ASMs (P > .05; Figure 4C). SFP was 0.4 (−0.1‐0.9) months when treated with B6 (n = 39), 1.4 (−0.9‐3.7) months with VPA (n = 43), 0.8 (−0.7‐2.3) months with ZNS (n = 17), 6.9 (−9.1‐22.9) months with ACTH (n = 6), and 0.2 (−0.4‐0.8) months with PB (n = 5). Mean SFP using ACTH was significantly longer than using B6 or VPA.
Kaplan‐Meier curve analysis of generalized ES showed significantly better seizure outcome when treated with ACTH compared to B6 or ZNS (Figure 4D). Around five months from the start of treatment, the probability of seizure control by ACTH was almost the same as that by CBZ, but the probability by CBZ decreased after 5 months. KMC for VPA had the characteristics of early relapse but few relapses in the chronic stage. KMC of generalized ES in FS‐GES group had similar characteristics as those in GES group.
3.6. Characteristics of pharmacoresistance of drugs in FS‐GES + FS group
Mean SFPs of generalized ES were not significantly different among drugs (P > .05; Figure 4E). Mean SFP of generalized ES was 0.1 (−0.1‐0.3) months when treated with B6 (n = 8), 1.5 (−3.3‐6.3) months with VPA (n = 4), 6.0 months with ZNS (n = 1), 2.0 (−0.7‐4.7) months with ACTH (n = 6), and 0.2 (−2.3‐2.7) months with PB (n = 2).
Mean SFPs of focal seizures were not significantly different among drugs (P > .05; Figure 5A). Mean SFP of focal seizures was 3.8 (−3.3‐10.8) months when treated with B6 (n = 8), 0.0 (0.0‐0.0) months with VPA (n = 4), 0.0 months with ZNS (n = 1), 13.3 (−17.0‐43.6) months with ACTH (n = 6), and 0.0 (0.0‐0.0) months with PB (n = 2). Mean SFP was not different between generalized ES and focal seizures (P > .05; Figure 5B).
FIGURE 5.
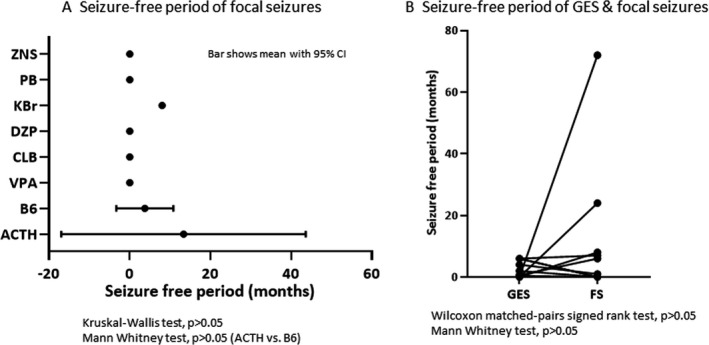
Seizure outcome of generalized epileptic spasms (GES) and focal seizures (FS) at the last observation of first treatment in FS‐GES + FS group (focal seizures at epilepsy onset evolving to generalized ES and focal seizures). A, Seizure‐free period of focal seizures. Bars show mean with 95% confidence interval of SFP after the start of first treatment for ES. B, Seizure‐free periods of GES and focal seizures in individual patients. ACTH, ACTH therapy; B6, vitamin B6 (pyridoxal phosphate hydrate); CLB, clobazam; DZP, diazepam; KBr, potassium bromide; PB, phenobarbital; VPA, valproate; ZNS, zonisamide
Kaplan‐Meier curve analysis of generalized ES when treated with ACTH showed significantly better seizure outcome compared to B6 (P < .03; Figure 4F).
3.7. Characteristics of pharmacoresistance to drugs in FS‐FES group
Mean SFPs of FES were not significantly different among drugs (P > .05; Figure 4G). Mean SFP was 0.0 (0.0‐0.0) months when treated with B6 (n = 3), 1.0 month with ZNS (n = 1), 0.0 months with ACTH (n = 1), and 0.0 (0.0‐0.0) months with PB (n = 2).
Kaplan‐Meier curve analysis of focal ES showed no significant difference among drugs (P > .05; Figure 4H).
3.8. Significance of semiology and etiology for predicting treatment response
We found that 17 of 27 patients (63.0%) with TSC in GES group became seizure‐free from generalized ES, although one of six patients with TSC in FS‐GES group became seizure‐free from generalized ES at the last observation (P = .07). Nineteen of twenty‐five patients (76.0%) with chromosome abnormalities in GES group became seizure‐free from generalized ES, and that 104 of 171 patients (60.8%) with unknown etiology in GES group became seizure‐free from generalized ES at the last observation. SFR was not different by the etiology in GES group (P = .34).
4. DISCUSSION
Current treatment strategies for ISs recommend standard treatments including ACTH, VGB, and corticosteroids, regardless of subclassification of ES and evolution of seizure type. 3 However, these standard treatments are ineffective in 45%–61% of patients with ISs. 3 Furthermore, the proportions of patients achieving freedom from seizures after receiving the first, second, and third regimens were reported to be 15.1%, 21.9%, and 10.3%, respectively. 11 These data suggest that the drugs for the initial treatment are not necessarily selected appropriately. Application of precision medicine approach to the initial treatment is expected to improve seizure and cognitive outcomes.
Patients with typical ISs usually develop generalized ES at infantile‐onset and show neither evolution to other seizure types nor coexistence of other seizure types until 1 year of age. Our data of patients with intractable ISs revealed that apart from the typical generalized ES at onset, focal ES was found in approximately 1.5% of patients, coexistence of generalized ES and focal seizures in approximately 5.0%, and evolution from focal seizures to generalized ES in 26.0% (Figure 1). The existence of atypical ISs requires consideration of the precision medicine approach, considering subclassification of ES.
Data of all patients with ISs irrespective of seizure type and evolution suggested significant superiority of ACTH over B6, VPA, ZNS, PB, CBZ, and CZP (Figure 2). However, when IS patients were divided into four groups, SFP of generalized ES was significantly shorter in FS‐GES + FS group (2.0 months) than in GES group (25.8 months; P < .02), and ACTH did not show significantly better SFP compared with other oral ASMs in FS‐GES + FS group. These data suggest that pharmacoresistance of generalized ES is different between GES group and FS‐GES + FS group and that ACTH cannot be considered the standard treatment for generalized ES in FS‐GES + FS group. Recent reports recommend levetiracetam, topiramate, ZNS, VPA, and benzodiazepines (CZP or nitrazepam) as the second‐line ASMs for ES. 12 Ketogenic diet was reported to be at least as effective as ACTH in patients with prior treatment with VGB. 13 , 14 Furthermore, resection surgery for patients with focal ES is highlighted. 15 Three types of pathophysiology for ES have been reported: West syndrome, developmental and epileptic encephalopathies (DEE), and focal epilepsy. 16 For atypical ISs with combined generalized ES and focal seizures, precision medicine including second‐line oral ASMs, ketogenic diet, and surgical intervention should be considered, because standard treatment with ACTH has poor long‐term seizure outcome.
In patients with generalized ES combined with focal seizures (FS‐GES + FS group), mean SFP of generalized ES by the first treatment was short, ranging from 0 to 2 months (Figure 4). In this group, mean SFP of generalized ES was significantly shorter than that in GES group (Figure 3). Etiologies in FS‐GES + FS group include a higher proportion of structural abnormalities (such as hypoxic encephalopathy, TSC, and intracranial hemorrhage) compared to that in GES group. These structural abnormalities seem to be causally related to DEEs, and ES are documented often along with other seizure types in patients with DEEs. 16 In FS‐GES + FS group, pathophysiology related to DEEs may contribute to the pharmacoresistance of generalized ES. The longest SFP in our study was 6 months, when ACTH, VPA, or ZNS was used as the first treatment. These findings suggest that further studies of second‐line oral ASMs (including VPA and ZNS) may contribute to establish precision medicine for generalized ES in FS‐GES + FS group. If the initial response to the first standard treatment is poor, immediate switching to the second treatment would be necessary. Especially, when VGB fails to achieve seizure control, ketogenic diet is recommended because ketogenic diet has been reported to be as effective as ACTH. 13
ILAE classifies focal ES as one subtype of focal seizures. 2 We found eight patients with focal ES (8/489 patients, 1.6%) in patients with ISs. SFR of focal ES was 0%, and mean SFP was 0.1 month in FS‐FES group, while KMC showed early relapse (Figure 3). Many drugs including ACTH had little effect in controlling focal ES (Figure 4G,H). Seizure outcome of patients with focal ES treated by the first treatment is extremely poor. In the context of focal epilepsies, focal ES is sometimes associated with focal brain lesions. 15 Surgical treatments for refractory ES including hemispherectomy, resection, and tuberectomy achieved ILAE class I outcome in 71% of patients with ES. 16 Univariate analyses revealed that concordance between MRI and interictal discharges and continuous discharges on electrocorticography were important factors associated with a favorable surgical outcome and that 82% of 64 children with drug‐resistant spasms had favorable outcome. 17 Clinicians should not hesitate to start presurgical evaluation for patients in FS‐FES group, even during the first treatment. In FS‐FES group, all three patients with confirmed gene mutation had mutated CDKL5. In patients with CDKL5 mutations, no effective drugs have been reported, and the effect of ACTH is temporary. 18 An open‐label drug trial provided class III evidence for the long‐term safety and efficacy of cannabidiol (CBD) treatment in patients with CDKL5 deficiency disorder. 19 Further studies on CBD therapy in patients with focal ES after focal seizure onset are needed.
In both GES and FS‐GES groups, generalized ES are the common treatment target, although evolution of epilepsy differs depending on whether focal seizures exist before the start of treatment for generalized ES. SFR and mean SFP of generalized ES were not significantly different between GES and FS‐GES groups (Figure 2A,B). Mean SFP was significantly different among drugs for the first treatment, and SFP for ACTH therapy was better than those for B6, VPA, and ZNS in both groups (Figure 4). KMC of generalized ES also showed similar characteristics of early rapid relapse followed by slow relapse over the long term in both groups. These common features in the two groups suggest that pharmacoresponse characteristics of generalized ES are not affected by evolution from preceding focal seizures. In patients with generalized ES without preceding focal seizures, standard strategy using first‐line treatment with ACTH and others may be considered. According to the ILAE recommendation for the management of ISs, ACTH is preferred for short‐term control of ES other than TSC. 20 ACTH as recommended by ILAE is effective for patients in GES and FS‐GES groups, that is, patients with generalized ES only, independent of preceding seizure evolution.
At the start of the first treatment for ES, etiology cannot be confirmed in many patients, although etiology seems to be one of crucial factors to predict seizure outcome. In GES group, SFR at the final observation was not different by the major etiologies. This may suggest that semiology of ES is important, compared with etiology in GES group. We need prospective study to confirm importance of semiology and etiology about prediction of effective treatments for individual patients, after confirmation of seizure types with ictal video EEG monitoring.
4.1. Limitations
Our study has several limitations. First, our data were collected retrospectively from 526 patients at 11 NHO hospitals that treat many patients with intractable epilepsy. Consequently, our study population contained a larger proportion of severe cases with unfavorable seizure outcome than general patient population with ISs. These may have contributed to the low rate of seizure control after the first treatment. Second, our data were collected mainly in the era before VGB was launched in Japan. Third, diagnosis of seizure types at the onset of ES was determined by clinical semiology of seizures without video‐EEG monitoring data in 6.4% of the patients. Fourth, we have not considered composition of etiological factors in four groups in the relationship with seizure outcome. Difference of etiologies in four groups might also affect seizure outcome. Fifth, numbers of patients in FS‐FS + GES group and FS‐FES group were too low to evaluate the efficacy of ASMs.
5. CLINICAL RELEVANCE
Our study is the first nationwide multicenter study that reveals the difference of pharmacoresistance between generalized ES and focal ES subclassified according to the revised ILAE classification of seizure types. The findings show characteristic intractability of focal ES and generalized ES combined with focal seizures. These results are expected to stimulate further research for the provision of useful precision medicine also for patients with atypical ISs.
CONFLICT OF INTERESTS
Author YT received academic donation from Eisai. The remaining authors have no conflict of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Figure S1
ACKNOWLEDGMENTS
This study was funded mainly by NHO in Japan and in part by Grants‐in‐aid for Scientific Research I Nos. 15K09634, 18K07865, and 21K07788; Health and Labor Sciences Research Grants for Comprehensive Research on Disability Health and Welfare; grant for Research on Rare and Intractable Disease; AMED under Grant Number JP18lk0201069s0502; and grants from Japan Epilepsy Research Foundation. The authors thank all doctors who treated the patients in NHO hospitals as well as the patients and their guardians who agreed to participate in this research.
Takahashi Y, Ota A, Tohyama J, et al. Different pharmacoresistance of focal epileptic spasms, generalized epileptic spasms, and generalized epileptic spasms combined with focal seizures. Epilepsia Open.2022;7:85–97. 10.1002/epi4.12560
REFERENCES
- 1. Lux AL, Osborne JP. A proposal for case definitions and outcome measures in studies of infantile spasms and West syndrome: consensus statement of the West Delphi Group. Epilepsia. 2004;45:1416–28. [DOI] [PubMed] [Google Scholar]
- 2. Fisher RS, Cross JH, D’Souza S, French JA, Haut SR, Higurashi N, et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia. 2017;58(4):531–42. [DOI] [PubMed] [Google Scholar]
- 3. Riikonen R. Infantile spasms: outcome in clinical studies. Pediatr Neurol. 2020;108:54–64. [DOI] [PubMed] [Google Scholar]
- 4. Knupp KG, Coryell J, Nickels KC, Ryan N, Leister E, Loddenkemper T, et al. Response to treatment in a prospective national infantile spasms cohort. Ann Neurol. 2016;79(3):475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Callaghan FJ, Edwards SW, Alber FD, Hancock E, Johnson AL, Kennedy CR, et al. Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): a randomized, multicenter, open‐label trial. Lancet Neurol. 2017;16(1):33–42. [DOI] [PubMed] [Google Scholar]
- 6. Hahn J, Park G, Kang HC, Lee JS, Kim HD, Kim SH, et al. Optimized treatment for infantile spasms: vigabatrin versus prednisolone versus combination therapy. Clin Med. 2019;8:1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Riikonen R. A long‐term follow‐up study of 214 children with the syndrome of infantile spasms. Neuropediatrics. 1982;13(1):14–23. [DOI] [PubMed] [Google Scholar]
- 8. Strasser L, Downes M, Kung J, Cross JH, De Haan M. Prevalence and risk factors for autism spectrum disorder in epilepsy: a systematic review and meta‐analysis. Dev Med Child Neurol. 2018;60(1):19–29. [DOI] [PubMed] [Google Scholar]
- 9. Tsuji T, Okumura A, Ozawa H, Ito M, Watanabe K. Current treatment of West syndrome in Japan. J Child Neurol. 2007;22:560–4. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi Y, Nishimura S, Takao E, Kasai R, Enokida K, Kubota M. Study in 157 patients with non‐herpetic acute limbic encephalitis: treatment in acute stage & outcome. Neuroinfection. 2016;21:121–7. [Google Scholar]
- 11. Mao L, Kessi M, Peng P, He F, Zhang C, Yang L, et al. The patterns of response of eleven regimens for infantile spasms. Sci Rep. 2020;10(1):11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song JM, Hahn J, Kim SH, Chang MJ. Efficacy of treatments for infantile spasms: a systematic review. Clin Neuropharmacol. 2017;40(2):63–84. [DOI] [PubMed] [Google Scholar]
- 13. Dressler A, Benninger F, Trimmel‐Schwahofer P, Gröppel G, Porsche B, Abraham K, et al. Efficacy and tolerability of the ketogenic diet versus high‐dose adrenocorticotropic hormone for infantile spasms: a single‐center parallel‐cohort randomized controlled trial. Epilepsia. 2019;60(3):441–51. [DOI] [PubMed] [Google Scholar]
- 14. Prezioso G, Carlone G, Zaccara G, Verrotti A. Efficacy of ketogenic diet for infantile spasms: a systematic review. Acta Neurol Scand. 2018;137(1):4–11. [DOI] [PubMed] [Google Scholar]
- 15. Chugani HT, Ilyas M, Kumar A, Juhász C, Kupsky WJ, Sood S, et al. Surgical treatment for refractory epileptic spasms: the Detroit series. Epilepsia. 2015;56:1941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fusco L, Serino D, Santarone ME. Three different scenarios for epileptic spasms. Epilepsy Behav. 2020;113:107531. [DOI] [PubMed] [Google Scholar]
- 17. Liu Y, Zhou W, Hong B, Wang H, Lin J, Sun Z, et al. Analysis of surgical strategies for children with epileptic spasms. Epileptic Disord. 2021;23:85–93. [DOI] [PubMed] [Google Scholar]
- 18. Kobayashi Y, Tohyama J, Takahashi Y, Goto T, Haginoya K, Inoue T, et al. Clinical manifestations and epilepsy treatment in Japanese patients with pathogenic CDKL5 variants. Brain Dev. 2021;43:505–14. [DOI] [PubMed] [Google Scholar]
- 19. Devinsky O, Verducci C, Thiele EA, Laux LC, Patel AD, Filloux F, et al. Open‐label use of highly purified CBD (Epidiolex®) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav. 2018;86:131–7. [DOI] [PubMed] [Google Scholar]
- 20. Wilmshurst JM, Gaillard WD, Vinayan KP, Tsuchida TN, Plouin P, Van Bogaert P, et al. Summary of recommendations for the management of infantile seizures: Task Force Report for the ILAE Commission of Pediatrics. Epilepsia. 2015;56:1185–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1